
Therapeutic Targets and Tumor Microenvironment in Colorectal Cancer



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Definition
3. Angiogenesis and Hypoxia
4. Cancer-Associated Fibroblasts
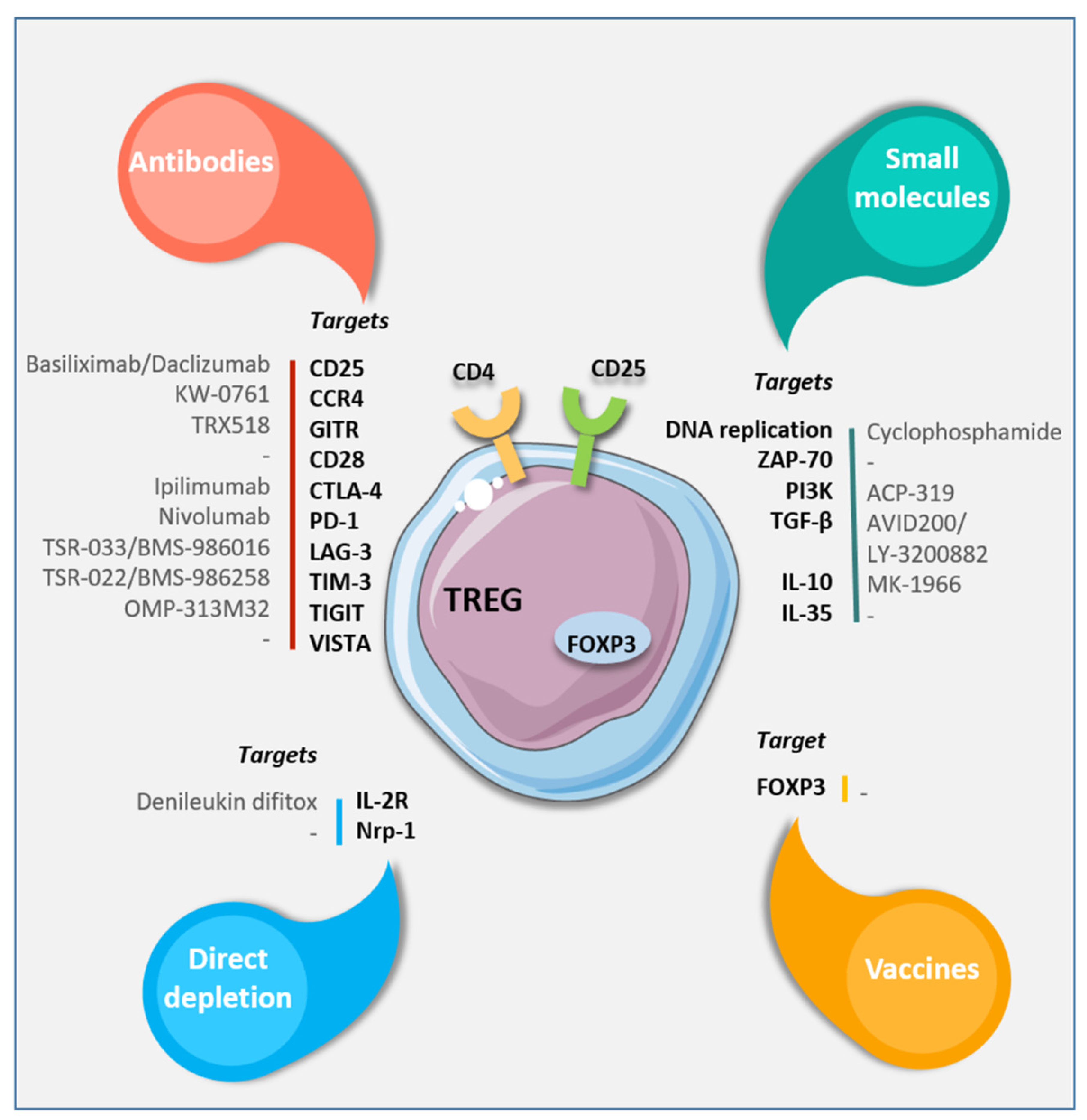
5. Regulatory T Cells
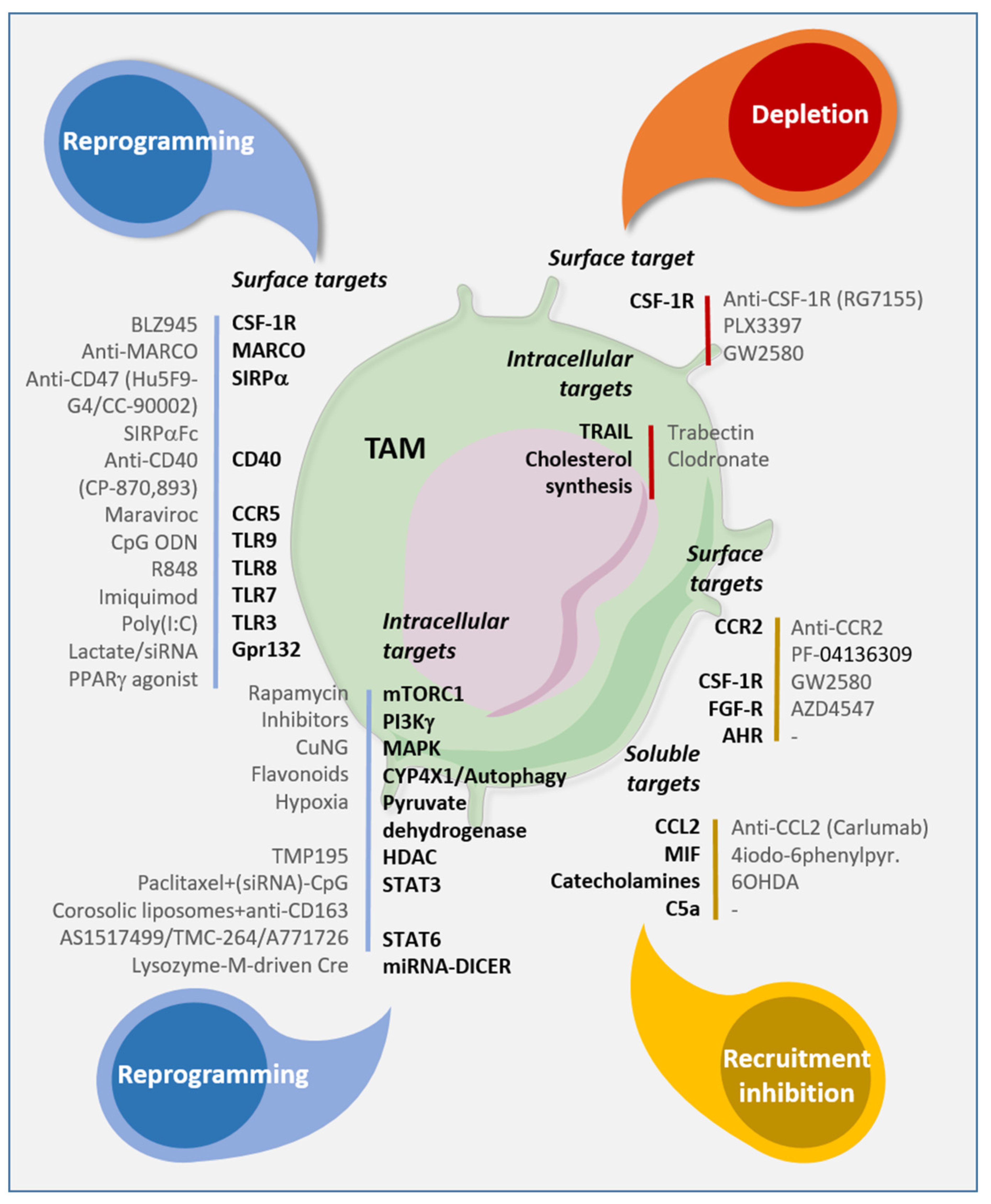
6. Tumor-Associated Macrophages
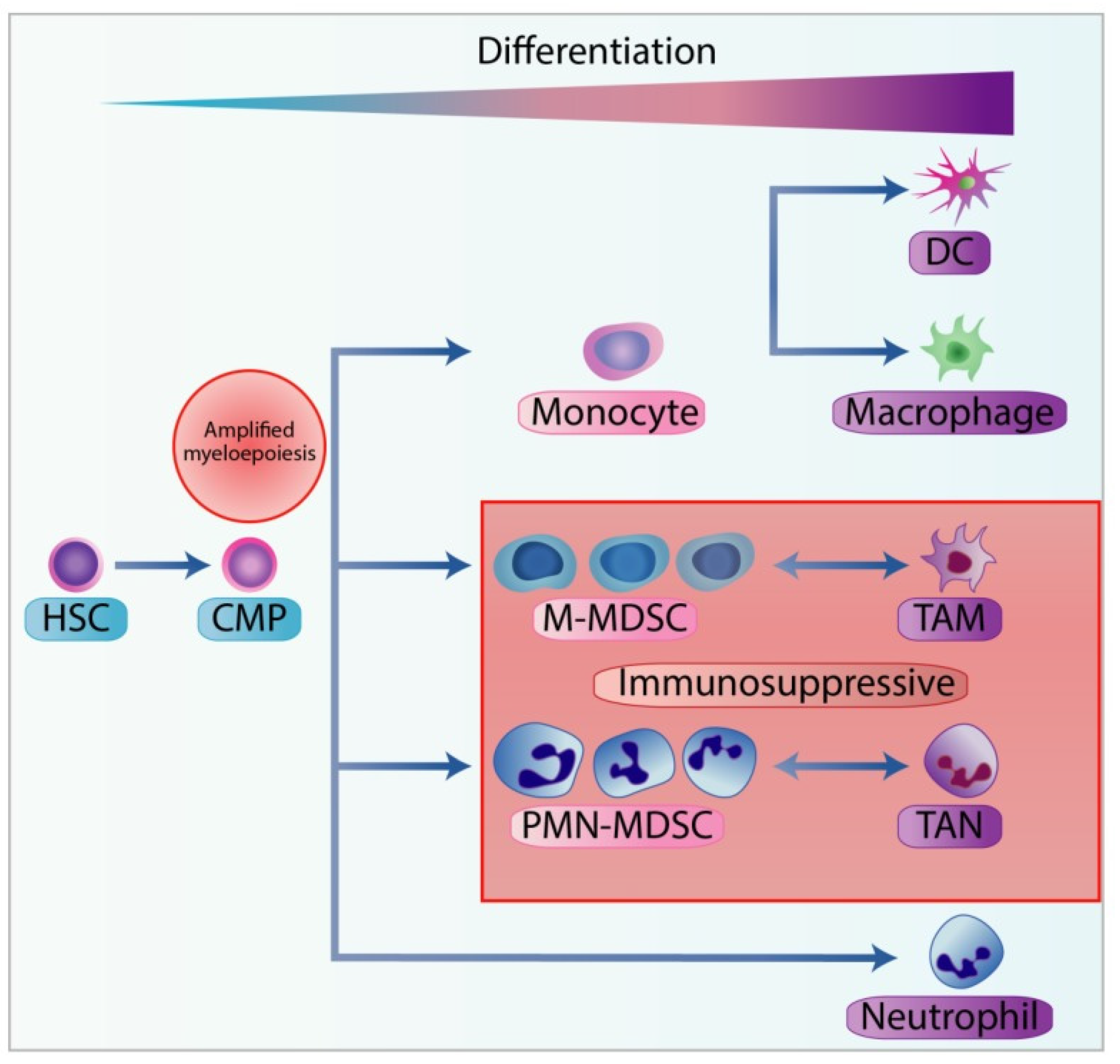
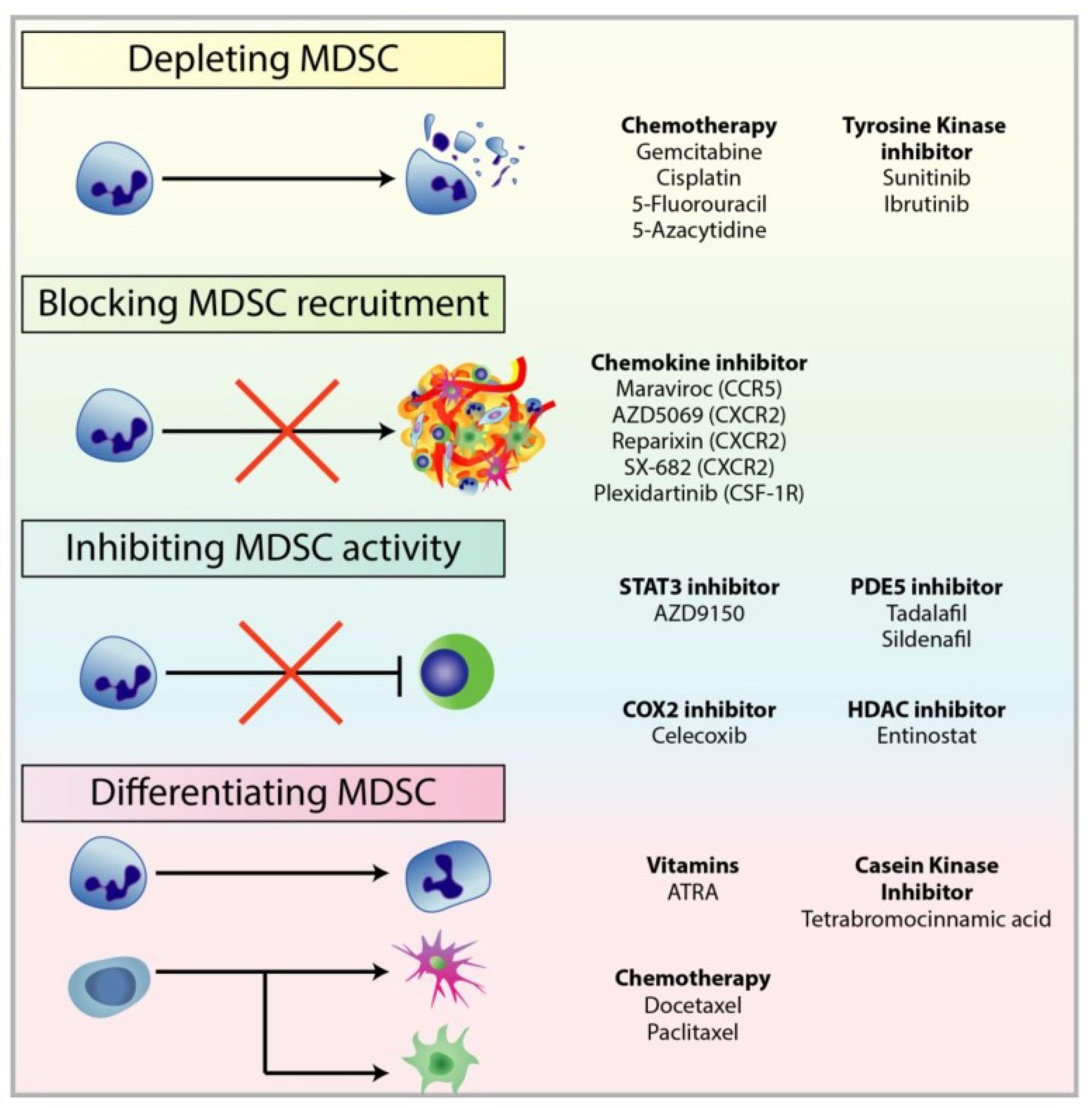
7. Myeloid-Derived Suppressor Cells
8. Emerging Treatments and Others
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Brenner, H.; Kloor, M.; Pox, C.P. Colorectal cancer. Lancet 2014, 383, 1490–1502. [Google Scholar] [CrossRef]
- Gallo, G.; Sena, G.; Vescio, G.; Papandrea, M.; Sacco, R.; Trompetto, M.; Sammarco, G. The prognostic value of KRAS and BRAF in stage I-III colorectal cancer. A systematic review. Ann. Ital. Chir. 2019, 90, 127–137. [Google Scholar] [PubMed]
- O’Connell, J.B.; Maggard, M.A.; Ko, C.Y. Colon cancer survival rates with the new American Joint Committee on Cancer sixth edition staging. J. Natl. Cancer Inst. 2004, 96, 1420–1425. [Google Scholar] [CrossRef]
- Fong, Y.; Fortner, J.; Sun, R.L.; Brennan, M.F.; Blumgart, L.H. Clinical score for predicting recurrence after hepatic resection for metastatic colorectal cancer: Analysis of 1001 consecutive cases. Ann. Surg. 1999, 230, 309–318. [Google Scholar] [CrossRef]
- Van Cutsem, E.; Cervantes, A.; Nordlinger, B.; Arnold, D.; ESMO Guidelines Working Group. Metastatic colorectal cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2014, 3, III1–III9. [Google Scholar] [CrossRef]
- Tarallo, S.; Ferrero, G.; Gallo, G.; Francavilla, A.; Clerico, G.; Realis Luc, A.; Manghi, P.; Thomas, A.M.; Vineis, P.; Segata, N.; et al. Altered Fecal Small RNA Profiles in Colorectal Cancer Reflect Gut Microbiome Composition in Stool Samples. Msystems 2019, 4, e00289–e00319. [Google Scholar] [CrossRef]
- Li, I.; Nabet, B.Y. Exosomes in the tumor microenvironment as mediators of cancer therapy resistance. Mol. Cancer 2019, 18, 32. [Google Scholar] [CrossRef] [PubMed]
- Fang, H.; Declerck, Y.A. Targeting the tumor microenvironment: From understanding pathways to effective clinical trials. Cancer Res. 2013, 73, 4965–4977. [Google Scholar] [CrossRef]
- Fridman, W.H.; Miller, I.; Sautès-Fridman, C.; Byrne, A.T. Therapeutic Targeting of the Colorectal Tumor Stroma. Gastroenterology 2020, 158, 303–321. [Google Scholar] [CrossRef]
- Xiang, W.; Shi, R.; Kang, X.; Zhang, X.; Chen, P.; Zhang, L.; Hou, A.; Wang, R.; Zhao, Y.; Zhao, K.; et al. Monoacylglycerol lipase regulates cannabinoid receptor 2-dependent macrophage activation and cancer progression. Nat. Commun. 2018, 9, 2574. [Google Scholar] [CrossRef] [PubMed]
- Liotta, L.A.; Kohn, E.C. The microenvironment of the tumour-host interface. Nature 2001, 411, 375–379. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J. Tumor angiogenesis: Therapeutic implications. N. Engl. J. Med. 1971, 285, 1182–1186. [Google Scholar]
- Sammarco, G.; Gallo, G.; Vescio, G.; Picciariello, A.; De Paola, G.; Trompetto, M.; Currò, G.; Ammendola, M. Mast Cells, microRNAs and Others: The Role of Translational Research on Colorectal Cancer in the Forthcoming Era of Precision Medicine. J. Clin. Med. 2020, 9, 2852. [Google Scholar] [CrossRef] [PubMed]
- Pellino, G.; Gallo, G.; Pallante, P.; Capasso, R.; De Stefano, A.; Maretto, I.; Malapelle, U.; Qiu, S.; Nikolaou, S.; Barina, A.; et al. Noninvasive Biomarkers of Colorectal Cancer: Role in Diagnosis and Personalised Treatment Perspectives. Gastroenterol. Res. Pract. 2018, 2018, 2397863. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.H.; Malietzis, G.; Askari, A.; Bernardo, D.; Al-Hassi, H.O.; Clark, S.K. Is right-sided colon cancer different to left-sided colorectal cancer?—A systematic review. Eur. J. Surg. Oncol. 2015, 41, 300–308. [Google Scholar] [CrossRef] [PubMed]
- Guinney, J.; Dienstmann, R.; Wang, X.; de Reyniès, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef] [PubMed]
- Sadanandam, A.; Lyssiotis, C.A.; Homicsko, K.; Collisson, E.A.; Gibb, W.J.; Wullschleger, S.; Ostos, L.C.; Lannon, W.A.; Grotzinger, C.; Del Rio, M.; et al. A colorectal cancer classification system that associates cellular phenotype and responses to therapy. Nat. Med. 2013, 19, 619–625. [Google Scholar] [CrossRef]
