
Evaluation of Host Cell Impurity Effects on the Performance of Sterile Filtration Processes for Therapeutic Viruses


Abstract
:
1. Introduction
2. Materials and Methods
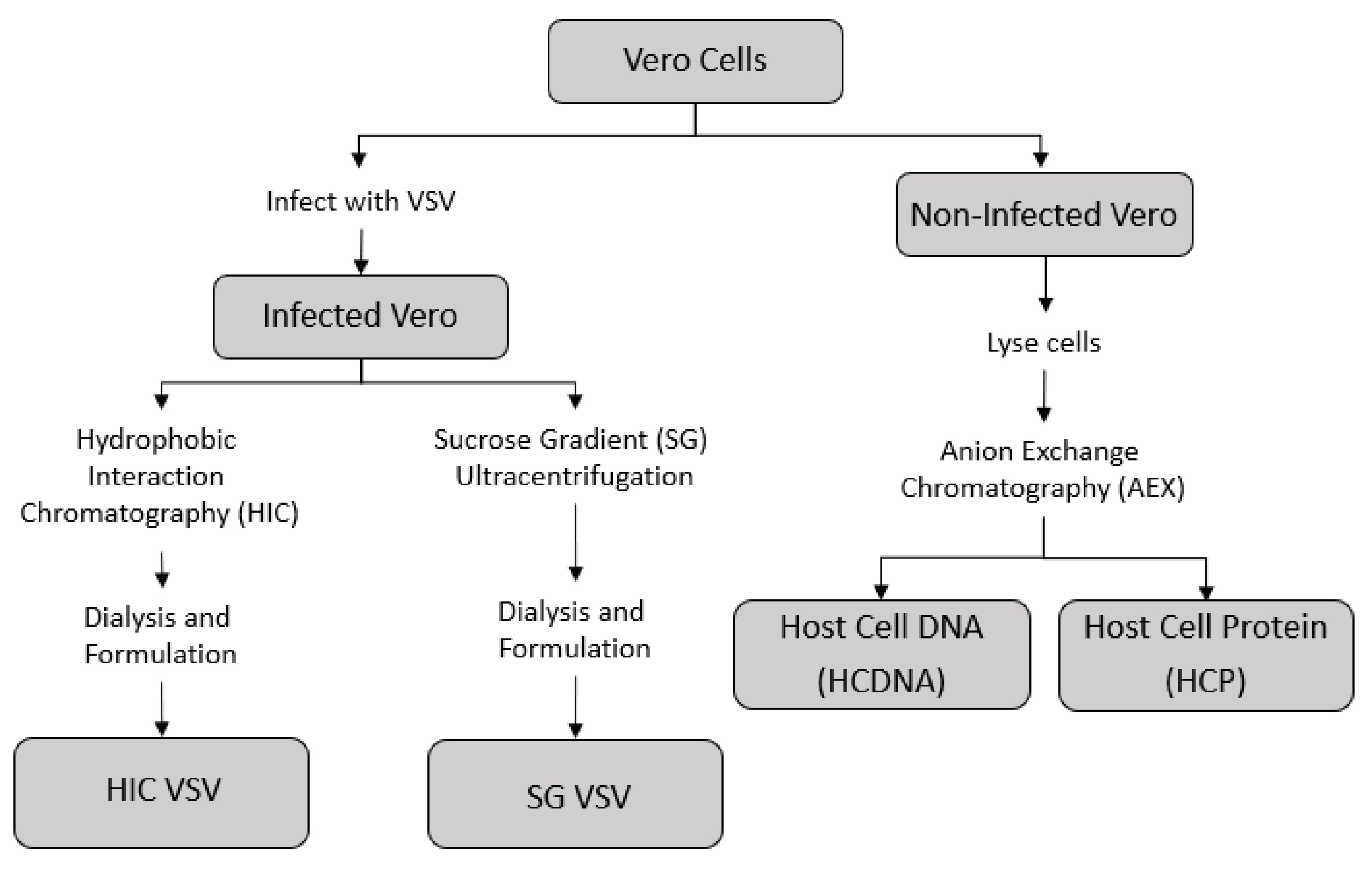
2.1. Production of VSV and Isolated Host Cell Impurities
2.2. Sterile Filtration
2.3. Static Adsorption
2.4. Assays
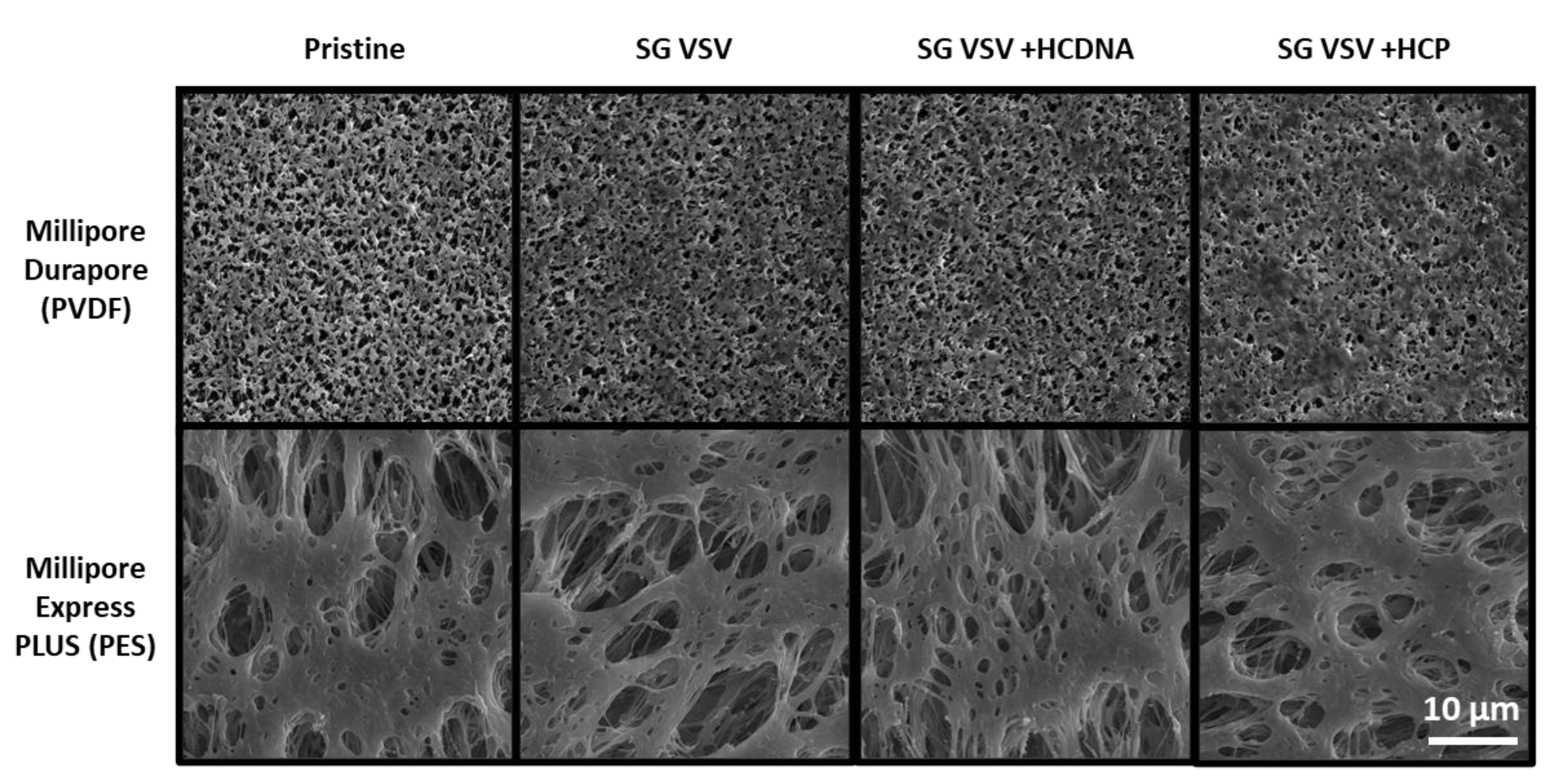
2.5. Scanning Electron Microscopy
3. Results and Discussion
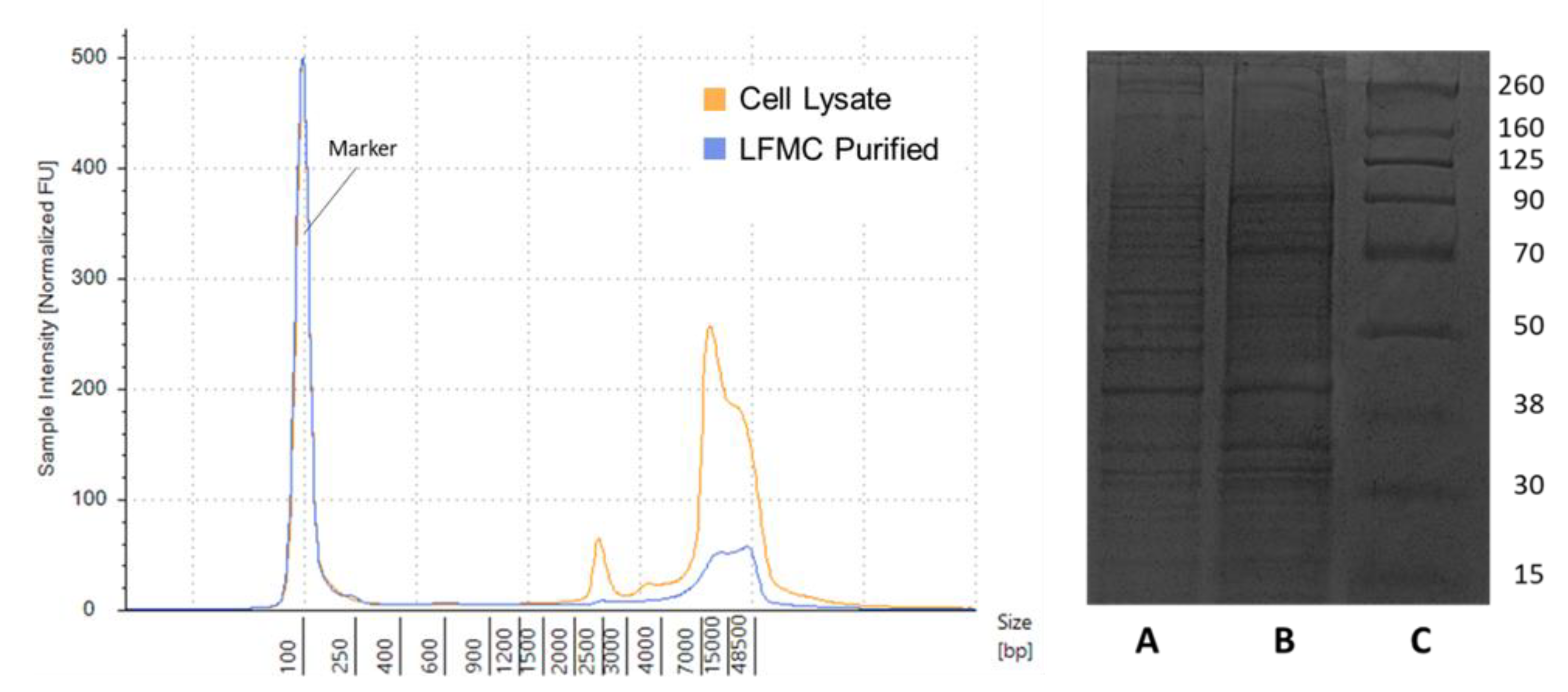
3.1. Production of Purified VSV Batches and Host Cell Impurities
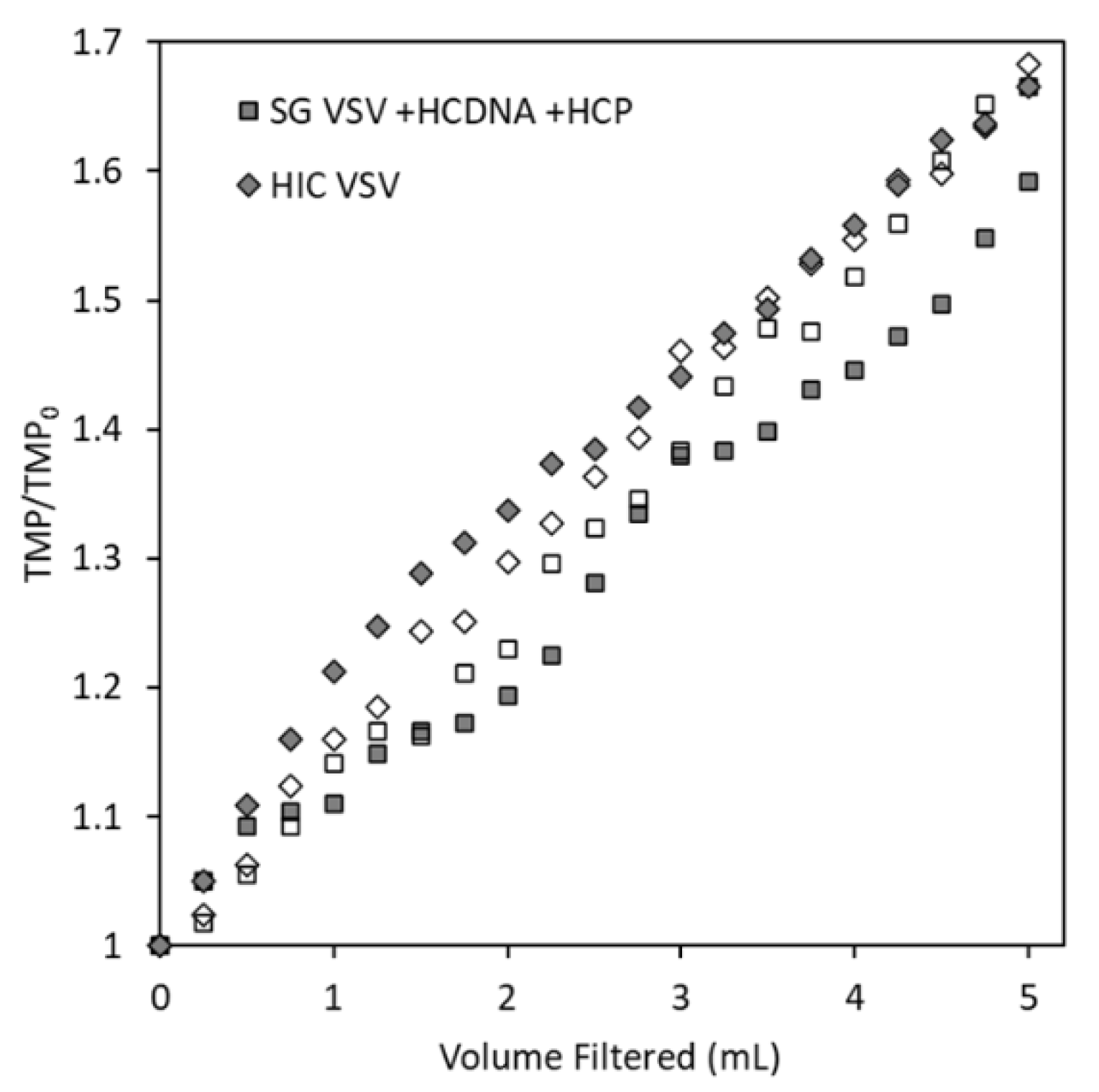
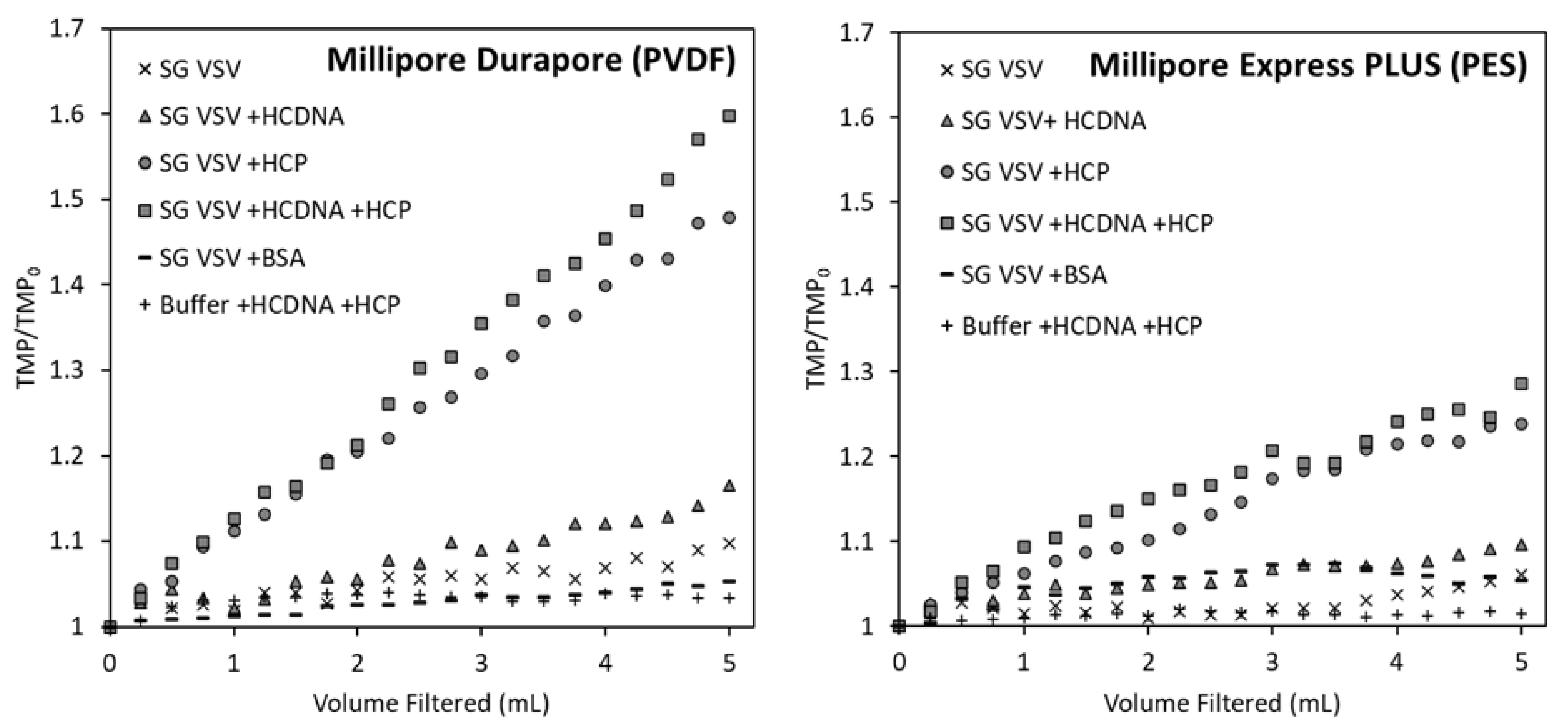
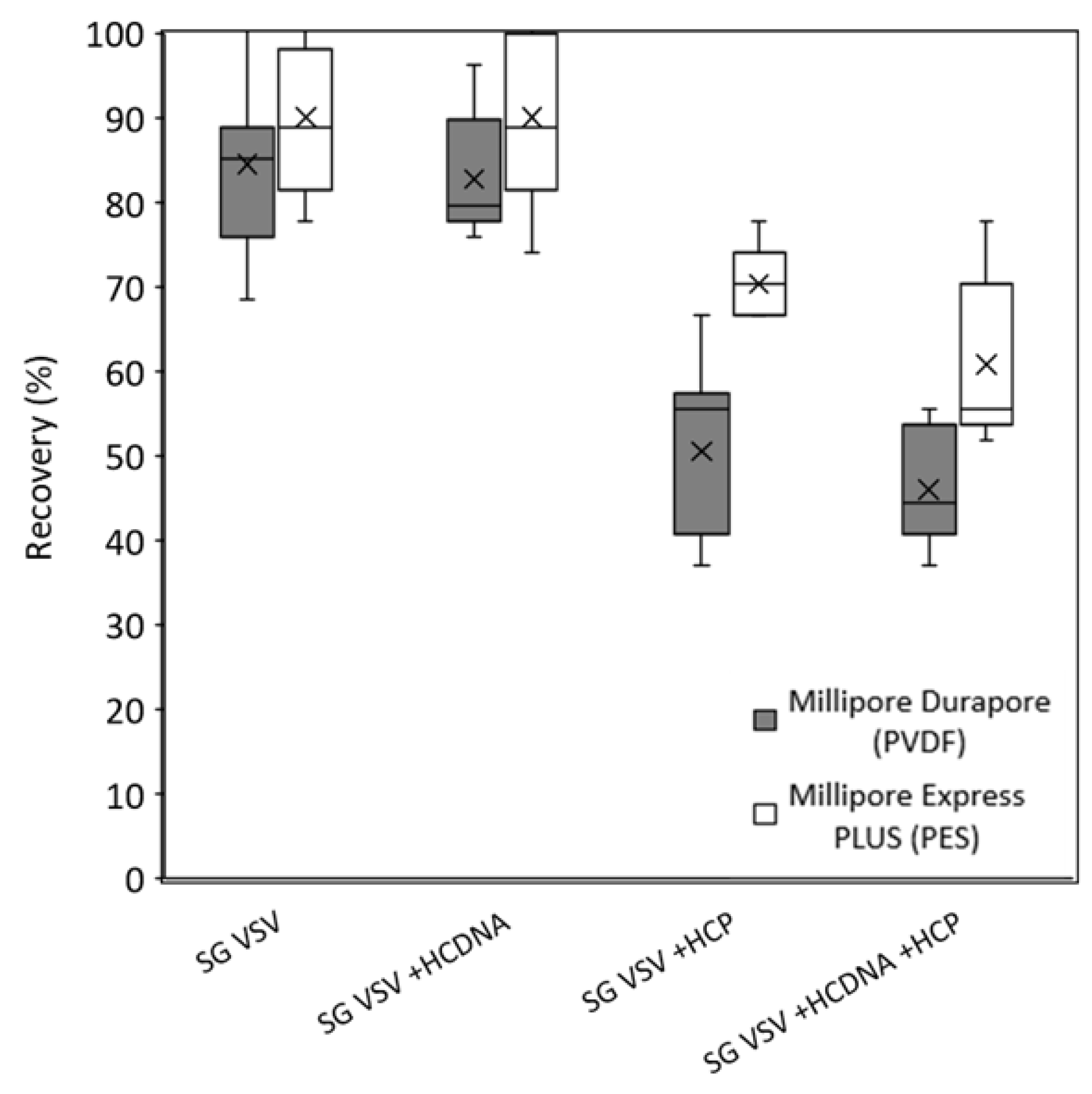
3.2. Effect of Host Cell Impurities on Sterile Filtration
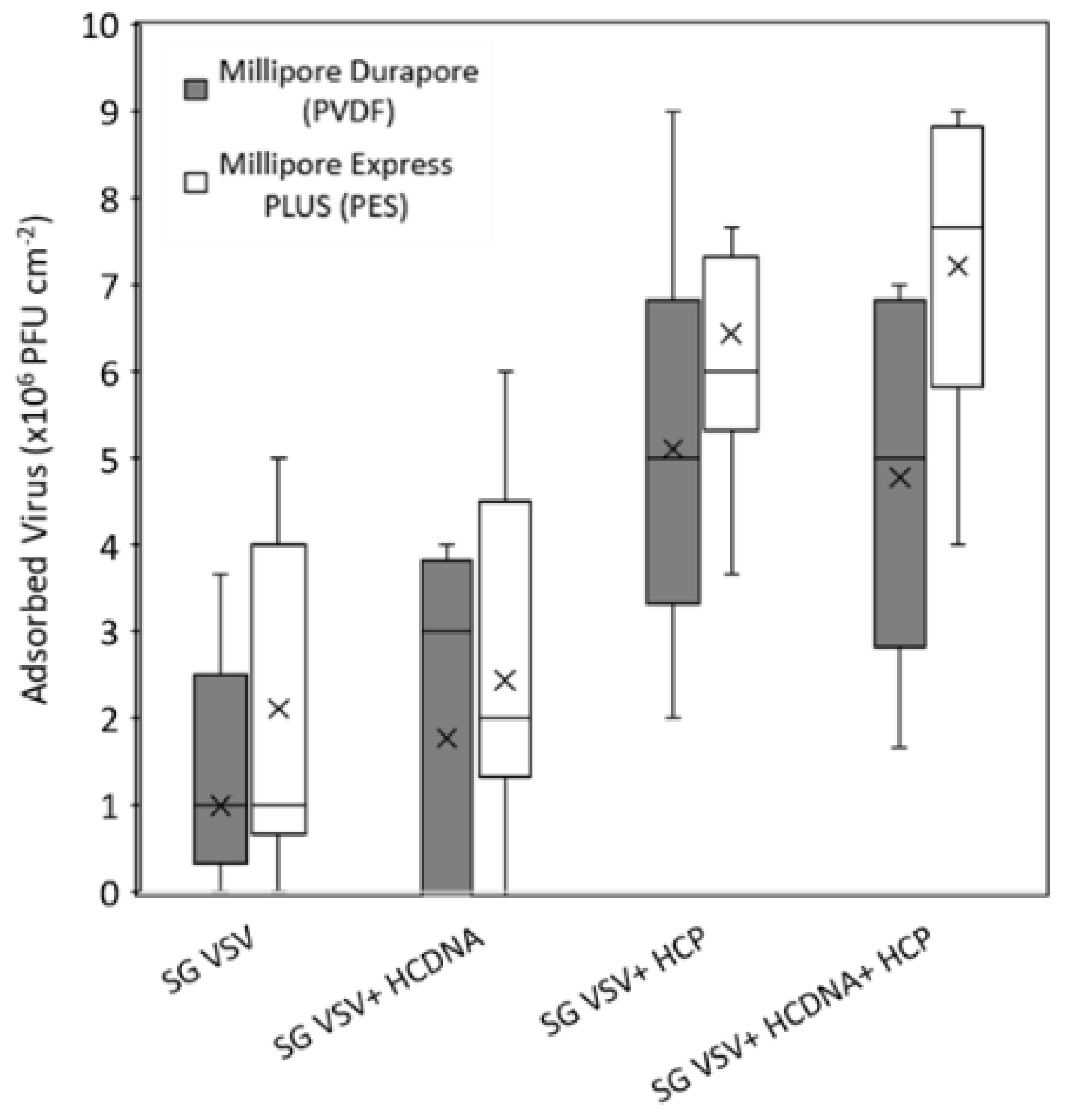
3.3. Adsorption of VSV, Host Cell Protein, and DNA to Microfiltration Membranes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ungerechts, G.; Bossow, S.; Leuchs, B.; Holm, P.S.; Rommelaere, J.; Coffey, M.; Coffin, R.; Bell, J.; Nettelbeck, D.M. Moving Oncolytic Viruses into the Clinic: Clinical-Grade Production, Purification, and Characterization of Diverse Oncolytic Viruses. Mol. Ther.-Methods Clin. Dev. 2016, 3, 16018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reale, A.; Vitiello, A.; Conciatori, V.; Parolin, C.; Calistri, A.; Palù, G. Perspectives on Immunotherapy via Oncolytic Viruses. Infect. Agent. Cancer 2019, 14, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russell, L.; Peng, K.-W. The Emerging Role of Oncolytic Virus Therapy against Cancer. Chin. Clin. Oncol. 2018, 7, 16–26. [Google Scholar] [CrossRef]
- Regules, J.A.; Beigel, J.H.; Paolino, K.M.; Voell, J.; Castellano, A.R.; Hu, Z.; Muñoz, P.; Moon, J.E.; Ruck, R.C.; Bennett, J.W.; et al. A Recombinant Vesicular Stomatitis Virus Ebola Vaccine. N. Engl. J. Med. 2017, 376, 330–341. [Google Scholar] [CrossRef] [PubMed]
- Kinch, M.S.; Griesenauer, R.H. 2017 in Review: FDA Approvals of New Molecular Entities. Drug Discov. Today 2018, 23, 1469–1473. [Google Scholar] [CrossRef] [PubMed]
- Tomás, H.A.; Rodrigues, A.F.; Carrondo, M.J.T.; Coroadinha, A.S. LentiPro26: Novel Stable Cell Lines for Constitutive Lentiviral Vector Production. Sci. Rep. 2018, 8, 5271. [Google Scholar] [CrossRef] [Green Version]
