
Effects of Hydration and Temperature on the Microstructure and Transport Properties of Nafion Polyelectrolyte Membrane: A Molecular Dynamics Simulation

 ,
,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
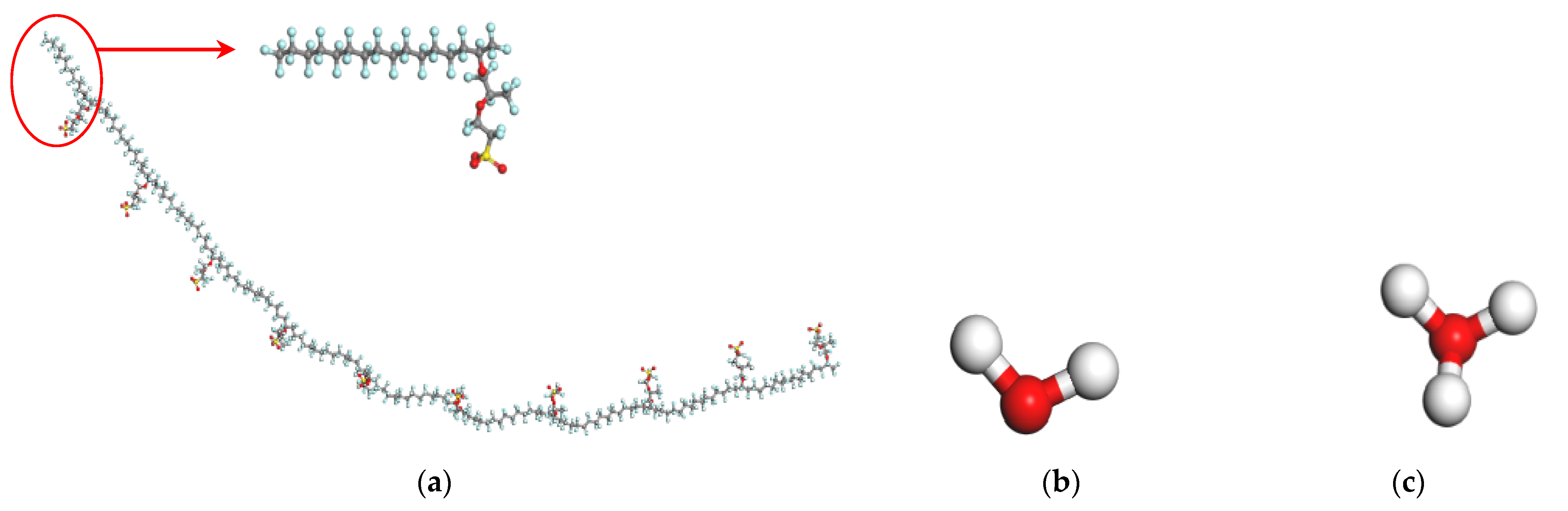
2.1. Atomistic Models and Amorphous Cell Construction
2.2. Force Field
2.3. Molecular Dynamic Simulation
- (a)
- MD simulation for 100 ps at 300 K in NVT ensemble.
- (b)
- MD simulation for 100 ps at 300 K in NPT ensemble.
- (c)
- The final structure of NPT MD simulation is heated from 300 K to 600 K, MD simulation is performed for 50 ps for every 100 K increase in temperature in the NVT ensemble.
- (d)
- The final structure of procedure (c) is cooled from 600 K to 300 K, MD simulation is performed for 50 ps for every 100 K decrease in temperature in NVT ensemble.
- (e)
- MD simulation for 100 ps at 300 K in NPT ensemble.