- Alderdice, M.; Richman, S.D.; Gollins, S.; Stewart, J.P.; Hurt, C.; Adams, R.; McCorry, A.M.; Roddy, A.C.; Vimalachandran, D.; Isella, C.; et al. Prospective patient stratification into robust cancer-cell intrinsic subtypes from colorectal cancer biopsies. J. Pathol. 2018, 245, 19–28. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Paget, S. The distribution of secondary growths in cancer of the breast. 1889. Cancer Metastasis Rev. 1989, 8, 98–101. [Google Scholar]
- Trédan, O.; Galmarini, C.M.; Patel, K.; Tannock, I.F. Drug resistance and the solid tumor microenvironment. J. Natl. Cancer Inst. 2000, 99, 1441–1454. [Google Scholar] [CrossRef]
- Leung, D.W.; Cachianes, G.; Kuang, W.J.; Goeddel, D.V.; Ferrara, N. Vascular endothelial growth factor is a secreted angiogenic mitogen. Science 1989, 246, 1306–1309. [Google Scholar] [CrossRef] [PubMed]
- Senger, D.R.; Galli, S.J.; Dvorak, A.M.; Perruzzi, C.A.; Harvey, V.S.; Dvorak, H.F. Tumor cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science 1983, 219, 983–985. [Google Scholar] [CrossRef]
- Yang, J.C.; Haworth, L.; Sherry, R.M.; Hwu, P.; Schwartzentruber, D.J.; Topalian, S.L.; Steinberg, S.M.; Chen, H.X.; Rosenberg, S.A. A randomized trial of bevacizumab, an anti-vascular endothelial growth factor antibody, for metastatic renal cancer. N. Engl. J. Med. 2003, 349, 427–434. [Google Scholar] [CrossRef]
- Kabbinavar, F.F.; Hambleton, J.; Mass, R.D.; Hurwitz, H.I.; Bergsland, E.; Sarkar, S. Combined analysis of efficacy: The addition of bevacizumab to fluorouracil/leucovorin improves survival for patients with metastatic colorectal cancer. J. Clin. Oncol. 2005, 23, 3706–3712. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; Kohne, C.H.; Hitre, E.; Zaluski, J.; Chang Chien, C.R.; Makhson, A.; D’Haens, G.; Pinter, T.; Lim, R.; Bodoky, G.; et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N. Engl. J. Med. 2009, 360, 1408–1417. [Google Scholar] [CrossRef] [PubMed]
- Douillard, J.Y.; Oliner, K.S.; Siena, S.; Tabernero, J.; Burkes, R.; Barugel, M.; Humblet, Y.; Bodoky, G.; Cunningham, D.; Jassem, J.; et al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N. Engl. J. Med. 2013, 369, 1023–1034. [Google Scholar] [CrossRef] [PubMed]
- Khambata-Ford, S.; Garrett, C.R.; Meropol, N.J.; Basik, M.; Harbison, C.T.; Wu, S.; Wong, T.W.; Huang, X.; Takimoto, C.H.; Godwin, A.K.; et al. Expression of epiregulin and amphiregulin and K-ras mutation status predict disease control in metastatic colorectal cancer patients treated with cetuximab. J. Clin. Oncol. 2007, 25, 3230–3237. [Google Scholar] [CrossRef]
- Stintzing, S.; Modest, D.P.; Rossius, L.; Lerch, M.M.; von Weikersthal, L.F.; Decker, T.; Kiani, A.; Vehling-Kaiser, U.; Al-Batran, S.E.; Heintges, T.; et al. FOLFIRI plus cetuximab versus FOLFIRI plus bevacizumab for metastatic colorectal cancer (FIRE-3): A post-hoc anal- ysis of tumour dynamics in the final RAS wild-type subgroup of this randomised open-label phase 3 trial. Lancet Oncol. 2016, 17, 1426–1434. [Google Scholar] [CrossRef]
- Tabernero, J.; Yoshino, T.; Cohn, A.L.; Obermannova, R.; Bodoky, G.; Garcia-Carbonero, R.; Ciuleanu, T.E.; Portnoy, D.C.; Van Cutsem, E.; Grothey, A.; et al. Ramucirumab versus placebo in combination with second-line FOLFIRI in patients with metastatic colorectal carcinoma that progressed during or after first-line therapy with bevacizumab, oxaliplatin, and a fluoropyrimidine (RAISE): A randomised, double-blind, multicentre, phase 3 study. Lancet Oncol. 2015, 16, 499–508. [Google Scholar]
- Tabernero, J.; Van Cutsem, E.; Lakomy, R.; Prausova, J.; Ruff, P.; van Hazel, G.A.; Moiseyenko, V.M.; Ferry, D.R.; Mc-Kendrick, J.J.; Soussan-Lazard, K.; et al. Aflibercept versus placebo in combination with fluoro- uracil, leucovorin and irinotecan in the treatment of previously treated metastatic colorectal cancer: Pre-specified subgroup analyses from the VELOUR trial. Eur. J. Cancer 2014, 50, 320–331. [Google Scholar] [CrossRef]
- Tew, W.P.; Gordon, M.; Murren, J.; Dupont, J.; Pezzulli, S.; Aghajanian, C.; Sabbatini, P.; Mendelson, D.; Schwartz, L.; Gettinger, S.; et al. Phase 1 study of aflibercept administered subcutane- ously to patients with advanced solid tumors. Clin. Cancer Res. 2010, 16, 358–366. [Google Scholar] [CrossRef]
- Garon, E.B.; Ciuleanu, T.E.; Arrieta, O.; Prabhash, K.; Syrigos, K.N.; Goksel, T.; Park, K.; Gorbunova, V.; Kowalyszyn, R.D.; Pikiel, J.; et al. Ramucirumab plus docetaxel versus placebo plus docetaxel for second-line treatment of stage IV non-small-cell lung cancer after disease progression on platinum-based therapy (REVEL): A multicentre, double-blind, randomised phase 3 trial. Lancet 2014, 384, 665–673. [Google Scholar] [CrossRef]