- Valkama, A.J.; Leinonen, H.M.; Lipponen, E.M.; Turkki, V.; Malinen, J.; Heikura, T.; Ylä-Herttuala, S.; Lesch, H.P. Optimization of Lentiviral Vector Production for Scale-up in Fixed-Bed Bioreactor. Gene Ther. 2018, 25, 39–46. [Google Scholar] [CrossRef] [Green Version]
- Bandeira, V.; Peixoto, C.; Rodrigues, A.F.; Cruz, P.E.; Alves, P.M.; Coroadinha, A.S.; Carrondo, M.J.T. Downstream Processing of Lentiviral Vectors: Releasing Bottlenecks. Hum. Gene Ther. Methods 2012, 23, 255–263. [Google Scholar] [CrossRef] [Green Version]
- Nestola, P.; Peixoto, C.; Silva, R.R.J.S.; Alves, P.M.; Mota, J.P.B.; Carrondo, M.J.T. Improved Virus Purification Processes for Vaccines and Gene Therapy: Improved Virus Purification Processes for Vaccines. Biotechnol. Bioeng. 2015, 112, 843–857. [Google Scholar] [CrossRef]
- Kramberger, P.; Urbas, L.; Štrancar, A. Downstream Processing and Chromatography Based Analytical Methods for Production of Vaccines, Gene Therapy Vectors, and Bacteriophages. Hum. Vaccines Immunother. 2015, 11, 1010–1021. [Google Scholar] [CrossRef]
- Nestola, P.; Martins, D.L.; Peixoto, C.; Roederstein, S.; Schleuss, T.; Alves, P.M.; Mota, J.P.B.; Carrondo, M.J.T. Evaluation of Novel Large Cut-Off Ultrafiltration Membranes for Adenovirus Serotype 5 (Ad5) Concentration. PLoS ONE 2014, 9, e115802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- FDA. Sterile Drug Products Produced by Aseptic Processing—Current Good Manufacturing Practice; Center for Drug Evaluation and Research, Food and Drug Administration: Rockville, MD, USA, 2004. [Google Scholar]
- Kon, T.C.; Onu, A.; Berbecila, L.; Lupulescu, E.; Ghiorgisor, A.; Kersten, G.F.; Cui, Y.-Q.; Amorij, J.-P.; Van der Pol, L. Influenza Vaccine Manufacturing: Effect of Inactivation, Splitting and Site of Manufacturing. Comparison of Influenza Vaccine Production Processes. PLoS ONE 2016, 11, e0150700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chou, A.-H.; Liu, C.-C.; Chang, C.-P.; Guo, M.-S.; Hsieh, S.-Y.; Yang, W.-H.; Chao, H.-J.; Wu, C.-L.; Huang, J.-L.; Lee, M.-S.; et al. Pilot Scale Production of Highly Efficacious and Stable Enterovirus 71 Vaccine Candidates. PLoS ONE 2012, 7, e34834. [Google Scholar] [CrossRef] [PubMed]
