The Biochemical Properties and Functions of CALM and AP180 in Clathrin Mediated Endocytosis

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
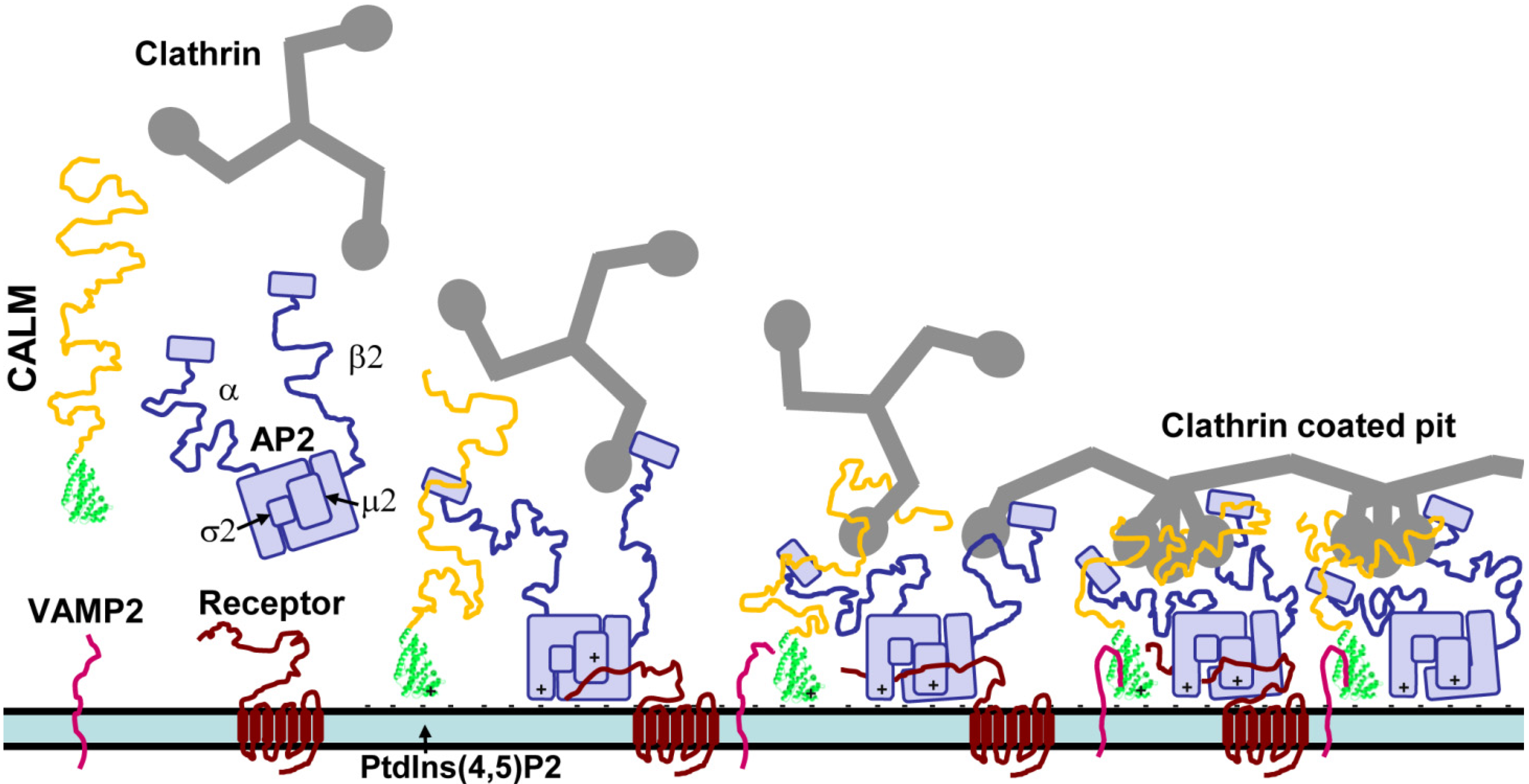
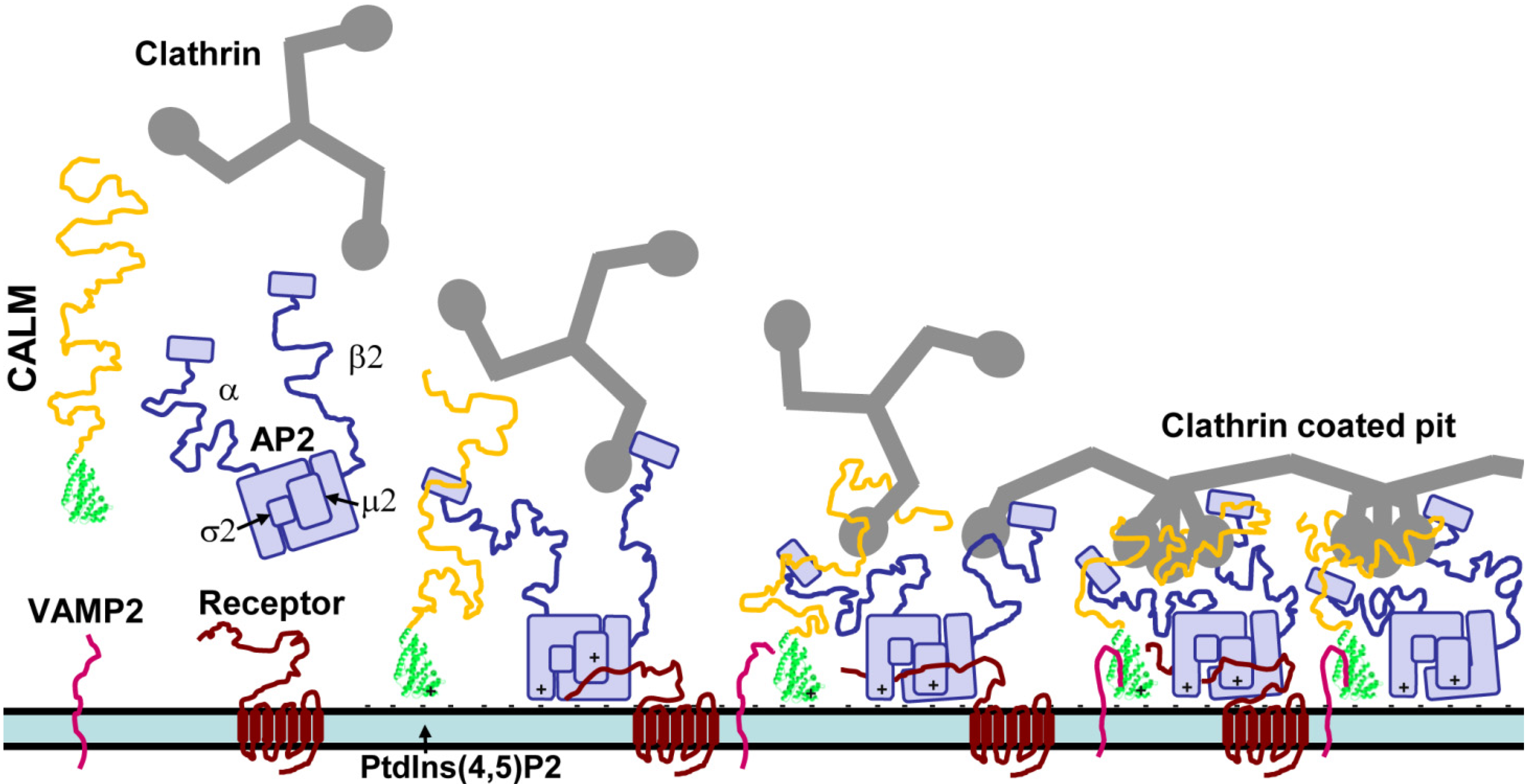
2. The Early Stages of CME
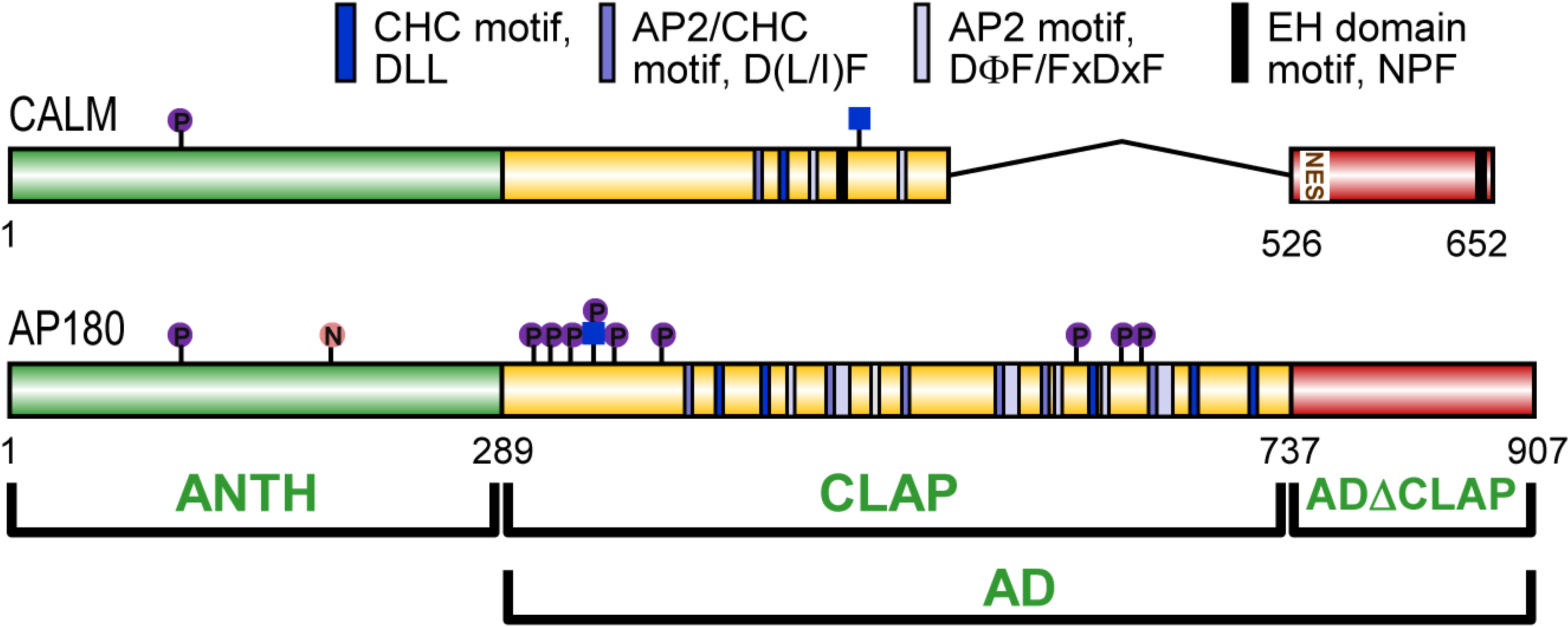
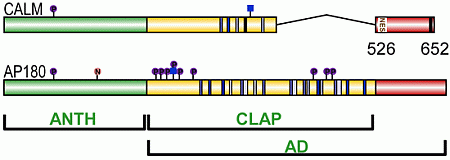
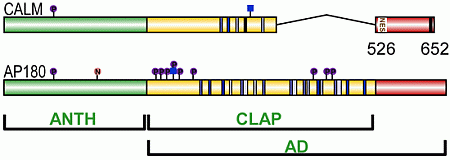
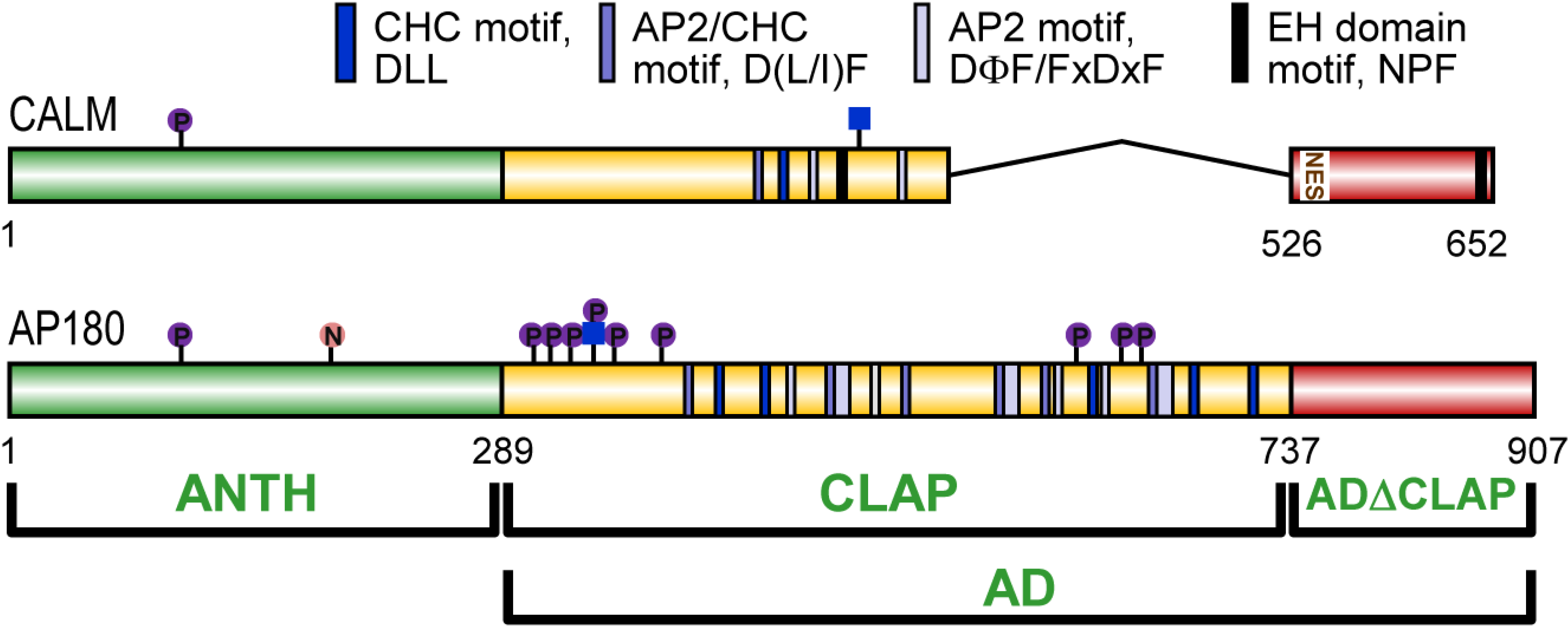
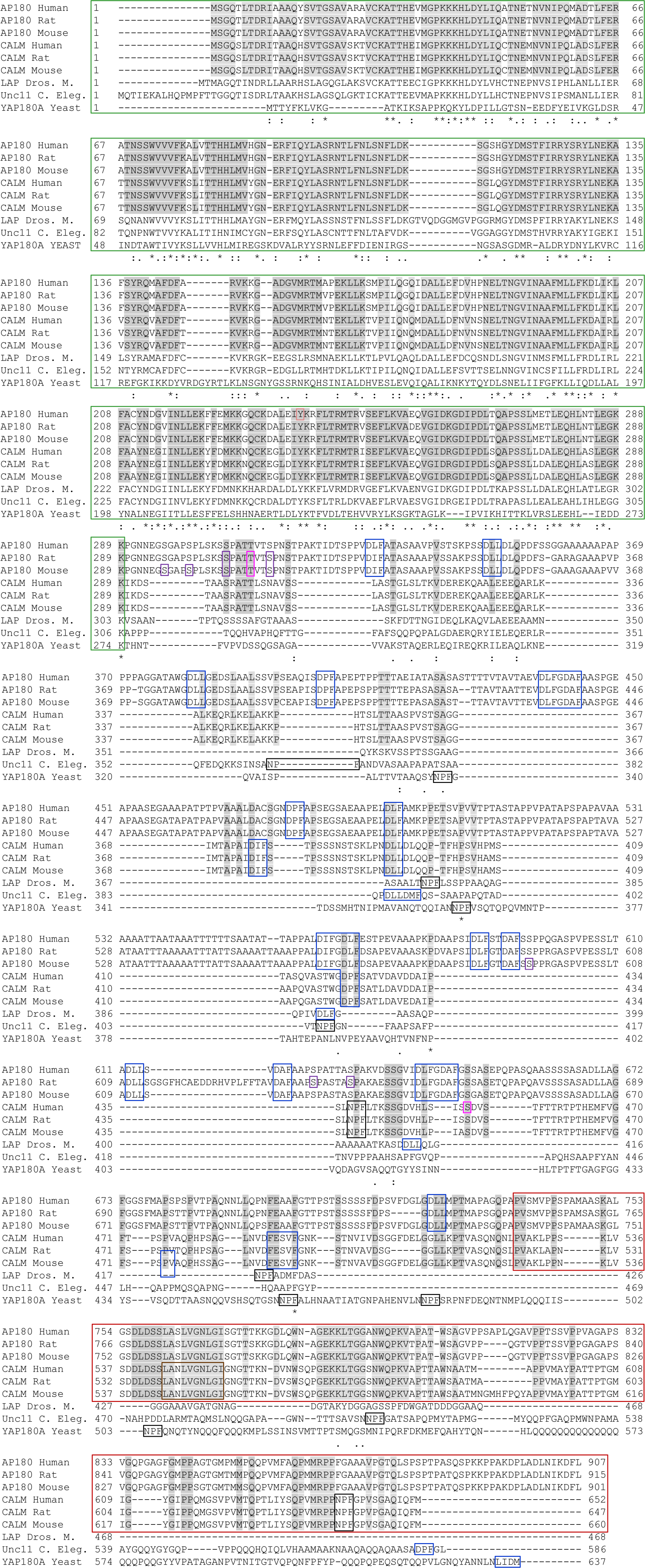
3. CALM and AP180 Domain Structure and Sequence Similarities
3.1. A Structured ANTH Domain
3.2. A Disordered Assembly Domain with Short Protein Binding Motifs
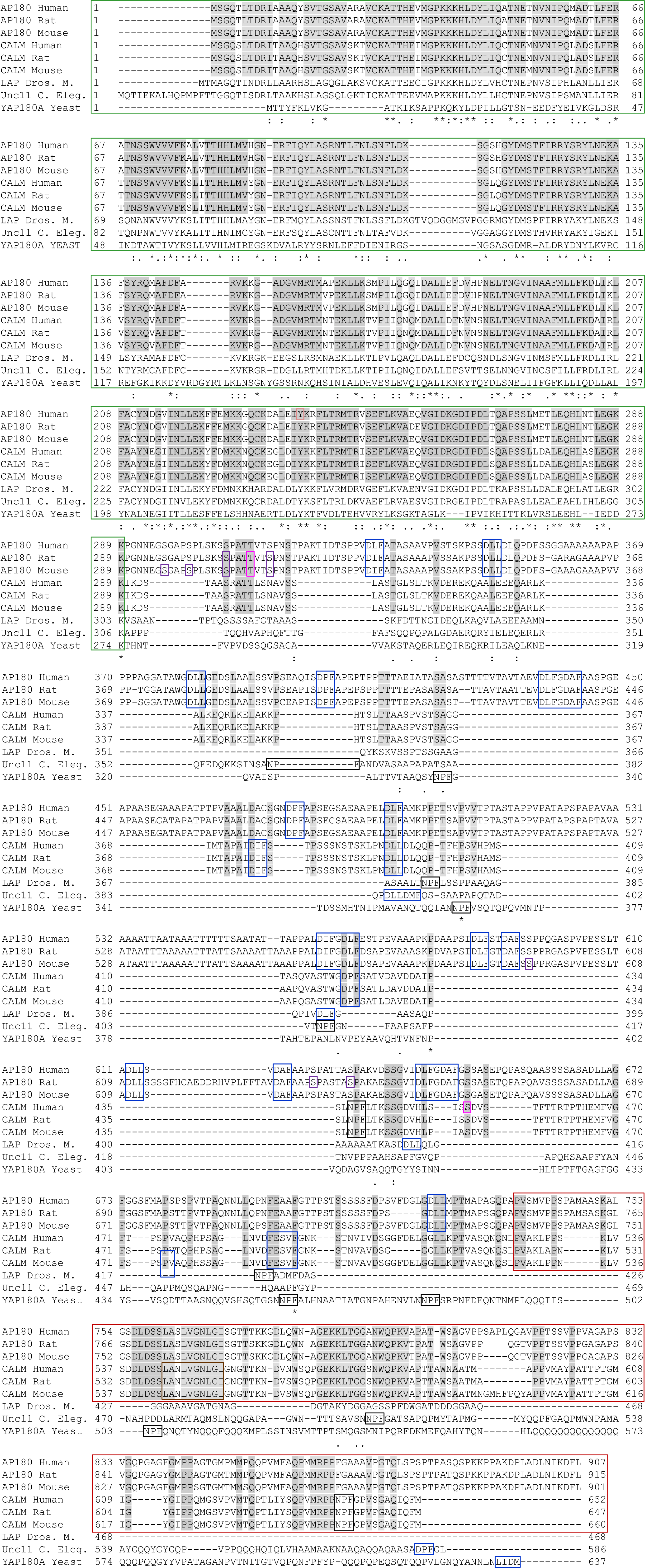
4. The Function of the CALM and AP180 Assembly Domain
4.1. Clathrin Binding and Assembly
4.2. Mechanism of Clathrin Assembly
5. The Cargo Sorting Function of the CALM and AP180 ANTH Domains
6. Role of CALM in Receptor Uptake
7. Post-Translational Modifications of CALM and AP180
8. CALM and AP180 in Disease
8.1. Leukemia
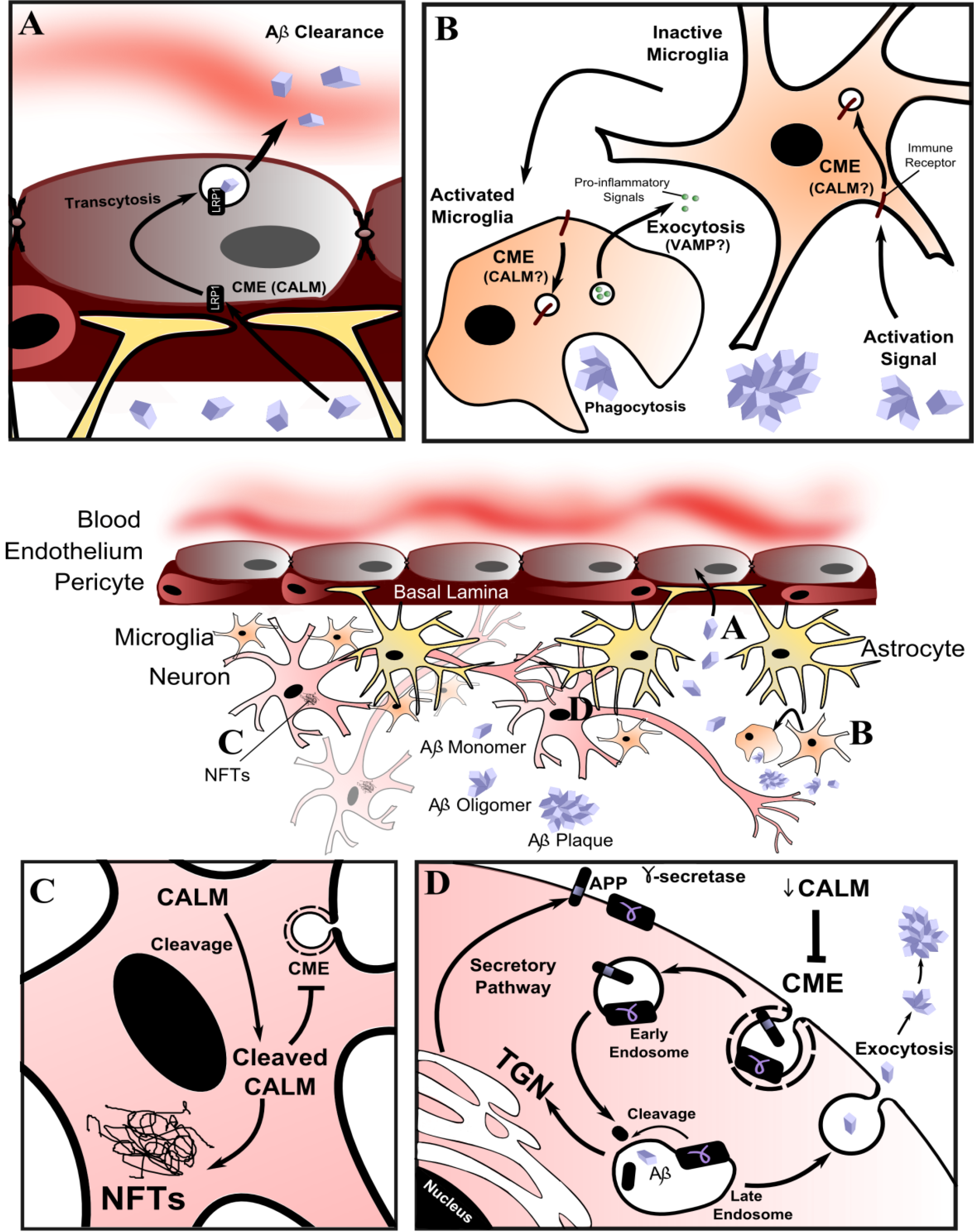
8.2. Alzheimer’s Disease
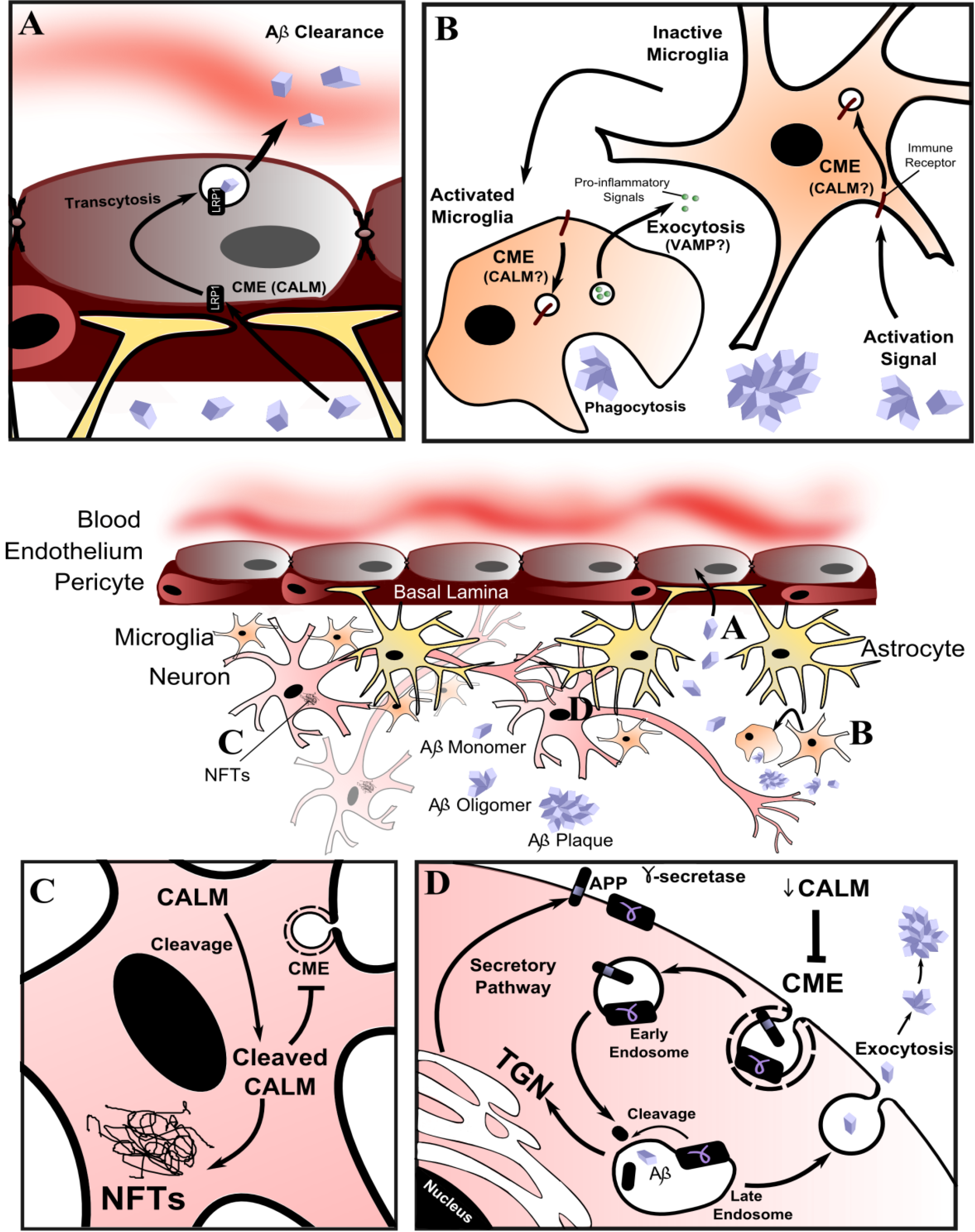
9. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- McMahon, H.T.; Boucrot, E. Molecular mechanism and physiological functions of clathrin-mediated endocytosis. Nat. Rev. Mol. Cell Biol. 2011, 12, 517–533. [Google Scholar] [CrossRef]
- Kirchhausen, T.; Owen, D.; Harrison, S.C. Molecular structure, function, and dynamics of clathrin-mediated membrane traffic. Cold Spring Harb. Perspect. Biol. 2014, 6. [Google Scholar] [CrossRef]
- Saheki, Y.; de Camilli, P. Synaptic vesicle endocytosis. Cold Spring Harb. Perspect. Biol. 2012, 4. [Google Scholar] [CrossRef]
- Kelly, B.T.; Owen, D.J. Endocytic sorting of transmembrane protein cargo. Curr. Opin. Cell Biol. 2011, 23, 404–412. [Google Scholar] [CrossRef]
- Poudel, K.R.; Bai, J. Synaptic vesicle morphology: A case of protein sorting? Curr. Opin. Cell Biol. 2014, 26C, 28–33. [Google Scholar] [CrossRef]
- Godlee, C.; Kaksonen, M. From uncertain beginnings: Initiation mechanisms of clathrin-mediated endocytosis. J. Cell Biol. 2013, 203, 717–725. [Google Scholar] [CrossRef]
- Gaidarov, I.; Chen, Q.; Falck, J.R.; Reddy, K.K.; Keen, J.H. A functional phosphatidylinositol 3,4,5-trisphosphate/phosphoinositide binding domain in the clathrin adaptor AP-2 alpha subunit. Implications for the endocytic pathway. J. Biol. Chem. 1996, 271, 20922–20929. [Google Scholar]