3. Results and Discussion
3.1. Density
3.2. Radial Distribution Functions and Coordination Numbers
3.3. Sizes and Connectivity of Water Clusters
3.4. Mean Square Displacements and Diffusion Coefficients
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Sazali, N.; Salleh, W.N.W.; Jamaludin, A.S.; Razali, M.N.M. New perspectives on fuel cell technology: A brief review. Membranes 2020, 10, 99. [Google Scholar] [CrossRef]
- Budak, Y.; Devrim, Y. Investigation of micro-combined heat and power application of PEM fuel cell systems. Energy Convers. Manag. 2018, 160, 486–494. [Google Scholar] [CrossRef]
- Ishimoto, T.; Koyama, M. A review of molecular-level mechanism of membrane degradation in the polymer electrolyte fuel cell. Membranes 2012, 2, 395–414. [Google Scholar] [CrossRef] [Green Version]
- Goh, J.; Rahim, A.A.; Masdar, M.; Shyuan, L. Enhanced performance of polymer electrolyte membranes via modification with ionic liquids for fuel cell applications. Membranes 2021, 11, 395. [Google Scholar] [CrossRef]
- Bunkin, N.F.; Gorelik, V.S.; Kozlov, V.A.; Shkirin, A.V.; Suyazov, N.V. Phase states of water near the surface of a polymer membrane. Phase microscopy and luminescence spectroscopy experiments. J. Exp. Theor. Phys. 2014, 119, 924–932. [Google Scholar] [CrossRef]
- Hink, S.; Wagner, N.; Bessler, W.G.; Roduner, E. Impedance spectroscopic investigation of proton conductivity in Nafion using transient electrochemical atomic force microscopy (AFM). Membranes 2012, 2, 237–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, D.R.; Sun, X. Water-sorption and transport properties of Nafion 117 H. J. Appl. Polym. Sci. 1993, 50, 1445–1452. [Google Scholar] [CrossRef]
- Zawodzinski, T.A.; Davey, J.; Valerio, J.; Gottesfeld, S. The water content dependence of electro-osmotic drag in proton-conducting polymer electrolytes. Electrochim. Acta 1995, 40, 297–302. [Google Scholar] [CrossRef]
- Okada, T.; Xie, G.; Meeg, M. Simulation for water management in membranes for polymer electrolyte fuel cells. Electrochim. Acta 1998, 43, 2141–2155. [Google Scholar] [CrossRef]
- Cheng, R.H.; Cai, H.H.; Huang, Y.R.; Cui, X.H.; Chen, Z.; Chen, H.Y.; Ding, S.W. A broad-range variable-temperature solid state NMR spectral and relaxation investigation of the water state in Nafion 117. Phys. Chem. Chem. Phys. 2021, 23, 10899–10908. [Google Scholar] [CrossRef]
- Bahlakeh, G.; Nikazar, M.; Hafezi, M.-J.; Hasani-Sadrabadi, M.M. Investigation of the effects of methanol presence on characteristics of sulfonated aromatic electrolyte membranes: Molecular dynamics simulations. J. Power Sources 2013, 243, 935–945. [Google Scholar] [CrossRef]