- Wilke, H.; Muro, K.; Van Cutsem, E.; Oh, S.C.; Bodoky, G.; Shimada, Y.; Hironaka, S.; Sugimoto, N.; Lipatov, O.; Kim, T.Y.; et al. Ramucirumab plus paclitaxel versus placebo plus paclitaxel in patients with previously treated advanced gastric or gastro-oesophageal junction adenocarcinoma (RAINBOW): A double-blind, randomised phase 3 trial. Lancet Oncol. 2014, 15, 1224–1235. [Google Scholar] [CrossRef]
- Leow, C.C.; Coffman, K.; Inigo, I.; Breen, S.; Czapiga, M.; Soukharev, S.; Gingles, N.; Peterson, N.; Fazenbaker, C.; Woods, R.; et al. MEDI3617, a human anti-angiopoietin 2 monoclonal antibody, inhibits angiogenesis and tumor growth in human tumor xenograft models. Int. J. Oncol. 2012, 40, 1321–1330. [Google Scholar] [CrossRef]
- Kloepper, J.; Riedemann, L.; Amoozgar, Z.; Seano, G.; Susek, K.; Yu, V.; Dalvie, N.; Amelung, R.L.; Datta, M.; Song, J.W.; et al. Ang-2/VEGF bispecific antibody reprograms macrophages and resident microglia to anti-tumor phenotype and prolongs glioblastoma survival. Proc. Natl. Acad. Sci. USA 2016, 113, 4476–4481. [Google Scholar] [CrossRef]
- Rankin, E.B.; Giaccia, A.J. Hypoxic control of metastasis. Science 2016, 352, 175–180. [Google Scholar] [CrossRef]
- Rankin, E.B.; Nam, J.M.; Giaccia, A.J. Hypoxia: Signaling the metastatic cascade. Trends Cancer 2016, 2, 295–304. [Google Scholar] [CrossRef]
- Ziello, J.E.; Jovin, I.S.; Huang, Y. Hypoxia-inducible factor (HIF)-1 regulatory pathway and its potential for therapeutic intervention in malignancy and ischemia. Yale J. Biol. Med. 2007, 80, 51–60. [Google Scholar]
- Duffy, A.G.; Melillo, G.; Turkbey, B.; Allen, D.; Choyke, P.L.; Chen, C.; Raffeld, M.; Doroshow, J.H.; Murgo, A.; Kummar, S. A pilot trial of oral topotecan (TPT) in patients with refractory advanced solid neoplasms expressing HIF-1α. J. Clin. Oncol. 2010, 28, e13518. [Google Scholar] [CrossRef]
- Jeong, W.; Rapisarda, A.; Park, S.R.; Kinders, R.J.; Chen, A.; Melillo, G.; Turkbey, B.; Steinberg, S.M.; Choyke, P.; Doroshow, J.H.; et al. Pilot trial of EZN-2968, an antisense oligonucleotide inhibitor of hypoxia-inducible factor-1 alpha (HIF-1α), in patients with refractory solid tumors. Cancer Chemother. Pharmacol. 2014, 73, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Son, G.M.; Kwon, M.S.; Shin, D.H.; Shin, N.; Ryu, D.; Kang, C.D. Comparisons of cancer-associated fibroblasts in the intratumoral stroma and invasive front in colorectal cancer. Medicine 2019, 98, e15164. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Song, E. Turning foes to friends: Targeting cancer-associated fibroblasts. Nat. Rev. Drug Discov. 2019, 18, 99–115. [Google Scholar] [CrossRef]
- Mayo, L.D.; Dixon, J.E.; Durden, D.L.; Tonks, N.K.; Donner, D.B. PTEN protects p53 from Mdm2 and sensitizes cancer cells to chemotherapy. J. Biol. Chem. 2002, 277, 5484–5489. [Google Scholar] [CrossRef]
- Paauwe, M.; Schoonderwoerd, M.J.A.; Helderman, R.; Harryvan, T.J.; Groenewoud, A.; van Pelt, G.W.; Bor, R.; Hemmer, D.M.; Versteeg, H.H.; Snaar-Jagalska, B.E.; et al. Endoglin Expression on Cancer-Associated Fibroblasts Regulates Invasion and Stimulates Colorectal Cancer Metastasis. Clin. Cancer Res. 2018, 24, 6331–6344. [Google Scholar] [CrossRef]
- Lu, C.; Vickers, M.F.; Kerbel, R.S. Interleukin 6: A fibro- blast-derived growth inhibitor of human melanoma cells from early but not advanced stages of tumor progression. Proc. Natl. Acad. Sci. USA 1992, 89, 9215–9219. [Google Scholar] [CrossRef]
- Calon, A.; Tauriello, D.V.; Batlle, E. TGF-beta in CAF-mediated tumor growth and metastasis. Semin. Cancer Biol. 2014, 25, 15–22. [Google Scholar] [CrossRef]
- Albrengues, J.; Bourget, I.; Pons, C.; Butet, V.; Hofman, P.; Tartare-Deckert, S.; Feral, C.C.; Meneguzzi, G.; Gaggioli, C. LIF mediates proinvasive activation of stromal fibroblasts in cancer. Cell Rep. 2014, 7, 1664–1678. [Google Scholar] [CrossRef]
- Sugimoto, H.; Mundel, T.M.; Kieran, M.W.; Kalluri, R. Identification of fibroblast heterogeneity in the tumor microenvironment. Cancer Biol. Ther. 2006, 5, 1640–1646. [Google Scholar] [CrossRef]
- Albrengues, J.; Bertero, T.; Grasset, E.; Bonan, S.; Maiel, M.; Bourget, I.; Philippe, C.; Herraiz Serrano, C.; Benamar, S.; Croce, O.; et al. Epigenetic switch drives the conversion of fibroblasts into proinvasive cancer-associated fibroblasts. Nat. Commun. 2015, 6, 10204. [Google Scholar] [CrossRef]