- Rapoport, I.; Miyazaki, M.; Boll, W.; Duckworth, B.; Cantley, L.C.; Shoelson, S.; Kirchhausen, T. Regulatory interactions in the recognition of endocytic sorting signals by AP-2 complexes. EMBO J. 1997, 16, 2240–2250. [Google Scholar] [CrossRef]
- Cocucci, E.; Aguet, F.; Boulant, S.; Kirchhausen, T. The first five seconds in the life of a clathrin-coated pit. Cell 2012, 150, 495–507. [Google Scholar] [CrossRef]
- Miller, S.E.; Sahlender, D.A.; Graham, S.C.; Honing, S.; Robinson, M.S.; Peden, A.A.; Owen, D.J. The molecular basis for the endocytosis of small R-SNAREs by the clathrin adaptor CALM. Cell 2011, 147, 1118–1131. [Google Scholar] [CrossRef]
- Koo, S.J.; Markovic, S.; Puchkov, D.; Mahrenholz, C.C.; Beceren-Braun, F.; Maritzen, T.; Dernedde, J.; Volkmer, R.; Oschkinat, H.; Haucke, V. SNARE motif-mediated sorting of synaptobrevin by the endocytic adaptors clathrin assembly lymphoid myeloid leukemia (CALM) and AP180 at synapses. Proc. Natl. Acad. Sci. USA 2011, 108, 13540–13545. [Google Scholar] [CrossRef]
- Zhang, B.; Koh, Y.H.; Beckstead, R.B.; Budnik, V.; Ganetzky, B.; Bellen, H.J. Synaptic vesicle size and number are regulated by a clathrin adaptor protein required for endocytosis. Neuron 1998, 21, 1465–1475. [Google Scholar] [CrossRef]
- Nonet, M.L.; Holgado, A.M.; Brewer, F.; Serpe, C.J.; Norbeck, B.A.; Holleran, J.; Wei, L.; Hartwieg, E.; Jorgensen, E.M.; Alfonso, A. UNC-11, a Caenorhabditis elegans AP180 homologue, regulates the size and protein composition of synaptic vesicles. Mol. Biol. Cell 1999, 10, 2343–2360. [Google Scholar] [CrossRef]
- Meyerholz, A.; Hinrichsen, L.; Groos, S.; Esk, P.C.; Brandes, G.; Ungewickell, E.J. Effect of clathrin assembly lymphoid myeloid leukemia protein depletion on clathrin coat formation. Traffic 2005, 6, 1225–1234. [Google Scholar] [CrossRef]
- Petralia, R.S.; Wang, Y.X.; Indig, F.E.; Bushlin, I.; Wu, F.; Mattson, M.P.; Yao, P.J. Reduction of AP180 and CALM produces defects in synaptic vesicle size and density. Neuromol. Med. 2013, 15, 49–60. [Google Scholar] [CrossRef]
- Owen, D.J.; Collins, B.M.; Evans, P.R. Adaptors for clathrin coats: Structure and function. Annu. Rev. Cell Dev. Biol. 2004, 20, 153–191. [Google Scholar] [CrossRef]
- Honing, S.; Ricotta, D.; Krauss, M.; Spate, K.; Spolaore, B.; Motley, A.; Robinson, M.; Robinson, C.; Haucke, V.; Owen, D.J. Phosphatidylinositol-(4,5)-bisphosphate regulates sorting signal recognition by the clathrin-associated adaptor complex AP2. Mol. Cell 2005, 18, 519–531. [Google Scholar] [CrossRef]