- Zheng, C.; Geng, F.; Rao, Z. Proton mobility and thermal conductivities of fuel cell polymer membranes: Molecular dynamics simulation. Comput. Mater. Sci. 2017, 132, 55–61. [Google Scholar] [CrossRef]
- Yana, J.; Nimmanpipug, P.; Chirachanchai, S.; Gosalawit, R.; Dokmaisrijan, S.; Vannarat, S.; Vilaithong, T.; Lee, V.S. Molecular dynamics simulations of Krytox-Silica–Nafion composite for high temperature fuel cell electrolyte membranes. Polymer 2010, 51, 4632–4638. [Google Scholar] [CrossRef]
- Devanathan, R.; Idupulapati, N.; Dupuis, M. Molecular modeling of the morphology and transport properties of two direct methanol fuel cell membranes: Phenylated sulfonated poly (ether ether ketone ketone) versus Nafion. J. Mater. Res. 2012, 27, 1927–1938. [Google Scholar] [CrossRef]
- Rao, Z.; Zheng, C.; Geng, F. Proton conduction of fuel cell polymer membranes: Molecular dynamics simulation. Comput. Mater. Sci. 2018, 142, 122–128. [Google Scholar] [CrossRef]
- Wang, R.; Liu, S.; Wang, L.; Li, M.; Gao, C. Understanding of nanophase separation and hydrophilic morphology in Nafion and SPEEK membranes: A combined experimental and theoretical studies. Nanomaterials 2019, 9, 869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sengupta, S.; Lyulin, A.V. Molecular modeling of structure and dynamics of Nafion protonation states. J. Phys. Chem. B 2019, 123, 6882–6891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; He, Y.L.; Tao, W.Q. The temperature effect on the diffusion processes of water and proton in the proton exchange membrane using molecular dynamics simulation. Numer. Heat Transf. Part A 2013, 65, 216–228. [Google Scholar] [CrossRef]
- Cui, S.; Liu, J.; Selvan, M.E.; Keffer, D.J.; Edwards, B.J.; Steele, W.V. A molecular dynamics study of a Nafion polyelectrolyte membrane and the aqueous phase structure for proton transport. J. Phys. Chem. B 2007, 111, 2208–2218. [Google Scholar] [CrossRef] [PubMed]
- Cui, S.; Liu, J.; Selvan, M.E.; Paddison, S.; Keffer, D.J.; Edwards, B. Comparison of the hydration and diffusion of protons in perfluorosulfonic acid membranes with molecular dynamics simulations. J. Phys. Chem. B 2008, 112, 13273–13284. [Google Scholar] [CrossRef]
- Jang, S.S.; Molinero, V.; Çaǧın, T.; Iii, W.A.G. Nanophase-segregation and transport in Nafion 117 from molecular dynamics simulations: Effect of monomeric sequence. J. Phys. Chem. B 2004, 108, 3149–3157. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Zhang, H.; Li, Z.-Z.; He, Y.-L.; Tao, W.-Q. Experimental and numerical study on thermal conductivity of proton exchange membrane. J. Nanosci. Nanotechnol. 2015, 15, 3087–3091. [Google Scholar] [CrossRef]
- Devanathan, R.; Dupuis, M. Insight from molecular modelling: Does the polymer side chain length matter for transport properties of perfluorosulfonic acid membranes? Phys. Chem. Chem. Phys. 2012, 14, 11281. [Google Scholar] [CrossRef]