- Shoaebargh, S.; Gough, I.; Fe Medina, M.; Smith, A.; van der Heijden, J.; Lichty, B.D.; Bell, J.C.; Latulippe, D.R. Sterile Filtration of Oncolytic Viruses: An Analysis of Effects of Membrane Morphology on Fouling and Product Recovery. J. Membr. Sci. 2018, 548, 239–246. [Google Scholar] [CrossRef]
- Ausubel, L.J.; Hall, C.; Sharma, A.; Shakeley, R.; Lopez, P.; Quezada, V.; Couture, S.; Laderman, K.; McMahon, R.; Huang, P.; et al. Production of CGMP-Grade Lentiviral Vectors. BioProcess Int. 2012, 10, 32–43. [Google Scholar]
- Truran, R.; Buckley, R.; Radcliffe, P.; Miskin, J.; Mitrophanous, K. Virus Purification. U.S. Patent US 9,169,491 B2, 27 October 2015. [Google Scholar]
- Mundle, S.T.; Hernandez, H.; Hamberger, J.; Catalan, J.; Zhou, C.; Stegalkina, S.; Tiffany, A.; Kleanthous, H.; Delagrave, S.; Anderson, S.F. High-Purity Preparation of HSV-2 Vaccine Candidate ACAM529 Is Immunogenic and Efficacious In Vivo. PLoS ONE 2013, 8, e57224. [Google Scholar] [CrossRef] [Green Version]
- Tang, V.A.; Renner, T.M.; Varette, O.; Le Boeuf, F.; Wang, J.; Diallo, J.-S.; Bell, J.C.; Langlois, M.-A. Single-Particle Characterization of Oncolytic Vaccinia Virus by Flow Virometry. Vaccine 2016, 34, 5082–5089. [Google Scholar] [CrossRef]
- Besnard, L.; Fabre, V.; Fettig, M.; Gousseinov, E.; Kawakami, Y.; Laroudie, N.; Scanlan, C.; Pattnaik, P. Clarification of Vaccines: An Overview of Filter Based Technology Trends and Best Practices. Biotechnol. Adv. 2016, 34, 1–13. [Google Scholar] [CrossRef]
- Dishari, S.K.; Micklin, M.R.; Sung, K.-J.; Zydney, A.L.; Venkiteshwaran, A.; Earley, J.N. Effects of Solution Conditions on Virus Retention by the Viresolve® NFP Filter. Biotechnol. Prog. 2015, 31, 1280–1286. [Google Scholar] [CrossRef] [Green Version]
- Arkhangelsky, E.; Gitis, V. Effect of Transmembrane Pressure on Rejection of Viruses by Ultrafiltration Membranes. Sep. Purif. Technol. 2008, 62, 619–628. [Google Scholar] [CrossRef]
- Wickramasinghe, S.R.; Stump, E.D.; Grzenia, D.L.; Husson, S.M.; Pellegrino, J. Understanding Virus Filtration Membrane Performance. J. Membr. Sci. 2010, 365, 160–169. [Google Scholar] [CrossRef]
- Konz, J.O.; Lee, A.L.; Lewis, J.A.; Sagar, S.L. Development of a Purification Process for Adenovirus: Controlling Virus Aggregation to Improve the Clearance of Host Cell DNA. Biotechnol. Prog. 2005, 21, 466–472. [Google Scholar] [CrossRef] [PubMed]