- Owen, D.J.; Vallis, Y.; Pearse, B.M.; McMahon, H.T.; Evans, P.R. The structure and function of the beta2-adaptin appendage domain. EMBO J. 2000, 19, 4216–4227. [Google Scholar] [CrossRef]
- Motley, A.; Bright, N.A.; Seaman, M.N.; Robinson, M.S. Clathrin-mediated endocytosis in AP-2-depleted cells. J. Cell Biol. 2003, 162, 909–918. [Google Scholar] [CrossRef]
- Owen, D.J.; Vallis, Y.; Noble, M.E.; Hunter, J.B.; Dafforn, T.R.; Evans, P.R.; McMahon, H.T. A structural explanation for the binding of multiple ligands by the alpha-adaptin appendage domain. Cell 1999, 97, 805–815. [Google Scholar] [CrossRef]
- Schmid, E.M.; Ford, M.G.; Burtey, A.; Praefcke, G.J.; Peak-Chew, S.Y.; Mills, I.G.; Benmerah, A.; McMahon, H.T. Role of the AP2 beta-appendage hub in recruiting partners for clathrin-coated vesicle assembly. PLoS Biol. 2006, 4, e262:1532–e246:1548. [Google Scholar]
- Thomas, D.C.; Roth, M.G. The basolateral targeting signal in the cytoplasmic domain of glycoprotein G from vesicular stomatitis virus resembles a variety of intracellular targeting motifs related by primary sequence but having diverse targeting activities. J. Biol. Chem. 1994, 269, 15732–15739. [Google Scholar]
- Mishra, S.K.; Hawryluk, M.J.; Brett, T.J.; Keyel, P.A.; Dupin, A.L.; Jha, A.; Heuser, J.E.; Fremont, D.H.; Traub, L.M. Dual-engagement regulation of protein interactions with the AP-2 adaptor alpha appendage. J. Biol. Chem. 2004, 279, 46191–46203. [Google Scholar] [CrossRef]
- Praefcke, G.J.; Ford, M.G.; Schmid, E.M.; Olesen, L.E.; Gallop, J.L.; Peak-Chew, S.Y.; Vallis, Y.; Babu, M.M.; Mills, I.G.; McMahon, H.T. Evolving nature of the AP2 alpha-appendage hub during clathrin-coated vesicle endocytosis. EMBO J. 2004, 23, 4371–4383. [Google Scholar] [CrossRef]
- Borner, G.H.; Antrobus, R.; Hirst, J.; Bhumbra, G.S.; Kozik, P.; Jackson, L.P.; Sahlender, D.A.; Robinson, M.S. Multivariate proteomic profiling identifies novel accessory proteins of coated vesicles. J. Cell Biol. 2012, 197, 141–160. [Google Scholar] [CrossRef]
- Prasad, K.; Lippoldt, R.E. Molecular characterization of the AP180 coated vesicle assembly protein. Biochemistry 1988, 27, 6098–6104. [Google Scholar] [CrossRef]
- Itoh, T.; De Camilli, P. BAR, F-BAR (EFC) and ENTH/ANTH domains in the regulation of membrane-cytosol interfaces and membrane curvature. Biochim. Biophys. Acta 2006, 1761, 897–912. [Google Scholar] [CrossRef]
