
The Non-Coding RNA Landscape in IgA Nephropathy—Where Are We in 2021?


Abstract
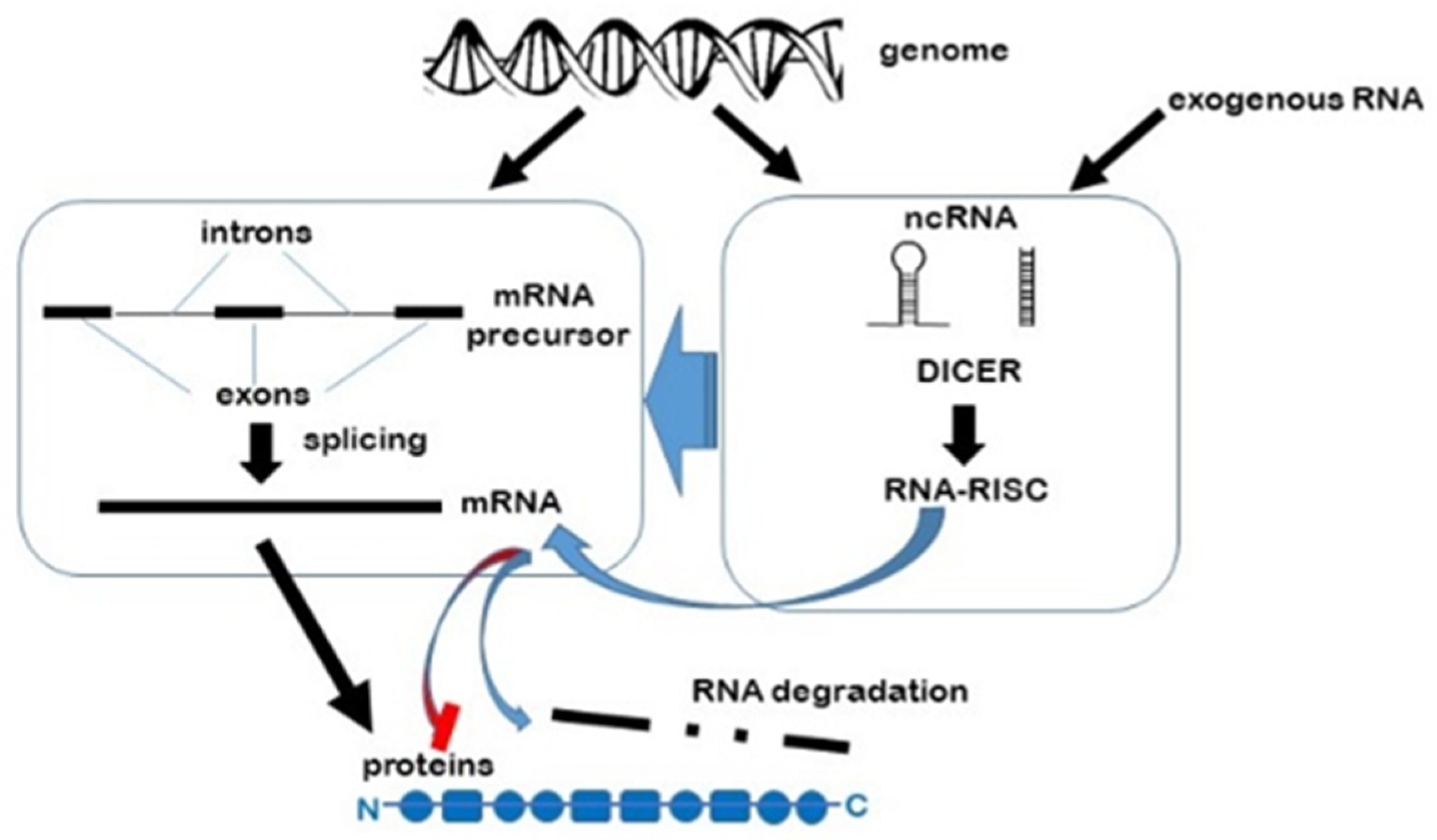
1. Non-Coding RNA
2. RNA Interference
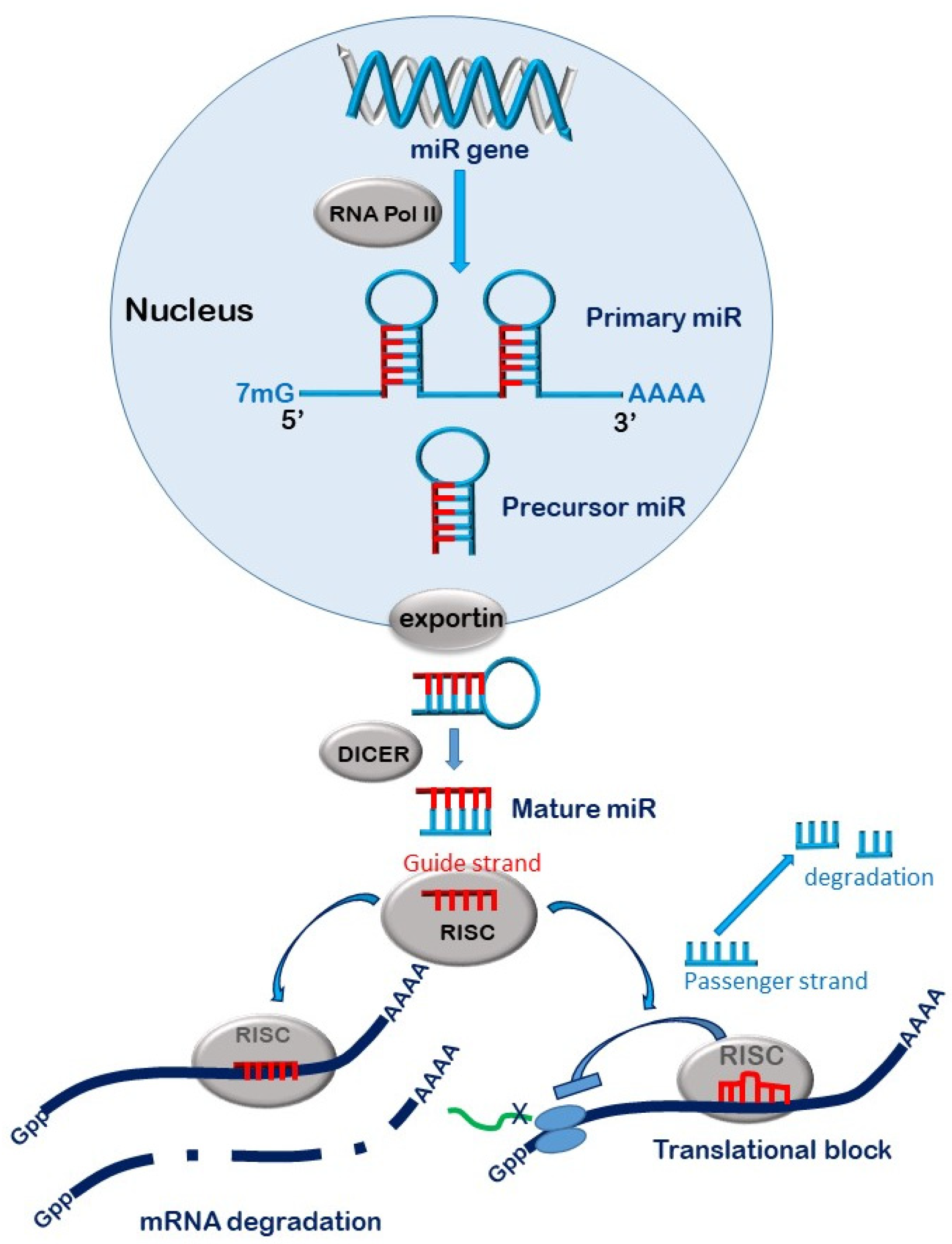
3. MicroRNAs
MiR Biogenesis
4. IgA Nephropathy
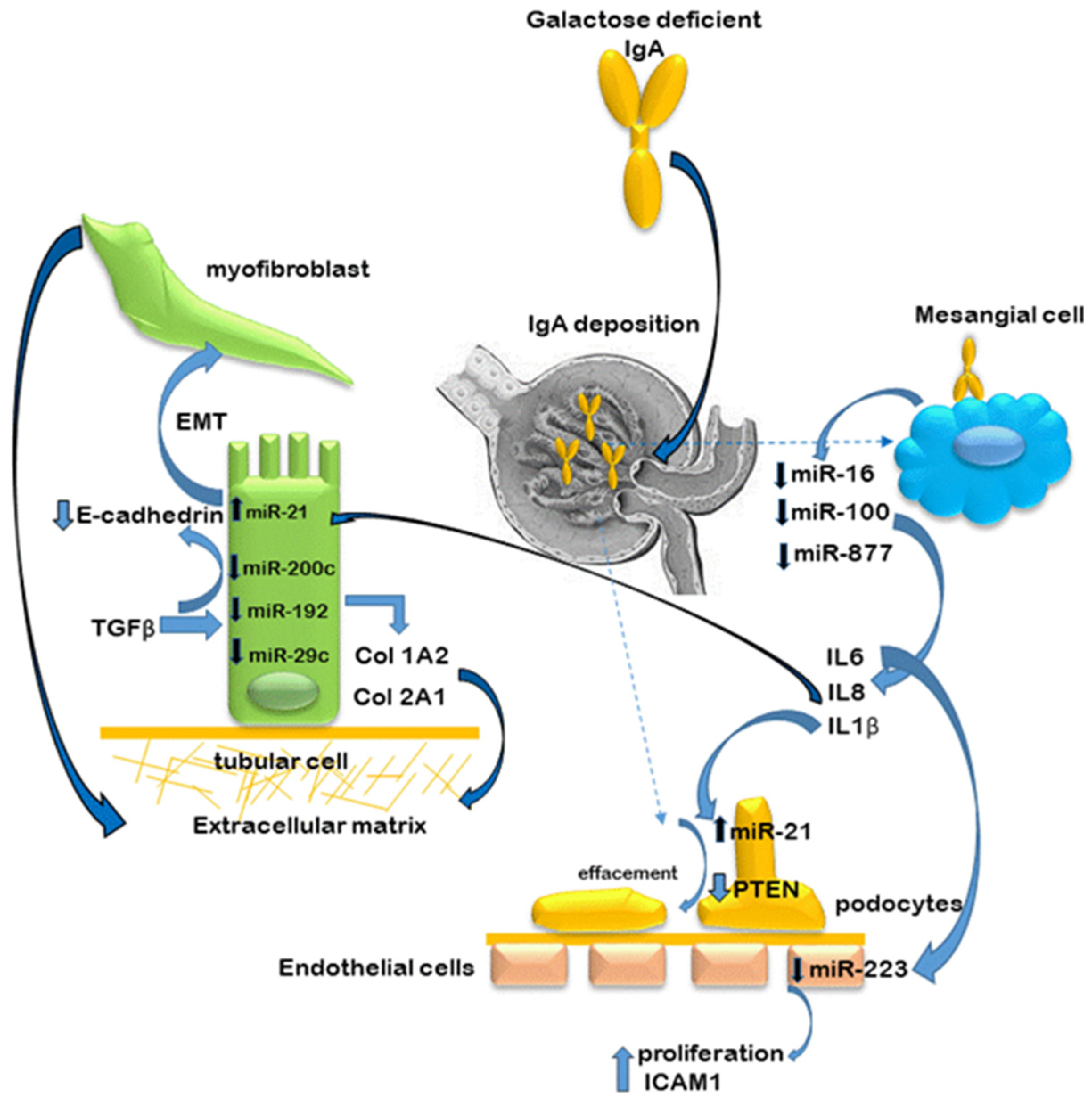
5. MiRs in IgA Nephropathy
5.1. Kidney Tissue and Cultured Kidney Cells
5.2. Extrarenal miR Expression
5.3. Peripheral Blood Mononuclear Cells
5.4. B Cells
5.5. T Cells
5.6. Erythrocytes
6. Extracellular miRs
6.1. Serum and Plasma
6.2. Urine
7. MiRs and Genetic Susceptibility to IgAN
8. miRs as Biomarkers in IgAN
- (1)
- Biologically plausible, with a clear role in mediating the disease of interest;
- (2)
- Stable at room temperature, to minimise degradation between collection and processing;
- (3)
- Readily measurable with available and affordable techniques;
- (4)
- Minimally invasive to collect;
- (5)
- Generalisable across populations;
- (6)
- Validated prospectively and independently.
- (7)
- Sensitive; its presence should reliably diagnose the disease;
- (8)
- Specific; its absence should reliably exclude the disease.