- Cheng, C.-H.; Hong, C.-W. Investigation of atomistic scale transport phenomena of the proton exchange membrane fuel cell. J. Fuel Cell Sci. Technol. 2006, 4, 474–480. [Google Scholar] [CrossRef]
- Kreuer, K. On the development of proton conducting materials for technological applications. Solid State Ion. 1997, 97, 1–15. [Google Scholar] [CrossRef]
- Li, Z.-Z.; Chen, L.; Tao, W.-Q. Molecular dynamics simulation of water permeation through the Nafion membrane. Numer. Heat Transfer Part A Appl. 2016, 70, 1232–1241. [Google Scholar] [CrossRef]
- Venkatnathan, A.; Devanathan, R.; Dupuis, M. Atomistic simulations of hydrated Nafion and temperature effects on hydronium ion mobility. J. Phys. Chem. B 2007, 111, 7234–7244. [Google Scholar] [CrossRef]
- Sunda, A.P.; Venkatnathan, A. Atomistic simulations of structure and dynamics of hydrated Aciplex polymer electrolyte membrane. Soft Matter 2012, 8, 10827–10836. [Google Scholar] [CrossRef]
- Oh, K.S.; Kim, N.H.; Park, S. Behaviour of water molecules in Nafion 117 for polymer electrolyte membrane fuel cell by molecular dynamics simulation. Mol. Simul. 2008, 34, 1237–1244. [Google Scholar] [CrossRef]
- Vishnyakov, A.; Neimark, A.V. Molecular simulation study of Nafion membrane solvation in water and methanol. J. Phys. Chem. B 2000, 104, 4471–4478. [Google Scholar] [CrossRef]
- Wang, Q.F.; Keffer, D.J.; Deng, S.X.; Mays, J. Structure and proton transport in proton exchange membranes based on cross-linked sulfonated poly (1, 3-cyclohexadiene) with varying local acid environment. Polymer 2013, 54, 2299–2307. [Google Scholar] [CrossRef]
- Han, K.W.; Ko, K.H.; Abu-Hakmeh, K.; Bae, C.; Sohn, Y.J.; Jang, S.S. Molecular dynamics simulation study of a polysulfone-based anion exchange membrane in comparison with the proton exchange membrane. J. Phys. Chem. C 2014, 118, 12577–12587. [Google Scholar] [CrossRef]
- Tse, Y.-L.S.; Herring, A.; Kim, K.; Voth, G.A. Molecular dynamics simulations of proton transport in 3M and Nafion perfluorosulfonic acid membranes. J. Phys. Chem. C 2013, 117, 8079–8091. [Google Scholar] [CrossRef]
- Savage, J.; Tse, Y.-L.S.; Voth, G.A. Proton transport mechanism of perfluorosulfonic acid membranes. J. Phys. Chem. C 2014, 118, 17436–17445. [Google Scholar] [CrossRef]
- Hristov, I.H.; Paddison, S.J.; Paul, R. Molecular modeling of proton transport in the short-side-chain perfluorosulfonic acid ionomer. J. Phys. Chem. B 2008, 112, 2937–2949. [Google Scholar] [CrossRef] [PubMed]
- Avci, A.H.; Messana, D.A.; Santoro, S.; Tufa, R.A.; Curcio, E.; di Profio, G.; Fontananova, E. Energy harvesting from brines by reverse electrodialysis using Nafion membranes. Membranes 2020, 10, 168. [Google Scholar] [CrossRef]