- Orimo, A.; Weinberg, R.A. Stromal fibroblasts in cancer: A novel tumor-promoting cell type. Cell Cycle 2006, 5, 1597–1601. [Google Scholar] [CrossRef]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef]
- Neuzillet, C.; Tijeras-Raballand, A.; Cohen, R.; Cros, J.; Faivre, S.; Raymond, E.; de Gramont, A. Targeting the TGFβ pathway for cancer therapy. Pharmacol. Ther. 2015, 147, 22–31. [Google Scholar] [CrossRef]
- Smith, A.L.; Robin, T.P.; Ford, H.L. Molecular pathways: Targeting the TGF-β pathway for cancer therapy. Clin. Cancer Res. 2012, 18, 4514–4521. [Google Scholar] [CrossRef]
- Akhurst, R.J.; Hata, A. Targeting the TGFbeta signalling pathway in disease. Nat. Rev. Drug Discov. 2012, 11, 790–811. [Google Scholar] [CrossRef]
- Gorelik, L.; Flavell, R.A. Immune-mediated eradication of tumors through the blockade of transforming growth factor-beta signaling in T cells. Nat. Med. 2001, 7, 1118–1122. [Google Scholar] [CrossRef]
- Calon, A.; Lonardo, E.; Berenguer-Llergo, A.; Espinet, E.; Hernando-Momblona, X.; Iglesias, M.; Sevillano, M.; Palomo-Ponce, S.; Tauriello, D.V.; Byrom, D.; et al. Stromal gene expression defines poor-prognosis subtypes in colorectal cancer. Nat. Genet. 2015, 47, 320–329. [Google Scholar] [CrossRef]
- Gasteiger, G.; Hemmers, S.; Firth, M.A.; Le Floc’h, A.; Huse, M.; Sun, J.C.; Rudensky, A.Y. IL-2–dependent tuning of NK cell sensitivity for target cells is controlled by regulatory T cells. J. Exp. Med. 2013, 210, 1167–1178. [Google Scholar] [CrossRef]
- Grossman, W.J.; Verbsky, J.W.; Barchet, W.; Colonna, M.; Atkinson, J.P.; Ley, T.J. Human T regulatory cells can use the perforin pathway to cause autologous target cell death. Immunity 2004, 21, 589–601. [Google Scholar] [CrossRef] [PubMed]
- Von Boehmer, H. Mechanisms of suppression by suppressor T cells. Nat. Immunol. 2005, 6, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Laplagne, C.; Domagala, M.; Le Naour, A.; Quemerais, C.; Hamel, D.; Fournié, J.J.; Couderc, B.; Bousquet, C.; Ferrand, A.; Poupot, M. Latest Advances in Targeting the Tumor Microenvironment for Tumor Suppression. Int. J. Mol. Sci. 2019, 20, 4719. [Google Scholar] [CrossRef]
- Karabon, L.; Markiewicz, M.; Kosmaczewska, A.; Partyka, A.; Pawlak-Adamska, E.; Tomkiewicz, A.; Ciszak, L.; Jagoda, K.; Dzierzak-Mietla, M.; Kyrcz-Krzemien, S.; et al. Pretransplant donor and recipient CTLA-4 mRNA and protein levels as a prognostic marker for aGvHD in allogeneic hema-topoietic stem cell transplantation. Immunol. Lett. 2015, 165, 52–59. [Google Scholar] [CrossRef]
- Rech, A.J.; Mick, R.; Martin, S.; Recio, A.; Aqui, N.A.; Powell, D.J.; Colligon, T.A.; Trosko, J.A.; Leinbach, L.I.; Pletcher, C.H.; et al. CD25 blockade depletes and selectively reprograms regulatory T cells in concert with immunotherapy in cancer patients. Sci. Transl. Med. 2012, 4, 134ra62. [Google Scholar] [CrossRef]
- Marangoni, F.; Zhang, R.; Mani, V.; Thelen, M.; Ali Akbar, N.J.; Warner, R.D.; Äijö, T.; Zappulli, V.; Martinez, G.J.; Turka, L.A.; et al. Tumor Tolerance-Promoting Function of Regulatory T Cells Is Optimized by CD28, but Strictly Dependent on Calcineurin. J. Immunol. 2018, 200, 3647–3661. [Google Scholar] [CrossRef]
- De Groot, A.E.; Pienta, K.J. Epigenetic control of macrophage polarization: Implications for targeting tumor-associated macro-phages. Oncotarget 2018, 9, 20908–20927. [Google Scholar] [CrossRef] [PubMed]
- Prasmickaite, L.; Tenstad, E.M.; Pettersen, S.; Jabeen, S.; Egeland, E.V.; Nord, S.; Pandya, A.; Haugen, M.H.; Kristensen, V.N.; Børresen-Dale, A.L.; et al. Basal-like breast cancer engages tumor-supportive macrophages via secreted factors induced by extracellular S100A4. Mol. Oncol. 2018, 12, 1540–1558. [Google Scholar] [CrossRef]
- Alipoor, S.D.; Mortaz, E.; Varahram, M.; Movassaghi, M.; Kraneveld, A.D.; Garssen, J.; Adcock, I.M. The potential biomarkers and immunological effects of tumor-derived exosomes in lung cancer. Front. Immunol. 2018, 9, 819. [Google Scholar] [CrossRef]
- Mantovani, A.; Sica, A.; Locati, M. New vistas on macrophage differentiation and activation. Eur. J. Immunol. 2007, 37, 14–16. [Google Scholar] [CrossRef]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef]
- Tzeng, H.T.; Su, C.C.; Chang, C.P.; Lai, W.W.; Su, W.C.; Wang, Y.C. Rab37 in lung cancer mediates exocytosis of soluble ST2 and thus skews macrophages towards tumor-suppressing phenotype. Int. J. Cancer 2018, 143, 1753–1763. [Google Scholar] [CrossRef]