9. RNAi Therapeutics in IgAN
10. In Summary
Author Contributions
Funding
Conflicts of Interest
References
- Zhang, P.; Wu, W.; Chen, W.; Chen, M. Non-coding RNAs and their integrated networks. J. Integr. Bioinform. 2019, 16. [Google Scholar] [CrossRef]
- Cech, T.R.; Steitz, J.A. The noncoding RNA revolution—Trashing old rules to forge new ones. Cell 2014, 157, 77–94. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Meng, X.; Liao, Q.; Chen, M. Versatile interactions and bioinformatics analysis of non-coding RNA. Brief. Bioinform. 2019, 20, 1781–1794. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef]
- Waterhouse, P.M.; Graham, M.W.; Wang, M.-B. Virus resistance and gene silencing in plants can be induced by simultaneous expression of sense and antisense RNA. Proc. Natl. Acad. Sci. USA 1998, 95, 13959–13964. [Google Scholar] [CrossRef] [PubMed]
- Russo, J.; Harrington, A.W.; Steiniger, M. Antisense transcription of retrotransposons in drosophila: An origin of endogenous small interfering RNA precursors. Genetics 2016, 202, 107–121. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Bajic, V.B.; Zhang, Z. On the classification of long non-coding RNAs. RNA Biol. 2013, 10, 924–933. [Google Scholar] [CrossRef]
- Zamecnik, P.C.; Stephenson, M.L. Inhibition of Rous sarcoma virus replication and cell transformation by a specific oligonucleotide. Proc. Natl. Acad. Sci. USA 1978, 75, 280–284. [Google Scholar] [CrossRef]
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998, 391, 806–811. [Google Scholar] [CrossRef] [PubMed]
- Murchison, E.P.; Hannon, G.J. miRNAs on the move: miRNA biogenesis and the RNAi machinery. Curr. Opin. Cell Biol. 2004, 16, 223–229. [Google Scholar] [CrossRef]
- Olina, A.V.; Kulbachinskij, A.V.; Aravin, A.A.; Esyunina, D.W. Argonaute proteins and mechanisms of RNA interference in eukaryotes and prokaryotes. Biochemistry 2018, 83, 483–497. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Jansson, M.D.; Lund, A.H. MicroRNA and cancer. Mol. Oncol. 2012, 6, 590–610. [Google Scholar] [CrossRef]
- Vegter, E.L.; van der Meer, P.; de Windt, L.J.; Pinto, Y.M.; Voors, A.A. MicroRNAs in heart failure: From biomarker to target therapy. Eur. J. Heart Fail. 2016, 18, 457–468. [Google Scholar] [CrossRef]
- Trionfini, P.; Benigni, A.; Remuzzi, G. MicroRNAs in kidney physiology and disease. Nat. Rev. Nephrol. 2015, 11, 23–33. [Google Scholar] [CrossRef]
- Lee, Y.; Ahn, C.; Han, J.; Choi, H.; Kim, Y.; Yim, J.; Lee, J.; Provost, P.; Radmark, O.; Kim, J.; et al. The nuclear RNase Drosha initiates microRNA processing. Nature 2003, 425, 415–419. [Google Scholar] [CrossRef] [PubMed]
- Yi, R.; Qin, Y.; Macara, I.G.; Cullen, B.R. Exportin-5 mediates the nuclear export of pre-microRNAs and short hairpin RNAs. Genes Dev. 2003, 17, 3011–3016. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.; Cullen, B.R. Structural requirements for pre-microRNA binding and nuclear export by exportin 5. Nucleic Acids Res. 2004, 32, 4776–4785. [Google Scholar] [CrossRef]
- Zhang, H.; Kolb, F.A.; Brondani, V.; Billy, E.; Filipowicz, W. Human Dicer preferentially cleaves ds RNA at their termini without a requirement for ATP. EMBO J. 2002, 21, 5875–5885. [Google Scholar] [CrossRef]
- Pratt, A.J.; MacRae, I.J. The RNA induced silencing complex: A versatile gene-silencing machine. J. Biol. Chem. 2009, 284, 17897–17901. [Google Scholar] [CrossRef]
- Khvorova, A.; Reynolds, A.; Jayasena, S.D. Functional siRNAs and miRNAs exhibit strand bias. Cell 2003, 115, 209–216. [Google Scholar] [CrossRef]
- Liu, J.; Carmell, M.A.; Rivas, F.V.; Marsden, C.G.; Thomson, J.M.; Song, J.J.; Hammond, S.M.; Joshua-Tor, L.; Hannon, G.J. Argonaute 2 is the catalytic engine of mammalian RNAi. Science 2004, 305, 804–813. [Google Scholar] [CrossRef] [PubMed]
- Mullany, L.E.; Hendrrick, J.S.; Wolff, R.K.; Slattery, M.L. MicroRNA seed region length impact on target messenger RNA expression and survival in colorectal cancer. PLoS ONE 2016, 11, e0154177. [Google Scholar] [CrossRef]
- Yekya, S.; Shih, I.H.; Bartel, D.P. MicroRNA-directed cleavage of HOXB8 mRNA. Science 2004, 304, 594–596. [Google Scholar]
- Jonas, S.; Izaurralde, E. Towards a molecular understanding of microRNA-mediated gene silencing. Nat. Rev. Genet. 2015, 16, 1346–1350. [Google Scholar] [CrossRef]
- Xu, W.; San Lucas, A.; Wang, Z.; Liu, Y. Identifying microRNA targets in different gene regions. BMC Bioinform. 2014, 15, 1–11. [Google Scholar] [CrossRef]
- Marakova, J.A.; Shkurnikov, M.U.; Wicklein, D.; Lange, T.; Samatov, T.R.; Turchinovich, A.A.; Tonevitsky, A.G. Intracellular and extracellular microRNA: An update on localisation and biological role. Prog. Histochem. Cytochem. 2016, 51, 33–49. [Google Scholar]
- Hsu, P.W.; Huang, H.D.; Hsu, S.D.; Lin, L.-Z.; Tsou, A.-P.; Tseng, C.-P.; Stadler, P.F.; Washietl, S.; Hofacker, I.L. miRNAMap: Genomic maps of microRNA genes and their target genes in mammalian genomes. Nucleic Acids Res. 2006, 34, D135–D139. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, A.; Griffiths-Jones, J.; Ashurst, J.L.; Bradley, A. Identification of mammalian microRNA host genes and transcription units. Genome Res. 2004, 14, 1902–1910. [Google Scholar] [CrossRef]
- Lagos-Quintana, M.; Rauhut, R.; Lendeckel, W.; Tuschl, T. Identification of novel genes coding for small expressed FRNAs. Science 2001, 294, 853–858. [Google Scholar] [CrossRef] [PubMed]