- Morgan, J.R.; Prasad, K.; Hao, W.; Augustine, G.J.; Lafer, E.M. A conserved clathrin assembly motif essential for synaptic vesicle endocytosis. J. Neurosci. 2000, 20, 8667–8676. [Google Scholar]
- Reider, A.; Wendland, B. Endocytic adaptors—Social networking at the plasma membrane. J. Cell Sci. 2011, 124, 1613–1622. [Google Scholar] [CrossRef]
- Taylor, M.J.; Perrais, D.; Merrifield, C.J. A high precision survey of the molecular dynamics of mammalian clathrin-mediated endocytosis. PLoS Biol. 2011, 9, e1000604:1–e1000604:23. [Google Scholar]
- Farley, J.; Auerbach, S. Protein kinase C activation induces conductance changes in Hermissenda photoreceptors like those seen in associative learning. Nature 1986, 319, 220–223. [Google Scholar] [CrossRef]
- Meloty-Kapella, L.; Shergill, B.; Kuon, J.; Botvinick, E.; Weinmaster, G. Notch ligand endocytosis generates mechanical pulling force dependent on dynamin, epsins, and actin. Dev. Cell 2012, 22, 1299–1312. [Google Scholar] [CrossRef]
- Aguet, F.; Antonescu, C.N.; Mettlen, M.; Schmid, S.L.; Danuser, G. Advances in analysis of low signal-to-noise images link dynamin and AP2 to the functions of an endocytic checkpoint. Dev. Cell 2013, 26, 279–291. [Google Scholar] [CrossRef]
- Daumke, O.; Roux, A.; Haucke, V. BAR domain scaffolds in dynamin-mediated membrane fission. Cell 2014, 156, 882–892. [Google Scholar] [CrossRef]
- Von Kleist, L.; Stahlschmidt, W.; Bulut, H.; Gromova, K.; Puchkov, D.; Robertson, M.J.; MacGregor, K.A.; Tomilin, N.; Pechstein, A.; Chau, N.; et al. Role of the clathrin terminal domain in regulating coated pit dynamics revealed by small molecule inhibition. Cell 2011, 146, 471–484. [Google Scholar] [CrossRef]
- Mao, Y.; Chen, J.; Maynard, J.A.; Zhang, B.; Quiocho, F.A. A novel all helix fold of the AP180 amino-terminal domain for phosphoinositide binding and clathrin assembly in synaptic vesicle endocytosis. Cell 2001, 104, 433–440. [Google Scholar] [CrossRef]
- Ford, M.G.; Pearse, B.M.; Higgins, M.K.; Vallis, Y.; Owen, D.J.; Gibson, A.; Hopkins, C.R.; Evans, P.R.; McMahon, H.T. Simultaneous binding of PtdIns(4,5)P2 and clathrin by AP180 in the nucleation of clathrin lattices on membranes. Science 2001, 291, 1051–1055. [Google Scholar] [CrossRef]
- McMahon, H.T. Endocytosis: An assembly protein for clathrin cages. Curr. Biol. 1999, 9, R332–R335. [Google Scholar] [CrossRef]
