Theoretical Evaluation of Graphene Membrane Performance for Hydrogen Separation Using Molecular Dynamic Simulation




Abstract
:
1. Introduction
2. MD Simulation Methodology
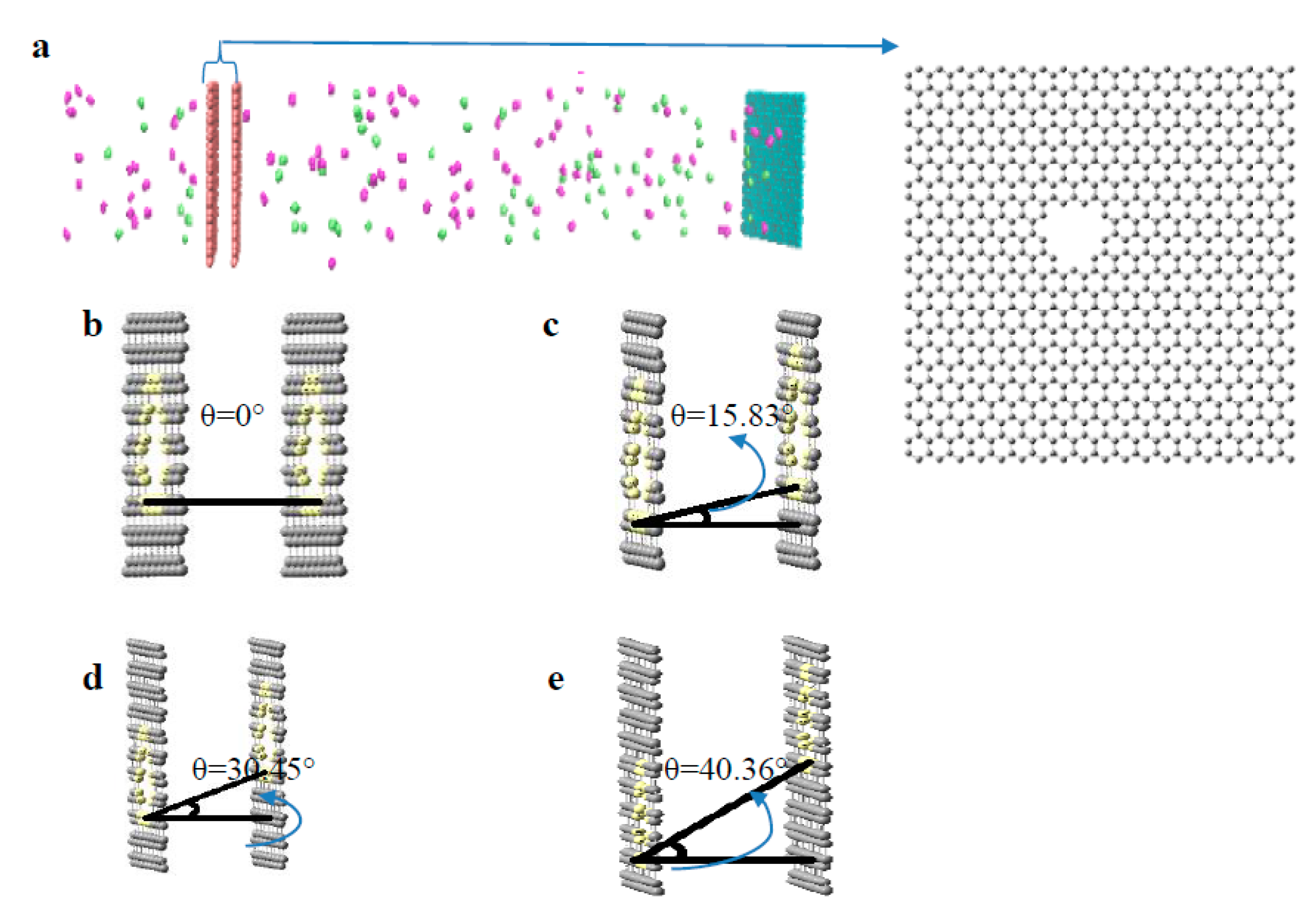
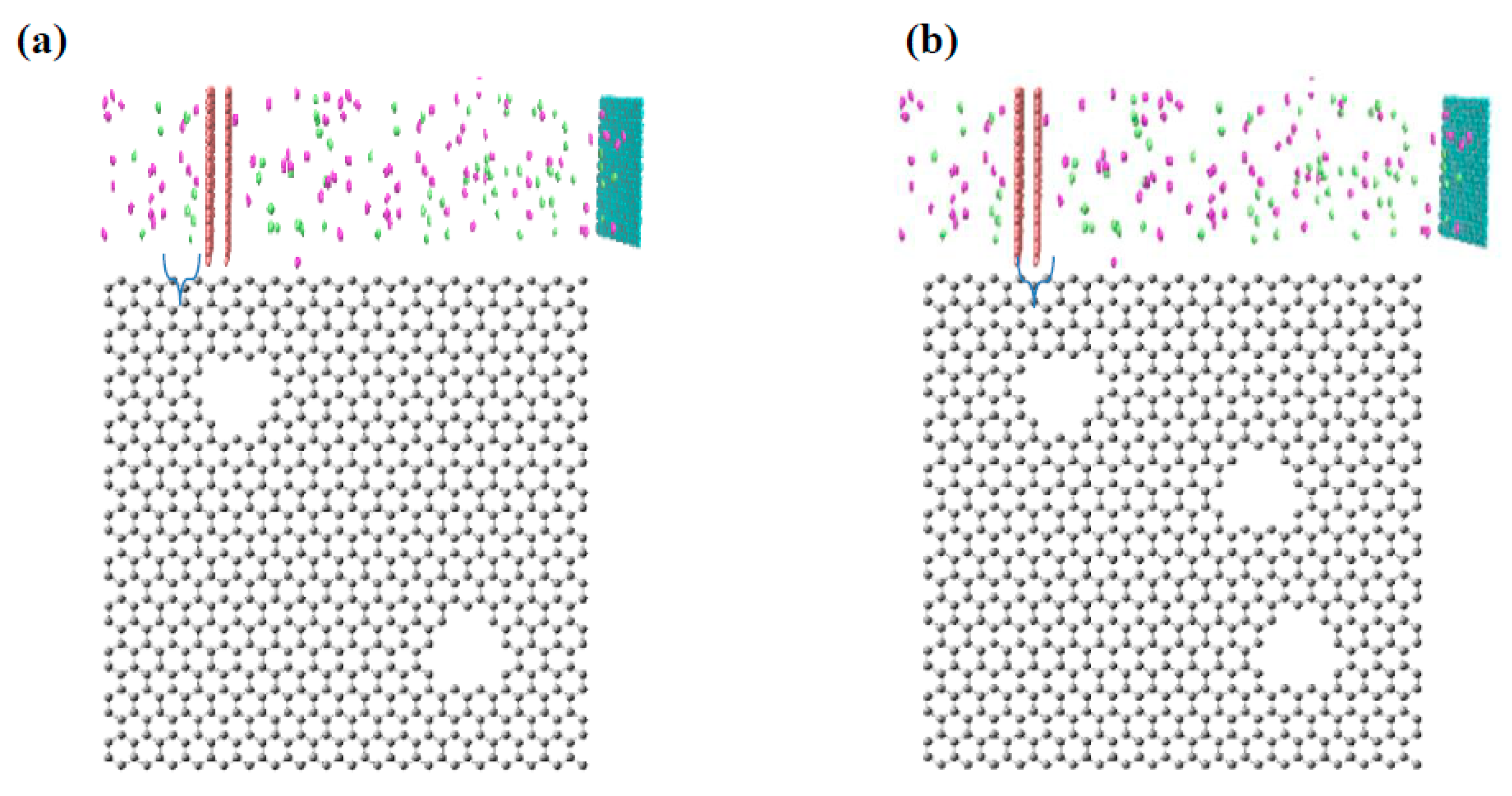
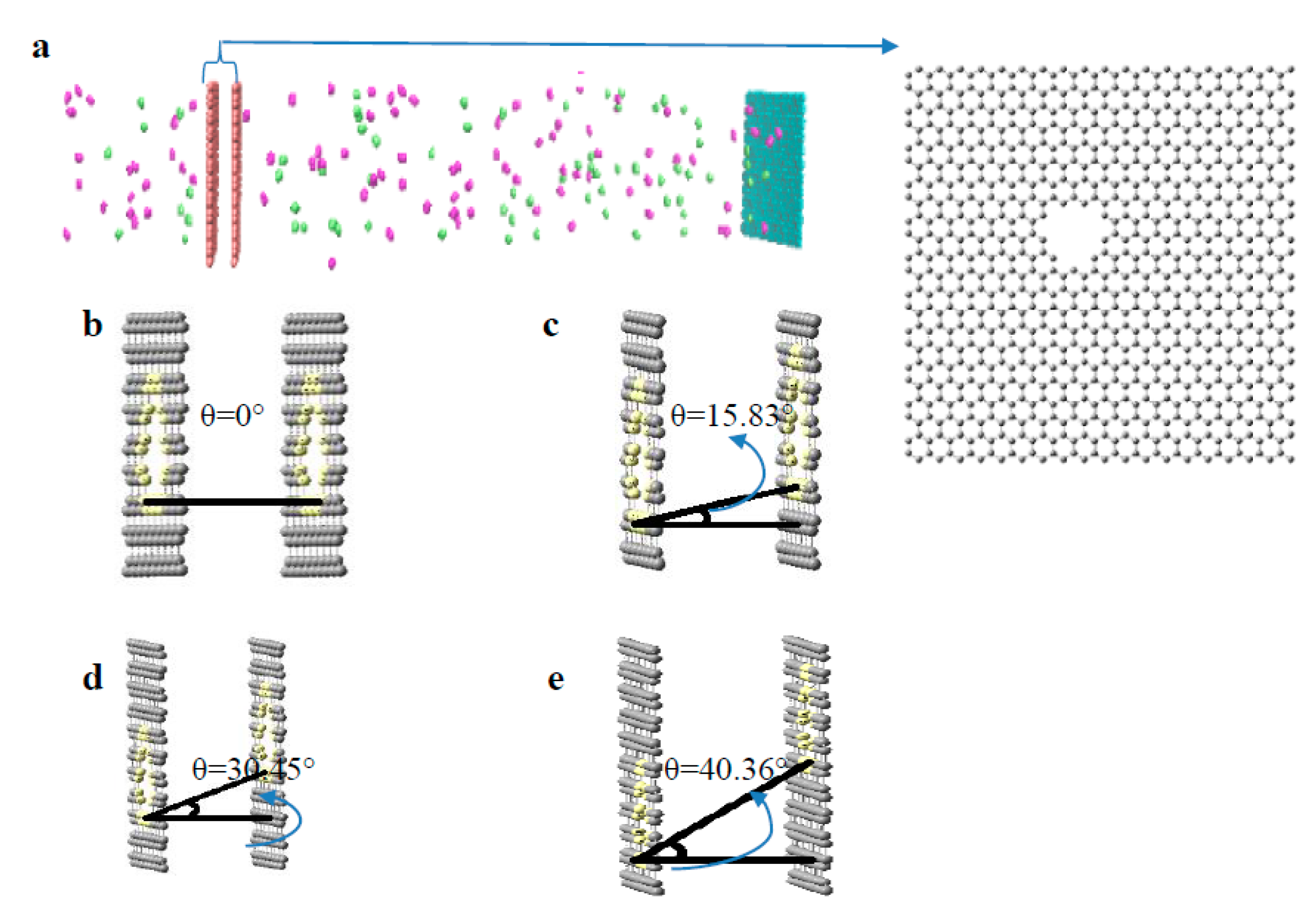
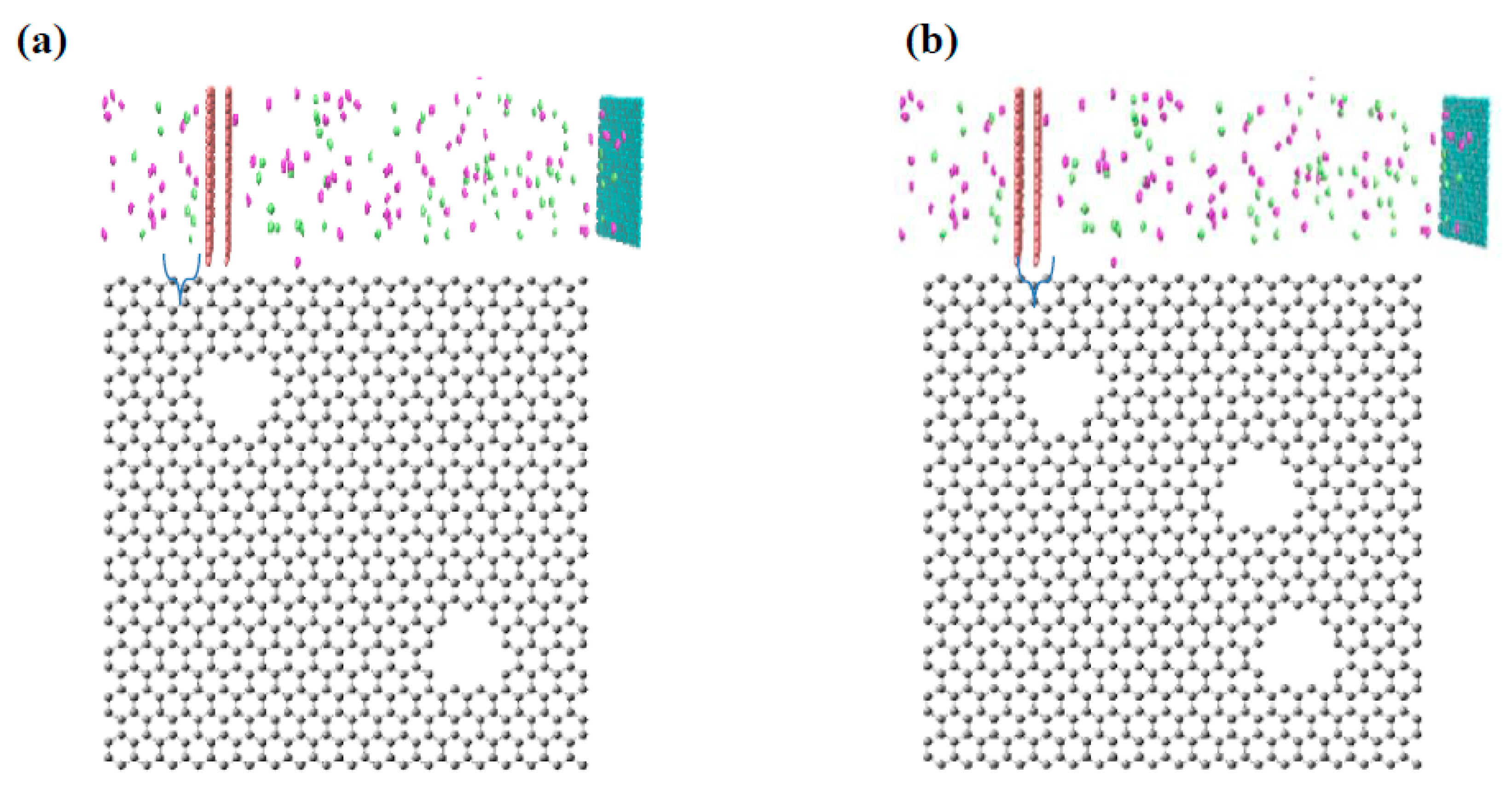
2.1. Preparation of Virtual Membrane Cell
2.2. Solution Procedure
3. Results and Discussions
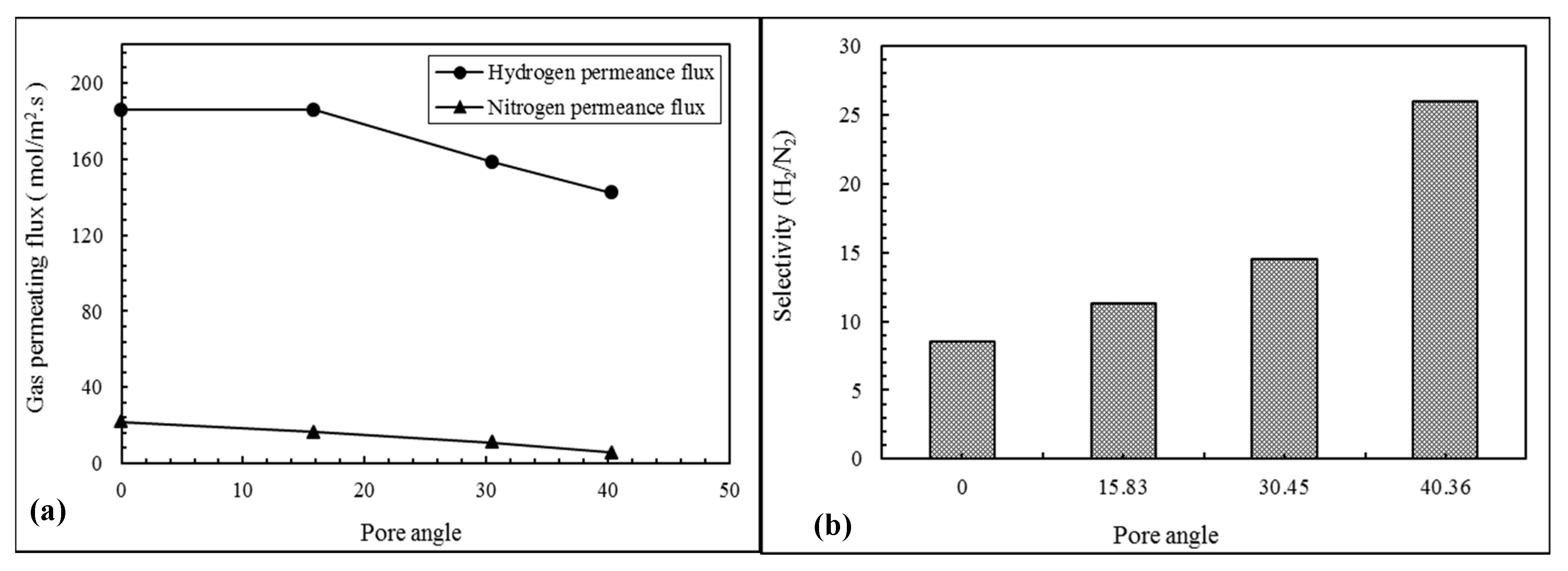
3.1. Evaluation of Pore Angle Effects on NPG Membrane Performance
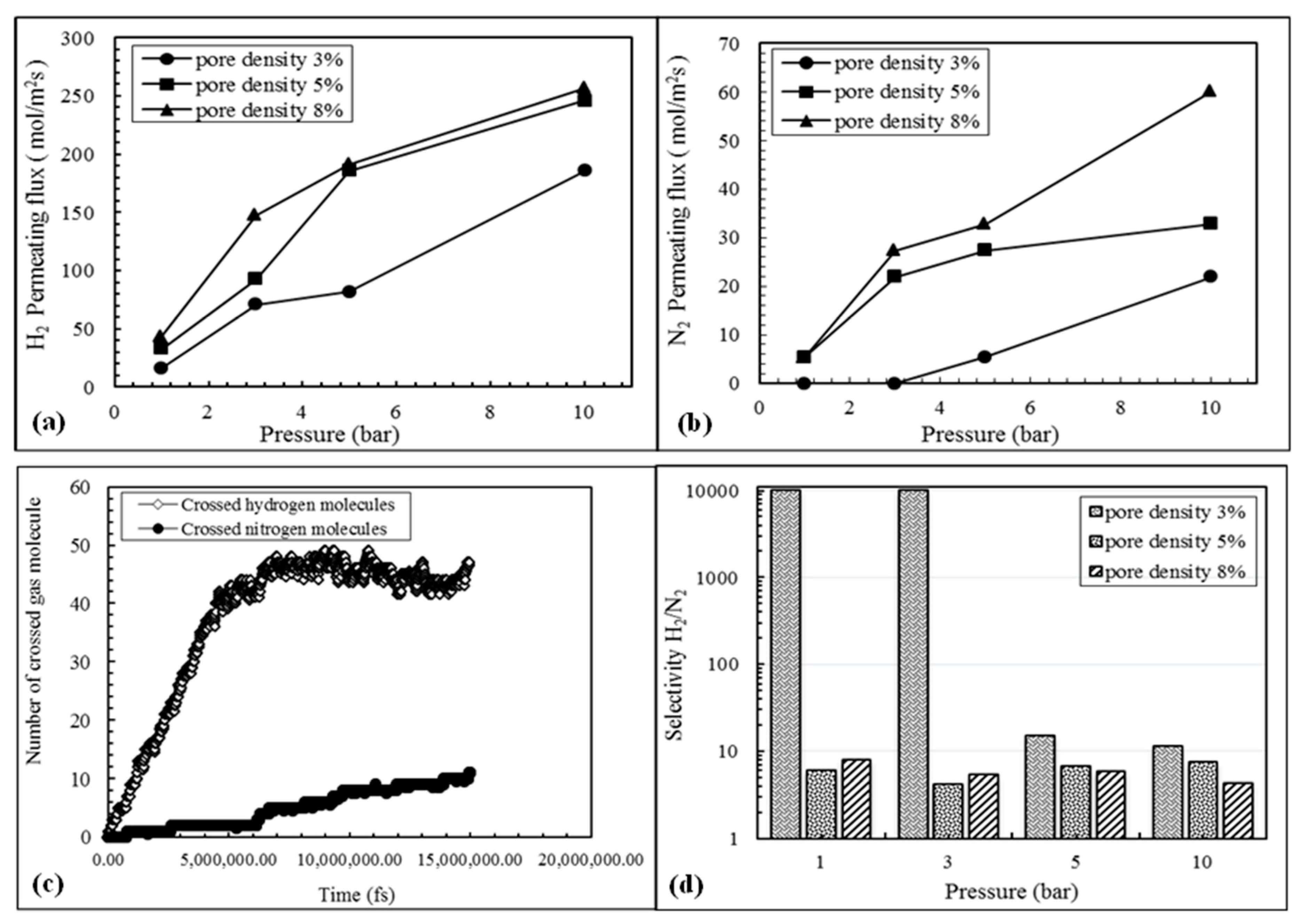
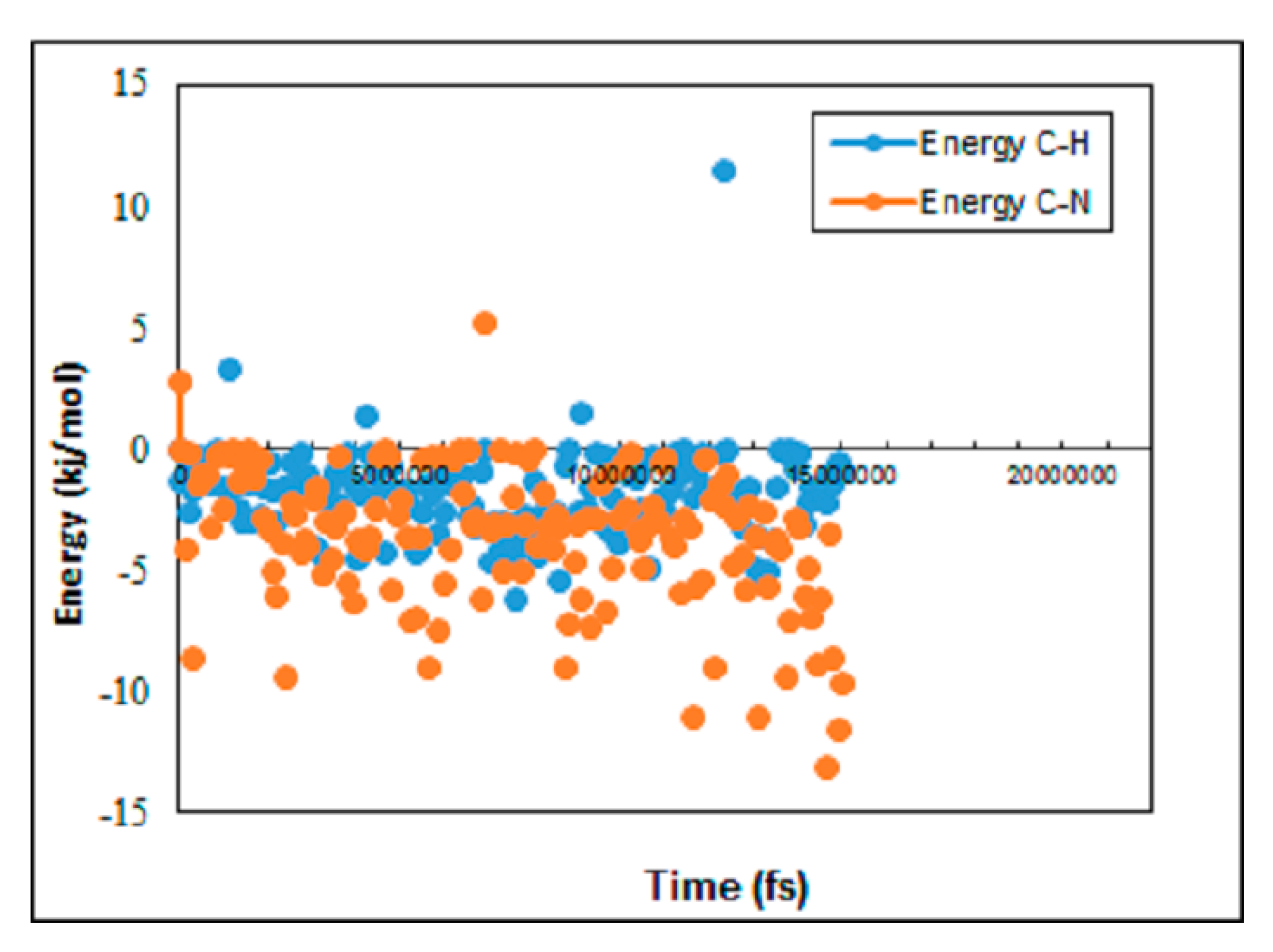
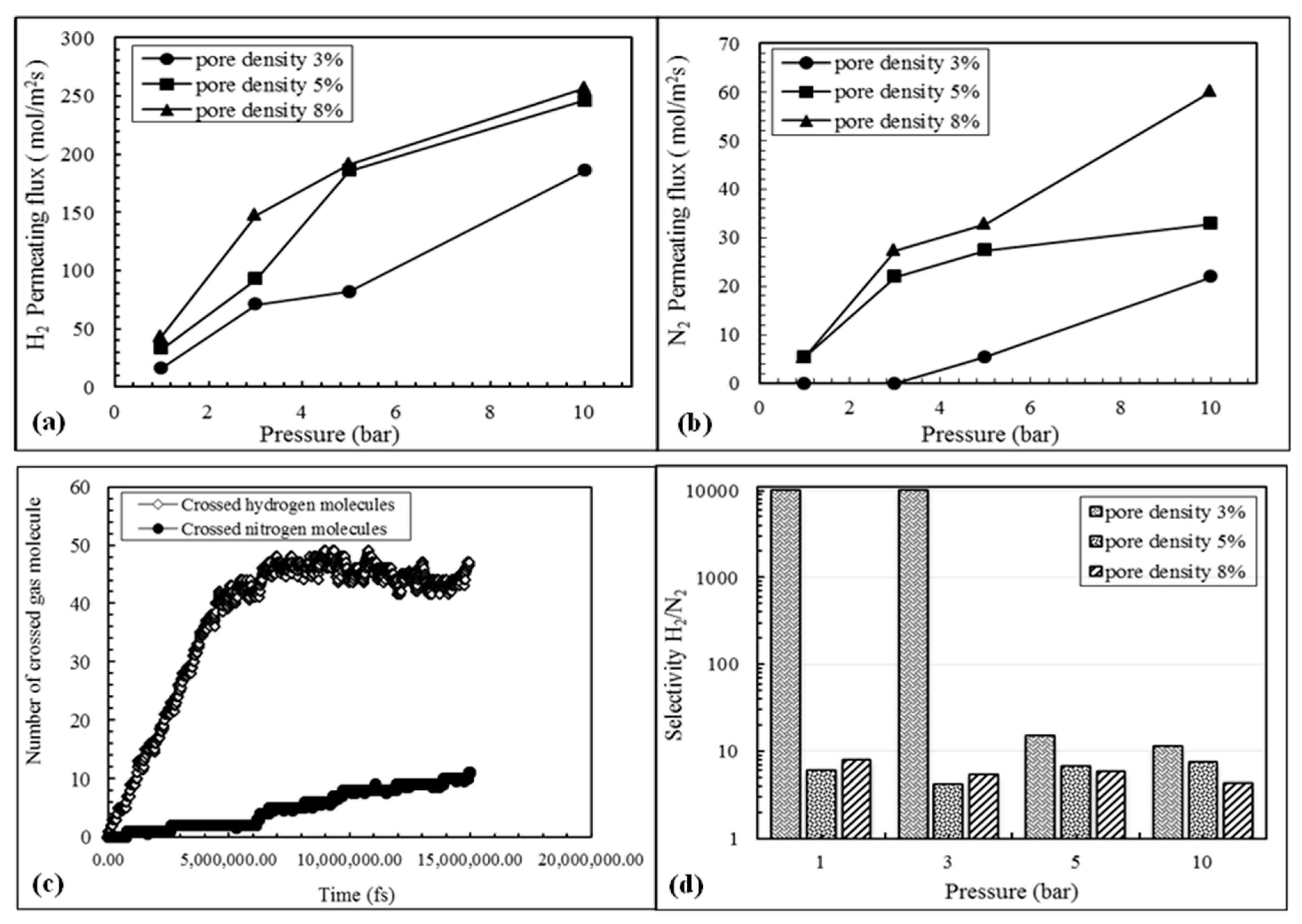
3.2. Evaluation of Pore Density Effects on NPG Membrane Performance
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Gin, D.L.; Noble, R.D. Designing the next generation of chemical separation membranes. Science 2011, 332, 674–676. [Google Scholar] [CrossRef] [PubMed]