- Lau, N.C.; Lim, L.P.; Wienstein, E.G.; Bartel, D.P. An abundant class of tiny RNAs with probable regulatory role in Caenorhabditis elegans. Science 2001, 294, 858–862. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.C.; Ambrose, V. An extensive class of small RNA in Caenorhabditis. Science 2001, 294, 862–864. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Jeon, K.; Lee, J.T.; Kim, S.; Kim, V.N. MicroRNA maturation: Stepwise processing and subcellular location. EMBO J. 2002, 21, 4663–4670. [Google Scholar] [CrossRef]
- Gantier, M.P.; McCoy, C.E.; Rusinova, I.; Saulep, D.; Wang, D.; Xu, D.; Irving, A.T.; Behlke, M.A.; Hertzog, P.J.; Mackay, F.; et al. Analysis of microRNA turnover in mammalian cells following Dicer 1 ablation. Nucleic Acids Res. 2011, 39, 5692–5703. [Google Scholar] [CrossRef]
- Lagos-Quintana, M.; Rauhut, R.; Meyer, J.; Borkhardt, A.; Tuschl, T. New microRNAs from mouse and human. RNA 2003, 9, 175–179. [Google Scholar] [CrossRef]
- Pasquinelli, A.E.; Reinhart, B.J.; Slack, F.; Martindale, M.Q.; Kuroda, M.I.; Maller, B.; Hayward, D.C.; Ball, E.E.; Degnan, B.; Müller, P.; et al. Conservation of sequences and temporal expression of Let-7 heterochronic regulatory RNA. Nature 2000, 408, 86–89. [Google Scholar] [CrossRef]
- Lim, L.P.; Lau, N.C.; Garret-Engele, P.; Grimson, A.; Schelter, J.M.; Castle, J.; Bartel, D.P.; Linsley, P.S.; Johnson, J.M. MicroRNA analysis shows that some microRNAs down regulate large numbers of target mRNAs. Nature 2005, 433, 769–773. [Google Scholar] [CrossRef]
- Joplin, C. Liver specific microRNA-122: Biogenesis and function. RNA Biol. 2012, 9, 137–142. [Google Scholar] [CrossRef]
- Schratt, G.M.; Tuebing, F.; Nigh, E.A.; Kane, C.G.; Sabatini, M.E.; Kiebler, M.; Greenberg, M.E. A brain-specific microRNA regulates dendritic spine development. Nature 2006, 439, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, S.; Scheele, C.; Yfanti, C.; Akerstrom, T.; Nielsen, A.R.; Pedersen, B.K.; Laye, M.J. Muscle-specific microRNAs are regulated by endurance exercise in human skeletal muscle. J. Physiol. 2010, 588, 4029–4037. [Google Scholar] [CrossRef]
- Suzuki, H.; Kiryluk, K.; Novak, J.; Moldoveanu, Z.; Herr, A.B.; Renfrow, M.B.; Wyatt, R.S.; Scolari, F.; Mestecky, J. The pathophysiology of IgA nephropathy. J. Am. Soc. Nephrol. 2011, 22, 1795–1803. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Koo, S.; White, N.; Peralta, E.; Esau, C.; Dean, N.M.; Perera, R.J. Development of a micro-array to detect human and mouse microRNAs and characterisation of expression in human organs. Nucleic Acids Res. 2004, 32, e188. [Google Scholar] [CrossRef] [PubMed]
- Rudnicki, M.; Perco, P.; D’haene, B.; Leierer, J.; Heinzel, A.; Muhlberger, I.; Scweibert, N.; Sunzenauer, J.; Regele, H.; Kronbichler, A.; et al. Renal microRNA- and RNA-profiles in progressive chronic kidney disease. Eur. J. Clin. Investig. 2016, 46, 213–226. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Sui, W.; Yan, H.; Huang, H.; Huang, Y. Microarray analysis of micro ribonucleic acid expression in primary immunoglobulin A nephropathy. Saudi Med. J. 2008, 29, 1388–1393. [Google Scholar] [PubMed]
- Tan, K.; Chen, J.; Li, W.; Chen, Y.; Sui, W.; Zhang, Y.; Dai, Y. Genome-wide analysis of microRNAs expression profiling in patients with primary IgA nephropathy. Genome 2013, 56, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Kwan, B.C.; Lai, F.M.; Choi, P.C.; Chow, K.M.; Li, P.; Szeto, C.C. Intra-renal expression of microRNAs in patients with IgA nephropathy. Lab. Investig. 2010, 90, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Kwan, B.C.-H.; Lai, F.M.-M.; Chow, K.-M.; Li, P.K.-T.; Szeto, C.C. Elevated levels of miR-146a and miR-155 in kidney biopsy and urine from patients with IgA nephropathy. Dis. Markers 2011, 30, 171–179. [Google Scholar] [CrossRef]
- Hennino, M.; Buob, D.; van der Hauwaert, C.; Gnemmi, V.; Jomaa, Z.; Pottier, N.; Savary, G.; Drumez, E.; Noel, C.; Cauffiez, C.; et al. miR-21 renal expression is associated with fibrosis and renal survival in patients with IgA nephropathy. Sci. Rep. 2016, 6, 27209. [Google Scholar] [CrossRef]
- Bao, H.; Hu, S.; Zhang, C.; Shi, S.; Qin, W.; Zeng, C.; Zen, K.; Liu, Z. Inhibition of miRNA-21 prevents fibrogenic activation in podocytes and tubular cells in IgA nephropathy. Biochem. Biophys. Res. Commun. 2014, 444, 455–460. [Google Scholar] [CrossRef]
- Pawluczyk, I.Z.A.; Didangelos, A.; Barbour, S.J.; Er, L.; Becker, J.U.; Martin, R.; Taylor, S.; Bhachu, J.S.; Lyons, E.G.; Jenkins, R.; et al. Differential expression of microRNAs in IgA Nephropathy: miR-150-5p, a potential mediator and marker of disease progression. Kidney Int. 2021, 99, 1127–1139. [Google Scholar] [CrossRef]
- Baker, M.A.; Davis, S.J.; Liu, P.; Pan, X.; Williams, A.M.; Iczkowski, K.A.; Gallagher, S.T.; Bishop, K.; Regner, K.R.; Liu, Y.; et al. Tissue specific microRNA expression patterns in four types of kidney disease. J. Am. Soc. Nephrol. 2017, 28, 2985–2992. [Google Scholar] [CrossRef]
- Bao, H.; Chen, H.; Zhu, X.; Zhang, M.; Yao, G.; Yu, Y.; Qin, W.; Zeng, C.; Zen, K.; Liu, Z. miR-223 downregulation promotes glomerular endothelial cell activation by upregulating importin α4 and α5 in IgA nephropathy. Kidney Int. 2014, 85, 624–635. [Google Scholar] [CrossRef]
- Guo, Y.; Liao, Y. miR-200bc/429 cluster alleviates inflammation in IgA nephropathy by targeting TWEAK/Fn14. Int. Immunopharmacol. 2017, 52, 150–155. [Google Scholar] [CrossRef]
