
FluB-RAM and FluB-RANS: Genome Rearrangement as Safe and Efficacious Live Attenuated Influenza B Virus Vaccines

 ,
,  , ,
, ,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Eggs
2.2. Recombinant Plasmids
2.3. Rescue of FluB-RAM and FluB-RANS Viruses with Rearranged Genomes
2.4. Stability of FluB-RAM and FluB-RANS Viruses through Serial Passages in ECEs
2.5. Virus Growth Kinetics
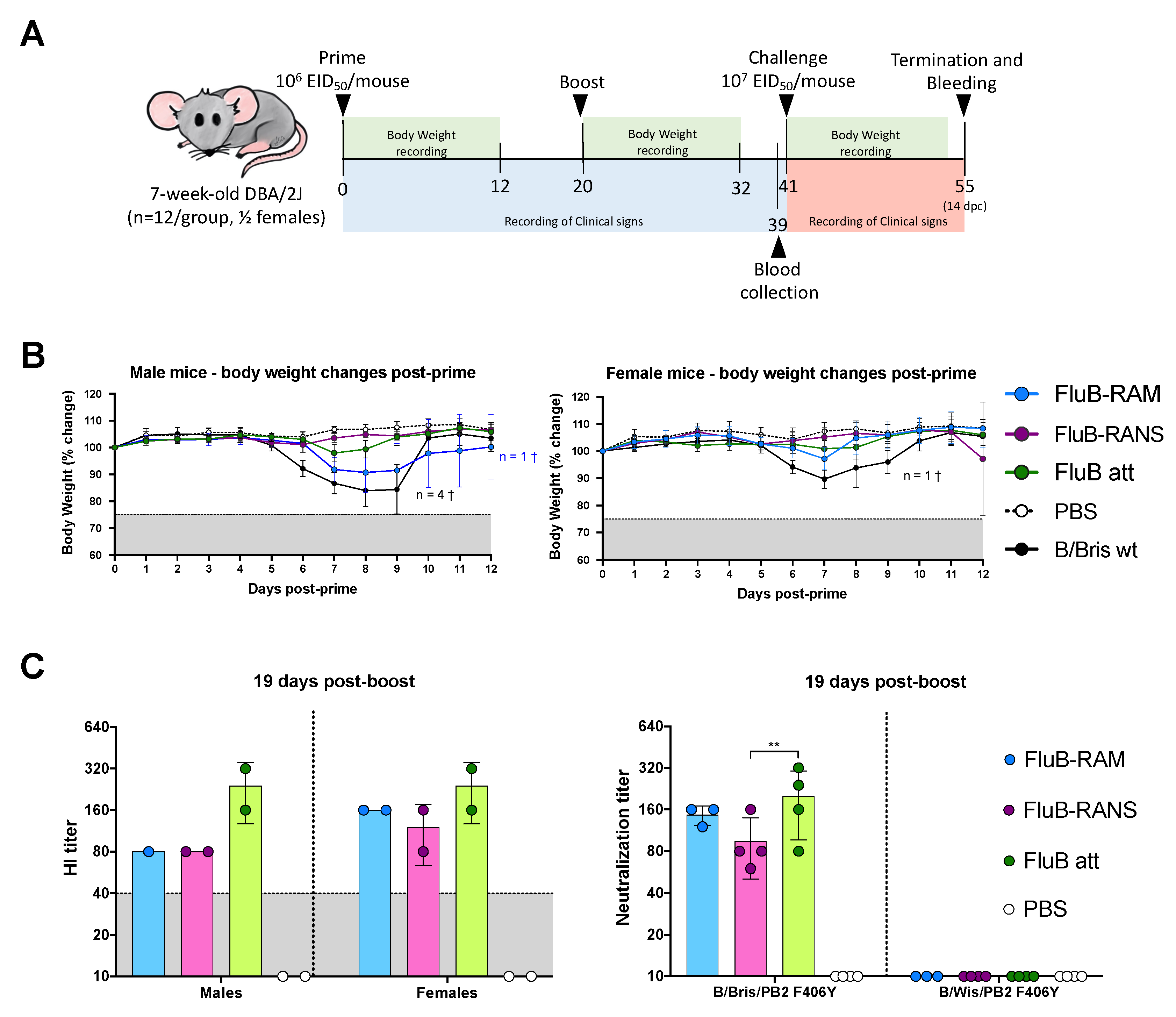
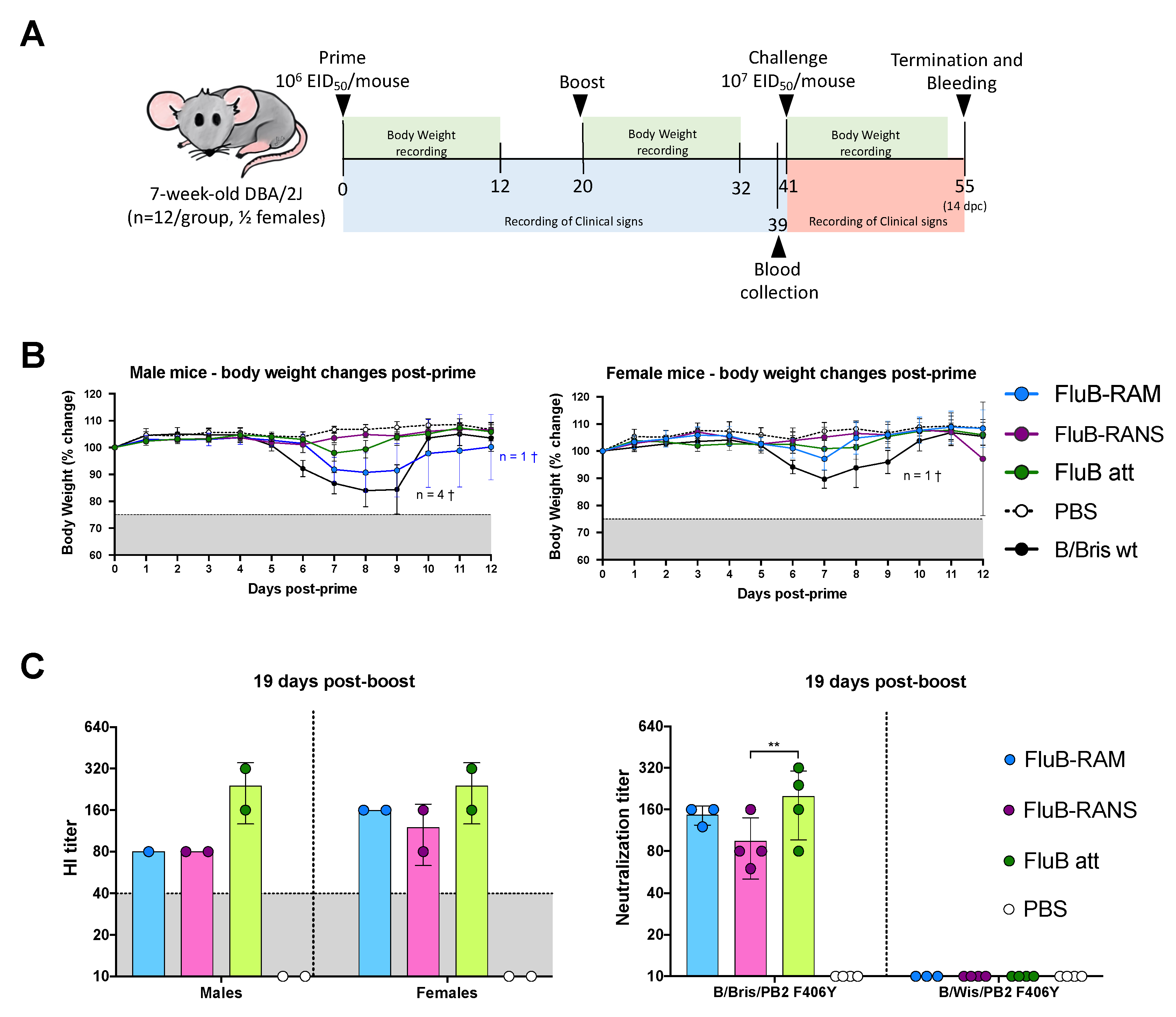
2.6. Mouse Studies
2.7. Vaccine Safety
2.8. Vaccine Efficacy
2.9. Hemagglutination Inhibition (HI) Assay
2.10. Virus Neutralization (VN) Assay
2.11. Microarray for IgG and IgA Determination
3. Results
