
Guiding the Immune Response to a Conserved Epitope in MSP2, an Intrinsically Disordered Malaria Vaccine Candidate

 , , , and
, , , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Surface Plasmon Resonance
2.2. MD Simulations
2.3. Peptide Synthesis
2.4. Protein Preparation and Mice Immunisation Experiments
2.5. ELISA
2.6. Merozoite ELISA
3. Results
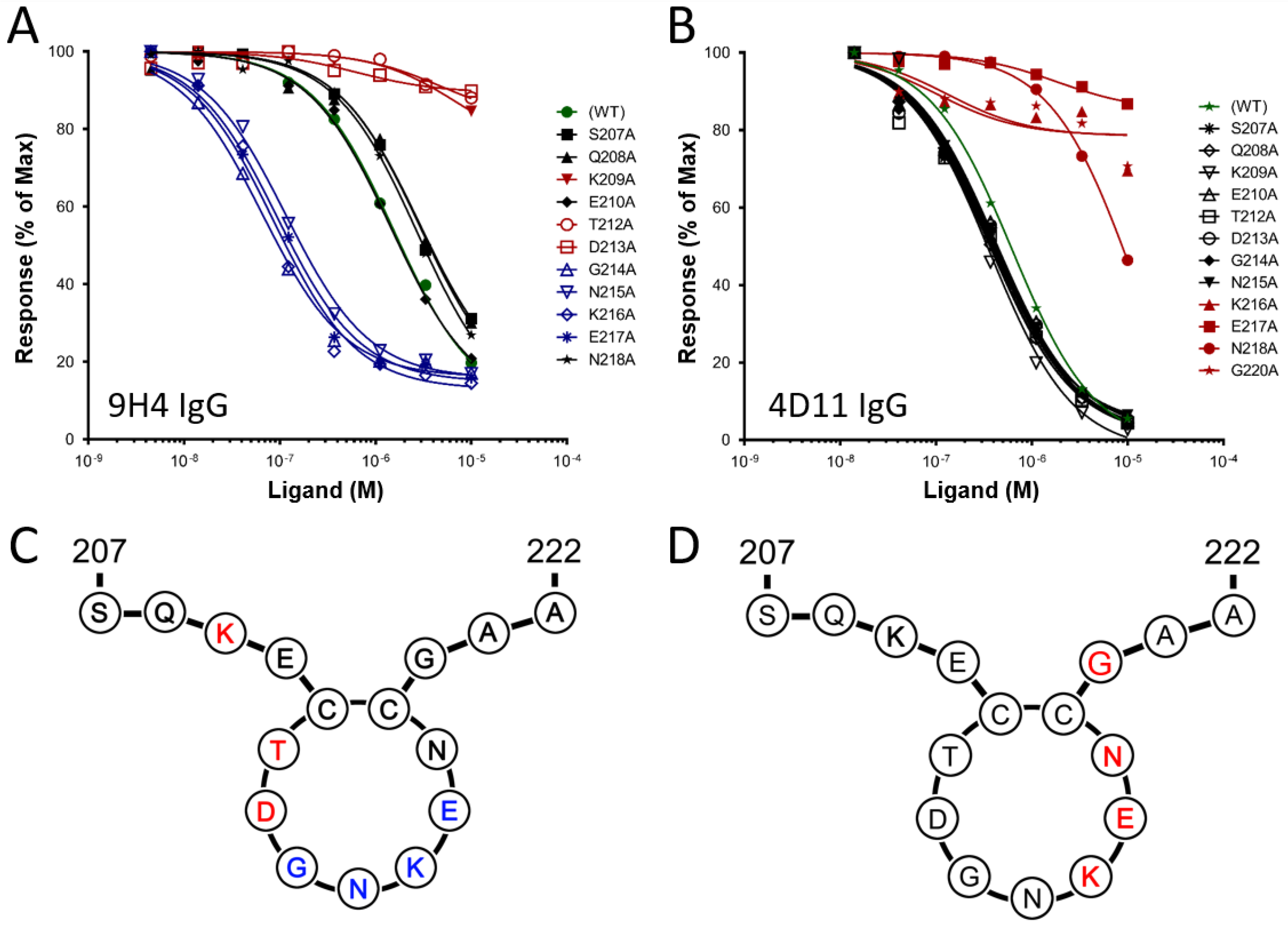
3.1. Alanine Scans of mAb 9H4 and 4D11 Epitopes
