
Regulation of K+ Conductance by a Hydrogen Bond in Kv2.1, Kv2.2, and Kv1.2 Channels


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Transfection
2.2. Molecular Biology
2.3. Electrophysiology
2.4. Western Blot
2.5. Statistics
3. Results
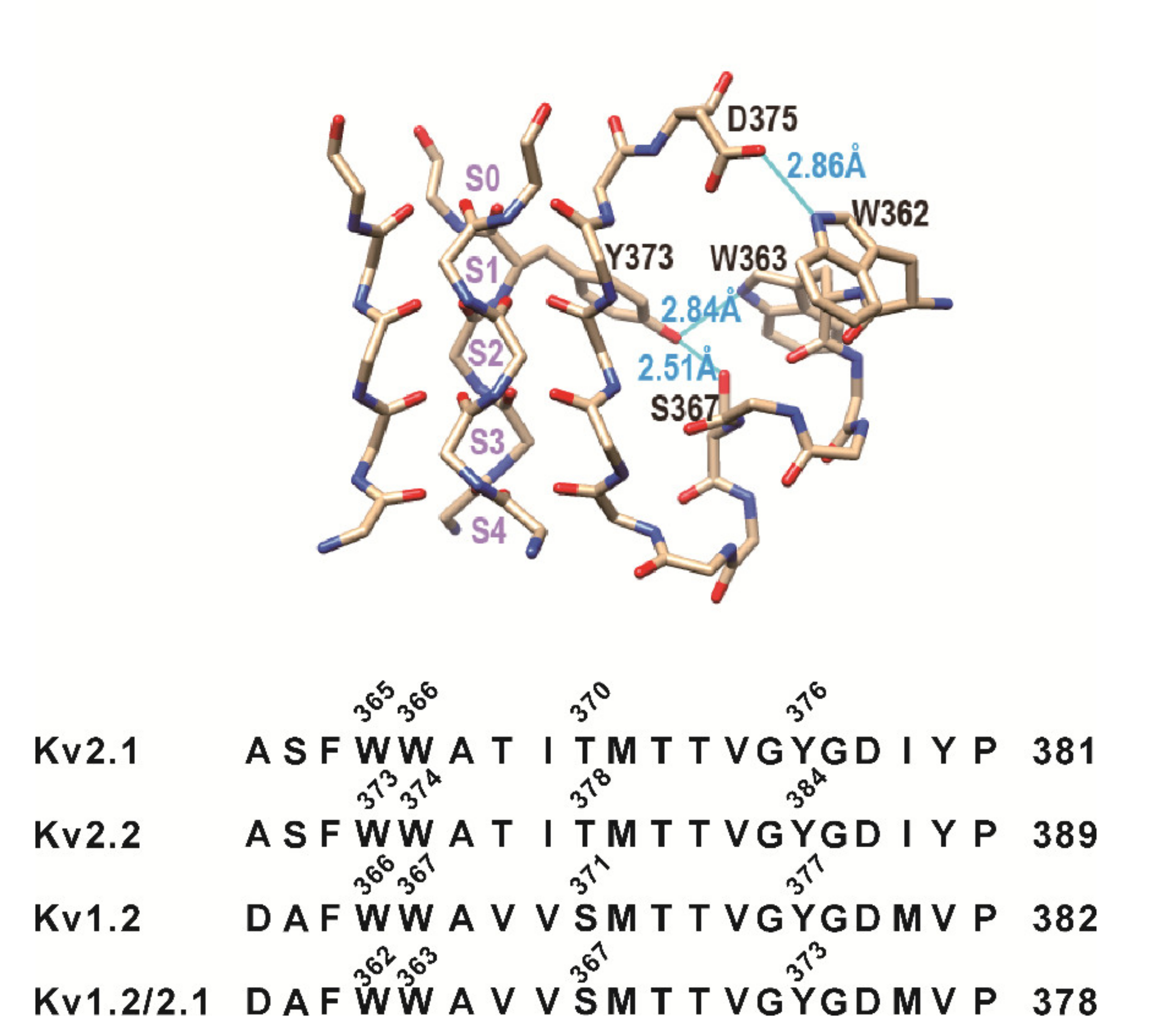
3.1. Mutation of the Residues Equivalent to W434 and T439 in Shaker Channels Did Not Change Slow Inactivation of Kv2.1, Kv2.2, and Kv1.2 Channels
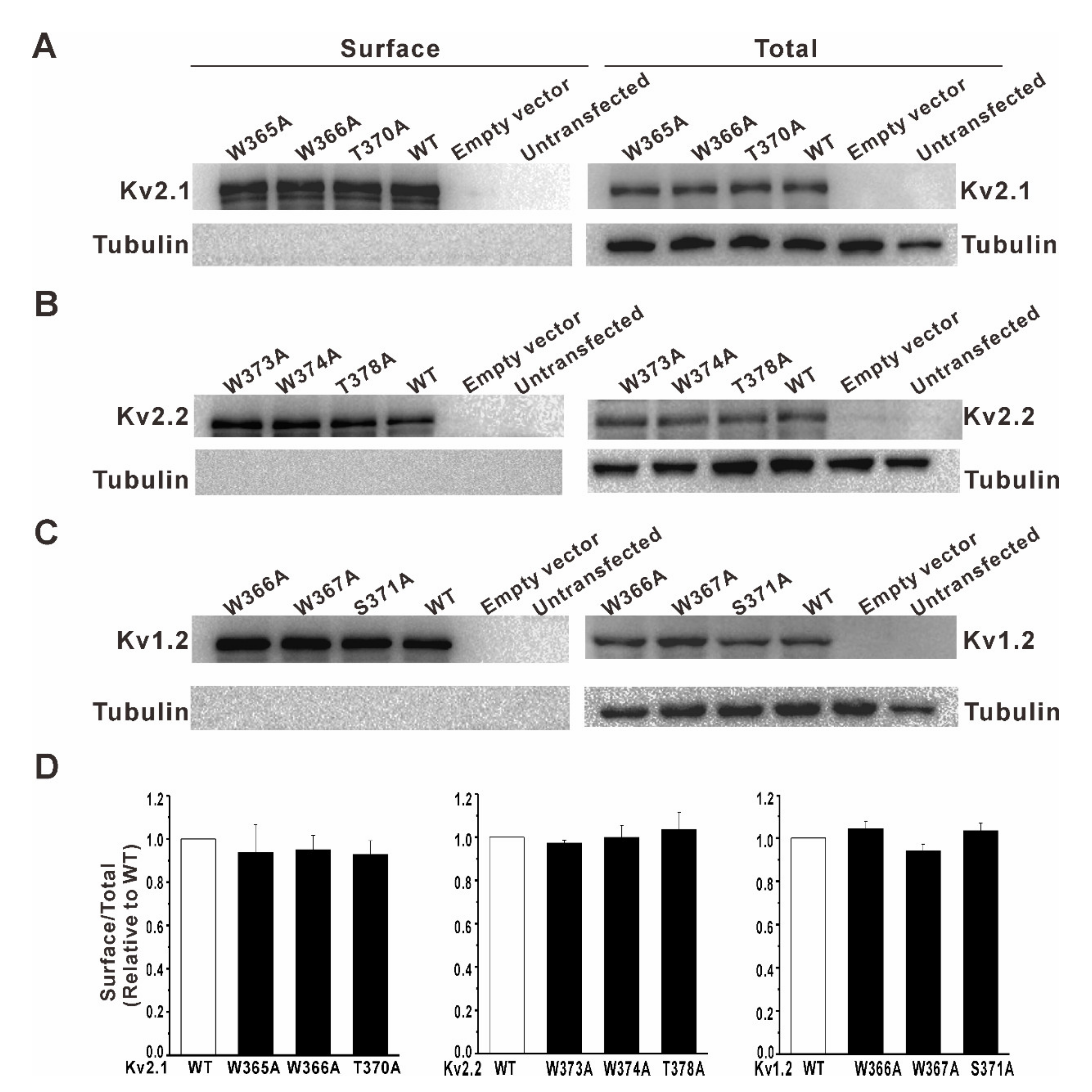
3.2. Mutation of a Conserved Tryptophan Residue Resulted in the Loss of Function of the Kv2.1, Kv2.2, and Kv1.2 Channels in HEK293 Cells
3.3. Y376F (Kv2.1), Y384F (Kv2.2), and Y377F (Kv1.2) Mutations Resulted in Complete Loss of K+ Conductance of the Kv Channels Expressed in the HEK293 Cells
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gutman, G.A.; Chandy, K.G.; Grissmer, S.; Lazdunski, M.; McKinnon, D.; Pardo, L.A.; Robertson, G.A.; Rudy, B.; Sanguinetti, M.C.; Stuhmer, W.; et al. International Union of Pharmacology. LIII. Nomenclature and molecular relationships of voltage-gated potassium channels. Pharmacol. Rev. 2005, 57, 473–508. [Google Scholar] [CrossRef]
- Shah, N.H.; Aizenman, E. Voltage-gated potassium channels at the crossroads of neuronal function, ischemic tolerance, and neurodegeneration. Transl. Stroke Res. 2014, 5, 38–58. [Google Scholar] [CrossRef] [PubMed]
- Hayabuchi, Y. The Action of Smooth Muscle Cell Potassium Channels in the Pathology of Pulmonary Arterial Hypertension. Pediatric Cardiol. 2017, 38, 1–14. [Google Scholar] [CrossRef]
- Hoshi, T.; Zagotta, W.N.; Aldrich, R.W. Two types of inactivation in Shaker K+ channels: Effects of alterations in the carboxy-terminal region. Neuron 1991, 7, 547–556. [Google Scholar] [CrossRef]
- Liu, Y.; Jurman, M.E.; Yellen, G. Dynamic rearrangement of the outer mouth of a K+ channel during gating. Neuron 1996, 16, 859–867. [Google Scholar] [CrossRef]