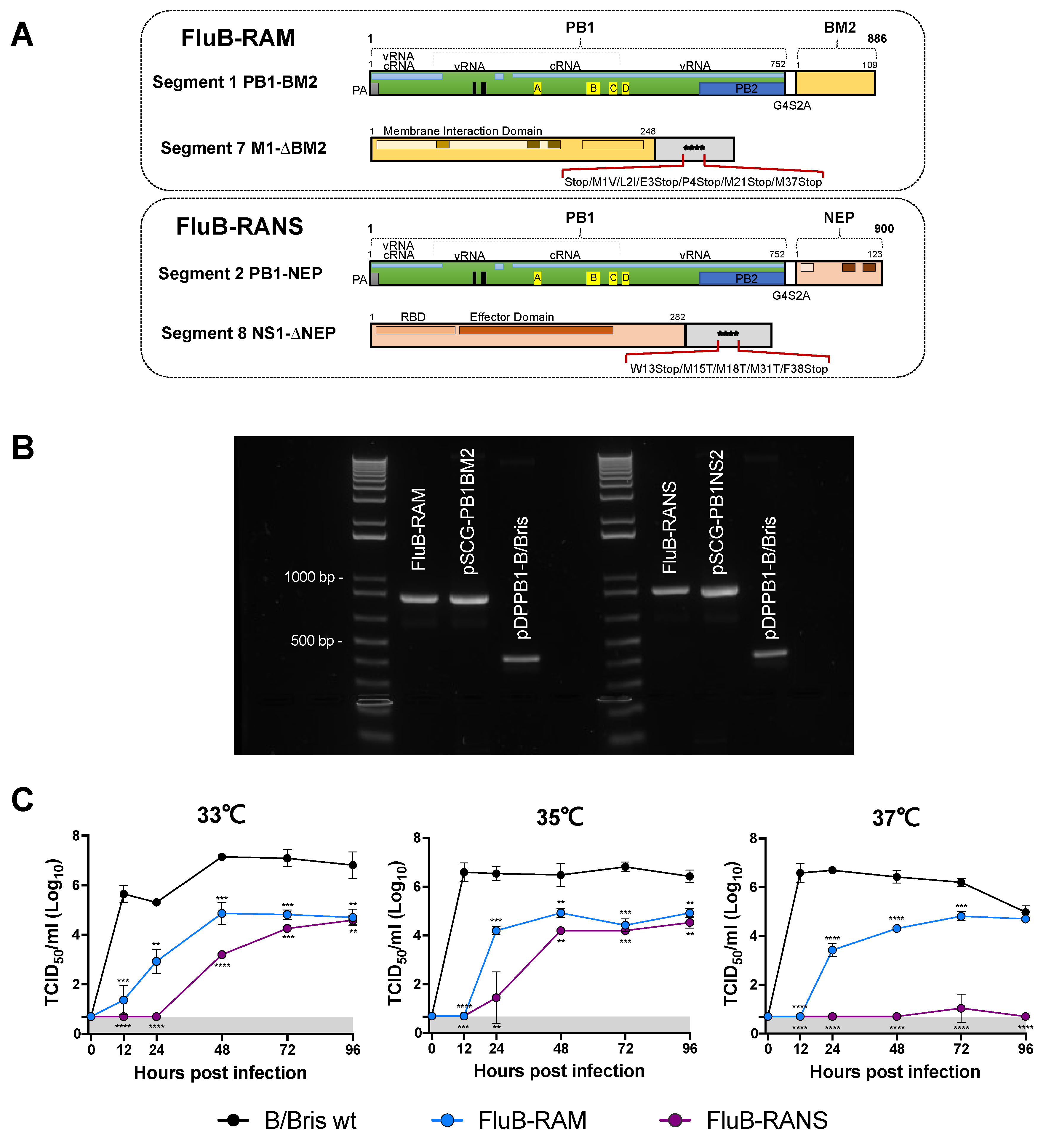
3.1. FluB-RAM and FluB-RANS Viruses with Rearranged Genomes
3.2. FluB-RAM and FluB-RANS Viruses Are Attenuated In Vitro
3.3. FluB-RAM and FluB-RANS Viruses Show Differences in Attenuation
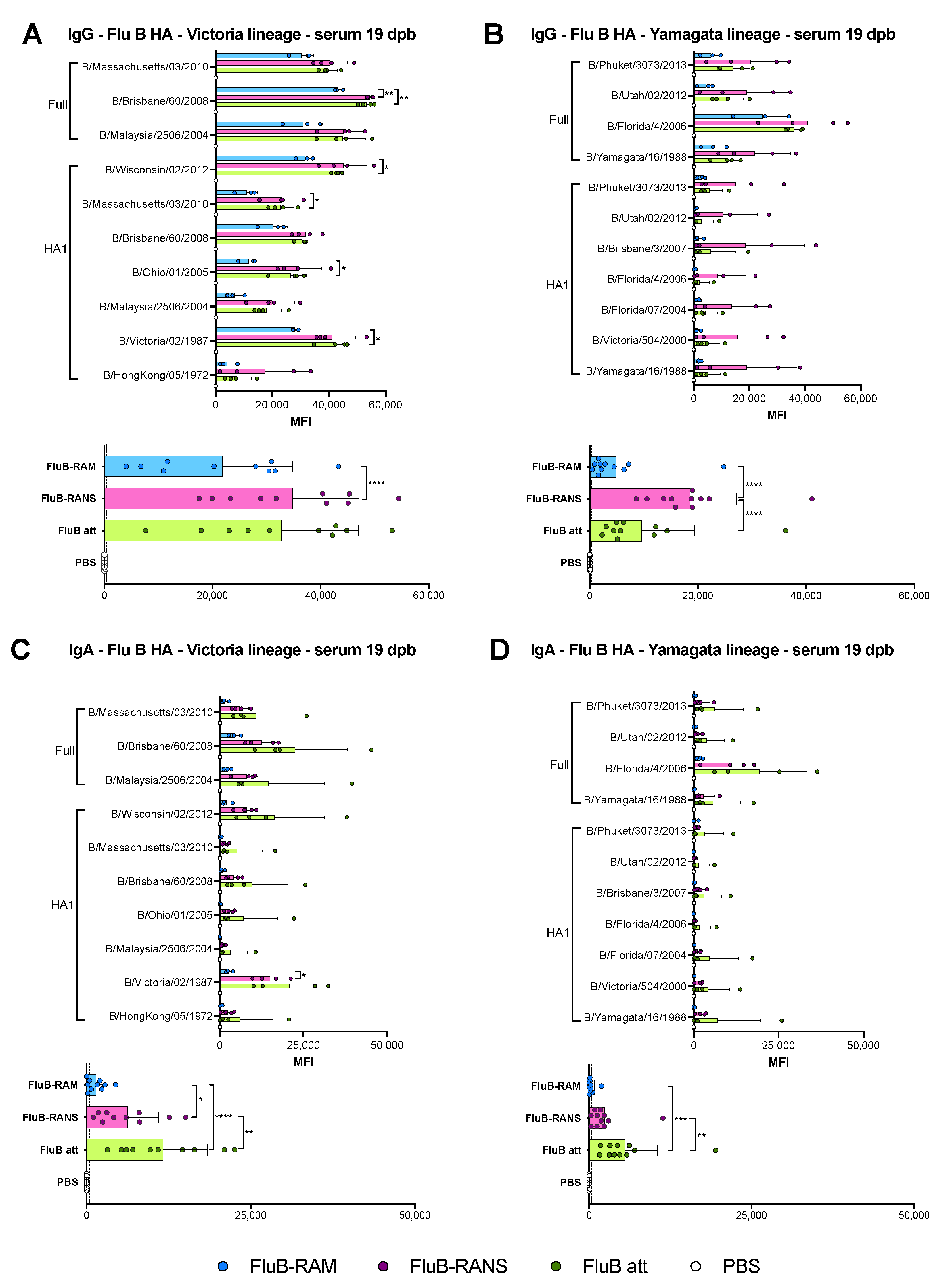
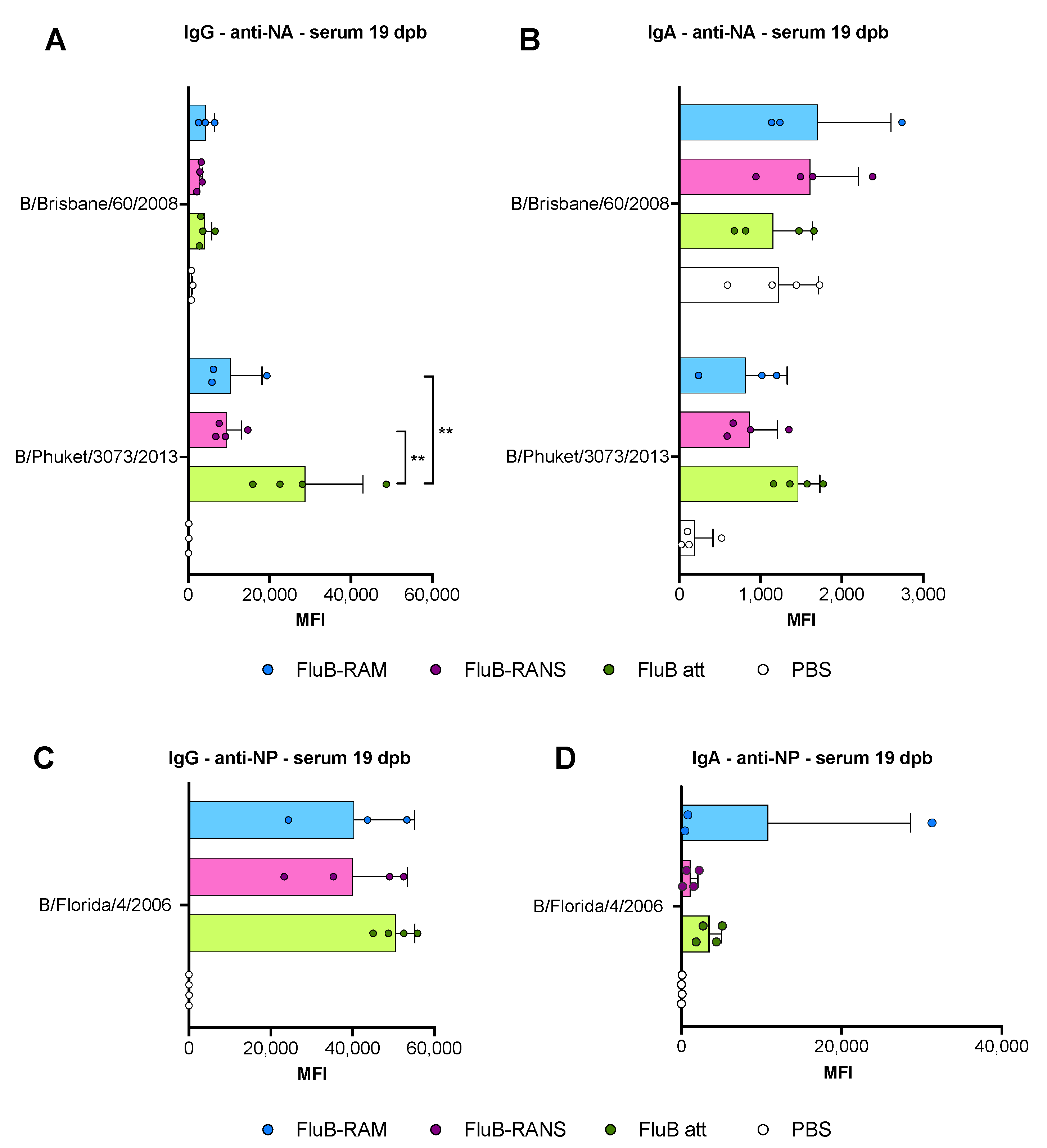
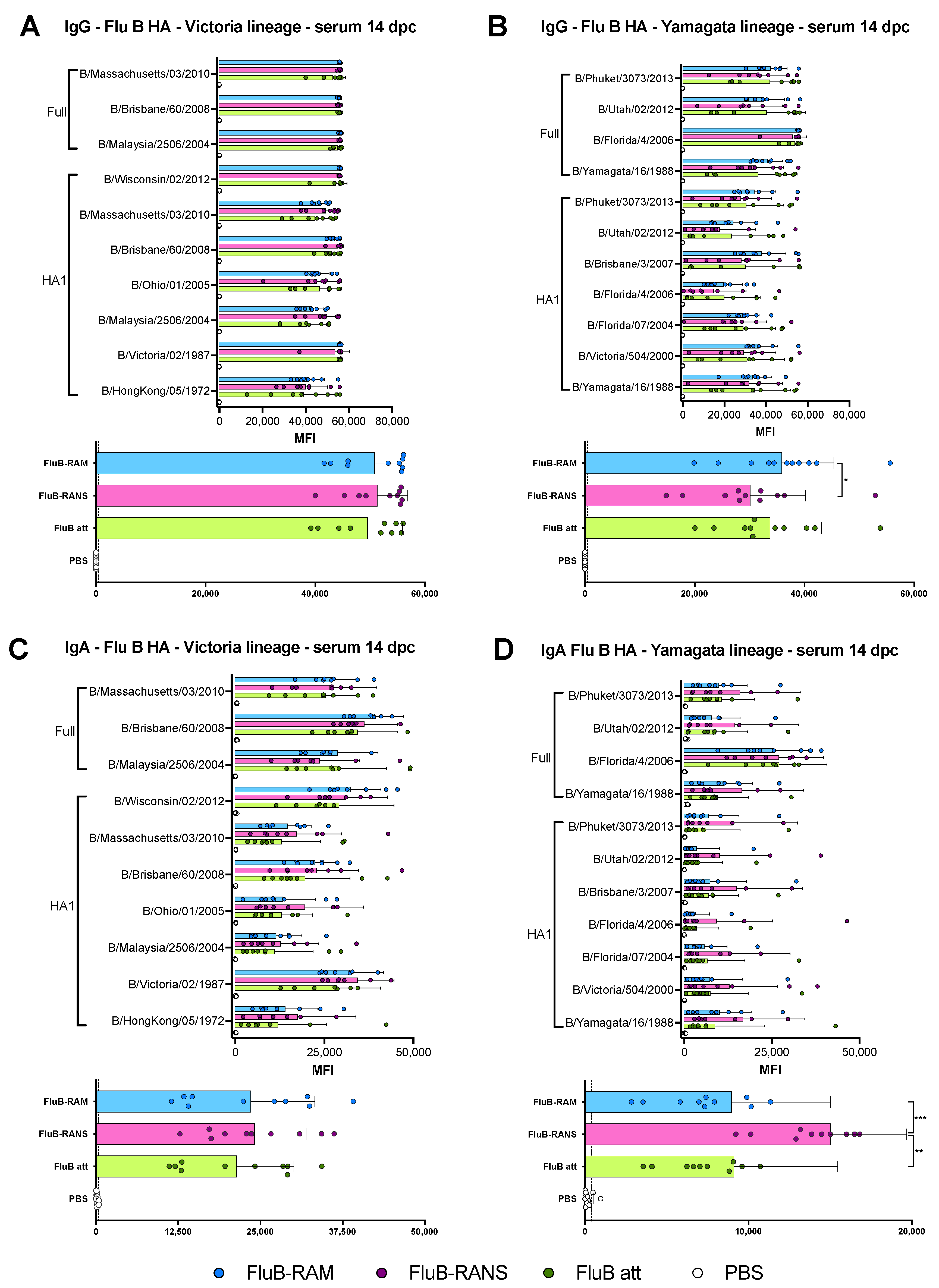
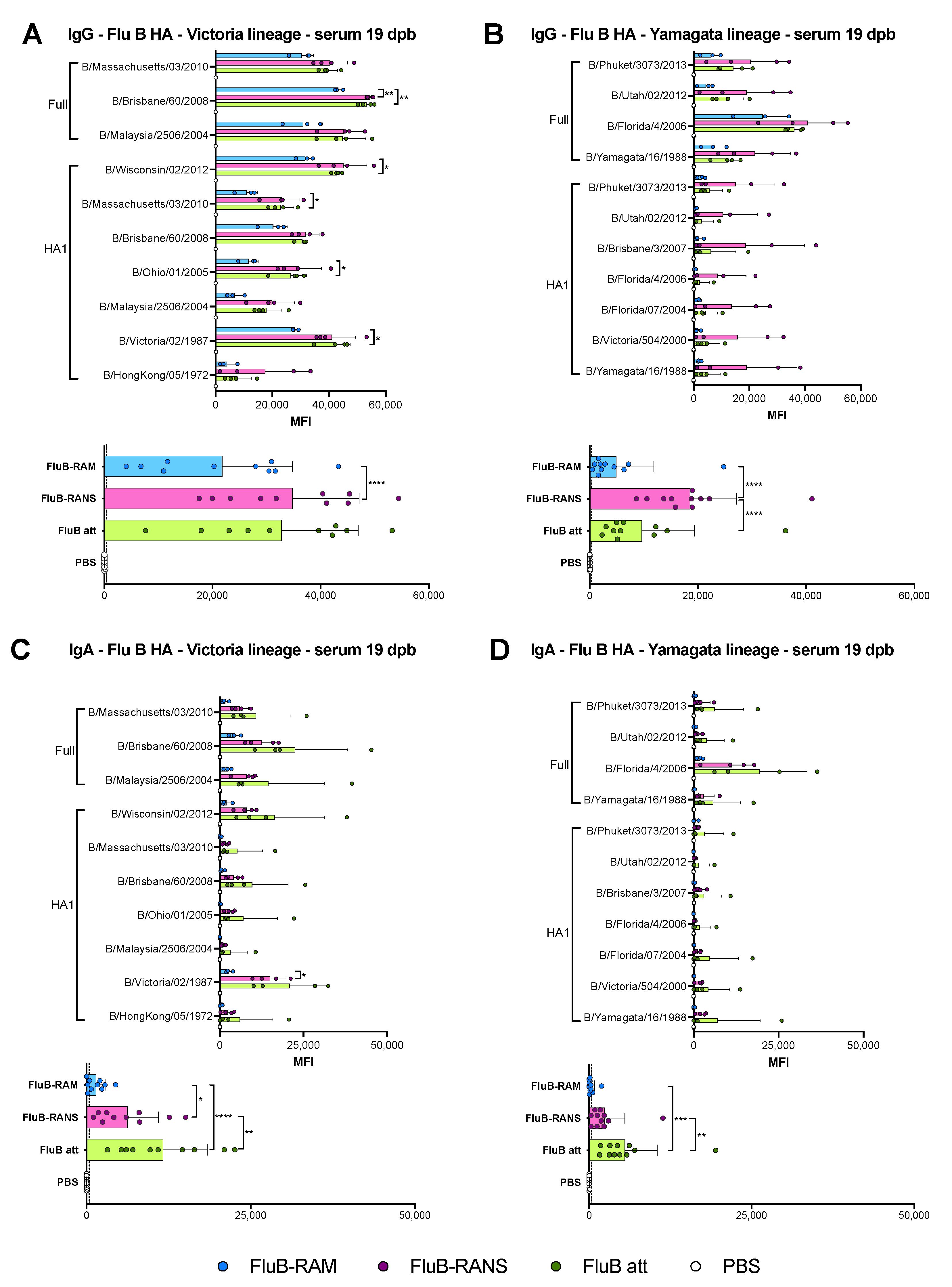
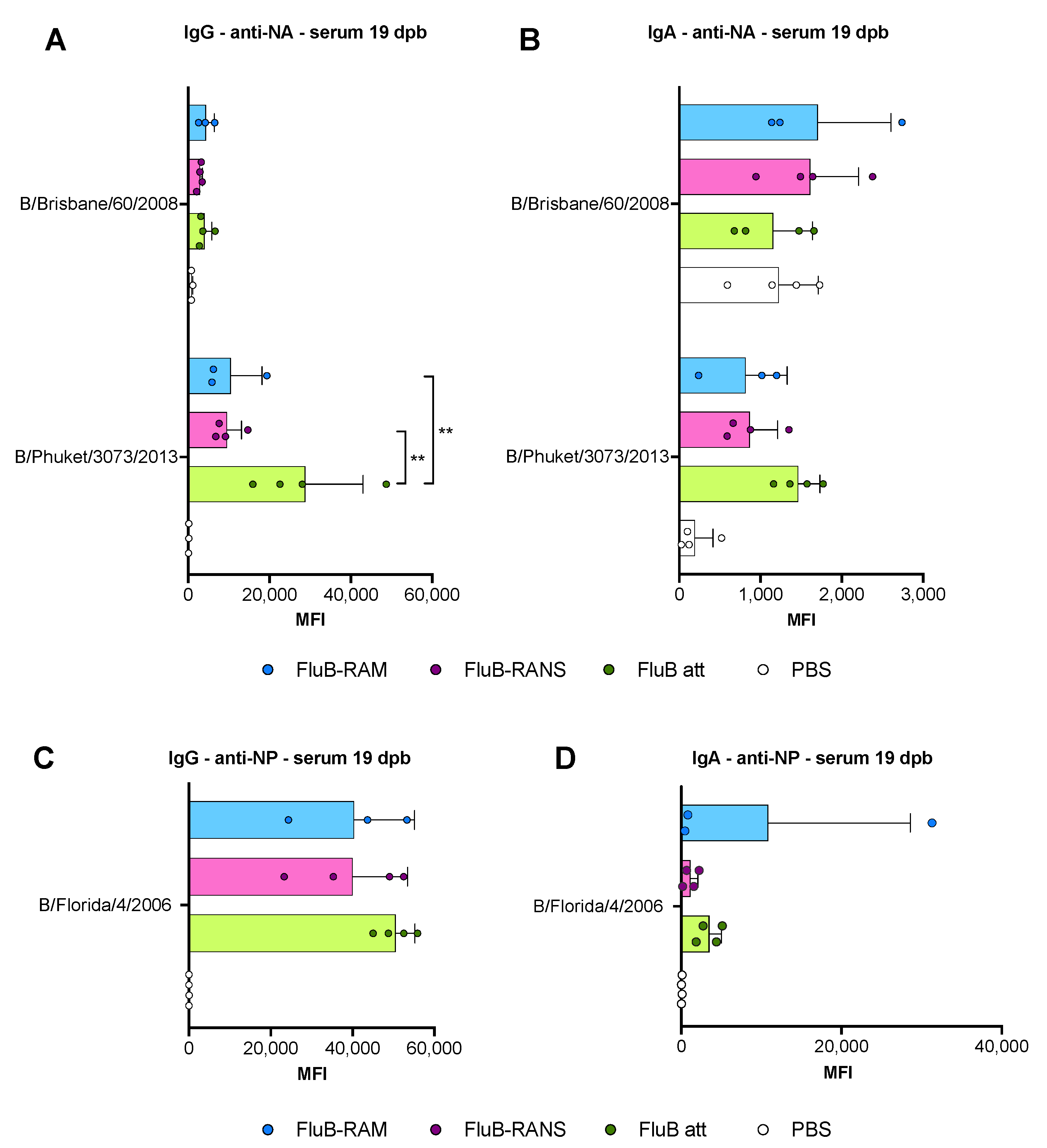
3.4. Qualitative Differences in Humoral Responses among Different Vaccine Groups
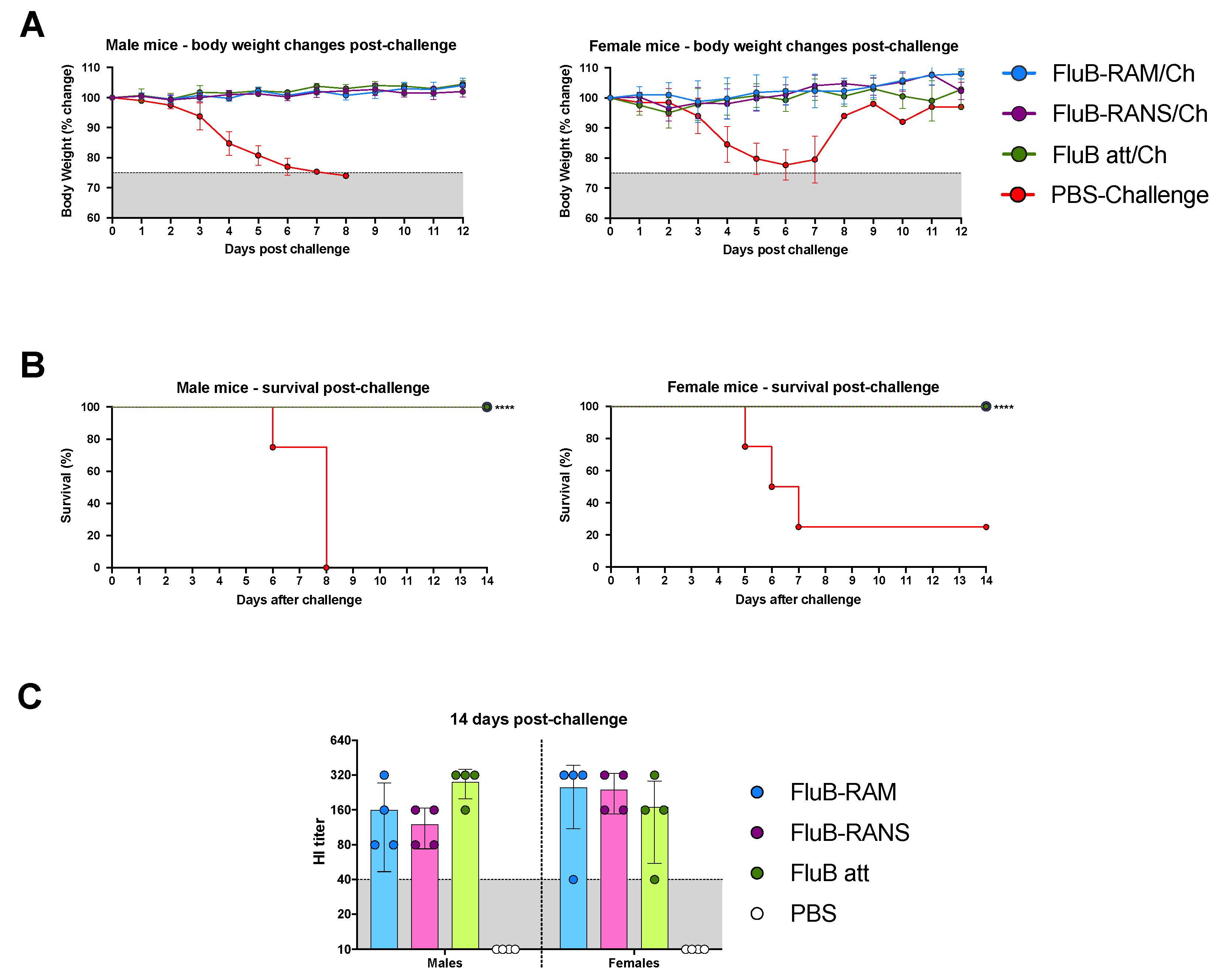
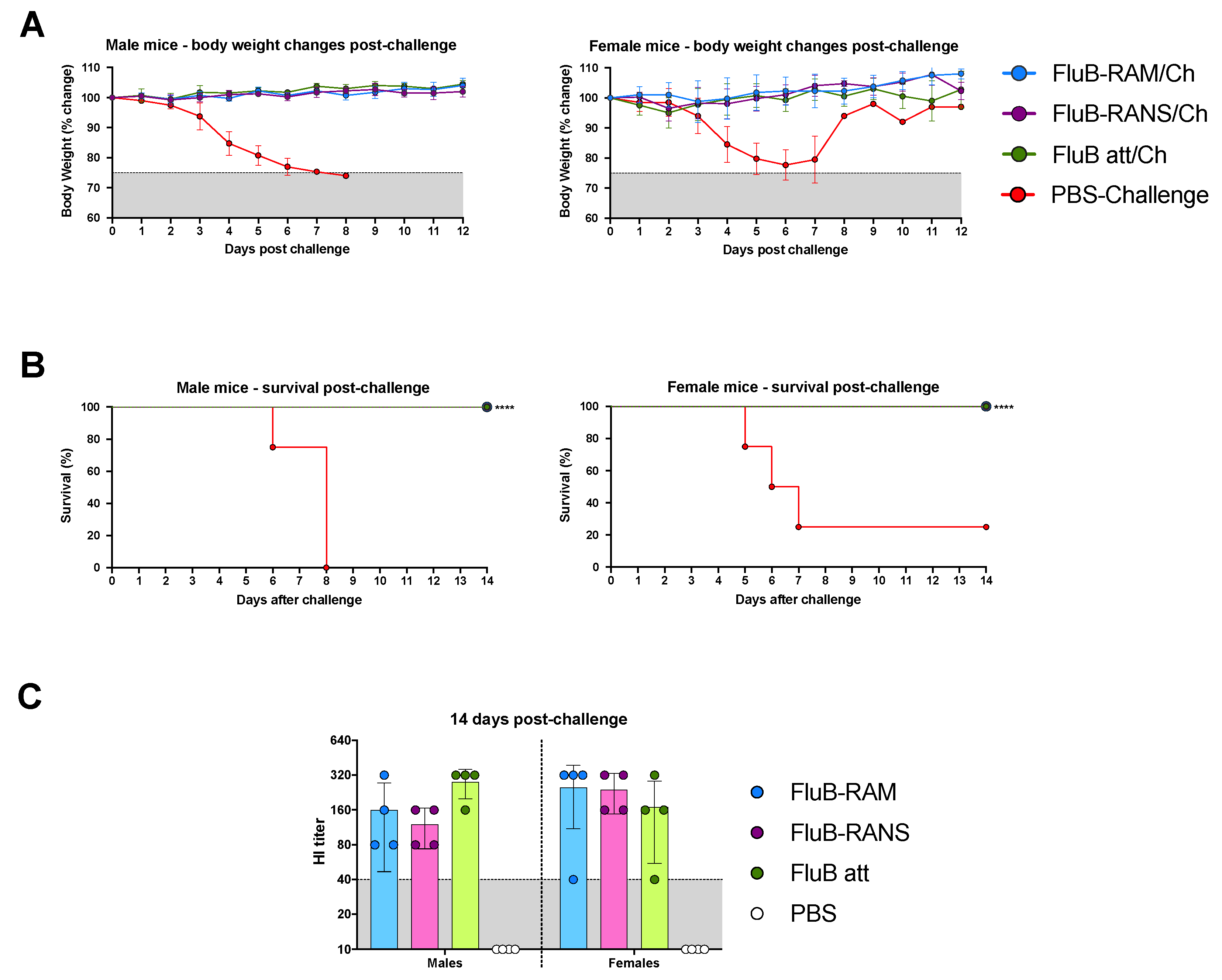
3.5. FluB-RAM and FluB-RANS Effectively Protect Mice against Lethal IBV Challenge
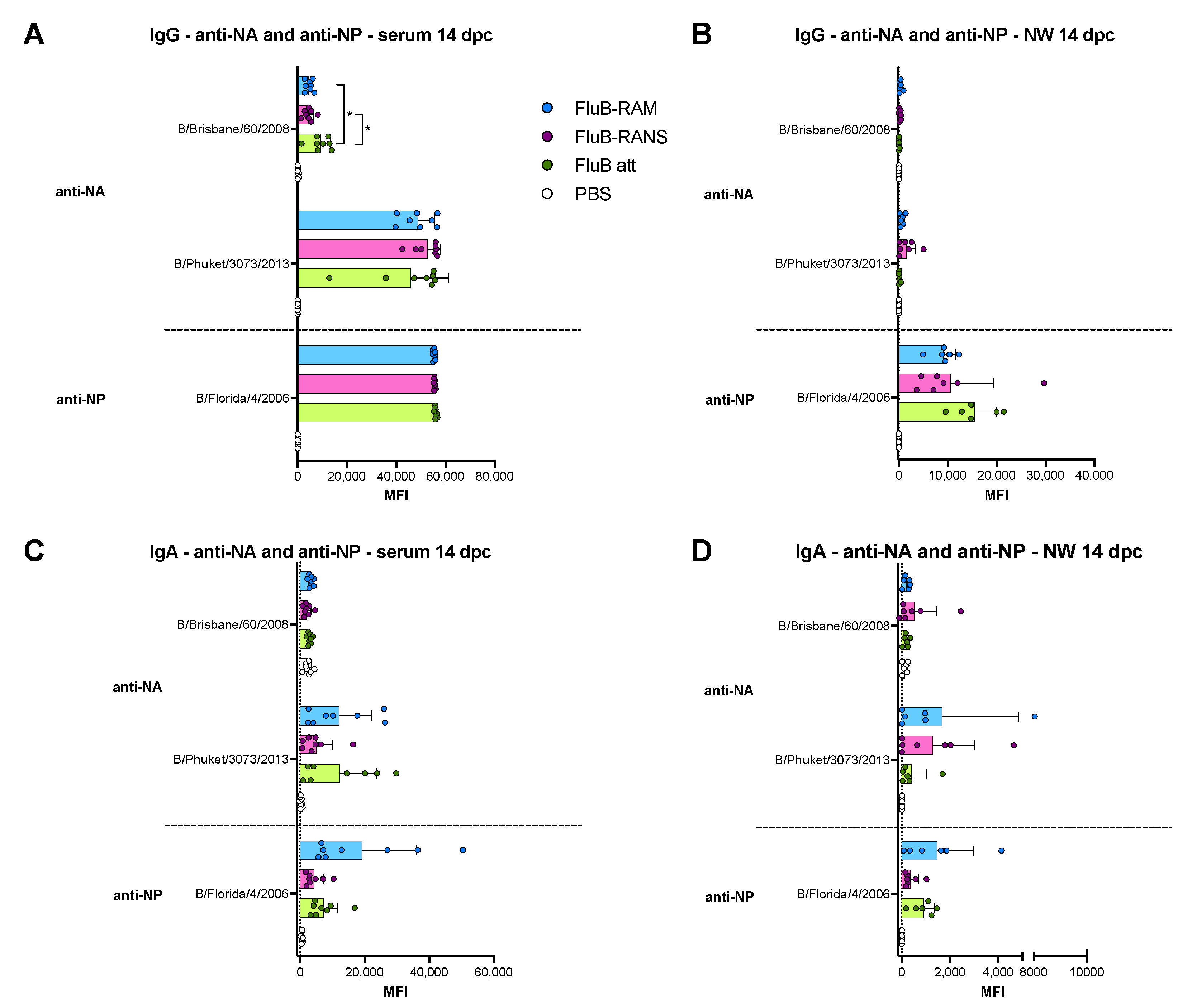