- Wright, J.F.; Le, T.; Prado, J.; Bahr-Davidson, J.; Smith, P.H.; Zhen, Z.; Sommer, J.M.; Pierce, G.F.; Qu, G. Identification of Factors That Contribute to Recombinant AAV2 Particle Aggregation and Methods to Prevent Its Occurrence during Vector Purification and Formulation. Mol. Ther. 2005, 12, 171–178. [Google Scholar] [CrossRef] [PubMed]
- van Voorthuizen, E.M.; Ashbolt, N.J.; Schäfer, A.I. Role of Hydrophobic and Electrostatic Interactions for Initial Enteric Virus Retention by MF Membranes. J. Membr. Sci. 2001, 194, 69–79. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.-K.; Liu, B.Y.H. A Filtration Model of Microporous Membrane Filters in Liquids. KSME J. 1994, 8, 78–87. [Google Scholar] [CrossRef]
- Cliver, D.O. Virus Interactions with Membrane Filters. Biotechnol. Bioeng. 1968, 10, 877–889. [Google Scholar] [CrossRef]
- Meltzer, T.H.; Jornitz, M.W. The Sterilizing Filter and Its Pore Size Rating. Am. Pharm. Rev. 2003, 6, 1–6. [Google Scholar]
- Loh, S.; Beuscher, U.; Poddar, T.K.; Porter, A.G.; Wingard, J.M.; Husson, S.M.; Wickramasinghe, S.R. Interplay among Membrane Properties, Protein Properties and Operating Conditions on Protein Fouling during Normal-Flow Microfiltration. J. Membr. Sci. 2009, 332, 93–103. [Google Scholar] [CrossRef]
- Ho, C.-C.; Zydney, A.L. Protein Fouling of Asymmetric and Composite Microfiltration Membranes. Ind. Eng. Chem. Res. 2001, 40, 1412–1421. [Google Scholar] [CrossRef]
- Mocé-Llivina, L.; Jofre, J.; Muniesa, M. Comparison of Polyvinylidene Fluoride and Polyether Sulfone Membranes in Filtering Viral Suspensions. J. Virol. Methods 2003, 109, 99–101. [Google Scholar] [CrossRef]
- Lukasik, J.; Scott, T.M.; Andryshak, D.; Farrah, S.R. Influence of Salts on Virus Adsorption to Microporous Filters. Appl. Environ. Microbiol. 2000, 66, 2914–2920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kissmann, J.; Ausar, S.F.; Rudolph, A.; Braun, C.; Cape, S.P.; Sievers, R.E.; Federspiel, M.J.; Joshi, S.B.; Middaugh, C.R. Stabilization of Measles Virus for Vaccine Formulation. Hum. Vaccines 2008, 4, 350–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cruz, P.E.; Silva, A.C.; Roldao, A.; Carmo, M.; Carrondo, M.J.T.; Alves, P.M. Screening of Novel Excipients for Improving the Stability of Retroviral and Adenoviral Vectors. Biotechnol. Prog. 2006, 22, 568–576. [Google Scholar] [CrossRef] [PubMed]