- Harel, A.; Wu, F.; Mattson, M.P.; Morris, C.M.; Yao, P.J. Evidence for CALM in directing VAMP2 trafficking. Traffic 2008, 9, 417–429. [Google Scholar] [CrossRef]
- Burston, H.E.; Maldonado-Baez, L.; Davey, M.; Montpetit, B.; Schluter, C.; Wendland, B.; Conibear, E. Regulators of yeast endocytosis identified by systematic quantitative analysis. J. Cell Biol. 2009, 185, 1097–1110. [Google Scholar] [CrossRef]
- Wen, Y.; Stavrou, I.; Bersuker, K.; Brady, R.J.; De, L.A.; O’Halloran, T.J. AP180-mediated trafficking of Vamp7B limits homotypic fusion of Dictyostelium contractile vacuoles. Mol. Biol. Cell 2009, 20, 4278–4288. [Google Scholar] [CrossRef]
- Kalthoff, C.; Alves, J.; Urbanke, C.; Knorr, R.; Ungewickell, E.J. Unusual structural organization of the endocytic proteins AP180 and epsin 1. J. Biol. Chem. 2002, 277, 8209–8216. [Google Scholar] [CrossRef]
- Murphy, J.E.; Pleasure, I.T.; Puszkin, S.; Prasad, K.; Keen, J.H. Clathrin assembly protein AP-3. The identity of the 155K protein, AP 180, and NP185 and demonstration of a clathrin binding domain. J. Biol. Chem. 1991, 266, 4401–4408. [Google Scholar]
- Morgan, J.R.; Prasad, K.; Jin, S.; Augustine, G.J.; Lafer, E.M. Eps15 homology domain-NPF motif interactions regulate clathrin coat assembly during synaptic vesicle recycling. J. Biol. Chem. 2003, 278, 33583–33592. [Google Scholar]
- Graham, M.E.; Thaysen-Andersen, M.; Bache, N.; Craft, G.E.; Larsen, M.R.; Packer, N.H.; Robinson, P.J. A novel post-translational modification in nerve terminals: O-linked N-acetylglucosamine phosphorylation. J. Proteome Res. 2011, 10, 2725–2733. [Google Scholar] [CrossRef]
- Morris, S.A.; Schroder, S.; Plessmann, U.; Weber, K.; Ungewickell, E. Clathrin assembly protein AP180: Primary structure, domain organization and identification of a clathrin binding site. EMBO J. 1993, 12, 667–675. [Google Scholar]
- Zhou, S.; Tannery, N.H.; Yang, J.; Puszkin, S.; Lafer, E.M. The synapse-specific phosphoprotein F1–20 is identical to the clathrin assembly protein AP-3. J. Biol. Chem. 1993, 268, 12655–12662. [Google Scholar]
- Kozlowski, L.P. Calculation of protein isoelectric point. Available online: http://isoelectric.ovh.org (accessed on 1 May 2014).
- Wendland, B.; Steece, K.E.; Emr, S.D. Yeast epsins contain an essential N-terminal ENTH domain, bind clathrin and are required for endocytosis. EMBO J. 1999, 18, 4383–4393. [Google Scholar] [CrossRef]
- ClustalW2. Available online: http://www.ebi.ac.uk/Tools/msa/clustalw2/ (accessed on 1 May 2014).