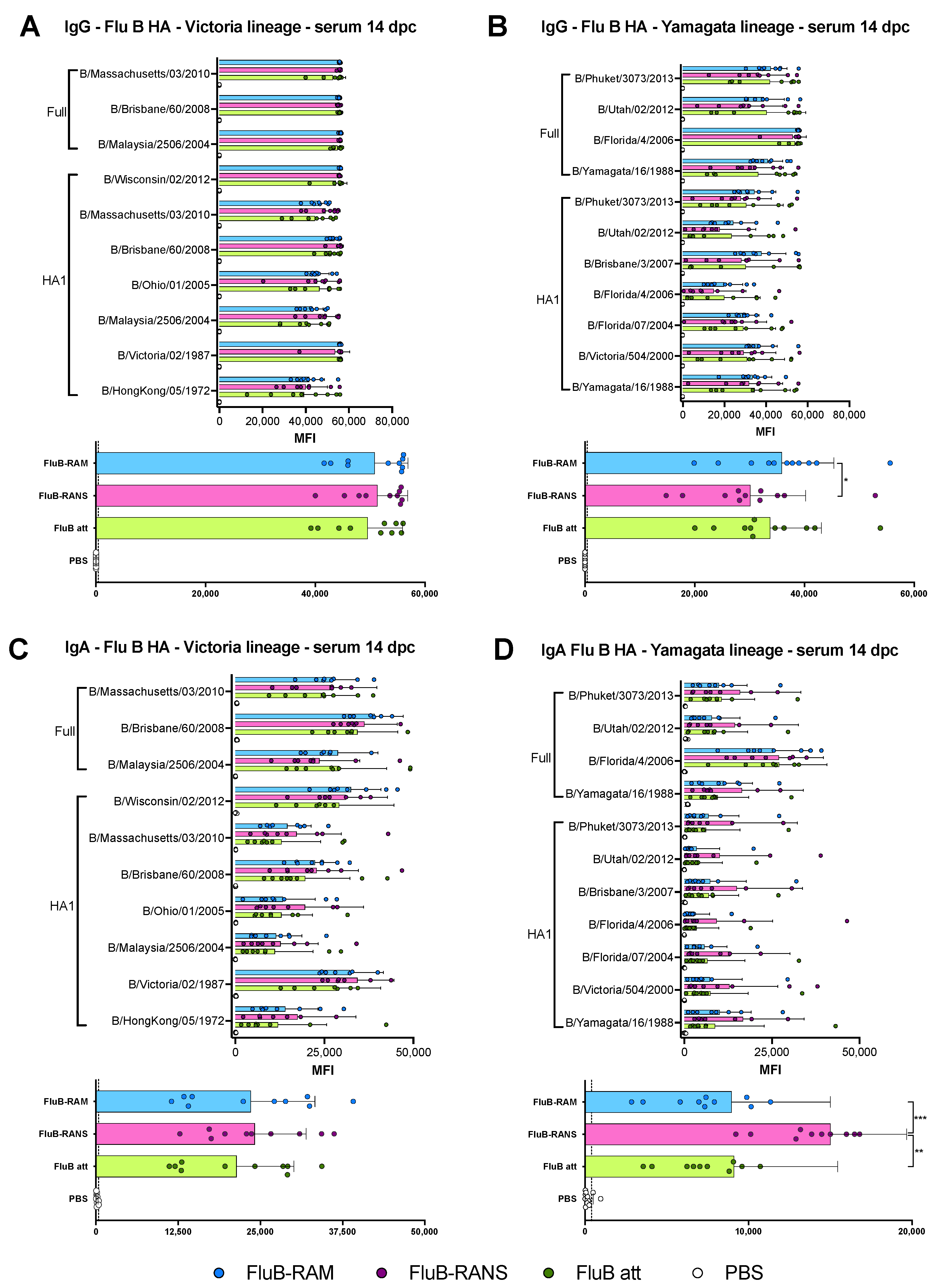
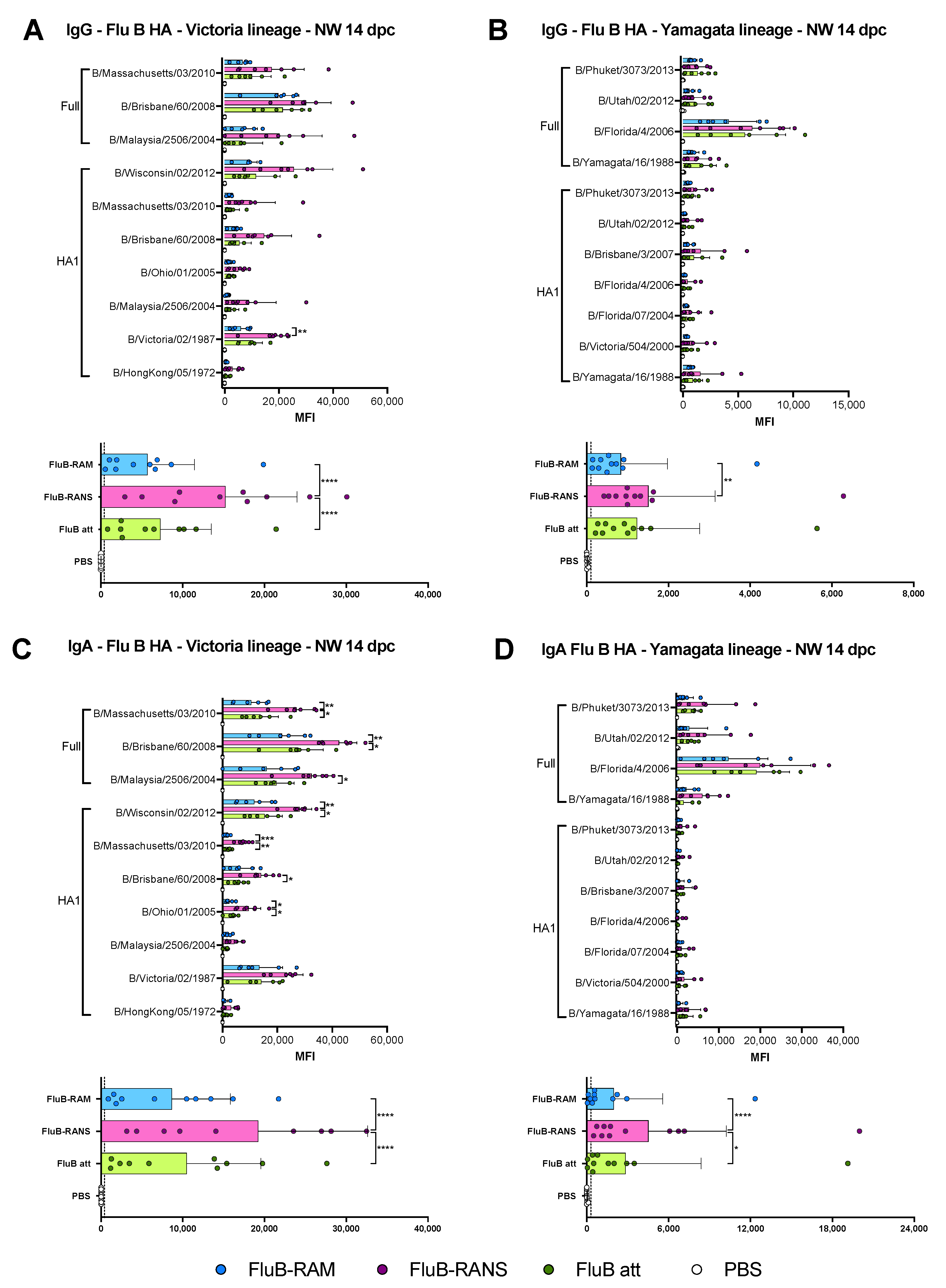
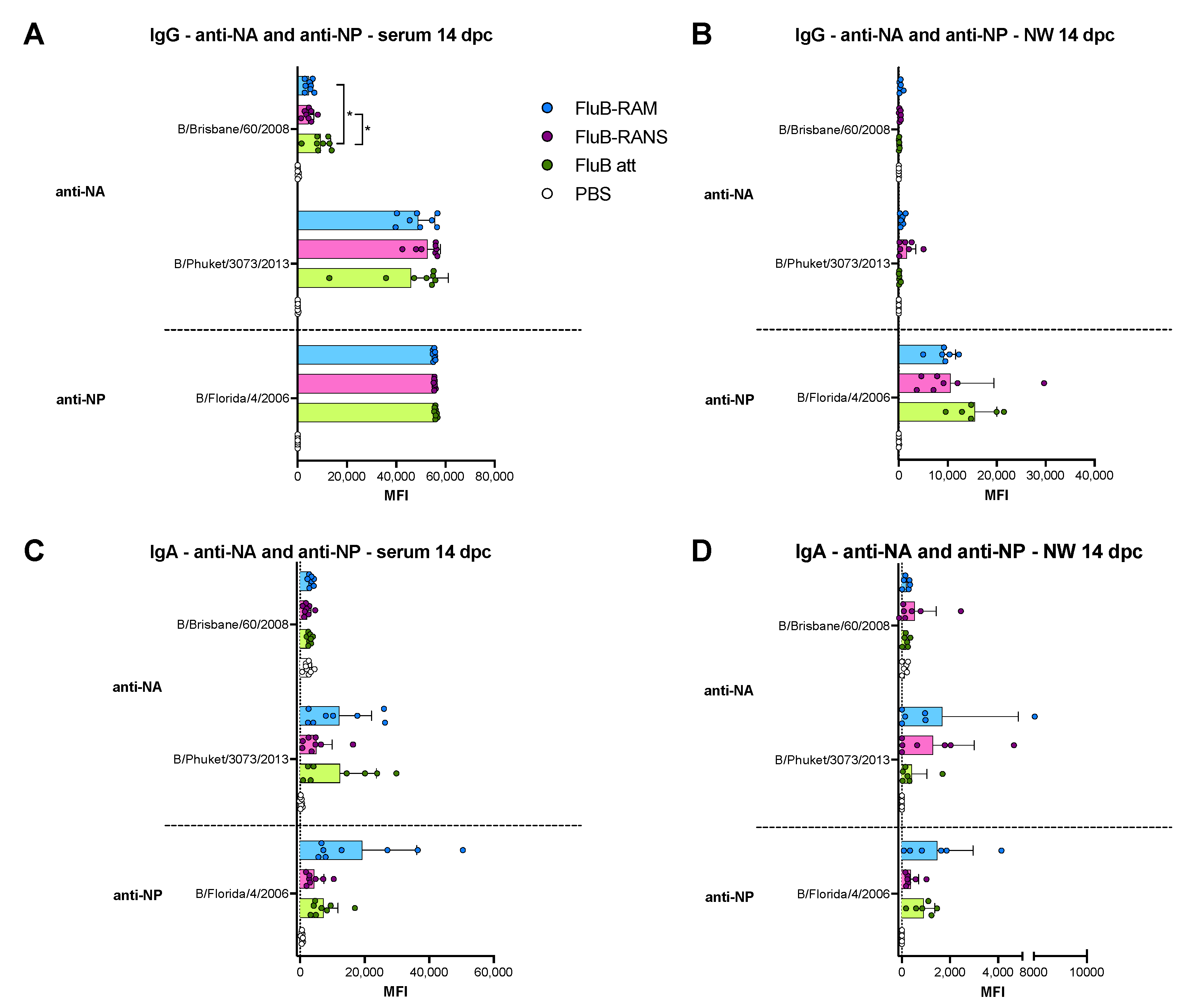
3.6. Qualitative Differences in Humoral and Mucosal Responses among Different Vaccine Groups at 14 dpc
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Francis, T., Jr. A new type of virus from epidemic influenza. Science 1940, 92, 405–408. [Google Scholar] [CrossRef] [PubMed]
- Ritchey, M.B.; Palese, P.; Kilbourne, E.D. RNAs of influenza A, B, and C viruses. J. Virol. 1976, 18, 738–744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Betakova, T.; Nermut, M.V.; Hay, A.J. The NB protein is an integral component of the membrane of influenza B virus. J. Gen. Virol. 1996, 77, 2689–2694. [Google Scholar] [CrossRef]