- Xing, L.-N.; Wang, H.; Yin, P.-H.; Liu, Y.-J.; Chi, Y.-F.; Wang, Y.-M.; Peng, W. Reduced miR-29b-3p expression up-regulate CDK6 and contributes to IgA nephropathy. Int. J. Clin. Exp. Med. 2014, 7, 5275–5281. [Google Scholar]
- Liang, Y.; Zhao, G.; Tang, L.; Zhang, J.; Li, T.; Liu, Z. MiR-100-3p and miR-877-3p regulate overproduction of IL8 and IL1β in mesangial cells activated by secretory IgA from IgA nephropathy patients. Exp. Cell Res. 2016, 347, 312–321. [Google Scholar] [CrossRef]
- Liang, Y.; Zhang, J.; Zhou, Y.; Xing, G.; Zhao, G.; Liu, Z. Proliferation and cytokine production of human mesangial cells stimulated by secretory IgA isolated from patients with IgA nephropathy. Cell Physiol. Biochem. 2015, 36, 1793–1808. [Google Scholar] [CrossRef] [PubMed]
- Wyatt, R.J.; Julian, B.A. IgA nephropathy. N. Engl. J. Med. 2013, 368, 2402–2414. [Google Scholar] [CrossRef] [PubMed]
- Silva, F.G.; Chande, P.; Pirani, C.L.; Hardy, M.A. Disappearance of glomerular mesangial IgA deposits after renal allograft transplantation. Transplantation 1982, 33, 241–246. [Google Scholar] [PubMed]
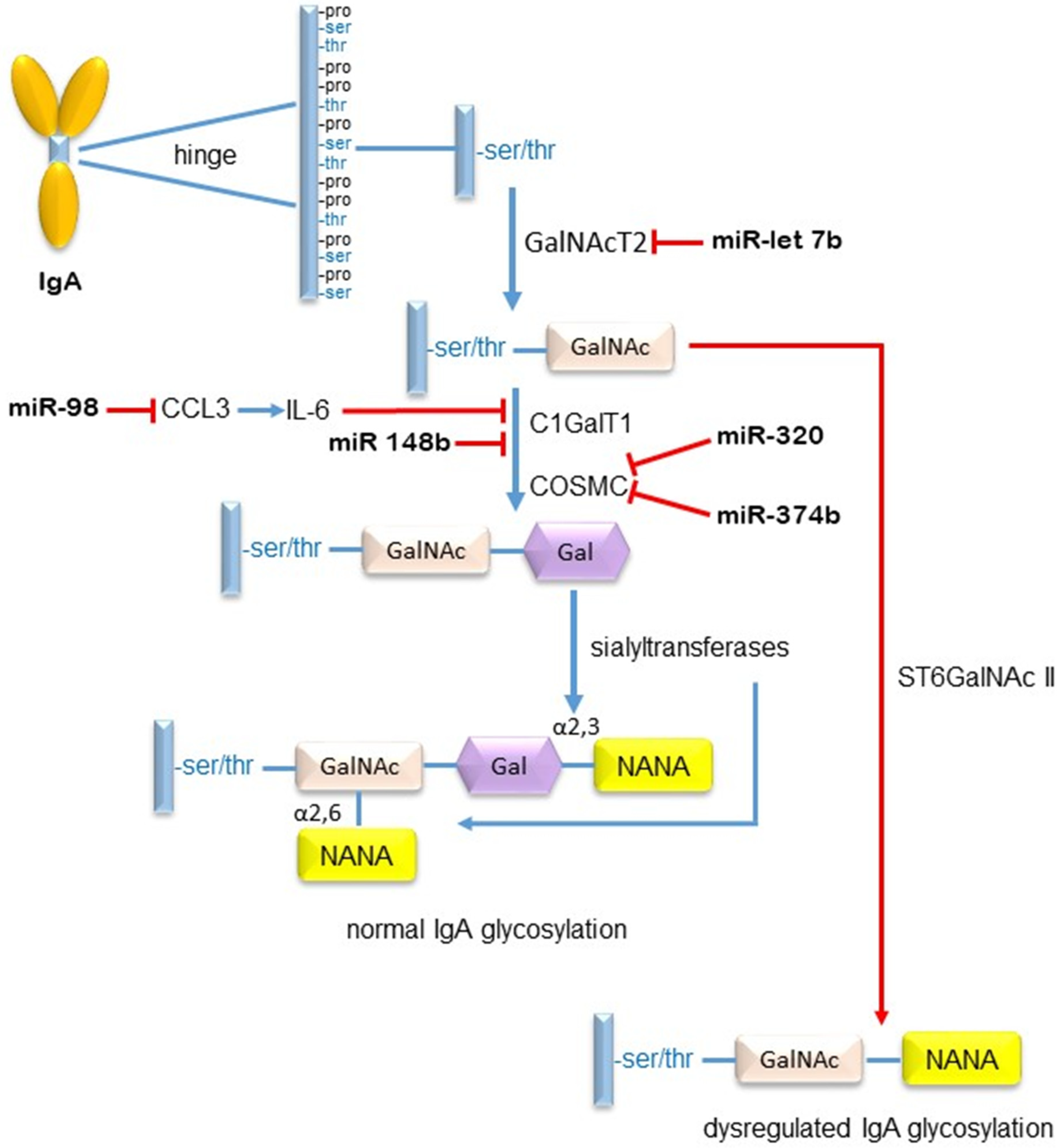
- Serino, G.; Sallusto, F.; Cox, S.N.; Pesce, F.; Schena, F.P. Abnormal miR-148b expression promotes aberrant glycosylation of IgA in IgA nephropathy. J. Soc. Nephrol. 2012, 23, 814–824. [Google Scholar] [CrossRef] [PubMed]
- Li, G.S.; Zhang, H.; Lv, J.C.; Shen, Y.; Wang, H.Y. Variants of C1GalT1 gene are associated with the genetic susceptibility to IgA nephropathy. Kidney Int. 2007, 71, 448–453. [Google Scholar] [CrossRef] [PubMed]
- Wen, L.; Zhao, Z.; Xiao, J.; Wang, Z.; He, X.; Birn, H. Renal miR-148b is associated with megalin down regulation in IgA nephropathy. Biosci Rep. 2018, 38, BSR20181578. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Liiiao, Y.; Wang, L.; Lin, Y.; Ye, Z.; Zeng, X.; Liu, X.; Wei, F.; Yang, N. Small RNA deep sequencing reveals novel miRNAs in peripheral blood mononuclear cells from patients with IgA nephropathy. Mol. Med. Rep. 2020, 22, 3378–3386. [Google Scholar] [CrossRef]
- Liu, D.; Xia, M.; Liu, Y.; Tan, X.; He, L.; Liu, Y.; Chen, G.; Liu, H. The upregulation of miR-98-5p affects the glycosylation of IgA1 through cytokines in IgA nephropathy. Int. Immunopharmacol. 2020, 82, 106362. [Google Scholar] [CrossRef]
- Hu, S.; Bao, H.; Xu, X.; Zhou, X.; Qin, W.; Zeng, C.; Liu, Z. Increased miR-374b promotes cell proliferation and the production of aberrant glycosylated IgA1 in B cells of IgA nephropathy. FEBS Lett. 2015, 589, 4019–4025. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Shi, J.; Zhao, Y. MiR-320 promotes B cell proliferation and the production of aberrant glycosylated IgA1 in IgA nephropathy. J. Cell Biochem. 2017, 119, 4607–4614. [Google Scholar] [CrossRef] [PubMed]
- Ruszkowski, J.; Lisowska, K.A.; Pindel, M.; Heleniak, Z.; Debska-Slizien, A.; Witkowski, J.M. T cells in IgA nephropathy: Role in pathogenesis, clinical significance and potential therapeutic target. Clin. Exp. Nephrol. 2019, 23, 291–303. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Zhang, X.Y.; Peng, W.; Wei, M.; Qui, W. MicroRNA-155-induced T lymphocyte subgroup drifting in IgA nephropathy. Int. Urol. Nephrol. 2017, 49, 353–361. [Google Scholar] [CrossRef]
- Jin, L.W.; Ye, H.Y.; Xu, X.Y.; Zheng, Y.; Chen, Y. MiR-133a/133b inhibits Treg differentiation in IgA nephropathy through targeting fork head box (FOXP3). Biomed. Pharmacother. 2018, 101, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.-Y.; Meng, S.J.; Shi, S.-F.; Liu, L.-J.; Lv, J.-C.; Zhu, L.; Zhang, H. MicroRNA-21-5p participates in IgA nephropathy by driving T helper cell polarisation. J. Nephrol. 2020, 33, 551–560. [Google Scholar] [CrossRef] [PubMed]
- Duan, Z.Y.; Cai, G.Y.; Li, J.J.; Bu, R.; Chen, X.M. Urinary Erythrocyte-Derived miRNAs: Emerging Role in IgA Nephropathy. Kidney Blood Press. Res. 2017, 42, 738–748. [Google Scholar] [CrossRef]
- Mitchell, P.S.; Parkin, R.K.; Kroh, E.M.; Fritz, B.R.; Wyman, S.K.; Pogosova-Agadjanyan, E.L.; Peterson, A.; Noteboom, J.; O’Briant, K.C.; Allen, A.; et al. Circulating microRNA as stable blood-based markers for cancer detection. Proc. Natl. Acad. Sci. USA 2008, 105, 0513–10518. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Ba, Y.; Ma, L.; Cai, X.; Yin, Y.; Wang, K.; Guo, J.; Zhang, Y.; Chen, J.; Guo, X.; et al. Characterisation of microRNAs in serum: A novel class of biomarkers for diagnosis of cancer and other diseases. Cell Res. 2008, 18, 997–1006. [Google Scholar] [CrossRef] [PubMed]
- Turchinovich, A.; Weiz, L.; Burwinkel, B. Extracellular miRNAs: The mystery of their origin and function. Trends Biochem. Sci. 2012, 37, 460–465. [Google Scholar] [CrossRef]
- Turchinovich, A.; Weiz, L.; Langheinz, A.; Burwinkel, B. Characterisation of extracellular circulating microRNA. Nucleic Acids Res. 2011, 39, 7223–7233. [Google Scholar] [CrossRef]
- Vickers, K.C.; Palmisano, B.T.; Shoucri, B.M.; Shamburek, R.D.; Remaley, A.T. MicroRNAs are transported in plasma and delivered to recipient cells by high density lipoproteins. Nat. Cell Biol. 2011, 13, 423–433. [Google Scholar] [CrossRef]