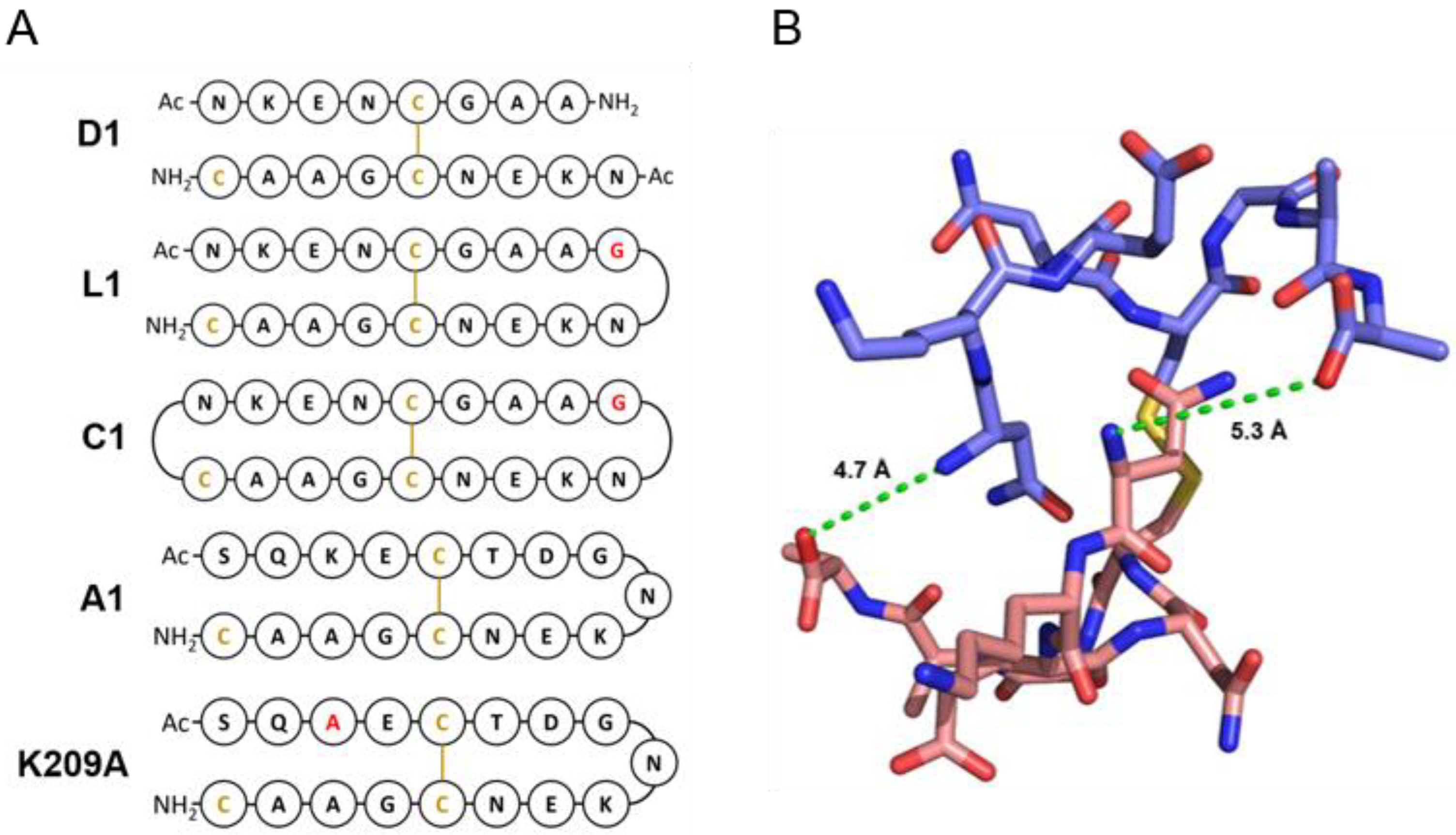
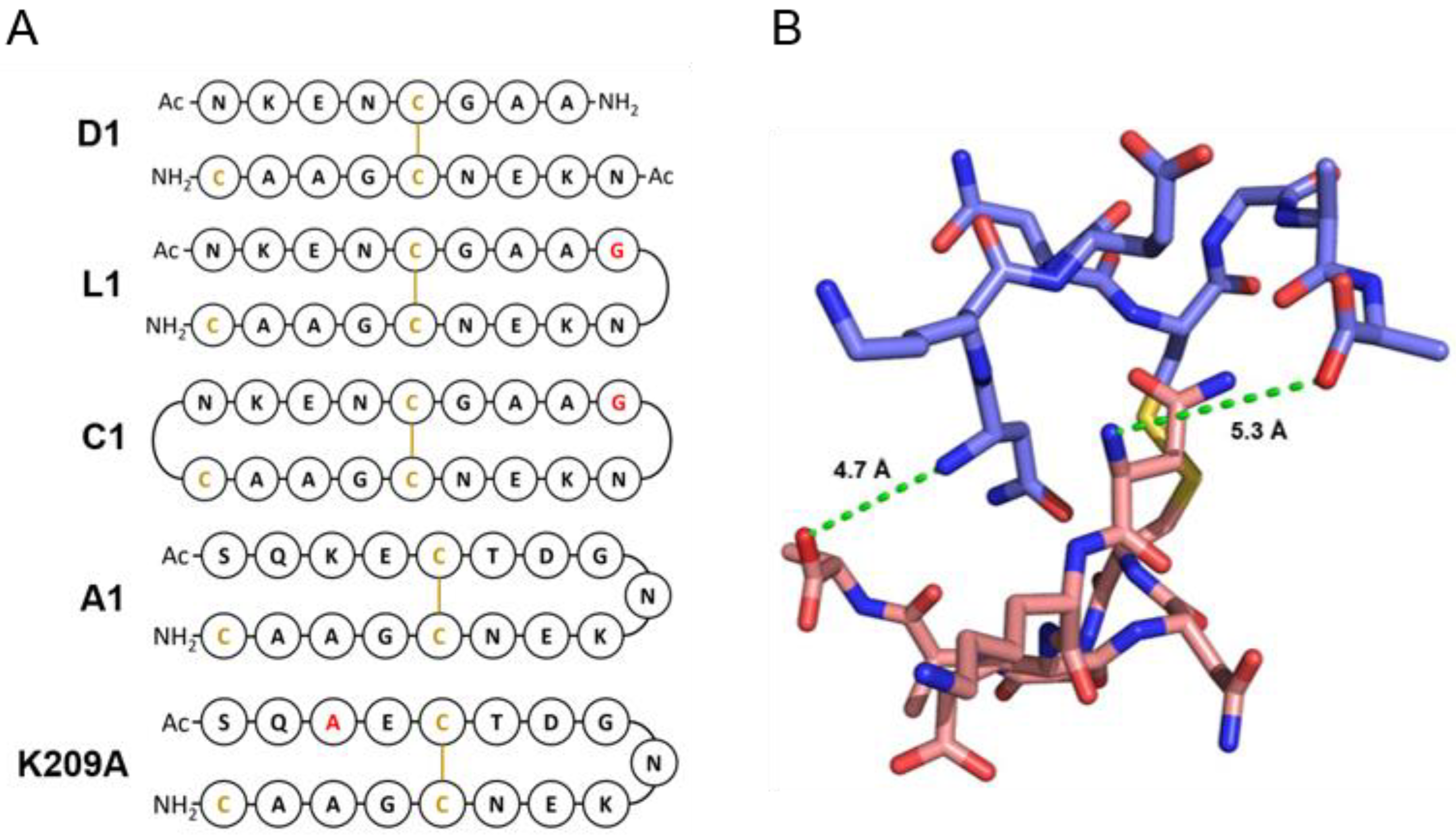
3.2. Design of Peptides for Immunisation
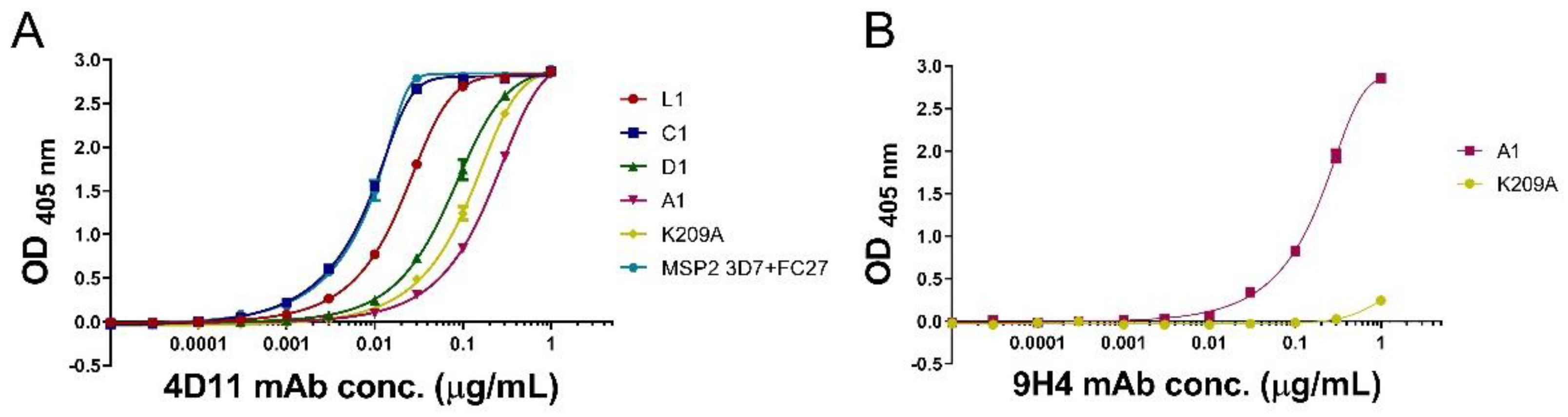