- Racaniello, V.R.; Palese, P. Influenza B virus genome: Assignment of viral polypeptides to RNA segments. J. Virol. 1979, 29, 361–373. [Google Scholar] [CrossRef] [Green Version]
- Shaw, M.W.; Choppin, P.W.; Lamb, R.A. A previously unrecognized influenza B virus glycoprotein from a bicistronic mRNA that also encodes the viral neuraminidase. Proc. Natl. Acad. Sci. USA 1983, 80, 4879–4883. [Google Scholar] [CrossRef] [Green Version]
- Briedis, D.J.; Lamb, R.A.; Choppin, P.W. Influenza B virus RNA segment 8 codes for two nonstructural proteins. Virology 1981, 112, 417–425. [Google Scholar] [CrossRef]
- Horvath, C.M.; Williams, M.A.; Lamb, R.A. Eukaryotic coupled translation of tandem cistrons: Identification of the influenza B virus BM2 polypeptide. EMBO J. 1990, 9, 2639–2647. [Google Scholar] [CrossRef]
- Bodewes, R.; de Mutsert, G.; van der Klis, F.R.; Ventresca, M.; Wilks, S.; Smith, D.J.; Koopmans, M.; Fouchier, R.A.; Osterhaus, A.D.; Rimmelzwaan, G.F. Prevalence of antibodies against seasonal influenza A and B viruses in children in Netherlands. Clin. Vaccine Immunol. 2011, 18, 469–476. [Google Scholar] [CrossRef] [Green Version]
- Beran, J.; Wertzova, V.; Honegr, K.; Kaliskova, E.; Havlickova, M.; Havlik, J.; Jirincova, H.; Van Belle, P.; Jain, V.; Innis, B.; et al. Challenge of conducting a placebo-controlled randomized efficacy study for influenza vaccine in a season with low attack rate and a mismatched vaccine B strain: A concrete example. BMC Infect. Dis. 2009, 9, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frey, S.; Vesikari, T.; Szymczakiewicz-Multanowska, A.; Lattanzi, M.; Izu, A.; Groth, N.; Holmes, S. Clinical efficacy of cell culture-derived and egg-derived inactivated subunit influenza vaccines in healthy adults. Clin. Infect. Dis. 2010, 51, 997–1004. [Google Scholar] [CrossRef] [PubMed]
- FluView: Influenza Associated Pediatric Mortality. 2004–2020. Available online: https://gis.cdc.gov/GRASP/Fluview/PedFluDeath.html (accessed on 14 July 2021).