- Wright, J. Product-Related Impurities in Clinical-Grade Recombinant AAV Vectors: Characterization and Risk Assessment. Biomedicines 2014, 2, 80–97. [Google Scholar] [CrossRef]
- Reiter, K.; Suzuki, M.; Olano, L.R.; Narum, D.L. Host Cell Protein Quantification of an Optimized Purification Method by Mass Spectrometry. J. Pharm. Biomed. Anal. 2019, 174, 650–654. [Google Scholar] [CrossRef]
- Hebben, M. Downstream Bioprocessing of AAV Vectors: Industrial Challenges & Regulatory Requirements. Cell Gene Ther. Insights 2018, 4, 131–146. [Google Scholar] [CrossRef]
- Doneanu, C.; Xenopoulos, A.; Fadgen, K.; Murphy, J.; Skilton, S.J.; Prentice, H.; Stapels, M.; Chen, W. Analysis of Host-Cell Proteins in Biotherapeutic Proteins by Comprehensive Online Two-Dimensional Liquid Chromatography/Mass Spectrometry. mAbs 2012, 4, 24–44. [Google Scholar] [CrossRef] [Green Version]
- Nogal, B.; Chhiba, K.; Emery, J.C. Select Host Cell Proteins Coelute with Monoclonal Antibodies in Protein a Chromatography. Biotechnol. Prog. 2012, 28, 454–458. [Google Scholar] [CrossRef]
- Kornecki, M.; Mestmäcker, F.; Zobel-Roos, S.; Heikaus de Figueiredo, L.; Schlüter, H.; Strube, J. Host Cell Proteins in Biologics Manufacturing: The Good, the Bad, and the Ugly. Antibodies 2017, 6, 13. [Google Scholar] [CrossRef] [Green Version]
- Kelly, S.T.; Zydney, A.L. Protein Fouling during Microfiltration: Comparative Behavior of Different Model Proteins. Biotechnol. Bioeng. 1997, 55, 91–100. [Google Scholar] [CrossRef]
- Higuchi, A.; Komuro, A.; Hirano, K.; Yoon, B.O.; Hara, M.; Hirasaki, T.; Nishimoto, Y.; Yokogi, M.; Manabe, S.-I. Permeation of γ-Globulin through Microporous Membranes in the Presence of Trace DNA. J. Membr. Sci. 2001, 186, 9–18. [Google Scholar] [CrossRef]
- Tracey, E.M.; Davis, R.H. Protein Fouling of Track-Etched Polycarbonate Microfiltration Membranes. J. Colloid Interface Sci. 1994, 167, 104–116. [Google Scholar] [CrossRef]
- Kelly, S.T.; Senyo Opong, W.; Zydney, A.L. The Influence of Protein Aggregates on the Fouling of Microfiltration Membranes during Stirred Cell Filtration. J. Membr. Sci. 1993, 80, 175–187. [Google Scholar] [CrossRef]