- Conway, A.E.; Scotland, P.B.; Lavau, C.P.; Wechsler, D.S. A CALM-derived nuclear export signal is essential for CALM-AF10-mediated leukemogenesis. Blood 2013, 121, 4758–4768. [Google Scholar] [CrossRef]
- Suzuki, M.; Yamagata, K.; Shino, M.; Aikawa, Y.; Akashi, K.; Watanabe, T.; Kitabayashi, I. Nuclear export signal within CALM is necessary for CALM-AF10-induced leukemia. Cancer Sci. 2014, 105, 315–323. [Google Scholar] [CrossRef]
- Vecchi, M.; Polo, S.; Poupon, V.; van de, L.J.W.; Benmerah, A.; di Fiore, P.P. Nucleocytoplasmic shuttling of endocytic proteins. J. Cell Biol. 2001, 153, 1511–1518. [Google Scholar] [CrossRef]
- Smith, C.M.; Chircop, M. Clathrin-mediated endocytic proteins are involved in regulating mitotic progression and completion. Traffic 2012, 13, 1628–1641. [Google Scholar] [CrossRef]
- Ye, W.; Lafer, E.M. Clathrin binding and assembly activities of expressed domains of the synapse-specific clathrin assembly protein AP-3. J. Biol. Chem. 1995, 270, 10933–10939. [Google Scholar] [CrossRef]
- Kim, J.A.; Kim, H.L. Cell-free expression and functional reconstitution of CALM in clathrin assembly. Exp. Mol. Med. 2001, 33, 89–94. [Google Scholar] [CrossRef]
- Kim, J.A.; Kim, S.R.; Jung, Y.K.; Woo, S.Y.; Seoh, J.Y.; Hong, Y.S.; Kim, H.L. Properties of GST-CALM expressed in E. coli. Exp. Mol. Med. 2000, 32, 93–99. [Google Scholar] [CrossRef]
- Tebar, F.; Bohlander, S.K.; Sorkin, A. Clathrin assembly lymphoid myeloid leukemia (CALM) protein: Localization in endocytic-coated pits, interactions with clathrin, and the impact of overexpression on clathrin-mediated traffic. Mol. Biol. Cell 1999, 10, 2687–2702. [Google Scholar] [CrossRef]
- Scotland, P.B.; Heath, J.L.; Conway, A.E.; Porter, N.B.; Armstrong, M.B.; Walker, J.A.; Klebig, M.L.; Lavau, C.P.; Wechsler, D.S. The PICALM protein plays a key role in iron homeostasis and cell proliferation. PLoS One 2012, 7, e44252:1–e44252:14. [Google Scholar]
- Ahle, S.; Ungewickell, E. Purification and properties of a new clathrin assembly protein. EMBO J. 1986, 5, 3143–3149. [Google Scholar]
- Ye, W.; Lafer, E.M. Bacterially expressed F1–20/AP-3 assembles clathrin into cages with a narrow size distribution: Implications for the regulation of quantal size during neurotransmission. J. Neurosci. Res. 1995, 41, 15–26. [Google Scholar] [CrossRef]
- Unanue, E.R.; Ungewickell, E.; Branton, D. The binding of clathrin triskelions to membranes from coated vesicles. Cell 1981, 26, 439–446. [Google Scholar] [CrossRef]
- Deak, F.; Schoch, S.; Liu, X.; Sudhof, T.C.; Kavalali, E.T. Synaptobrevin is essential for fast synaptic-vesicle endocytosis. Nat. Cell Biol. 2004, 6, 1102–1108. [Google Scholar] [CrossRef]
- Stevens, R.J.; Akbergenova, Y.; Jorquera, R.A.; Littleton, J.T. Abnormal synaptic vesicle biogenesis in Drosophila synaptogyrin mutants. J. Neurosci. 2012, 32, 18054–18067. [Google Scholar] [CrossRef]
- Sahlender, D.A.; Kozik, P.; Miller, S.E.; Peden, A.A.; Robinson, M.S. Uncoupling the functions of CALM in VAMP sorting and clathrin-coated pit formation. PLoS One 2013, 8, e64514:1–e64514:11. [Google Scholar]
- Zhuo, Y.; Ilangovan, U.; Schirf, V.; Demeler, B.; Sousa, R.; Hinck, A.P.; Lafer, E.M. Dynamic interactions between clathrin and locally structured elements in a disordered protein mediate clathrin lattice assembly. J. Mol. Biol. 2010, 404, 274–290. [Google Scholar] [CrossRef]
- Lindner, R.; Ungewickell, E. Clathrin-associated proteins of bovine brain coated vesicles. An analysis of their number and assembly-promoting activity. J. Biol. Chem. 1992, 267, 16567–16573. [Google Scholar]
- Scheele, U.; Alves, J.; Frank, R.; Duwel, M.; Kalthoff, C.; Ungewickell, E.J. Molecular and functional characterization of clathrin and AP-2 binding determinants within a disordered domain of auxilin. J. Biol. Chem. 2003, 278, 25357–25368. [Google Scholar]
- Ter Haar, E.; Harrison, S.C.; Kirchhausen, T. Peptide-in-groove interactions link target proteins to the beta-propeller of clathrin. Proc. Natl. Acad. Sci. USA 2000, 97, 1096–1100. [Google Scholar] [CrossRef]
- Willox, A.K.; Royle, S.J. Functional analysis of interaction sites on the N-terminal domain of clathrin heavy chain. Traffic 2012, 13, 70–81. [Google Scholar] [CrossRef]
- Evans, P.R.; Owen, D.J. Endocytosis and vesicle trafficking. Curr. Opin. Struct. Biol. 2002, 12, 814–821. [Google Scholar] [CrossRef]
- Hao, W.; Luo, Z.; Zheng, L.; Prasad, K.; Lafer, E.M. AP180 and AP-2 interact directly in a complex that cooperatively assembles clathrin. J. Biol. Chem. 1999, 274, 22785–22794. [Google Scholar] [CrossRef]
- Maldonado-Baez, L.; Dores, M.R.; Perkins, E.M.; Drivas, T.G.; Hicke, L.; Wendland, B. Interaction between Epsin/Yap180 adaptors and the scaffolds Ede1/Pan1 is required for endocytosis. Mol. Biol. Cell 2008, 19, 2936–2948. [Google Scholar] [CrossRef]
- Traub, L.M.; Bonifacino, J.S. Cargo recognition in clathrin-mediated endocytosis. Cold Spring Harb. Perspect. Biol. 2013, 5, a016790:1–a016790:23. [Google Scholar]
- Maritzen, T.; Koo, S.J.; Haucke, V. Turning CALM into excitement: AP180 and CALM in endocytosis and disease. Biol. Cell 2012, 104, 588–602. [Google Scholar] [CrossRef]
- Fasshauer, D.; Sutton, R.B.; Brunger, A.T.; Jahn, R. Conserved structural features of the synaptic fusion complex: SNARE proteins reclassified as Q- and R-SNAREs. Proc. Natl. Acad. Sci. USA 1998, 95, 15781–15786. [Google Scholar] [CrossRef]
- Sudhof, T.C.; Rizo, J. Synaptic vesicle exocytosis. Cold Spring Harb. Perspect. Biol. 2011, 3, 1–14. [Google Scholar]
- Koo, S.J.; Puchkov, D.; Haucke, V. AP180 and CALM: Dedicated endocytic adaptors for the retrieval of synaptobrevin 2 at synapses. Cell Logist. 2011, 1, 168–172. [Google Scholar] [CrossRef]
- Takamori, S.; Holt, M.; Stenius, K.; Lemke, E.A.; Gronborg, M.; Riedel, D.; Urlaub, H.; Schenck, S.; Brugger, B.; Ringler, P.; et al. Molecular anatomy of a trafficking organelle. Cell 2006, 127, 831–846. [Google Scholar] [CrossRef]
- Schoch, S.; Deak, F.; Konigstorfer, A.; Mozhayeva, M.; Sara, Y.; Sudhof, T.C.; Kavalali, E.T. SNARE function analyzed in synaptobrevin/VAMP knockout mice. Science 2001, 294, 1117–1122. [Google Scholar] [CrossRef]
- Bao, H.; Daniels, R.W.; Macleod, G.T.; Charlton, M.P.; Atwood, H.L.; Zhang, B. AP180 maintains the distribution of synaptic and vesicle proteins in the nerve terminal and indirectly regulates the efficacy of Ca2+-triggered exocytosis. J. Neurophysiol. 2005, 94, 1888–1903. [Google Scholar] [CrossRef]
- Vanlandingham, P.A.; Barmchi, M.P.; Royer, S.; Green, R.; Bao, H.; Reist, N.; Zhang, B. AP180 couples protein retrieval to clathrin-mediated endocytosis of synaptic vesicles. Traffic 2014, 15, 433–450. [Google Scholar] [CrossRef]
- Miller, S.E.; Collins, B.M.; McCoy, A.J.; Robinson, M.S.; Owen, D.J. A SNARE-adaptor interaction is a new mode of cargo recognition in clathrin-coated vesicles. Nature 2007, 450, 570–574. [Google Scholar] [CrossRef]
- Zhao, X.; Greener, T.; Al Hasani, H.; Cushman, S.W.; Eisenberg, E.; Greene, L.E. Expression of auxilin or AP180 inhibits endocytosis by mislocalizing clathrin: Evidence for formation of nascent pits containing AP1 or AP2 but not clathrin. J. Cell Sci. 2001, 114, 353–365. [Google Scholar]