- Palaga, T.; Wongchana, W.; Kueanjinda, P. Notch Signaling in Macrophages in the Context of Cancer Immunity. Front. Immunol. 2018, 9, 652. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.S.; Sansanaphongpricha, K.; Xie, Y.; Donnelly, C.R.; Luo, X.; Heath, B.R.; Zhao, X.; Bellile, E.; Hu, H.; Chen, H.; et al. Mitigating SOX2-potentiated Immune Escape of Head and Neck Squamous Cell Carcinoma with a STING-inducing Nanosatellite Vaccine. Clin. Cancer Res. 2018, 24, 4242–4255. [Google Scholar] [CrossRef] [PubMed]
- Savage, N.D.; de Boer, T.; Walburg, K.V.; Joosten, S.A.; van Meijgaarden, K.; Geluk, A.; Ottenhoff, T.H. Human anti-inflammatory macrophages induce Foxp3+ GITR+ CD25+ regulatory T cells, which suppress via membrane-bound TGFbeta-1. J. Immunol. 2008, 181, 2220–2226. [Google Scholar] [CrossRef]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef]
- Zhang, Q.W.; Liu, L.; Gong, C.Y.; Shi, H.S.; Zeng, Y.H.; Wang, X.Z.; Zhao, Y.W.; Wei, Y.Q. Prognostic significance of tumor-associated macrophages in solid tumor: A meta-analysis of the literature. PLoS ONE 2012, 7, e50946. [Google Scholar] [CrossRef]
- Bates, R.C.; Pursell, B.M.; Mercurio, A.M. Epithelial-mesenchymal transition and colorectal cancer: Gaining insights into tumor progression using LIM 1863 cells. Cells Tissues Organs 2007, 185, 29–39. [Google Scholar] [CrossRef]
- Pollard, J.W. Tumour-educated macrophages promote tumour progression and metastasis. Nat. Rev. Cancer 2004, 4, 71–78. [Google Scholar] [CrossRef]
- Aharinejad, S.; Abraham, D.; Paulus, P.; Abri, H.; Hofmann, M.; Grossschmidt, K.; Schäfer, R.; Stanley, E.R.; Hofbauer, R. Colony-stimulating factor-1 antisense treatment suppresses growth of human tumor xenografts in mice. Cancer Res. 2002, 62, 5317–5324. [Google Scholar]
- Bataille, F.; Rohrmeier, C.; Bates, R.; Weber, A.; Rieder, F.; Brenmoehl, J.; Strauch, U.; Farkas, S.; Fürst, A.; Hofstädter, F.; et al. Evidence for a role of epithelial mesenchymal transition during pathogenesis of fistulae in Crohn’s disease. Inflamm. Bowel Dis. 2008, 14, 1514–1527. [Google Scholar] [CrossRef]
- Guerriero, J.L. Macrophages: The road less traveled, changing anticancer therapy. Trends Mol. Med. 2018, 24, 472–489. [Google Scholar] [CrossRef]
- Sinha, P.; Chornoguz, O.; Clements, V.K.; Artemenko, K.A.; Zubarev, R.A.; Ostrand-Rosenberg, S. Myeloid-derived suppressor cells express the death receptor Fas and apoptose in response to T cell-expressed FasL. Blood 2011, 117, 5381–5390. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Sieminska, I.; Baran, J. Myeloid-Derived Suppressor Cells in Colorectal Cancer. Front. Immunol. 2020, 11, 1526. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Pan, P.Y.; Li, Q.; Sato, A.I.; Levy, D.E.; Bromberg, J.; Divino, C.M.; Chen, S.H. Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res. 2006, 66, 1123–1131. [Google Scholar] [CrossRef] [PubMed]
- Sinha, P.; Clements, V.K.; Ostrand-Rosenberg, S. Interleukin-13-regulated M2 macrophages in combination with myeloid suppressor cells block immune surveillance against metastasis. Cancer Res. 2005, 65, 11743–11751. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Bronte, V.; Chen, S.H.; Colombo, M.P.; Ochoa, A.; Ostrand-Rosenberg, S.; Schreiber, H. The terminology issue for myeloid-derived suppressor cells. Cancer Res. 2007, 67, 425. [Google Scholar] [CrossRef]
- Law, A.M.K.; Valdes-Mora, F.; Gallego-Ortega, D. Myeloid-Derived Suppressor Cells as a Therapeutic Target for Cancer. Cells 2020, 9, 561. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012, 12, 253–268. [Google Scholar] [CrossRef]
- Dumitru, C.A.; Moses, K.; Trellakis, S.; Lang, S.; Brandau, S. Neutrophils and granulocytic myeloid-derived suppressor cells: Immunophenotyping, cell biology and clinical relevance in human oncology. Cancer Immunol. Immunother. 2012, 61, 1155–1167. [Google Scholar] [CrossRef]
- Kusmartsev, S.; Gabrilovich, D.I. Effect of tumor-derived cytokines and growth factors on differentiation and immune suppressive features of myeloid cells in cancer. Cancer Metastasis Rev. 2006, 25, 323–331. [Google Scholar] [CrossRef]
- Meirow, Y.; Kanterman, J.; Baniyash, M. Paving the Road to Tumor Development and Spreading: Myeloid-Derived Suppressor Cells are Ruling the Fate. Front. Immunol. 2015, 6, 523. [Google Scholar] [CrossRef]