- Brandon, N.P.; Kurban, Z. Clean energy and the hydrogen economy. Philos. Trans. R. Soc. A 2017, 375, 20160400. [Google Scholar] [CrossRef] [PubMed]
- Iulianelli, A.; Liguori, S.; Huang, Y.; Basile, A. Model biogas steam reforming in a thin Pd-supported membrane reactor to generate clean hydrogen for fuel cells. J. Power Sources 2015, 273, 25–32. [Google Scholar] [CrossRef]
- Rahimpour, M.R.; Samimi, F.; Babapoor, A.; Tohidian, T.; Mohebi, S. Palladium membranes applications in reaction systems for hydrogen separation and purification: A review. Chem. Eng. Process. 2017, 121, 24–49. [Google Scholar] [CrossRef]
- De Vos, R.M.; Verweij, H. High-Selectivity, High-Flux silica membranes for gas separation. Science 1998, 279, 1710–1711. [Google Scholar] [CrossRef]
- Jabbari, A.; Ghasemzadeh, K.; Khajavi, P.; Assa, F.; Abdi, M.-A.; Babaluo, A.-A.; Basile, A. Surface modification of α-alumina support in synthesis of silica membrane for hydrogen purification. Int. J. Hydrogen Energy 2014, 39, 18585–18591. [Google Scholar] [CrossRef]
- Ghasemzadeh, K.; Aghaeinejad-Meybodi, A.; Vaezi, M.J.; Gholizadeh, A.; Abdi, M.A.; Babaluo, A.A.; Haghighi, M.; Basile, A. Hydrogen Production via Silica Membrane Reactor during Methanol, Steam Reforming Process: Experimental Study. R. Soc. Chem. Adv. 2015, 116, 95823–95832. [Google Scholar] [CrossRef]
- Lai, Z.; Bonilla, G.; Diaz, I.; Nery, J.G.; Sujaoti, K.; Amat, M.A.; Kokkoli, E.; Terasaki, O.; Thompson, R.W.; Tsapatsis, M.; et al. Microstructural optimization of a zeolite membrane for organic vapor separation. Science 2003, 300, 456–460. [Google Scholar]
- Yu, M.; Noble, R.D.; Falconer, J.L. Zeolite membranes: Microstructure Characterization and Permeation Mechanisms. Acc. Chem. Res. 2011, 44, 1196–1206. [Google Scholar] [CrossRef]