- Granseth, B.; Odermatt, B.; Royle, S.J.; Lagnado, L. Clathrin-mediated endocytosis: The physiological mechanism of vesicle retrieval at hippocampal synapses. J. Physiol. 2007, 585, 681–686. [Google Scholar] [CrossRef]
- Xiao, Q.; Gil, S.C.; Yan, P.; Wang, Y.; Han, S.; Gonzales, E.; Perez, R.; Cirrito, J.R.; Lee, J.M. Role of phosphatidylinositol clathrin assembly lymphoid-myeloid leukemia (PICALM) in intracellular amyloid precursor protein (APP) processing and amyloid plaque pathogenesis. J. Biol. Chem. 2012, 287, 21279–21289. [Google Scholar]
- Huang, F.; Khvorova, A.; Marshall, W.; Sorkin, A. Analysis of clathrin-mediated endocytosis of EGF receptor by RNA interference. J. Biol. Chem. 2004, 279, 16657–16661. [Google Scholar]
- Suzuki, M.; Tanaka, H.; Tanimura, A.; Tanabe, K.; Oe, N.; Rai, S.; Kon, S.; Fukumoto, M.; Takei, K.; Abe, T.; et al. The clathrin assembly protein PICALM is required for erythroid maturation and transferrin internalization in mice. PLoS One 2012, 7, e31854:1–e31854:12. [Google Scholar]
- Cousin, M.A.; Robinson, P.J. The dephosphins: Dephosphorylation by calcineurin triggers synaptic vesicle endocytosis. Trends Neurosci. 2001, 24, 659–665. [Google Scholar] [CrossRef]
- Tan, T.C.; Valova, V.A.; Malladi, C.S.; Graham, M.E.; Berven, L.A.; Jupp, O.J.; Hansra, G.; McClure, S.J.; Sarcevic, B.; Boadle, R.A.; et al. Cdk5 is essential for synaptic vesicle endocytosis. Nat. Cell Biol. 2003, 5, 701–710. [Google Scholar] [CrossRef]
- Anggono, V.; Smillie, K.J.; Graham, M.E.; Valova, V.A.; Cousin, M.A.; Robinson, P.J. Syndapin I is the phosphorylation-regulated dynamin I partner in synaptic vesicle endocytosis. Nat. Neurosci. 2006, 9, 752–760. [Google Scholar] [CrossRef]
- Murphy, J.E.; Hanover, J.A.; Froehlich, M.; DuBois, G.; Keen, J.H. Clathrin assembly protein AP-3 is phosphorylated and glycosylated on the 50-kDa structural domain. J. Biol. Chem. 1994, 269, 21346–21352. [Google Scholar]
- Keen, J.H.; Black, M.M. The phosphorylation of coated membrane proteins in intact neurons. J. Cell Biol. 1986, 102, 1325–1333. [Google Scholar] [CrossRef]
- Morris, S.A.; Mann, A.; Ungewickell, E. Analysis of 100–180-kDa phosphoproteins in clathrin-coated vesicles from bovine brain. J. Biol. Chem. 1990, 265, 3354–3357. [Google Scholar]
- Ballif, B.A.; Carey, G.R.; Sunyaev, S.R.; Gygi, S.P. Large-scale identification and evolution indexing of tyrosine phosphorylation sites from murine brain. J. Proteome Res. 2008, 7, 311–318. [Google Scholar] [CrossRef]
- Ballif, B.A.; Villen, J.; Beausoleil, S.A.; Schwartz, D.; Gygi, S.P. Phosphoproteomic analysis of the developing mouse brain. Mol. Cell Proteomics 2004, 2, 1093–1101. [Google Scholar]
- Munton, R.P.; Tweedie-Cullen, R.; Livingstone-Zatchej, M.; Weinandy, F.; Waidelich, M.; Longo, D.; Gehrig, P.; Potthast, F.; Rutishauser, D.; Gerrits, B.; et al. Qualitative and quantitative analyses of protein phosphorylation in naive and stimulated mouse synaptosomal preparations. Mol. Cell Proteomics 2007, 6, 283–293. [Google Scholar]
- Wisniewski, J.R.; Nagaraj, N.; Zougman, A.; Gnad, F.; Mann, M. Brain phosphoproteome obtained by a FASP-based method reveals plasma membrane protein topology. J. Proteome Res. 2010, 9, 3280–3289. [Google Scholar] [CrossRef]
- Wu, C.C.; MacCoss, M.J.; Howell, K.E.; Yates, J.R., III. A method for the comprehensive proteomic analysis of membrane proteins. Nat. Biotechnol. 2003, 21, 532–538. [Google Scholar] [CrossRef]
- Meggio, F.; Pinna, L.A. One-thousand-and-one substrates of protein kinase CK2? FASEB J. 2003, 17, 349–368. [Google Scholar] [CrossRef]
- Cousin, M.A.; Tan, T.C.; Robinson, P.J. Protein phosphorylation is required for endocytosis in nerve terminals. Potential role for the dephosphins dynamin I and synaptojanin, but not AP180 or amphiphysin. J. Neurochem. 2001, 76, 105–116. [Google Scholar] [CrossRef]
- Murakami, N.; Bolton, D.C.; Kida, E.; Xie, W.; Hwang, Y.W. Phosphorylation by Dyrk1A of clathrin coated vesicle-associated proteins: Identification of the substrate proteins and the effects of phosphorylation. PLoS One 2012, 7, e34845:1–e34845:17. [Google Scholar]
- Hahne, H.; Kuster, B. Discovery of O-GlcNAc-6-phosphate-modified proteins by re-analyzing large-scale phosphoproteomics data. Mol. Cell Proteomics 2012, 11, 1063–1069. [Google Scholar] [CrossRef]
- Graham, M.E.; Stone, R.S.; Robinson, P.J.; Payne, R.J. Synthesis and protein binding studies of a peptide fragment of clathrin assembly protein AP180 bearing an O-linked beta-n-acetylglucosaminyl-6-phosphate modification. Organ. Biomol. Chem. 2012, 10, 2545–2551. [Google Scholar]
- Khidekel, N.; Ficarro, S.B.; Clark, P.M.; Bryan, M.C.; Swaney, D.L.; Rexach, J.E.; Sun, Y.E.; Coon, J.J.; Peters, E.C.; Hsieh-Wilson, L.C. Probing the dynamics of O-GlcNAc glycosylation in the brain using quantitative proteomics. Nat. Chem. Biol. 2007, 3, 339–348. [Google Scholar]
- Trinidad, J.C.; Barkan, D.T.; Gulledge, B.F.; Thalhammer, A.; Sali, A.; Schoepfer, R.; Burlingame, A.L. Global identification and characterization of both O-GlcNAcylation and phosphorylation at the murine synapse. Mol. Cell Proteomics 2012, 11, 215–229. [Google Scholar]
- PhosphoSitePlus. Available online: http://www.phosphosite.org (accessed on 1 May 2014).
- Zhan, X.; Desiderio, D.M. The human pituitary nitroproteome: Detection of nitrotyrosyl-proteins with two-dimensional Western blotting, and amino acid sequence determination with mass spectrometry. Biochem. Biophys. Res. Commun. 2004, 325, 1180–1186. [Google Scholar] [CrossRef]
- Uhlmann, T.; Geoghegan, V.L.; Thomas, B.; Ridlova, G.; Trudgian, D.C.; Acuto, O. A method for large-scale identification of protein arginine methylation. Mol. Cell Proteomics 2012, 11, 1489–1499. [Google Scholar] [CrossRef]
- Guo, A.; Gu, H.; Zhou, J.; Mulhern, D.; Wang, Y.; Lee, K.A.; Yang, V.; Aguiar, M.; Kornhauser, J.; Jia, X.; et al. Immunoaffinity enrichment and mass spectrometry analysis of protein methylation. Mol. Cell Proteomics 2014, 13, 372–387. [Google Scholar] [CrossRef]
- Dreyling, M.H.; Martinez-Climent, J.A.; Zheng, M.; Mao, J.; Rowley, J.D.; Bohlander, S.K. The t(10;11)(p13;q14) in the U937 cell line results in the fusion of the AF10 gene and CALM, encoding a new member of the AP-3 clathrin assembly protein family. Proc. Natl. Acad. Sci. USA 1996, 93, 4804–4809. [Google Scholar]
- Sundstrom, C.; Nilsson, K. Establishment and characterization of a human histiocytic lymphoma cell line (U-937). Int. J. Cancer 1976, 17, 565–577. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, Z.J.; Brew, K.; Lee, E.Y.C. Mutational analysis of the catalytic subunit of muscle protein phosphatase-1. Biochemistry 1996, 35, 6276–6282. [Google Scholar] [CrossRef]
- Bohlander, S.K.; Muschinsky, V.; Schrader, K.; Siebert, R.; Schlegelberger, B.; Harder, L.; Schemmel, V.; Fonatsch, C.; Ludwig, W.D.; Hiddemann, W.; et al. Molecular analysis of the CALM/AF10 fusion: Identical rearrangements in acute myeloid leukemia, acute lymphoblastic leukemia and malignant lymphoma patients. Leukemia 2000, 14, 93–99. [Google Scholar] [CrossRef]
- Dreyling, M.H.; Schrader, K.; Fonatsch, C.; Schlegelberger, B.; Haase, D.; Schoch, C.; Ludwig, W.; Loffler, H.; Buchner, T.; Wormann, B.; et al. MLL and CALM are fused to AF10 in morphologically distinct subsets of acute leukemia with translocation t(10;11): Both rearrangements are associated with a poor prognosis. Blood 1998, 91, 4662–4667. [Google Scholar]