- Loots, E.; Isacoff, E.Y. Protein rearrangements underlying slow inactivation of the Shaker K+ channel. J. Gen. Physiol. 1998, 112, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Cordero-Morales, J.F.; Jogini, V.; Lewis, A.; Vasquez, V.; Cortes, D.M.; Roux, B.; Perozo, E. Molecular driving forces determining potassium channel slow inactivation. Nat. Struct. Mol. Biol. 2007, 14, 1062–1069. [Google Scholar] [CrossRef]
- Cuello, L.G.; Jogini, V.; Cortes, D.M.; Perozo, E. Structural mechanism of C-type inactivation in K+ channels. Nature 2010, 466, 203–208. [Google Scholar] [CrossRef]
- Cheng, W.W.; McCoy, J.G.; Thompson, A.N.; Nichols, C.G.; Nimigean, C.M. Mechanism for selectivity-inactivation coupling in KcsA potassium channels. Proc. Natl. Acad. Sci. USA 2011, 108, 5272–5277. [Google Scholar] [CrossRef] [PubMed]
- Cordero-Morales, J.F.; Jogini, V.; Chakrapani, S.; Perozo, E. A multipoint hydrogen-bond network underlying KcsA C-type inactivation. Biophys. J. 2011, 100, 2387–2393. [Google Scholar] [CrossRef]
- Perozo, E.; MacKinnon, R.; Bezanilla, F.; Stefani, E. Gating currents from a nonconducting mutant reveal open-closed conformations in Shaker K+ channels. Neuron 1993, 11, 353–358. [Google Scholar] [CrossRef]
- Yang, Y.; Yan, Y.; Sigworth, F.J. How does the W434F mutation block current in Shaker potassium channels? J. Gen. Physiol. 1997, 109, 779–789. [Google Scholar] [CrossRef] [PubMed]
- Molina, A.; Castellano, A.G.; Lopez-Barneo, J. Pore mutations in Shaker K+ channels distinguish between the sites of tetraethylammonium blockade and C-type inactivation. J. Physiol. 1997, 499 Pt 2, 361–367. [Google Scholar] [CrossRef]
- Molina, A.; Ortega-Saenz, P.; Lopez-Barneo, J. Pore mutations alter closing and opening kinetics in Shaker K+ channels. J. Physiol. 1998, 509 Pt 2, 327–337. [Google Scholar] [CrossRef]
- Pless, S.A.; Galpin, J.D.; Niciforovic, A.P.; Kurata, H.T.; Ahern, C.A. Hydrogen bonds as molecular timers for slow inactivation in voltage-gated potassium channels. eLife 2013, 2, e01289. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Barneo, J.; Hoshi, T.; Heinemann, S.H.; Aldrich, R.W. Effects of external cations and mutations in the pore region on C-type inactivation of Shaker potassium channels. Recept. Channels 1993, 1, 61–71. [Google Scholar] [PubMed]