- Xiao, J.; Yan, D.; Li, J.; Li, Q.; Sun, H. Hydronium ions diffusion behavior in Nafion membrane by mesoscopic simulation. AIP Adv. 2018, 8, 075303. [Google Scholar] [CrossRef] [Green Version]
- Sun, H. COMPASS: An ab initio force-field optimized for condensed-phase applications overview with details on alkane and benzene compounds. J. Phys. Chem. B 1998, 102, 7338–7364. [Google Scholar] [CrossRef]
- Bunte, S.W.; Sun, H. Molecular modeling of energetic materials: The parameterization and validation of nitrate esters in the COMPASS force field. J. Phys. Chem. B 2000, 104, 2477–2489. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Ren, Y.; Tian, A.M.; Sun, H. COMPASS force field for 14 inorganic molecules, He, Ne, Ar, Kr, Xe, H2, O2, N2, NO, CO, CO2, NO2, CS2, and SO2, in liquid phases. J. Phys. Chem. B 2000, 104, 4951–4957. [Google Scholar] [CrossRef]
- Kwon, S.H.; Kang, H.; Sohn, Y.-J.; Lee, J.; Shim, S.; Lee, S.G. Molecular dynamics simulation study on the effect of perfluorosulfonic acid side chains on oxygen permeation in hydrated ionomers of PEMFCs. Sci. Rep. 2021, 11, 1–10. [Google Scholar] [CrossRef]
- Huang, Y.-R.; Chien, C.-T.; Chen, C.-L. A molecular dynamics simulation based investigation of the proton conductivity of anhydrous pyrazole doped poly (vinylphosphonic acid) composite system. Polymers 2020, 12, 2906. [Google Scholar] [CrossRef]
- Li, X.; Zhao, Y.; Wang, S.; Xie, X. Molecular dynamics simulation study of a polynorbornene-based polymer: A prediction of proton exchange membrane design and performance. Int. J. Hydrogen Energy 2016, 41, 16254–16263. [Google Scholar] [CrossRef]
- Sunda, A.; Venkatnathan, A. Molecular dynamics simulations of triflic acid and triflate ion/water mixtures: A proton conducting electrolytic component in fuel cells. J. Comput. Chem. 2011, 32, 3319–3328. [Google Scholar] [CrossRef]
- Holz, M.; Heil, S.R.; Sacco, A. Temperature-dependent self-diffusion coefficients of water and six selected molecular liquids for calibration in accurate 1H NMR PFG measurements. Phys. Chem. Chem. Phys. 2000, 2, 4740–4742. [Google Scholar] [CrossRef]
- Easteal, A.J.; Price, W.E.; Woolf, L.A. Diaphragm cell for high-temperature diffusion measurements. Tracer diffusion coefficients for water to 363 K. J. Chem. Soc. Faraday Trans. 1 Phys. Chem. Condens. Phases 1989, 85, 1091–1097. [Google Scholar] [CrossRef]
- Tuckerman, M.E.; Chandra, A.; Marx, D. Structure and dynamics of OH−(aq). Acc. Chem. Res. 2006, 39, 151–158. [Google Scholar] [CrossRef]










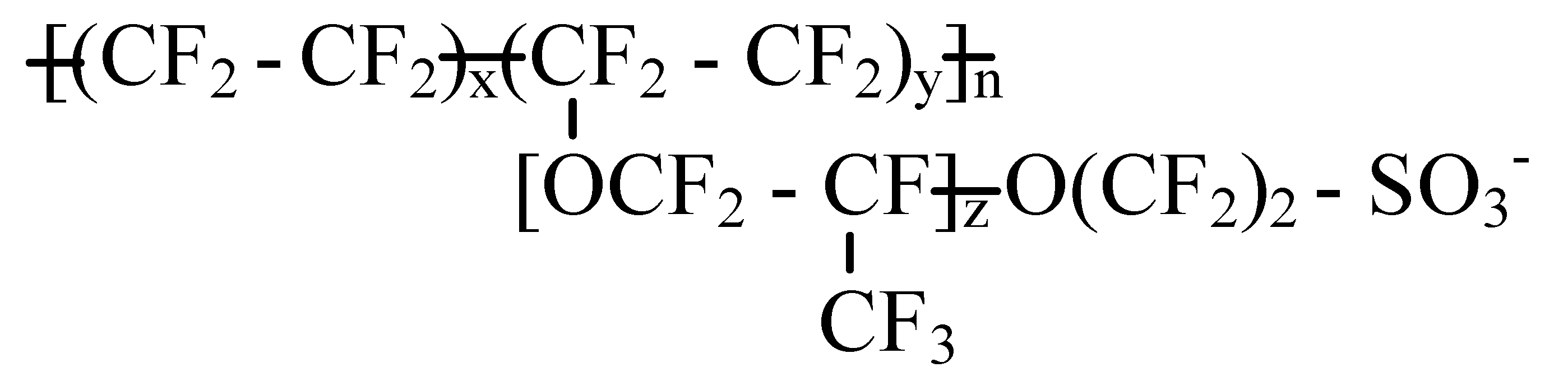{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| λ | No. of Nafion Chains | No. of Water Molecules | No. of Hydronium Ions |
|---|---|---|---|
| 3.5 | 4 | 100 | 40 |