- Turchinovich, A.; Samatov, T.R.; Tonevitsky, A.G.; Burwinkel, B. Circulating miRNAs: Cell-cell communication function. Front. Genet. 2013, 4, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Arroyo, J.D.; Chevillet, J.R.; Kroh, E.M.; Ruf, I.K.; Pritchard, C.C.; Gibson, D.F.; Mitchell, P.S.; Bennett, C.F.; Pogosova-Agadjanyan, E.L.; Stirewalt, D.L.; et al. Argonaute2 complexes carry a population of circulating microRNAs independent of vesicles in human plasma. Proc. Natl. Acad. Sci. USA 2011, 108, 5003–5008. [Google Scholar] [CrossRef]
- Gallo, A.; Tandom, M.; Alevizos, I.; Illei, G.G. The majority of microRNAs detectable in serum and saliva is concentrated in exosomes. PLoS ONE 2012, 7, e30679. [Google Scholar] [CrossRef]
- Wang, Z.; Xi, J.; Hao, X.; Deng, W.; Liu, J.; Wei, C.; Gao, Y.; Zhang, L.; Wang, H. Red blood cells release microparticles containing human argonaute 2 and miRs to targert genes of plasmodium falciparum. Emerg. Microbes Infect. 2017, 6, e75. [Google Scholar] [CrossRef]
- Pigati, L.; Yaddanapudi, S.C.S.; Lyengar, R.; Kim, D.-J.; Hearn, S.A.; Danforth, D.; Hastings, M.L.; Duelli, D.M. Selective release of microRNA species from normal and malignant mammary epithelial cells. PLoS ONE 2010, 5, e13515. [Google Scholar] [CrossRef]
- Iguchi, H.; Kosaka, N.; Ochiya, T. Secretory microRNA as a versatile communication tool. Commun. Integr. Biol. 2010, 3, 478–481. [Google Scholar] [CrossRef] [PubMed]
- Tian, T.; Zhu, Y.L.; Zhou, Y.Y.; Liang, G.F.; Wang, Y.Y.; Hu, F.H.; Xioa, Z.-D. Exosome uptake through clathrin-mediated endocytosis and micropinocytosis and mediating miR-21 delivery. J. Biol. Chem. 2014, 289, 22258–22267. [Google Scholar] [CrossRef] [PubMed]
- Soares, A.R.; Martins-Marques, B.; Ribeiro-Rodrigues, T.; Ferreira, J.V.; Catarino, S.; Pinho, M.J.; Zuzarte, M.; Anjo, S.A.; Manadas, B.P.G.; Sluijter, J.; et al. Gap junctional protein Cx43 is involved in the coomunication between extracellular vesicles and mammalian cells. Sci. Rep. 2015, 5, 13243. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, A.; Costa-Silva, B.; Shen, T.L.; Rodrigues, G.; Hashimoto, A.; Tesic, M.M.; Molina, H.; Kohsaka, S.; Di Giannatale, A.; Creder, S.; et al. Tumour exosome integrins determine organotropic metastasis. Nature 2015, 527, 329–335. [Google Scholar] [CrossRef]
- Batista, B.S.; Eng, W.S.; Pilobello, K.T.; Hendticks-Munoz, K.D.; Mahal, L.K. Identification of a conserved glycan signature for microvesicles. J. Proteome Res. 2011, 10, 4624–4633. [Google Scholar] [CrossRef]
- Chen, X.; Liang, H.; Zhang, I.; Zen, K.; Zhang, C.Y. Secreted microRNAs: A new form of intracellular communication. Trends Cell Biol. 2012, 22, 125–132. [Google Scholar] [CrossRef]
- Serino, G.; Pesce, F.; Sallusto, F.; De Palma, G.; Cox, S.N.; Curci, C.; Zaza, G.; Lai, K.N.; Leung, J.C.K.; Tang, S.C.W.; et al. In a retrospective international study, circulating miR-148b and let7b were found to be serum markers for detecting primary IgA nephropathy. Kidney Int. 2016, 89, 683–692. [Google Scholar] [CrossRef]
- Wu, J.; Zhang, H.; Wang, W.; Zhu, M.; Qi, L.; Wang, T.; Cheng, W.; Zhu, J.; Shan, X.; Huang, Z.; et al. Plasma microRNA signature of patients with IgA nephropathy. Gene 2018, 649, 80–86. [Google Scholar] [CrossRef]
- Lee, H.S.; Lee, M.S.; Lee, S.M.; Lee, S.Y.; Lee, E.S.; Lee, E.Y.; Park, S.Y.; Han, J.S.; Kim, S.; Lee, J.S. Histological grading of IgA nephropathy predicting renal outcome: Revisiting HS Lee’s glomerular grading systems. Nephrol. Dial. Transplant. 2005, 20, 342–348. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Wan, Q.; Li, T.; Qi, D.; Dong, X.; Xu, Y.; Chen, H.; Liu, H.; Huang, H.; Wei, C.; et al. Circulating miR-29a, possible use as a biomarker for monitoring IgA nephropathy. Iran. J. Kidney Dis. 2020, 14, 107–118. [Google Scholar]
- Working group of the International IgA Nephropathy Network of the Renal Pathology Society; Roberts, I.S.D.; Cook, H.T.; Troyanov, S.; Alpers, C.E.; Amore, A.; Barratt, J.; Berthoux, F.; Bonsib, S.; Bruijn, J.A.; et al. The Oxford classification of IgA nephropathy: Pathology definitions, correlations, and reproducibility. Kidney Int. 2009, 76, 546–556. [Google Scholar] [CrossRef]
- Fan, Q.; Lu, R.; Zhu, M.; Yan, Y.; Guo, X.; Qian, Y.; Zhang, L.; Dai, H.; Ni, Z.; Gu, L. Serum miR-192 is related to tubulointerstitial lesion and short-term disease progression in IgA nephropathy. Dis. Markers 2010, 28, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Kwan, B.C.-H.; Lai, F.M.-M.; Chow, K.M.; Li, P.K.-T.; Szeto, C.C. Expression of microRNAs in the urinary sediment of patients with IgA nephropathy. Dis. Markers 2010, 28, 79–86. [Google Scholar] [CrossRef]
- Wang, G.; Kwan, B.C.-H.; Lai, F.M.-M.; Chow, K.-M.; Li, P.K.-T.; Szeto, C.C. Urinary miR-21, miR-29, and miR-93: Novel biomarkers of fibrosis. Am. J. Nephrol. 2012, 36, 412–418. [Google Scholar] [CrossRef]
- Liang, S.; Cai, G.-Y.; Duan, Z.Y.; Liu, S.-W.; Wu, J.; Lv, Y.; Hou, K.; Li, Z.-X.; Zhang, X.-G.; Chen, X.-M. Urinary sediment miRs reflect tubulointerstitial damage and therapeutic response in IgA nephropathy. BMC Nephrol. 2017, 15, 63. [Google Scholar] [CrossRef]
- Duan, A.; Liu, L.; Lou, Y.; Zhang, D.; Li, H.; Chen, Y.; Cui, W.; Miao, L. Diagnostic value of urinary miR-152-5p in patients with IgA nephropathy with elevated proteinuria levels. Clin. Lab. 2019, 65. [Google Scholar] [CrossRef]
- Wang, N.; Bu, R.; Duan, Z.; Zhang, X.; Chen, P.; Li, Z.; Wu, J.; Cai, G.; Chen, X. Profiling and initial validation of urinary microRNAs as biomarkers in IgA nephropathy. PeerJ 2015, 3, e990. [Google Scholar] [CrossRef]