- Martin, R.K.; Saleem, S.J.; Folgosa, L.; Zellner, H.B.; Damle, S.R.; Nguyen, G.K.; Ryan, J.J.; Bear, H.D.; Irani, A.M.; Conrad, D.H. Mast cell histamine promotes the immunoregulatory activity of myeloid-derived suppressor cells. J. Leukoc. Biol. 2014, 96, 151–159. [Google Scholar] [CrossRef]
- Saleem, S.J.; Martin, R.K.; Morales, J.K.; Sturgill, J.L.; Gibb, D.R.; Graham, L.; Bear, H.D.; Manjili, M.H.; Ryan, J.J.; Conrad, D.H. Cutting edge: Mast cells critically augment myeloid-derived suppressor cell activity. J. Immunol. 2012, 189, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Burdan, F.; Chałas, A.; Szumiło, J. Cyklooksygenaza i prostanoidy–znaczenie biologiczne [Cyclooxygenase and prostanoids–biological implications]. Postepy Hig. Med. Dosw. Online 2006, 60, 129–141. [Google Scholar]
- Chun, E.; Lavoie, S.; Michaud, M.; Gallini, C.A.; Kim, J.; Soucy, G.; Odze, R.; Glickman, J.N.; Garrett, W.S. CCL2 Promotes Colorectal Carcinogenesis by Enhancing Polymorphonuclear Myeloid-Derived Suppressor Cell Population and Function. Cell Rep. 2015, 12, 244–257. [Google Scholar] [CrossRef]
- Geiger, R.; Rieckmann, J.C.; Wolf, T.; Basso, C.; Feng, Y.; Fuhrer, T.; Kogadeeva, M.; Picotti, P.; Meissner, F.; Mann, M.; et al. L-Arginine Modulates T Cell Metabolism and Enhances Survival and Anti-tumor Activity. Cell 2016, 167, 829–842. [Google Scholar] [CrossRef]
- Rodriguez, P.C.; Zea, A.H.; Culotta, K.S.; Zabaleta, J.; Ochoa, J.B.; Ochoa, A.C. Regulation of T cell receptor CD3zeta chain expression by L-arginine. J. Biol. Chem. 2002, 277, 21123–21129. [Google Scholar] [CrossRef]
- Rodriguez, P.C.; Quiceno, D.G.; Zabaleta, J.; Ortiz, B.; Zea, A.H.; Piazuelo, M.B.; Delgado, A.; Correa, P.; Brayer, J.; Sotomayor, E.M.; et al. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res. 2004, 64, 5839–5849. [Google Scholar] [CrossRef]
- Rodriguez, P.C.; Quiceno, D.G.; Ochoa, A.C. L-arginine availability regulates T-lymphocyte cell-cycle progression. Blood 2007, 109, 1568–1573. [Google Scholar] [CrossRef]
- Obermajer, N.; Muthuswamy, R.; Lesnock, J.; Edwards, R.P.; Kalinski, P. Positive feedback between PGE2 and COX2 redirects the differentiation of human dendritic cells toward stable myeloid-derived suppressor cells. Blood 2011, 118, 5498–5505. [Google Scholar] [CrossRef]
- Han, C.; Demetris, A.J.; Stolz, D.B.; Xu, L.; Lim, K.; Wu, T. Modulation of Stat3 Activation by the cytosolic phospholipase a 2 α and cyclooxygenase2-controlled prostaglandin E2 signaling pathway. J. Biol. Chem. 2006, 281, 24831–24846. [Google Scholar] [CrossRef]
- Corvinu, F.M.; Orth, C.; Moriggl, R.; Tsareva, S.A.; Wagner, S.; Pfitzner, E.B.; Baus, D.; Kaufmann, R.; Huber, L.A.; Zatloukal, K.; et al. Persistent STAT3 activation in colon cancer is associated with enhanced cell proliferation and tumor growth. Neoplasia 2005, 7, 545–555. [Google Scholar] [CrossRef]
- Lee, H.; Herrmann, A.; Deng, J.H.; Kujawski, M.; Niu, G.; Li, Z.; Forman, S.; Jove, R.; Pardoll, D.M.; Yu, H. Persistently activated Stat3 maintains constitutive NF-kappaB activity in tumors. Cancer Cell 2009, 15, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Ma, P.; Beatty, P.L.; McKolanis, J.; Brand, R.; Schoen, R.E.; Finn, O.J. Circulating myeloid derived suppressor cells (MDSC) that accumulate in premalignancy share phenotypic and functional characteristics with MDSC in cancer. Front. Immunol. 2019, 10, 1401. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; DeBusk, L.M.; Fukuda, K.; Fingleton, B.; Green-Jarvis, B.; Shyr, Y.; Matrisian, L.M.; Carbone, D.P.; Lin, P.C. Expansion of myeloid immune suppressor Gr+CD11b+ cells in tumor-bearing host directly promotes tumor angiogenesis. Cancer Cell 2004, 6, 409–421. [Google Scholar] [CrossRef] [PubMed]
- Mira, E.; Lacalle, R.A.; Buesa, J.M.; de Buitrago, G.G.; Jiménez-Baranda, S.; Gómez-Moutón, C.; Martínez-A, C.; Mañes, S. Secreted MMP9 promotes angiogenesis more efficiently than constitutive active MMP9 bound to the tumor cell surface. J. Cell Sci. 2004, 117, 1847–1857. [Google Scholar] [CrossRef]
- Yang, L.; Huang, J.; Ren, X.; Gorska, A.E.; Chytil, A.; Aakre, M.; Carbone, D.P.; Matrisian, L.M.; Richmond, A.; Lin, P.C.; et al. Abrogation of TGF beta signaling in mammary carcinomas recruits Gr-1+CD11b+ myeloid cells that promote metastasis. Cancer Cell 2008, 13, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Toh, B.; Wang, X.; Keeble, J.; Sim, W.J.; Khoo, K.; Wong, W.C.; Kato, M.; Prevost-Blondel, A.; Thiery, J.P.; Abastado, J.P. Mesenchymal transition and dissemination of cancer cells is driven by myeloid-derived suppressor cells infiltrating the primary tumor. PLoS Biol. 2011, 9, e1001162. [Google Scholar] [CrossRef]