| 7 | 4 | 240 | 40 |
| 10 | 4 | 360 | 40 |
| 13 | 4 | 480 | 40 |
| 16 | 4 | 600 | 40 |
| λ | CNs of Water(0–4.6 Å) | CNs of Hydronium Ion(0–4.25 Å) | ||||
|---|---|---|---|---|---|---|
| 298 K | 323 K | 353 K | 298 K | 323 K | 353 K | |
| 3.5 | 2.457 ± 0.075 | 2.375 ± 0.072 | 2.277 ± 0.082 | 2.316 ± 0.039 | 2.368 ± 0.054 | 2.387 ± 0.042 |
| 7 | 3.723 ± 0.078 | 3.504 ± 0.098 | 3.261 ± 0.066 | 2.028 ± 0.006 | 2.112 ± 0.013 | 2.206 ± 0.039 |
| 10 | 3.952 ± 0.085 | 3.866 ± 0.084 | 3.586 ± 0.078 | 1.888 ± 0.005 | 1.926 ± 0.007 | 2.005 ± 0.035 |
| 13 | 4.612 ± 0.080 | 4.477 ± 0.069 | 4.208 ± 0.069 | 1.749 ± 0.032 | 1.786 ± 0.057 | 1.862 ± 0.048 |
| 16 | 5.050 ± 0.074 | 4.872 ± 0.075 | 4.588 ± 0.066 | 1.570 ± 0.065 | 1.621 ± 0.025 | 1.654 ± 0.060 |
| λ | D of H2O (×10−5 cm2/s) | D of H3O+ (×10−6 cm2/s) | ||||
|---|---|---|---|---|---|---|
| 298 K | 323 K | 353 K | 298 K | 323 K | 353 K | |
| 3.5 | 0.132 ± 0.026 | 0.173 ± 0.028 | 0.319 ± 0.034 | 0.035 ± 0.014 | 0.085 ± 0.013 | 0.125 ± 0.018 |
| 7 | 0.458 ± 0.047 | 0.612 ± 0.061 | 0.829 ± 0.048 | 0.221 ± 0.041 | 0.435 ± 0.092 | 0.728 ± 0.118 |
| 10 | 0.964 ± 0.051 | 1.255 ± 0.070 | 1.645 ± 0.092 | 0.632 ± 0.021 | 0.826 ± 0.071 | 1.428 ± 0.151 |
| 13 | 1.288 ± 0.071 | 1.680 ± 0.055 | 2.036 ± 0.117 | 0.941 ± 0.103 | 1.821 ± 0.079 | 2.449 ± 0.177 |
| 16 | 1.594 ± 0.054 | 2.166 ± 0.043 | 2.925 ± 0.077 | 1.649 ± 0.149 | 2.449 ± 0.134 | 2.953 ± 0.226 |
| Bulk | 2.299 [45] | 3.983 [45] | 6.582 [46] | 93.1 [47] | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, G.; Yang, G.; Li, S.; Shen, Q.; Wang, H.; Li, Z.; Zhou, Y.; Ye, W. Effects of Hydration and Temperature on the Microstructure and Transport Properties of Nafion Polyelectrolyte Membrane: A Molecular Dynamics Simulation. Membranes 2021, 11, 695. https://doi.org/10.3390/membranes11090695
Zhang G, Yang G, Li S, Shen Q, Wang H, Li Z, Zhou Y, Ye W. Effects of Hydration and Temperature on the Microstructure and Transport Properties of Nafion Polyelectrolyte Membrane: A Molecular Dynamics Simulation. Membranes. 2021; 11(9):695. https://doi.org/10.3390/membranes11090695
Chicago/Turabian StyleZhang, Guoling, Guogang Yang, Shian Li, Qiuwan Shen, Hao Wang, Zheng Li, Yang Zhou, and Weiqiang Ye. 2021. "Effects of Hydration and Temperature on the Microstructure and Transport Properties of Nafion Polyelectrolyte Membrane: A Molecular Dynamics Simulation" Membranes 11, no. 9: 695. https://doi.org/10.3390/membranes11090695
APA StyleZhang, G., Yang, G., Li, S., Shen, Q., Wang, H., Li, Z., Zhou, Y., & Ye, W. (2021). Effects of Hydration and Temperature on the Microstructure and Transport Properties of Nafion Polyelectrolyte Membrane: A Molecular Dynamics Simulation. Membranes, 11(9), 695. https://doi.org/10.3390/membranes11090695