- FluView: National, Regional, and State Level Outpatient Illness and Viral Surveillance. 2019. Available online: https://gis.cdc.gov/grasp/fluview/fluportaldashboard.html (accessed on 14 July 2021).
- FluView: Influenza Hospitalization Surveillance Network. 2020. Available online: https://gis.cdc.gov/grasp/fluview/FluHospChars.htm (accessed on 14 July 2021).
- Arencibia, A.; Pinon, A.; Acosta, B.; Fernandez, L.; Mune, M.; Valdes, O.; Savon, C.; Oropesa, S.; Gonzalez, G.; Roque, R.; et al. Vaccine-mismatched influenza B/Yamagata lineage viruses in Cuba, 2012–2013 season. Infect. Genet. Evol. 2018, 58, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Loconsole, D.; De Robertis, A.L.; Morea, A.; Casulli, D.; Mallamaci, R.; Baldacci, S.; Centrone, F.; Bruno, V.; Quarto, M.; Accogli, M.; et al. High public-health impact in an influenza-B-mismatch season in southern italy, 2017–2018. Biomed. Res. Int. 2019, 2019, 4643260. [Google Scholar] [CrossRef] [Green Version]
- Noh, J.Y.; Choi, W.S.; Song, J.Y.; Lee, H.S.; Lim, S.; Lee, J.; Seo, Y.B.; Lee, J.S.; Wie, S.H.; Jeong, H.W.; et al. Significant circulation of influenza B viruses mismatching the recommended vaccine-lineage in South Korea, 2007–2014. Vaccine 2018, 36, 5304–5308. [Google Scholar] [CrossRef]
- Puzelli, S.; Di Martino, A.; Facchini, M.; Fabiani, C.; Calzoletti, L.; Di Mario, G.; Palmieri, A.; Affanni, P.; Camilloni, B.; Chironna, M.; et al. Co-circulation of the two influenza B lineages during 13 consecutive influenza surveillance seasons in Italy, 2004–2017. BMC Infect. Dis. 2019, 19, 990. [Google Scholar] [CrossRef]
- Heikkinen, T.; Ikonen, N.; Ziegler, T. Impact of influenza B lineage-level mismatch between trivalent seasonal influenza vaccines and circulating viruses, 1999–2012. Clin. Infect. Dis. 2014, 59, 1519–1524. [Google Scholar] [CrossRef] [Green Version]
- Ambrose, C.S.; Levin, M.J. The rationale for quadrivalent influenza vaccines. Hum. Vaccin. Immunother. 2012, 8, 81–88. [Google Scholar] [CrossRef] [Green Version]
- Korem, M.; Orenbuch-Harroch, E.; Ben-Chetrit, E.; Israel, S.; Cohen, M.J.; Sviri, S.; Levin, P.D.; Mandelboim, M.; Wolf, D.G. Intensive Care admissions and associated severity of influenza B Versus a during influenza B vaccine-mismatched seasons. Clin. Infect. Dis. 2019, 69, 1049–1052. [Google Scholar] [CrossRef] [PubMed]
- Palekar, R.; Rodriguez, A.; Avila, C.; Barrera, G.; Barrera, M.; Brenes, H.; Bruno, A.; El Omeiri, N.; Fasce, R.; Ferreira de Almeida, W.; et al. Patterns of influenza B circulation in Latin America and the Caribbean, 2010–2017. PLoS ONE 2019, 14, e0219595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grohskopf, L.A.; Alyanak, E.; Broder, K.R.; Walter, E.B.; Fry, A.M.; Jernigan, D.B. Prevention and Control of seasonal influenza with vaccines: Recommendations of the advisory committee on immunization practices—United States, 2019–2020 influenza Season. MMWR Recomm. Rep. 2019, 68, 1–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardenas-Garcia, S.; Caceres, C.J.; Rajao, D.; Perez, D.R. Reverse genetics for influenza B viruses and recent advances in vaccine development. Curr. Opin. Virol. 2020, 44, 191–202. [Google Scholar] [CrossRef]
- Hai, R.; Martinez-Sobrido, L.; Fraser, K.A.; Ayllon, J.; Garcia-Sastre, A.; Palese, P. Influenza B virus NS1-truncated mutants: Live-attenuated vaccine approach. J. Virol. 2008, 82, 10580–10590. [Google Scholar] [CrossRef] [Green Version]
- Wressnigg, N.; Voss, D.; Wolff, T.; Romanova, J.; Ruthsatz, T.; Mayerhofer, I.; Reiter, M.; Nakowitsch, S.; Humer, J.; Morokutti, A.; et al. Development of a live-attenuated influenza B DeltaNS1 intranasal vaccine candidate. Vaccine 2009, 27, 2851–2857. [Google Scholar] [CrossRef]
- Santos, J.J.S.; Finch, C.; Sutton, T.; Obadan, A.; Aguirre, I.; Wan, Z.; Lopez, D.; Geiger, G.; Gonzalez-Reiche, A.S.; Ferreri, L.; et al. Development of an Alternative modified live influenza B virus vaccine. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [Green Version]
- Moser, M.J.; Hatta, Y.; Gabaglia, C.; Sanchez, A.; Dias, P.; Sarawar, S.; Kawaoka, Y.; Hatta, M.; Neumann, G.; Bilsel, P. Single-replication BM2SR vaccine provides sterilizing immunity and cross-lineage influenza B virus protection in mice. Vaccine 2019, 37, 4533–4542. [Google Scholar] [CrossRef] [PubMed]
- Stech, J.; Garn, H.; Herwig, A.; Stech, O.; Dauber, B.; Wolff, T.; Mettenleiter, T.C.; Klenk, H.D. Influenza B virus with modified hemagglutinin cleavage site as a novel attenuated live vaccine. J. Infect. Dis. 2011, 204, 1483–1490. [Google Scholar] [CrossRef] [Green Version]
- Sun, W.; Kirkpatrick, E.; Ermler, M.; Nachbagauer, R.; Broecker, F.; Krammer, F.; Palese, P. Development of influenza B Universal vaccine candidates using the “mosaic” hemagglutinin approach. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [Green Version]
- Wan, Z.; Cardenas Garcia, S.; Liu, J.; Santos, J.; Carnaccini, S.; Geiger, G.; Ferreri, L.; Rajao, D.; Perez, D.R. Alternative strategy for a quadrivalent live attenuated influenza virus vaccine. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [Green Version]
- Pena, L.; Sutton, T.; Chockalingam, A.; Kumar, S.; Angel, M.; Shao, H.; Chen, H.; Li, W.; Perez, D.R. Influenza viruses with rearranged genomes as live-attenuated vaccines. J. Virol. 2013, 87, 5118–5127. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, B.; Blazejewska, P.; Hessmann, M.; Bruder, D.; Geffers, R.; Mauel, S.; Gruber, A.D.; Schughart, K. Host genetic background strongly influences the response to influenza a virus infections. PLoS ONE 2009, 4, e4857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pica, N.; Iyer, A.; Ramos, I.; Bouvier, N.M.; Fernandez-Sesma, A.; Garcia-Sastre, A.; Lowen, A.C.; Palese, P.; Steel, J. The DBA.2 mouse is susceptible to disease following infection with a broad, but limited, range of influenza A and B viruses. J. Virol. 2011, 85, 12825–12829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nogales, A.; Perez, D.R.; Santos, J.J.S.; Finch, C.; Martinez-Sobrido, L. Reverse Genetics of influenza B viruses. In Reverse Geneitcs of RNA Viruses: Methods and Protocols; Perez, D.R., Ed.; Methods in Molecular Biology; Springer Science and Business Media LLC: New York, NY, USA, 2017. [Google Scholar]
- Zhou, B.; Lin, X.; Wang, W.; Halpin, R.A.; Bera, J.; Stockwell, T.B.; Barr, I.G.; Wentworth, D.E. Universal influenza B virus genomic amplification facilitates sequencing, diagnostics and reverse genetics. J. Clin. Microbiol. 2014, 52, 1330–1337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mena, I.; Nelson, M.I.; Quezada-Monroy, F.; Dutta, J.; Cortes-Fernandez, R.; Lara-Puente, J.H.; Castro-Peralta, F.; Cunha, L.F.; Trovao, N.S.; Lozano-Dubernard, B.; et al. Origins of the 2009 H1N1 influenza pandemic in swine in Mexico. Elife 2016, 5, e16777. [Google Scholar] [CrossRef] [PubMed]