- Suzuki, E.; Kapoor, V.; Jassar, A.S.; Kaiser, L.R.; Albelda, S.M. Gemcitabine selectively eliminates splenic Gr-1+/CD11b+ myeloid suppressor cells in tumor-bearing animals and enhances antitumor immune activity. Clin. Cancer Res. 2005, 11, 6713–6721. [Google Scholar] [CrossRef]
- Sevko, A.; Michels, T.; Vrohlings, M.; Umansky, L.; Beckhove, P.; Kato, M.; Shurin, G.V.; Shurin, M.R.; Umansky, V. Antitumor effect of paclitaxel is mediated by inhibition of myeloid-derived suppressor cells and chronic inflammation in the spontaneous melanoma model. J. Immunol. 2013, 190, 2464–2471. [Google Scholar] [CrossRef]
- Eriksson, E.; Wenthe, J.; Irenaeus, S.; Loskog, A.; Ullenhag, G. Gemcitabine reduces MDSCs, tregs and TGFbeta-1 while restoring the teff/treg ratio in patients with pancreatic cancer. J. Transl. Med. 2016, 14, 282. [Google Scholar] [CrossRef]
- Vincent, J.; Mignot, G.; Chalmin, F.; Ladoire, S.; Bruchard, M.; Chevriaux, A.; Martin, F.; Apetoh, L.; Rébé, C.; Ghiringhelli, F. 5-Fluorouracil selectively kills tumor-associated myeloid-derived suppressor cells resulting in enhanced T cell-dependent antitumor immunity. Cancer Res. 2010, 70, 3052–3061. [Google Scholar] [CrossRef]
- Ko, J.S.; Zea, A.H.; Rini, B.I.; Ireland, J.L.; Elson, P.; Cohen, P.; Golshayan, A.; Rayman, P.A.; Wood, L.; Garcia, J.; et al. Sunitinib mediates reversal of myeloid-derived suppressor cell accumulation in renal cell carcinoma patients. Clin. Cancer Res. 2009, 15, 2148–2157. [Google Scholar] [CrossRef]
- Kodera, Y.; Katanasaka, Y.; Kitamura, Y.; Tsuda, H.; Nishio, K.; Tamura, T.; Koizumi, F. Sunitinib inhibits lymphatic endothelial cell functions and lymph node metastasis in a breast cancer model through inhibition of vascular endothelial growth factor receptor 3. Breast Cancer Res. 2011, 13, R66. [Google Scholar] [CrossRef]
- Weber, R.; Fleming, V.; Hu, X.; Nagibin, V.; Groth, C.; Altevogt, P.; Utikal, J.; Umansky, V. Myeloid-Derived Suppressor Cells Hinder the Anti-Cancer Activity of Immune Checkpoint Inhibitors. Front. Immunol. 2018, 9, 1310. [Google Scholar] [CrossRef]
- Katoh, H.; Wang, D.; Daikoku, T.; Sun, H.; Dey, S.K.; Dubois, R.N. CXCR2-expressing myeloid-derived suppressor cells are essential to promote colitis-associated tumorigenesis. Cancer Cell 2013, 24, 631–644. [Google Scholar] [CrossRef]
- Steele, C.W.; Karim, S.A.; Leach, J.D.G.; Bailey, P.; Upstill-Goddard, R.; Rishi, L.; Foth, M.; Bryson, S.; McDaid, K.; Wilson, Z.; et al. CXCR2 Inhibition Profoundly Suppresses Metastases and Augments Immunotherapy in Pancreatic Ductal Adenocarcinoma. Cancer Cell 2016, 29, 832–845. [Google Scholar] [CrossRef]
- Veltman, J.D.; Lambers, M.E.; van Nimwegen, M.; Hendriks, R.W.; Hoogsteden, H.C.; Aerts, J.G.; Hegmans, J.P. COX-2 inhibition improves immunotherapy and is associated with decreased numbers of myeloid-derived suppressor cells in mesothelioma. Celecoxib influences MDSC function. BMC Cancer 2010, 10, 464. [Google Scholar] [CrossRef]
- Serafini, P.; Meckel, K.; Kelso, M.; Noonan, K.; Califano, J.; Koch, W.; Dolcetti, L.; Bronte, V.; Borrello, I. Phosphodiesterase-5 inhibition augments endogenous antitumor immunity by reducing myeloid-derived suppressor cell function. J. Exp. Med. 2006, 203, 2691–2702. [Google Scholar] [CrossRef] [PubMed]
- Mirza, N.; Fishman, M.; Fricke, I.; Dunn, M.; Neuger, A.M.; Frost, T.J.; Lush, R.M.; Antonia, S.; Gabrilovich, D.I. All-trans-retinoic acid improves differentiation of myeloid cells and immune response in cancer patients. Cancer Res. 2006, 66, 9299–9307. [Google Scholar] [CrossRef]
- Orillion, A.; Hashimoto, A.; Damayanti, N.; Shen, L.; Adelaiye-Ogala, R.; Arisa, S.; Chintala, S.; Ordentlich, P.; Kao, C.; Elzey, B.; et al. Entinostat Neutralizes Myeloid-Derived Suppressor Cells and Enhances the Antitumor Effect of PD-1 Inhibition in Murine Models of Lung and Renal Cell Carcinoma. Clin. Cancer Res. 2017, 23, 5187–5201. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Skora, A.D.; Li, Z.; Liu, Q.; Tam, A.J.; Blosser, R.L.; Diaz, L.A., Jr.; Papadopoulos, N.; Kinzler, K.W.; Vogelstein, B.; et al. Eradication of metastatic mouse cancers resistant to immune checkpoint blockade by suppression of myeloid-derived cells. Proc. Natl. Acad. Sci. USA 2014, 111, 11774–11779. [Google Scholar] [CrossRef] [PubMed]