- Geim, A.; Novoselov, K. The rise of graphene. Nat. Mater. 2007, 6, 183–191. [Google Scholar] [CrossRef]
- Zeynali, R.; Ghasemzadeh, K.; Sarand, A.B.; Kheiri, F.; Basile, A. Performance evaluation of graphene oxide (GO) nanocomposite membrane for hydrogen separation: Effect of dip coating sol concentration. Sep. Purif. Technol. 2018, 200, 169–176. [Google Scholar] [CrossRef]
- Ghasemzadeh, K.; Aghaeinejad-Meybodi, A.; Basile, A. Hydrogen production as a green fuel in silica membrane reactor: Experimental analysis and artificial neural network modeling. Fuel 2018, 222, 114–124. [Google Scholar] [CrossRef]
- Ghasemzadeh, K.; Ahmadnejad, F.; Aghaeinejad-Meybodi, A.; Basile, A. Hydrogen production by a PdAg membrane reactor during glycerol steam reforming: ANN modeling study. Int. J. Hydrogen Energy 2018, 43, 7722–7730. [Google Scholar] [CrossRef]
- Ghasemzadeh, K.; Ghahremani, M.; Amiri, T.Y.; Basile, A. Performance evaluation of PdAg membrane reactor in glycerol steam reforming process: Development of the CFD model. Int. J. Hydrogen Energy 2019, 44, 1000–1009. [Google Scholar] [CrossRef]
- Ghasemzadeh, K.; Zeynali, R.; Basile, A.; Iulianelli, A. CFD analysis of a hybrid sorption-enhanced membrane reactor for hydrogen production during WGS reaction. Int. J. Hydrogen Energy 2017, 42, 26914–26923. [Google Scholar] [CrossRef]
- Ghasemzadeh, K.; Zeynali, R.; Bahadori, F.; Basile, A. CFD analysis of Pd-Ag membrane reactor performance during ethylbenzene dehydrogenation process. Int. J. Hydrogen Energy 2018, 43, 7675–7683. [Google Scholar] [CrossRef]
- Cohen-Tanugi, D.; Lin, L.C.; Grossman, J.C. Multilayer Nanoporous Graphene Membranes for Water Desalination. Nano Lett. 2016, 16, 1027–1033. [Google Scholar] [CrossRef] [Green Version]