- Moleirinho, M.G.; Silva, R.J.S.; Alves, P.M.; Carrondo, M.J.T.; Peixoto, C. Current Challenges in Biotherapeutic Particles Manufacturing. Expert Opin. Biol. Ther. 2020, 20, 451–465. [Google Scholar] [CrossRef] [Green Version]
- Felt, S.A.; Grdzelishvili, V.Z. Recent Advances in Vesicular Stomatitis Virus-Based Oncolytic Virotherapy: A 5-Year Update. J. Gen. Virol. 2017, 98, 2895–2911. [Google Scholar] [CrossRef]
- Ge, P.; Tsao, J.; Schein, S.; Green, T.J.; Luo, M.; Zhou, Z.H. Cryo-EM Model of the Bullet-Shaped Vesicular Stomatitis Virus. Science 2010, 327, 689–693. [Google Scholar] [CrossRef] [Green Version]
- van den Pol, A.N.; Davis, J.N. Highly Attenuated Recombinant Vesicular Stomatitis Virus VSV-12’GFP Displays Immunogenic and Oncolytic Activity. J. Virol. 2013, 87, 1019–1034. [Google Scholar] [CrossRef] [Green Version]
- Valkama, A.J.; Oruetxebarria, I.; Lipponen, E.M.; Leinonen, H.M.; Käyhty, P.; Hynynen, H.; Turkki, V.; Malinen, J.; Miinalainen, T.; Heikura, T.; et al. Development of Large-Scale Downstream Processing for Lentiviral Vectors. Mol. Ther.-Methods Clin. Dev. 2020, 17, 717–730. [Google Scholar] [CrossRef]
- McNally, D.J.; Piras, B.A.; Willis, C.M.; Lockey, T.D.; Meagher, M.M. Development and Optimization of a Hydrophobic Interaction Chromatography-Based Method of AAV Harvest, Capture, and Recovery. Mol. Ther.-Methods Clin. Dev. 2020, 19, 275–284. [Google Scholar] [CrossRef]
- Moerdyk-Schauwecker, M.; Hwang, S.-I.; Grdzelishvili, V.Z. Analysis of Virion Associated Host Proteins in Vesicular Stomatitis Virus Using a Proteomics Approach. Virol. J. 2009, 6, 166. [Google Scholar] [CrossRef] [Green Version]
- Kawka, K.; Wilton, A.N.; Madadkar, P.; Medina, M.F.C.; Lichty, B.D.; Ghosh, R.; Latulippe, D.R. Integrated Development of Enzymatic DNA Digestion and Membrane Chromatography Processes for the Purification of Therapeutic Adenoviruses. Sep. Purif. Technol. 2021, 254, 117503. [Google Scholar] [CrossRef]
- Soderquist, R.G.; Trumbo, M.; Hart, R.A.; Zhang, Q.; Flynn, G.C. Development of Advanced Host Cell Protein Enrichment and Detection Strategies to Enable Process Relevant Spike Challenge Studies. Biotechnol. Prog. 2015, 31, 983–989. [Google Scholar] [CrossRef] [PubMed]
- Shukla, A.A.; Jiang, C.; Ma, J.; Rubacha, M.; Flansburg, L.; Lee, S.S. Demonstration of Robust Host Cell Protein Clearance in Biopharmaceutical Downstream Processes. Biotechnol. Prog. 2008, 24, 615–622. [Google Scholar] [CrossRef] [PubMed]