- Rasmusson, R.L.; Morales, M.J.; Castellino, R.C.; Zhang, Y.; Campbell, D.L.; Strauss, H.C. C-type inactivation controls recovery in a fast inactivating cardiac K+ channel (Kv1.4) expressed in Xenopus oocytes. J. Physiol. 1995, 489 Pt 3, 709–721. [Google Scholar] [CrossRef]
- Fedida, D.; Maruoka, N.D.; Lin, S. Modulation of slow inactivation in human cardiac Kv1.5 channels by extra- and intracellular permeant cations. J. Physiol. 1999, 515 Pt 2, 315–329. [Google Scholar] [CrossRef]
- Doyle, D.A.; Morais Cabral, J.; Pfuetzner, R.A.; Kuo, A.; Gulbis, J.M.; Cohen, S.L.; Chait, B.T.; MacKinnon, R. The structure of the potassium channel: Molecular basis of K+ conduction and selectivity. Science 1998, 280, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Long, S.B.; Tao, X.; Campbell, E.B.; MacKinnon, R. Atomic structure of a voltage-dependent K+ channel in a lipid membrane-like environment. Nature 2007, 450, 376–382. [Google Scholar] [CrossRef]
- Al-Sabi, A.; Kaza, S.K.; Dolly, J.O.; Wang, J. Pharmacological characteristics of Kv1.1- and Kv1.2-containing channels are influenced by the stoichiometry and positioning of their α subunits. Biochem. J. 2013, 454, 101–108. [Google Scholar] [CrossRef]
- Abraham, M.J.; Fleming, K.L.; Raymond, S.; Wong, A.Y.C.; Bergeron, R. The sigma-1 receptor behaves as an atypical auxiliary subunit to modulate the functional characteristics of Kv1.2 channels expressed in HEK293 cells. Physiol. Rep. 2019, 7, e14147. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Yin, H.; Feng, N.; Wang, L.; Wang, X. Inhibitory effects of antidepressant fluoxetine on cloned Kv2.1 potassium channel expressed in HEK293 cells. Eur. J. Pharmacol. 2020, 878, 173097. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.; Sun, X.; Cao, K.; Wang, R. Endogenous KV channels in human embryonic kidney (HEK-293) cells. J. Neurosci. Res. 1998, 52, 612–617. [Google Scholar]
- He, B.; Soderlund, D.M. Human embryonic kidney (HEK293) cells express endogenous voltage-gated sodium currents and Nav1.7 sodium channels. Neurosci. Lett. 2010, 469, 268. [Google Scholar] [CrossRef] [PubMed]