- Plimpton, S. Fast Parallel Algorithms for Short-Range Molecular Dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Buch, V.J. Path integral simulations of mixed para-D2 and ortho-D2 clusters: The orientational effects. J. Chem. Phys. 1994, 100, 7610–7629. [Google Scholar] [CrossRef]
- Takaba, H.; Matsuda, E.; Nair, B.N.; Nakao, S.I. Molecular Modeling of Gas Permeation through an Amorphous Microporous Silica Membrane. J. Chem. Eng. Jpn. 2002, 35, 1312–1321. [Google Scholar] [CrossRef]
- Huailiang, D.; Jingyuan, L.; Jing, Z.; Gang, S.; Xiaoyi, L.; Yuliang, Z. Separation of Hydrogen and Nitrogen Gases with Porous Graphene Membrane. J. Phys. Chem. 2011, 47, 23261–23266. [Google Scholar]
- Steele, W.A. The Interaction of Gases with Solid Surfaces; Pergamon Press: Oxford, UK, 1974. [Google Scholar]
- Herzberg, G.; Monfils, A. The dissociation energies of the hydrogen, HD, and D2 Molecules. J. Mol. Spectrosc. 1960, 5, 482–498. [Google Scholar] [CrossRef]
- Vedeneyev, V.I.; Gurvich, L.V.; Kondrat’yev, V.N.; Medvedev, V.A.; Frankevich, Y.L. Bond Energies, Ionization Potentials and Electron Affinities; St. Martin’s Press: Manhattan, NY, USA, 1962. [Google Scholar]
- Rao, C.N.R. Understanding Chemistry; World Scientific: Singapore, 2010. [Google Scholar]
- Collins, J.P.; Schwartz, R.W.; Sehgal, R.; Ward, T.L.; Brinker, C.; Hagen, G.P.; Udovich, C.A. Catalytic Dehydrogenation of Propane in Hydrogen Permselective Membrane Reactors. Ind. Eng. Chem. Res. 1996, 35, 4398–4405. [Google Scholar] [CrossRef]
- Anderson, M.R.; Mattes, B.R.; Reiss, H.; Kaner, R.B. Conjugated Polymer Films for Gas Separations. Science 1991, 252, 1412–1415. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nouri, M.; Ghasemzadeh, K.; Iulianelli, A. Theoretical Evaluation of Graphene Membrane Performance for Hydrogen Separation Using Molecular Dynamic Simulation. Membranes 2019, 9, 110. https://doi.org/10.3390/membranes9090110
Nouri M, Ghasemzadeh K, Iulianelli A. Theoretical Evaluation of Graphene Membrane Performance for Hydrogen Separation Using Molecular Dynamic Simulation. Membranes. 2019; 9(9):110. https://doi.org/10.3390/membranes9090110
Chicago/Turabian StyleNouri, Mahdi, Kamran Ghasemzadeh, and Adolfo Iulianelli. 2019. "Theoretical Evaluation of Graphene Membrane Performance for Hydrogen Separation Using Molecular Dynamic Simulation" Membranes 9, no. 9: 110. https://doi.org/10.3390/membranes9090110
APA StyleNouri, M., Ghasemzadeh, K., & Iulianelli, A. (2019). Theoretical Evaluation of Graphene Membrane Performance for Hydrogen Separation Using Molecular Dynamic Simulation. Membranes, 9(9), 110. https://doi.org/10.3390/membranes9090110