- Tarrant, R.D.R.; Velez-Suberbie, M.L.; Tait, A.S.; Smales, C.M.; Bracewell, D.G. Host Cell Protein Adsorption Characteristics during Protein a Chromatography. Biotechnol. Prog. 2012, 28, 1037–1044. [Google Scholar] [CrossRef]
- Hogwood, C.E.M.; Tait, A.S.; Koloteva-Levine, N.; Bracewell, D.G.; Smales, C.M. The Dynamics of the CHO Host Cell Protein Profile during Clarification and Protein A Capture in a Platform Antibody Purification Process. Biotechnol. Bioeng. 2013, 110, 240–251. [Google Scholar] [CrossRef]
- Cutler, M.W.; Kang, Y.; Ouattara, A.A.; Syvertsen, K.E. Purification Processes for Isolating Purified Vesicular Stomatitis Virus from Cell Culture. U.S. Patent No. 7,393,631 B2, 1 July 2008. [Google Scholar]
- Moleirinho, M.G.; Rosa, S.; Carrondo, M.J.T.; Silva, R.J.S.; Hagner-McWhirter, Å.; Ahlén, G.; Lundgren, M.; Alves, P.M.; Peixoto, C. Clinical-Grade Oncolytic Adenovirus Purification Using Polysorbate 20 as an Alternative for Cell Lysis. Curr. Gene Ther. 2018, 18, 366–374. [Google Scholar] [CrossRef]
- Thomas, C.R.; Geer, D. Effects of Shear on Proteins in Solution. Biotechnol. Lett. 2011, 33, 443–456. [Google Scholar] [CrossRef] [Green Version]
- Iritani, E. A Review on Modeling of Pore-Blocking Behaviors of Membranes During Pressurized Membrane Filtration. Dry. Technol. 2013, 31, 146–162. [Google Scholar] [CrossRef]
- Ho, C.-C.; Zydney, A.L. Effect of Membrane Morphology on the Initial Rate of Protein Fouling during Microfiltration. J. Membr. Sci. 1999, 155, 261–275. [Google Scholar] [CrossRef]
- Hlavacek, M.; Bouchet, F. Constant Flowrate Blocking Laws and an Example of Their Application to Dead-End Microfiltration of Protein Solutions. J. Membr. Sci. 1993, 82, 285–295. [Google Scholar] [CrossRef]
- Hahn, R.G.; Hatlen, J.B.; Kenny, G.E. Comparative Poliovirus Permeability of Silver, Polycarbonate, and Cellulose Membrane Filters. Appl. Microbiol. 1970, 19, 317–320. [Google Scholar] [CrossRef] [PubMed]
- Taylor, N.; Ma, W.; Kristopeit, A.; Wang, S.C.; Zydney, A.L. Evaluation of a sterile filtration process for viral vaccines using a model nanoparticle suspension. Biotechnol. Bioeng. 2021, 118, 106–115. [Google Scholar]