- O’Connella, K.M.S.; Loftusa, R.; Tamkuna, M.M. Localization-dependent activity of the Kv2.1 delayed rectifier K+ channel. Proc. Natl. Acad. Sci. USA 2010, 107, 12351–12356. [Google Scholar] [CrossRef]
- Starkus, J.G.; Kuschel, L.; Rayner, M.D.; Heinemann, S.H. Ion conduction through C-type inactivated Shaker channels. Mol. Cell. Biochem. 2002, 238, 69–79. [Google Scholar] [CrossRef]
- Kiss, L.; LoTurco, J.; Korn, S.J. Contribution of the selectivity filter to inactivation in potassium channels. Biophys. J. 1999, 76, 253–263. [Google Scholar] [CrossRef]
- Starkus, J.G.; Kuschel, L.; Rayner, M.D.; Heinemann, S.H. Macroscopic Na+ currents in the “Nonconducting” Shaker potassium channel mutant W434F. J. Gen. Physiol. 1998, 112, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Yellen, G.; Sodickson, D.; Chen, T.Y.; Jurman, M.E. An engineered cysteine in the external mouth of a K+ channel allows inactivation to be modulated by metal binding. Biophys. J. 1994, 66, 1068–1075. [Google Scholar] [CrossRef]
- Wang, Z.; Wong, N.C.; Cheng, Y.; Kehl, S.J.; Fedida, D. Control of voltage-gated K+ channel permeability to NMDG+ by a residue at the outer pore. J. Gen. Physiol. 2009, 133, 361–374. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Zhang, X.; Liu, C.; Hu, C. Regulation of K+ Conductance by a Hydrogen Bond in Kv2.1, Kv2.2, and Kv1.2 Channels. Membranes 2021, 11, 190. https://doi.org/10.3390/membranes11030190
Zhang Y, Zhang X, Liu C, Hu C. Regulation of K+ Conductance by a Hydrogen Bond in Kv2.1, Kv2.2, and Kv1.2 Channels. Membranes. 2021; 11(3):190. https://doi.org/10.3390/membranes11030190
Chicago/Turabian StyleZhang, Yuchen, Xuefeng Zhang, Cuiyun Liu, and Changlong Hu. 2021. "Regulation of K+ Conductance by a Hydrogen Bond in Kv2.1, Kv2.2, and Kv1.2 Channels" Membranes 11, no. 3: 190. https://doi.org/10.3390/membranes11030190
APA StyleZhang, Y., Zhang, X., Liu, C., & Hu, C. (2021). Regulation of K+ Conductance by a Hydrogen Bond in Kv2.1, Kv2.2, and Kv1.2 Channels. Membranes, 11(3), 190. https://doi.org/10.3390/membranes11030190