- Szeto, C.-C.; Wang, G.; Ng, J.K.-C.; Kwan, B.C.-H.; Lai, F.M.-M.; Chow, K.-M.; Luk, C.C.-W.; Lai, K.-B.; Li, P.K.-T. Urinary miRNA profile for the diagnosis of IgA nephropathy. BMC Nephrol. 2019, 20, 77. [Google Scholar] [CrossRef]
- Duan, Z.-Y.; Cai, G.-Y.; Bu, R.; Yang, L.; Hou, K.; Chen, X.-M. Selection of urinary sediment miRNAs as specific biomarkers of IgA nephropathy. Sci. Rep. 2016, 6, 23498. [Google Scholar] [CrossRef] [PubMed]
- Min, Q.-H.; Chen, X.-M.; Zou, Y.-Q.; Zhang, J.; Li, J.; Wang, Y.; Li, S.Q.; Gao, Q.-F.; Sun, F.; Liu, F.; et al. Differential expression of urinary exosomal microRNAs in IgA nephropathy. J. Clin. Lab. Anal. 2018, 32, e22226. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Bai, F.; Qin, N.; Liu, W.; Sun, Q.; Zhou, Y.; Yang, J. Non-proximal renal tubule-derived urinary exosomal miR-200b as a biomarker of renal fibrosis. Nephron 2018, 139, 269–282. [Google Scholar] [CrossRef]
- Konta, T.; Ichikawa, K.; Suzuki, K.; Kudo, K.; Satoh, H.; Kamei, K.; Nishidate, E.; Kubota, I. A microarray analysis of urinary microRNA in renal diseases. Clin. Exp. Nephrol. 2014, 18, 711–717. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Su, Y.; Ma, C.; Jing, Z.; Yang, X.; Zhang, D.; Xie, M.; Li, W.; Wei, J. 3’UTR variants of TNS3, PHLDB1, NTN4 and GNG2 genes are associated with IgA nephropathy risk in Chinese Han population. Int. Immunopharmcol. 2019, 71, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Feng, S.; Shi, D.; Xu, R.; Yin, P.; Wang, M.; Mao, H.; Huang, F.; Li, Z.; Yu, X.; et al. Association of FCRL3 gene polymorphisms with IgA nephropathy in a Chinese Han population. DNA Cell Biol. 2019, 38, 1155–1165. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Hiuang, Y.; Zhang, X.; Chen, J.; Sheng, H. Association of miR-146a rs2910164 with childhood IgA nephropathy. Paediatr. Nephrol. 2014, 29, 1979–1986. [Google Scholar] [CrossRef]
- Yang, B.; Wei, W.; Shi, Y.; Huang, Z.; Cai, B.; Zhang, J.; Ying, B.; Wang, L. Genetic variation in miR-146a is not associated with susceptibility to IgA nephropathy in adults from a Chinese Han population. PLoS ONE 2015, 10, e0139554. [Google Scholar] [CrossRef] [PubMed]
- Forero, D.; González-Giraldo, Y.; Castro-Vega, L.; Barreto, G. qPCR-based methods for expression analysis of miRNAs. BioTechniques 2019, 67, 192–199. [Google Scholar] [CrossRef]
- Nahid, M.A.; Satoh, M.; Chan, E.K.L. Interleukin 1β-responsive miR-146a is critical for the cytokine-induced tolerance and cross-tolerance to toll-like receptor ligands. J. Innate Immun. 2015, 7, 428–440. [Google Scholar] [CrossRef]
- Ichii, O.; Otsuka, S.; Saski, N.; Namiki, Y.; Hashimoto, T.; Kon, T. Altered expression of microRNA miR-146a correlates with the development of chronic renal inflammation. Kidney Int. 2012, 81, 280–292. [Google Scholar] [CrossRef]
- KDIGO. KDIGO 2012 Clinical Practice Guideline for glomerulonephritis. Chapter 10: Immunoglobulin A nephropathy. Kidney Int. Suppl. 2012, 2, 209–217. [Google Scholar] [CrossRef]
- Working Group of the International IgA Nephropathy Network and the Renal Pathology Society; Cattran, D.C.; Coppo, R.; Cook, H.T.; Feehally, J.; Roberts, I.S.; Troyanov, S.; Alpers, C.E.; Amore, A.; Barratt, J.; et al. The Oxford classification of IgA nephropathy: Rationale, clinicopathological correlations, and classification. Kidney Int. 2009, 76, 534–545. [Google Scholar] [CrossRef]
- Trimarchi, H.; Barratt, J.; Cattran, D.C.; Cook, H.T.; Coppo, R.; Haas, M.; Liu, Z.H.; Roberts, I.S.; Yuzawa, Y.; Zhang, H.; et al. IgAN Classification Working Group of the International IgA Nephropathy Network and the Renal Pathology Society; Conference Participants. Oxford Classification of IgA nephropathy 2016: An update from the IgA Nephropathy Classification Working Group. Kidney Int. 2017, 91, 1014–1021. [Google Scholar] [CrossRef] [PubMed]
- Barbour, S.J.; Reich, H. Risk Stratification of Patients with IgA Nephropathy. Am. J. Kidney Dis. 2012, 59, 865–873. [Google Scholar] [CrossRef]
- Radford, M., Jr.; Donadio, J., Jr.; Bergstralh, E.; Grande, J. Predicting renal outcome in IgA nephropathy. J. Am. Soc. Nephrol. 1997, 8, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Reich, H.; Troyanov, S.; Scholey, J.; Cattran, D. Remission of Proteinuria Improves Prognosis in IgA Nephropathy. J. Am. Soc. Nephrol. 2007, 18, 3177–3183. [Google Scholar] [CrossRef]
- Nam, K.; Kie, J.; Lee, M.; Chang, T.I.; Kang, E.W.; Kim, D.W.; Lim, B.J.; Park, J.T.; Kwon, Y.E.; Kim, Y.L.; et al. Optimal Proteinuria Target for Renoprotection in Patients with IgA Nephropathy. PLoS ONE 2014, 9, e101935. [Google Scholar] [CrossRef] [PubMed]
- Berthoux, F.; Mohey, H.; Laurent, B.; Mariat, C.; Afiani, A.; Thibaudin, L. Predicting the Risk for Dialysis or Death in IgA Nephropathy. J. Am. Soc. Nephrol. 2011, 22, 752–761. [Google Scholar] [CrossRef] [PubMed]
- Wakai, K.; Kawamura, T.; Endoh, M.; Kojima, M.; Tomino, Y.; Tamakoshi, A.; Ohno, Y.; Inaba, Y.; Sakai, H. A scoring system to predict renal outcome in IgA nephropathy: From a nationwide prospective study. Nephrol. Dial. Transplant. 2006, 21, 2800–2808. [Google Scholar] [CrossRef] [PubMed]
- Geddes, C.; Rauta, V.; Gronhagen-Riska, C.; Bartosik, L.P.; Jardine, A.G.; Ibels, L.S.; Pei, Y.; Cattran, D.C. A tricontinental view of IgA nephropathy. Nephrol. Dial. Transplant. 2003, 18, 1541–1548. [Google Scholar] [CrossRef] [PubMed]