- Okada, Y.; Feng, Q.; Lin, Y.; Jiang, Q.; Li, Y.; Coffield, V.M.; Su, L.; Xu, G.; Zhang, Y. hDOT1L links histone methylation to leukemogenesis. Cell 2005, 121, 167–178. [Google Scholar] [CrossRef]
- Chen, L.; Deshpande, A.J.; Banka, D.; Bernt, K.M.; Dias, S.; Buske, C.; Olhava, E.J.; Daigle, S.R.; Richon, V.M.; Pollock, R.M.; et al. Abrogation of MLL-AF10 and CALM-AF10-mediated transformation through genetic inactivation or pharmacological inhibition of the H3K79 methyltransferase Dot1l. Leukemia 2013, 27, 813–822. [Google Scholar] [CrossRef]
- Stoddart, A.; Tennant, T.R.; Fernald, A.A.; Anastasi, J.; Brodsky, F.M.; le Beau, M.M. The clathrin-binding domain of CALM-AF10 alters the phenotype of myeloid neoplasms in mice. Oncogene 2012, 31, 494–506. [Google Scholar] [CrossRef]
- Harold, D.; Abraham, R.; Hollingworth, P.; Sims, R.; Gerrish, A.; Hamshere, M.L.; Pahwa, J.S.; Moskvina, V.; Dowzell, K.; Williams, A.; et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer's disease. Nat. Genet. 2009, 41, 1088–1093. [Google Scholar] [CrossRef]
- Jun, G.; Naj, A.C.; Beecham, G.W.; Wang, L.S.; Buros, J.; Gallins, P.J.; Buxbaum, J.D.; Ertekin-Taner, N.; Fallin, M.D.; Friedland, R.; et al. Meta-analysis confirms CR1, CLU, and PICALM as alzheimer disease risk loci and reveals interactions with APOE genotypes. Arch. Neurol. 2010, 67, 1473–1484. [Google Scholar] [CrossRef]
- Schnetz-Boutaud, N.C.; Hoffman, J.; Coe, J.E.; Murdock, D.G.; Pericak-Vance, M.A.; Haines, J.L. Identification and confirmation of an exonic splicing enhancer variation in exon 5 of the Alzheimer disease associated PICALM gene. Ann. Hum. Genet. 2012, 76, 448–453. [Google Scholar] [CrossRef]
- Barral, S.; Bird, T.; Goate, A.; Farlow, M.R.; Az-Arrastia, R.; Bennett, D.A.; Graff-Radford, N.; Boeve, B.F.; Sweet, R.A.; Stern, Y.; et al. Genotype patterns at PICALM, CR1, BIN1, CLU, and APOE genes are associated with episodic memory. Neurology 2012, 78, 1464–1471. [Google Scholar] [CrossRef]
- Yao, P.J.; Zhu, M.; Pyun, E.I.; Brooks, A.I.; Therianos, S.; Meyers, V.E.; Coleman, P.D. Defects in expression of genes related to synaptic vesicle trafficking in frontal cortex of Alzheimer’s disease. Neurobiol. Dis. 2003, 12, 97–109. [Google Scholar] [CrossRef]
- Allen, M.; Zou, F.; Chai, H.S.; Younkin, C.S.; Crook, J.; Pankratz, V.S.; Carrasquillo, M.M.; Rowley, C.N.; Nair, A.A.; Middha, S.; et al. Novel late-onset Alzheimer disease loci variants associate with brain gene expression. Neurology 2012, 79, 221–228. [Google Scholar] [CrossRef]
- Karch, C.M.; Jeng, A.T.; Nowotny, P.; Cady, J.; Cruchaga, C.; Goate, A.M. Expression of novel Alzheimer’s disease risk genes in control and Alzheimer’s disease brains. PLoS One 2012, 7, e50976:1–e50976:9. [Google Scholar]
- Thomas, R.S.; Lelos, M.J.; Good, M.A.; Kidd, E.J. Clathrin-mediated endocytic proteins are upregulated in the cortex of the Tg2576 mouse model of Alzheimer’s disease-like amyloid pathology. Biochem. Biophys. Res. Commun. 2011, 415, 656–661. [Google Scholar]
- Ando, K.; Brion, J.P.; Stygelbout, V.; Suain, V.; Authelet, M.; Dedecker, R.; Chanut, A.; Lacor, P.; Lavaur, J.; Sazdovitch, V.; et al. Clathrin adaptor CALM/PICALM is associated with neurofibrillary tangles and is cleaved in Alzheimer’s brains. Acta Neuropathol. 2013, 125, 861–878. [Google Scholar] [CrossRef]
- Baig, S.; Joseph, S.A.; Tayler, H.; Abraham, R.; Owen, M.J.; Williams, J.; Kehoe, P.G.; Love, S. Distribution and expression of picalm in Alzheimer disease. J. Neuropathol. Exp. Neurol. 2010, 69, 1071–1077. [Google Scholar] [CrossRef]
- Parikh, I.; Fardo, D.W.; Estus, S. Genetics of PICALM expression and Alzheimer’s disease. PLoS One 2014, 9, e91242:1–e91242:7. [Google Scholar]
- Castellano, J.M.; Deane, R.; Gottesdiener, A.J.; Verghese, P.B.; Stewart, F.R.; West, T.; Paoletti, A.C.; Kasper, T.R.; DeMattos, R.B.; Zlokovic, B.V.; et al. Low-density lipoprotein receptor overexpression enhances the rate of brain-to-blood Abeta clearance in a mouse model of beta-amyloidosis. Proc. Natl. Acad. Sci. USA 2012, 109, 15502–15507. [Google Scholar] [CrossRef]
- Harasaki, K.; Lubben, N.B.; Harbour, M.; Taylor, M.J.; Robinson, M.S. Sorting of major cargo glycoproteins into clathrin-coated vesicles. Traffic 2005, 6, 1014–1026. [Google Scholar] [CrossRef]
- Lambert, J.C.; Ibrahim-Verbaas, C.A.; Harold, D.; Naj, A.C.; Sims, R.; Bellenguez, C.; DeStafano, A.L.; Bis, J.C.; Beecham, G.W.; Grenier-Boley, B.; et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nat. Genet. 2013, 45, 1452–1458. [Google Scholar] [CrossRef]
- Zhang, B.; Gaiteri, C.; Bodea, L.G.; Wang, Z.; McElwee, J.; Podtelezhnikov, A.A.; Zhang, C.; Xie, T.; Tran, L.; Dobrin, R.; et al. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell 2013, 153, 707–720. [Google Scholar] [CrossRef]
- Treusch, S.; Hamamichi, S.; Goodman, J.L.; Matlack, K.E.; Chung, C.Y.; Baru, V.; Shulman, J.M.; Parrado, A.; Bevis, B.J.; Valastyan, J.S.; et al. Functional links between Abeta toxicity, endocytic trafficking, and Alzheimer’s disease risk factors in yeast. Science 2011, 334, 1241–1245. [Google Scholar] [CrossRef]
- Kanatsu, K.; Morohashi, Y.; Suzuki, M.; Kuroda, H.; Watanabe, T.; Tomita, T.; Iwatsubo, T. Decreased CALM expression reduces Abeta42 to total Abeta ratio through clathrin-mediated endocytosis of gamma-secretase. Nat. Commun. 2014, 5, 3386:1–3386:12. [Google Scholar]
- Yao, P.J.; Coleman, P.D. Reduced O-glycosylated clathrin assembly protein AP180: Implication for synaptic vesicle recycling dysfunction in Alzheimer’s disease. Neurosci. Lett. 1998, 252, 33–36. [Google Scholar] [CrossRef]
- Yao, P.J.; Morsch, R.; Callahan, L.M.; Coleman, P.D. Changes in synaptic expression of clathrin assembly protein AP180 in Alzheimer's disease analysed by immunohistochemistry. Neuroscience 1999, 94, 389–394. [Google Scholar] [CrossRef]
- Wu, F.; Matsuoka, Y.; Mattson, M.P.; Yao, P.J. The clathrin assembly protein AP180 regulates the generation of amyloid-beta peptide. Biochem. Biophys. Res. Commun. 2009, 385, 247–250. [Google Scholar] [CrossRef]
- Goes, F.S.; Hamshere, M.L.; Seifuddin, F.; Pirooznia, M.; Belmonte-Mahon, P.; Breuer, R.; Schulze, T.; Nothen, M.; Cichon, S.; Rietschel, M.; et al. Genome-wide association of mood-incongruent psychotic bipolar disorder. Transl. Psychiatry 2012, 2, e180:1–e180:7. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Moshkanbaryans, L.; Chan, L.-S.; Graham, M.E. The Biochemical Properties and Functions of CALM and AP180 in Clathrin Mediated Endocytosis. Membranes 2014, 4, 388-413. https://doi.org/10.3390/membranes4030388
Moshkanbaryans L, Chan L-S, Graham ME. The Biochemical Properties and Functions of CALM and AP180 in Clathrin Mediated Endocytosis. Membranes. 2014; 4(3):388-413. https://doi.org/10.3390/membranes4030388
Chicago/Turabian StyleMoshkanbaryans, Lia, Ling-Shan Chan, and Mark E. Graham. 2014. "The Biochemical Properties and Functions of CALM and AP180 in Clathrin Mediated Endocytosis" Membranes 4, no. 3: 388-413. https://doi.org/10.3390/membranes4030388
APA StyleMoshkanbaryans, L., Chan, L.-S., & Graham, M. E. (2014). The Biochemical Properties and Functions of CALM and AP180 in Clathrin Mediated Endocytosis. Membranes, 4(3), 388-413. https://doi.org/10.3390/membranes4030388