- Mahler, H.-C.; Huber, F.; Kishore, R.S.K.; Reindl, J.; Rückert, P.; Müller, R. Adsorption Behavior of a Surfactant and a Monoclonal Antibody to Sterilizing-Grade Filters. J. Pharm. Sci. 2010, 99, 2620–2627. [Google Scholar] [CrossRef] [PubMed]
- Carneiro, F.A.; Bianconi, M.L.; Weissmüller, G.; Stauffer, F.; Da Poian, A.T. Membrane Recognition by Vesicular Stomatitis Virus Involves Enthalpy-Driven Protein-Lipid Interactions. J. Virol. 2002, 76, 3756–3764. [Google Scholar] [CrossRef] [Green Version]
- Kumar, R.; Ismail, A.F. Fouling control on microfiltration/ultrafiltration membranes: Effects of morphology, hydrophilicity, and charge. J. Appl. Polym. Sci. 2015, 132, 42042. [Google Scholar] [CrossRef]
- Ma, N.; Cao, J.; Li, H.; Zhang, Y.; Wang, H.; Meng, J. Surface grafting of zwitterionic and PEGylated cross-linked polymers toward PVDF membranes with ultralow protein adsorption. Polymer 2019, 167, 1–12. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Starting Solution | VSV Titer (PFU/mL) | Spike | Protein (µg/mL) | DNA (ng/mL) |
|---|---|---|---|---|
| HIC VSV | 2.2 ± 0.2 × 108 | N/A | 24.5 ± 5.2 | 20.7 ± 0.75 |
| SG VSV | 2.4 ± 0.4 × 108 | N/A | 1.24 ± 0.88 | BDL |
| +HCDNA | 1.26 ± 0.58 | 24.6 ± 1.4 | ||
| +HCP | 24.8 ± 4.2 | BDL | ||
| +HCDNA +HCP | 23.2 ± 3.5 | 23.2 ± 1.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wright, E.; Kawka, K.; Medina, M.F.C.; Latulippe, D.R. Evaluation of Host Cell Impurity Effects on the Performance of Sterile Filtration Processes for Therapeutic Viruses. Membranes 2022, 12, 359. https://doi.org/10.3390/membranes12040359
Wright E, Kawka K, Medina MFC, Latulippe DR. Evaluation of Host Cell Impurity Effects on the Performance of Sterile Filtration Processes for Therapeutic Viruses. Membranes. 2022; 12(4):359. https://doi.org/10.3390/membranes12040359
Chicago/Turabian StyleWright, Evan, Karina Kawka, Maria Fe C. Medina, and David R. Latulippe. 2022. "Evaluation of Host Cell Impurity Effects on the Performance of Sterile Filtration Processes for Therapeutic Viruses" Membranes 12, no. 4: 359. https://doi.org/10.3390/membranes12040359
APA StyleWright, E., Kawka, K., Medina, M. F. C., & Latulippe, D. R. (2022). Evaluation of Host Cell Impurity Effects on the Performance of Sterile Filtration Processes for Therapeutic Viruses. Membranes, 12(4), 359. https://doi.org/10.3390/membranes12040359