- De la Torre, P.; Pérez-Lorenzo, M.J.; Alcázar-Garrido, Á.; Flores, A.I. Cell-Based Nanoparticles Delivery Systems for Targeted Cancer Therapy: Lessons from Anti-Angiogenesis Treatments. Molecules 2020, 25, 715. [Google Scholar] [CrossRef] [PubMed]
- Shao, K.; Singha, S.; Clemente-Casares, X.; Tsai, S.; Yang, Y.; Santamaria, P. Nanoparticle-based immunotherapy for cancer. ACS Nano 2015, 9, 16–30. [Google Scholar] [CrossRef]
- Fan, Y.; Moon, J.J. Nanoparticle drug delivery systems designed to improve cancer vaccines and immunotherapy. Vaccines 2015, 3, 662–685. [Google Scholar] [CrossRef]
- He, C.; Duan, X.; Guo, N.; Chan, C.; Poon, C.; Weichselbaum, R.R.; Lin, W. Core-shell nanoscale coordination polymers combine chemotherapy and photodynamic therapy to potentiate checkpoint blockade cancer immunotherapy. Nat. Commun. 2016, 7, 12499–12511. [Google Scholar] [CrossRef]
- Mortezaee, K.; Khanlarkhani, N. Melatonin application in targeting oxidative-induced liver injuries: A review. J. Cell. Physiol. 2018, 233, 4015–4032. [Google Scholar] [CrossRef]
- Song, W.; Shen, L.; Wang, Y.; Liu, Q.; Goodwin, T.J.; Li, J.; Dorosheva, O.; Liu, T.; Liu, R.; Huang, L. Synergistic and low adverse effect cancer immunotherapy by immunogenic chemotherapy and locally expressed PD-L1 trap. Nat. Commun. 2018, 9, 2237–2248. [Google Scholar] [CrossRef]
- Verma, R.P.; Hansch, C. Matrix metalloproteinases (MMPs): Chemical-biological functions and (Q)SARs. Bioorg. Med. Chem. 2007, 15, 2223–2268. [Google Scholar] [CrossRef]
- Serra, R.; Gallelli, L.; Grande, R.; Amato, B.; De Caridi, G.; Sammarco, G.; Ferrari, F.; Butrico, L.; Gallo, G.; Rizzuto, A.; et al. Hemorrhoids and matrix metalloproteinases: A multicenter study on the predictive role of biomarkers. Surgery 2016, 159, 487–494. [Google Scholar] [CrossRef]
- Lucafò, M.; Pugnetti, L.; Bramuzzo, M.; Curci, D.; Di Silvestre, A.; Marcuzzi, A.; Bergamo, A.; Martelossi, S.; Villanacci, V.; Bozzola, A.; et al. Long Non-Coding RNA GAS5 and Intestinal MMP2 and MMP9 Expression: A Translational Study in Pediatric Patients with IBD. Int. J. Mol. Sci. 2019, 20, 5280. [Google Scholar] [CrossRef]
- Herszényi, L.; Hritz, I.; Lakatos, G.; Varga, M.Z.; Tulassay, Z. The behavior of matrix metalloproteinases and their inhibitors in colorectal cancer. Int. J. Mol. Sci. 2012, 13, 13240–13263. [Google Scholar] [CrossRef]
- Hilska, M.; Roberts, P.J.; Collan, Y.U.; Laine, V.J.O.; Kössi, J.; Hirsimäki, P.; Rahkonen, O.; Laato, M. Prognostic significance of matrix metalloproteinases-1, -2, -7 and -13 and tissue inhibitors of metalloproteinases-1, -2, -3 and -4 in colorectal cancer. Int. J. Cancer 2007, 121, 714–723. [Google Scholar] [CrossRef] [PubMed]
- Ribatti, D.; Vacca, A.; Nico, B.; Crivellato, E.; Roncali, L.; Dammacco, F. The role of mast cells in tumour angiogenesis. Br. J. Haematol. 2001, 115, 514–521. [Google Scholar] [CrossRef] [PubMed]
- Gulubova, M.; Vlaykova, T. Prognostic significance of mast cell number and microvascular density for the survival of patients with primary colorectal cancer. J. Gastroenterol. Hepatol. 2009, 24, 1265–1275. [Google Scholar] [CrossRef]
- Raeeszadeh-Sarmazdeh, M.; Do, L.D.; Hritz, B.G. Metalloproteinases and Their Inhibitors: Potential for the Development of New Therapeutics. Cells 2020, 9, 1313. [Google Scholar] [CrossRef]
- Ribatti, D. Mast cells as therapeutic target in cancer. Eur. J. Pharmacol. 2016, 778, 152–157. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gallo, G.; Vescio, G.; De Paola, G.; Sammarco, G. Therapeutic Targets and Tumor Microenvironment in Colorectal Cancer. J. Clin. Med. 2021, 10, 2295. https://doi.org/10.3390/jcm10112295
Gallo G, Vescio G, De Paola G, Sammarco G. Therapeutic Targets and Tumor Microenvironment in Colorectal Cancer. Journal of Clinical Medicine. 2021; 10(11):2295. https://doi.org/10.3390/jcm10112295
Chicago/Turabian StyleGallo, Gaetano, Giuseppina Vescio, Gilda De Paola, and Giuseppe Sammarco. 2021. "Therapeutic Targets and Tumor Microenvironment in Colorectal Cancer" Journal of Clinical Medicine 10, no. 11: 2295. https://doi.org/10.3390/jcm10112295
APA StyleGallo, G., Vescio, G., De Paola, G., & Sammarco, G. (2021). Therapeutic Targets and Tumor Microenvironment in Colorectal Cancer. Journal of Clinical Medicine, 10(11), 2295. https://doi.org/10.3390/jcm10112295