- Goto, M.; Wakai, K.; Kawamura, T.; Ando, M.; Endoh, M.; Tomino, Y. A scoring system to predict renal outcome in IgA nephropathy: A nationwide 10-year prospective cohort study. Nephrol. Dial. Transplant. 2009, 24, 3068–3074. [Google Scholar] [CrossRef]
- Arroyo, A.; Bomback, A.; Butler, B.; Radhakrishnan, J.; Herlitz, L.; Stokes, M.B.; D’Agati, V.; Markowitz, G.S.; Appel, G.B.; Canetta, P.A. Predictors of outcome for severe IgA Nephropathy in a multi-ethnic U.S. cohort. Clin. Nephrol. 2015, 84, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Inker, L.; Heerspink, H.; Tighiouart, H.; Levey, A.S.; Coresh, J.; Gansevoort, R.T.; Simon, A.L.; Ying, J.; Beck, G.J.; Wanner, C.; et al. GFR Slope as a Surrogate End Point for Kidney Disease Progression in Clinical Trials: A Meta-Analysis of Treatment Effects of Randomized Controlled Trials. J. Am. Soc. Nephrol. 2019, 30, 1735–1745. [Google Scholar] [CrossRef]
- Barbour, S.J.; Coppo, R.; Zhang, H.; Liu, Z.H.; Suzuki, Y.; Matsuzaki, K.; Katafuchi, R.; Er, L.; Espino-Hernandez, G.; Kim, S.J.; et al. Evaluating a New International Risk-Prediction Tool in IgA Nephropathy. JAMA Intern. Med. 2019, 179, 942. [Google Scholar] [CrossRef]
- Szeto, C.-C.; Kwan, B.C.-H.; Lai, K.-B.; Lai, F.M.-M.; Choi, P.C.-L.; Wang, G.; Chow, K.-M.; Li, P.K.-T. Micro-RNA expression in the urinary sediment of patients with chronic kidney diseases. Dis. Markers 2012, 33, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Selvaskandan, H.; Cheung, C.; Muto, M.; Barratt, J. New strategies and perspectives on managing IgA nephropathy. Clin. Exp. Nephrol. 2019, 23, 577–588. [Google Scholar] [CrossRef]
- Cheng, J.; Zhang, W.; Zhang, X.; He, Q.; Tao, X.; Chen, J. ACEI/ARB therapy for IgA nephropathy: A meta-analysis of randomised controlled trials. Int. J. Clin. Pract. 2009, 63, 880–888. [Google Scholar] [CrossRef]
- Rauen, T.; Eitner, F.; Fitzner, C.; Sommerer, C.; Zeier, M.; Otte, B.; Panzer, U.; Peters, H.; Benck, U.; Mertens, P.R.; et al. Intensive Supportive Care plus Immunosuppression in IgA Nephropathy. N. Engl. J. Med. 2015, 373, 2225–2236. [Google Scholar] [CrossRef]
- Editorial. Delivering the promise of RNA therapeutics. Nat. Med. 2019, 25(9), 1321. [Google Scholar] [CrossRef]
- Maillard, N.; Wyatt, R.; Julian, B.; Kiryluk, K.; Gharavi, A.; Fremeaux-Bacchi, V.; Novak, J. Current Understanding of the Role of Complement in IgA Nephropathy. J. Am. Soc. Nephrol. 2015, 26, 1503–1512. [Google Scholar] [CrossRef]
- Kusner, L.; Yucius, K.; Sengupta, M.; Sprague, A.G.; Desai, D.; Nguyen, T.; Charisse, K.; Kuchimanchi, S.; Kallanthottathil, R.; Fitzgerald, K.; et al. Investigational RNAi Therapeutic Targeting C5 Is Efficacious in Pre-clinical Models of Myasthenia Gravis. Mol. Ther. Methods Clin. Dev. 2019, 13, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Thurman, J.; Holers, V. The Central Role of the Alternative Complement Pathway in Human Disease. J. Immunol. 2006, 176, 1305–1310. [Google Scholar] [CrossRef] [PubMed]
- Hendrickson, D.; Hogan, D.; McCullough, H.; Liu, Z.H.; Suzuki, Y.; Matsuzaki, K.; Katafuchi, R.; Er, L.; Espino-Hernandez, G.; Kim, S.J.; et al. Concordant Regulation of Translation and mRNA Abundance for Hundreds of Targets of a Human microRNA. PLoS Biol. 2009, 7, e1000238. [Google Scholar] [CrossRef] [PubMed]
- Springer, A.; Dowdy, S. GalNAc-siRNA Conjugates: Leading the Way for Delivery of RNAi Therapeutics. Nucleic Acid Ther. 2018, 28, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Jaffe, G.; Sahni, J.; Fauser, S.; Geary, R.; Schneider, E.; McCaleb, M. Development of IONIS-FB-LRx to Treat Geographic Atrophy Associated with AMD. Investig. Ophthalmol. Vis. Sci. 2020, 61, 4305. [Google Scholar]
- Janas, M.; Harbison, C.; Perry, V.; Carito, B.; Sutherland, J.E.; Vaishnaw, A.K.; Keirstead, N.D.; Warner, G. The Nonclinical Safety Profile of GalNAc-conjugated RNAi Therapeutics in Subacute Studies. Toxicol. Pathol. 2018, 46, 735–745. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| RNA Type | Abbreviation | Full Name | Size (nt) |
|---|---|---|---|
| rRNA | ribosomal RNA | 120–4500 | |
| tRNA | transfer RNA | 76–90 | |
| snRNA | small nuclear RNA | 100–300 | |
| Housekeeping ncRNA | snoRNA | small nucleolar RNA | 60–400 |
| TERC | telomerase RNA | / | |
| tRF | tRNA-derived fragments | 16–28 | |
| tiRNA | tRNA halves | 29–50 | |
| miR | microRNA | 21–23 | |
| siRNA | small interfering RNA | 20–25 | |
| piRNA | piwi-intreracting RNA | 26–32 | |
| Regulatory ncRNA | eRNA | enhancer RNA | 50–2000 |
| lncRNA | long noncoding RNA | >200 | |
| circRNA | circular RNA | 100–10,000 | |
| Y RNA | Y RNA | / |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pawluczyk, I.Z.A.; Selvaskandan, H.; Barratt, J. The Non-Coding RNA Landscape in IgA Nephropathy—Where Are We in 2021? J. Clin. Med. 2021, 10, 2369. https://doi.org/10.3390/jcm10112369
Pawluczyk IZA, Selvaskandan H, Barratt J. The Non-Coding RNA Landscape in IgA Nephropathy—Where Are We in 2021? Journal of Clinical Medicine. 2021; 10(11):2369. https://doi.org/10.3390/jcm10112369
Chicago/Turabian StylePawluczyk, Izabella Z. A., Haresh Selvaskandan, and Jonathan Barratt. 2021. "The Non-Coding RNA Landscape in IgA Nephropathy—Where Are We in 2021?" Journal of Clinical Medicine 10, no. 11: 2369. https://doi.org/10.3390/jcm10112369
APA StylePawluczyk, I. Z. A., Selvaskandan, H., & Barratt, J. (2021). The Non-Coding RNA Landscape in IgA Nephropathy—Where Are We in 2021? Journal of Clinical Medicine, 10(11), 2369. https://doi.org/10.3390/jcm10112369