- Reed, L.J.; Muench, H. A simple method for estimating fifty percent endpoints. Am. J. Hyg. 1938, 27, 493–497. [Google Scholar]
- Nakajima, R.; Supnet, M.; Jasinskas, A.; Jain, A.; Taghavian, O.; Obiero, J.; Milton, D.K.; Chen, W.H.; Grantham, M.; Webby, R.; et al. Protein microarray analysis of the specificity and cross-reactivity of influenza virus hemagglutinin-specific antibodies. mSphere 2018, 3. [Google Scholar] [CrossRef] [Green Version]
- Mo, J.; Cardenas Garcia, S.; Santos, J.; Ferreri, L.; Caceres, C.J.; Geiger, G.; Perez, D.R.; Rajao, D. Mutation E48K in PB1 polymerase subunit improves stability of a candidate live attenuated influenza B virus vaccine. Vaccines 2021, 9, 800. [Google Scholar] [CrossRef]
- Sutton, T.C.; Obadan, A.; Lavigne, J.; Chen, H.; Li, W.; Perez, D.R. Genome rearrangement of influenza virus for anti-viral drug screening. Virus Res. 2014, 189, 14–23. [Google Scholar] [CrossRef] [Green Version]
- Fink, A.L.; Engle, K.; Ursin, R.L.; Tang, W.Y.; Klein, S.L. Biological sex affects vaccine efficacy and protection against influenza in mice. Proc. Natl. Acad. Sci. USA 2018, 115, 12477–12482. [Google Scholar] [CrossRef] [Green Version]
- Klein, S.L.; Hodgson, A.; Robinson, D.P. Mechanisms of sex disparities in influenza pathogenesis. J. Leukoc. Biol. 2012, 92, 67–73. [Google Scholar] [CrossRef] [Green Version]
- Engler, R.J.; Nelson, M.R.; Klote, M.M.; VanRaden, M.J.; Huang, C.Y.; Cox, N.J.; Klimov, A.; Keitel, W.A.; Nichol, K.L.; Carr, W.W.; et al. Half-vs. full-dose trivalent inactivated influenza vaccine (2004–2005): Age, dose, and sex effects on immune responses. Arch. Intern. Med. 2008, 168, 2405–2414. [Google Scholar] [CrossRef]
- Voigt, E.A.; Ovsyannikova, I.G.; Kennedy, R.B.; Grill, D.E.; Goergen, K.M.; Schaid, D.J.; Poland, G.A. Sex Differences in older adults’ immune responses to seasonal influenza vaccination. Front. Immunol. 2019, 10, 180. [Google Scholar] [CrossRef] [Green Version]
- Arsenovic-Ranin, N.; Petrovic, R.; Zivkovic, I.; Bufan, B.; Stoiljkovic, V.; Leposavic, G. Influence of aging on germinal centre reaction and antibody response to inactivated influenza virus antigens in mice: Sex-based differences. Biogerontology 2019, 20, 475–496. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.L.; Lau, Y.F.; Lamirande, E.W.; McCall, A.W.; Subbarao, K. Seasonal influenza infection and live vaccine prime for a response to the 2009 pandemic H1N1 vaccine. Proc. Natl. Acad. Sci. USA 2011, 108, 1140–1145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subbarao, K. Live attenuated cold-adapted influenza vaccines. Cold Spring Harb. Perspect. Med. 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene expression omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Region | IBV Strain | Lineage | Expression System | Catalog No. |
|---|---|---|---|---|---|
| HA | HA1 | B/Victoria/02/1987 | Victoria | HEK293 | 40163-V08H |
| HA | HA1 | B/Wisconsin/01/2012 | Victoria | HEK293 | 40462-V08H1 |
| HA | HA1 | B/Brisbane/60/2008 | Victoria | HEK293 | 40016-V08H1 |
| HA | HA1 | B/Ohio/01/2005 | Victoria | HEK293 | 40460-V08H1 |
| HA | HA1 | B/Massachusetts/03/2010 | Victoria | HEK293 | 40191-V08H1 |
| HA | HA1 | B/Malaysia/2506/2004 | Victoria | HEK293 | 11716-V08H1 |
| HA | HA1 | B/HongKong/05/1972 | Victoria | HEK293 | 40461-V08H1 |
| HA | HA1 | B/Yamagata/16/1988 | Yamagata | HEK293 | 40157-V08H1 |
| HA | HA1 | B/Victoria/504/2000 | Yamagata | HEK293 | 40391-V08H |
| HA | HA1 | B/Brisbane/3/2007 | Yamagata | HEK293 | 40431-V08H1 |
| HA | HA1 | B/Phuket/3073/2013 | Yamagata | HEK293 | 40498-V08H1 |
| HA | HA1 | B/Florida/07/2004 | Yamagata | HEK293 | 40432-V08H1 |
| HA | HA1 | B/Utah/02/2012 | Yamagata | HEK293 | 40463-V08H1 |
| HA | HA1 | B/Florida/4/2006 | Yamagata | HEK293 | 11053-V08H1 |
| HA | HA1+HA2 | B/Brisbane/60/2008 | Victoria | E. coli | 40016-V08B |
| HA | HA1+HA2 | B/Malaysia/2506/2004 | Victoria | HEK293 | 11716-V08H |
| HA | HA1+HA2 | B/Massachusetts/03/2010 | Victoria | Baculovirus | 40191-V08B |
| HA | HA1+HA2 | B/Florida/4/2006 | Yamagata | HEK293 | 11053-V08H |
| HA | HA1+HA2 | B/Phuket/3073/2013 | Yamagata | Baculovirus | 40498-V08B |
| HA | HA1+HA2 | B/Yamagata/16/1988 | Yamagata | Baculovirus | 40157-V08B |
| HA | HA1+HA2 | B/Utah/02/2012 | Yamagata | Baculovirus | 40463-V08B |
| HA | HA1+HA2 | B/Brisbane/60/2008 | Victoria | HEK293 | 40016-V08H |
| NA | NA | B/Brisbane/60/2008 | Victoria | HEK293 | 40203-VNAHC |
| NA | NA | B/Phuket/3073/2013 | Yamagata | Baculovirus | 40502-V07B |
| NP | NP | B/Florida/4/2006 | Yamagata | Baculovirus | 40438-V08B |
| FluB-RAM | FluB-RANS | |||
|---|---|---|---|---|
| Segment | Predicted Mutations | Egg Passage #6 | Predicted Mutations | Egg Passage #6 |
| PB1 | +BM2 ORF | +BM2 ORF | +BNS2 ORF | +BNS2 ORF M495I (g1485a) |
| PB2 | None | None | None | None |
| PA | None | None | None | S719P (t2184c) |
| HA | None | None | None | None |
| NP | None | None | None | A318T (g1012a) |
| NA | None | None | None | None |
| NB | None | None | None | None |
| BM1 | Stop (c774a) | g543a s a546g s Stop (c774a) | None | None |
| BM2 | M1V (a771g) L2I (c774a) E3Stop (g777a) P4Stop (c780t, c791g) M21Stop (a831t, t832a) M37Stop (a879t, t880a) | M1V (a771g) L2I (c774a) E3Stop (g777a) P4Stop (c780t, c791g) M21Stop (a831t, t832a) M37Stop (a879t, t880a) | None | None |
| NS1 | None | None | None | None |
| NS2/NEP | None | None | a734t (Splicing acceptor) W13Stop (g737a) M14T (t743c) M15T (t752c) M31T (t791c) F38Stop (t812a c813a) | a734t (Splicing acceptor) W13Stop (g737a) M14T (t743c) M15T (t752c) M31T (t791c) F38Stop (t812a c813a) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cardenas-Garcia, S.; Cáceres, C.J.; Jain, A.; Geiger, G.; Mo, J.-S.; Jasinskas, A.; Nakajima, R.; Rajao, D.S.; Davies, D.H.; Perez, D.R. FluB-RAM and FluB-RANS: Genome Rearrangement as Safe and Efficacious Live Attenuated Influenza B Virus Vaccines. Vaccines 2021, 9, 897. https://doi.org/10.3390/vaccines9080897
Cardenas-Garcia S, Cáceres CJ, Jain A, Geiger G, Mo J-S, Jasinskas A, Nakajima R, Rajao DS, Davies DH, Perez DR. FluB-RAM and FluB-RANS: Genome Rearrangement as Safe and Efficacious Live Attenuated Influenza B Virus Vaccines. Vaccines. 2021; 9(8):897. https://doi.org/10.3390/vaccines9080897
Chicago/Turabian StyleCardenas-Garcia, Stivalis, C. Joaquín Cáceres, Aarti Jain, Ginger Geiger, Jong-Suk Mo, Algimantas Jasinskas, Rie Nakajima, Daniela S. Rajao, D. Huw Davies, and Daniel R. Perez. 2021. "FluB-RAM and FluB-RANS: Genome Rearrangement as Safe and Efficacious Live Attenuated Influenza B Virus Vaccines" Vaccines 9, no. 8: 897. https://doi.org/10.3390/vaccines9080897
APA StyleCardenas-Garcia, S., Cáceres, C. J., Jain, A., Geiger, G., Mo, J.-S., Jasinskas, A., Nakajima, R., Rajao, D. S., Davies, D. H., & Perez, D. R. (2021). FluB-RAM and FluB-RANS: Genome Rearrangement as Safe and Efficacious Live Attenuated Influenza B Virus Vaccines. Vaccines, 9(8), 897. https://doi.org/10.3390/vaccines9080897