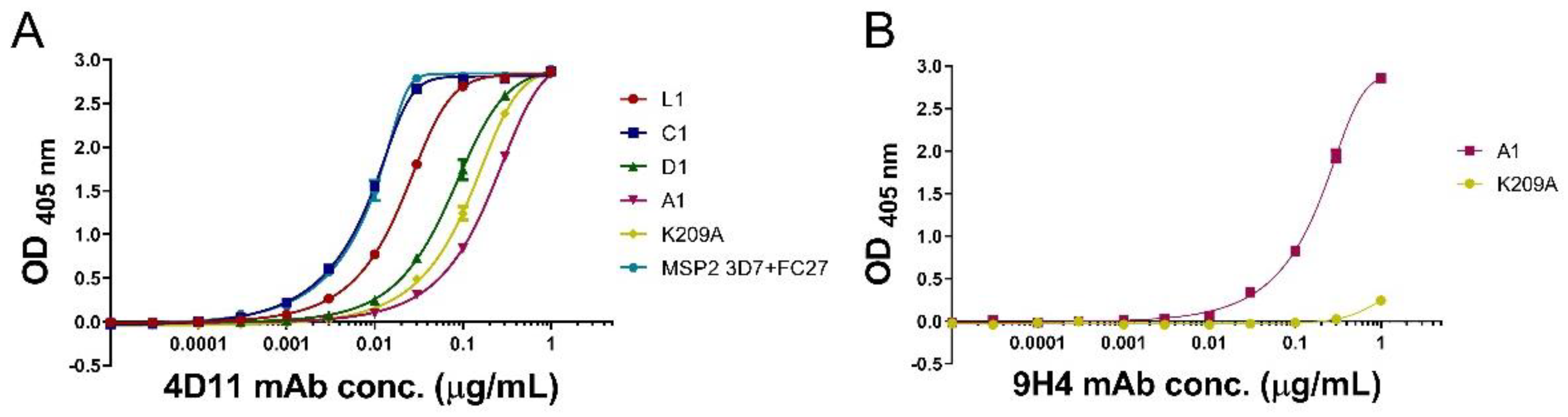
3.3. mAbs 4D11 and 9H4 Bind to Their Epitopes in Peptide-KLH Conjugates
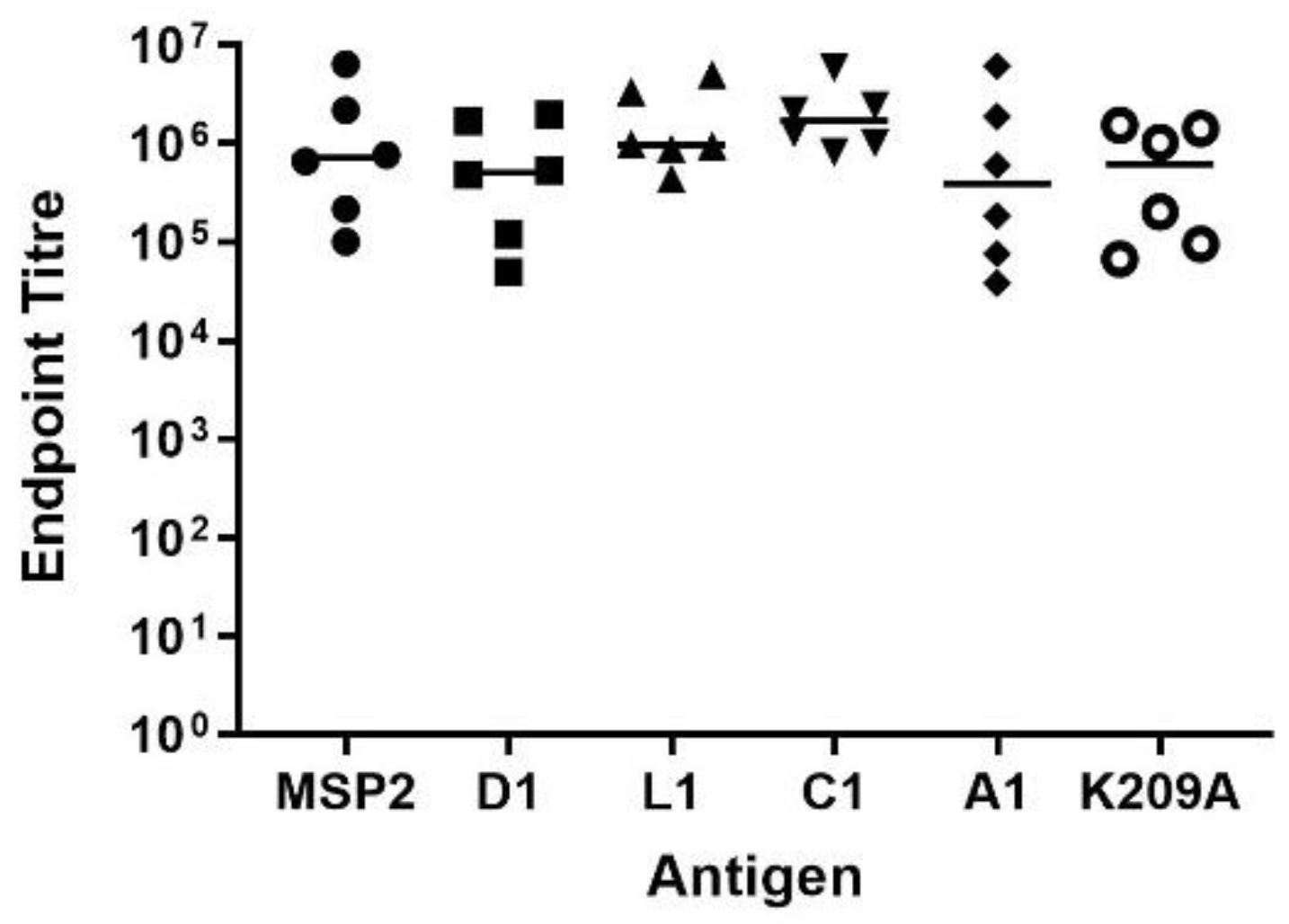
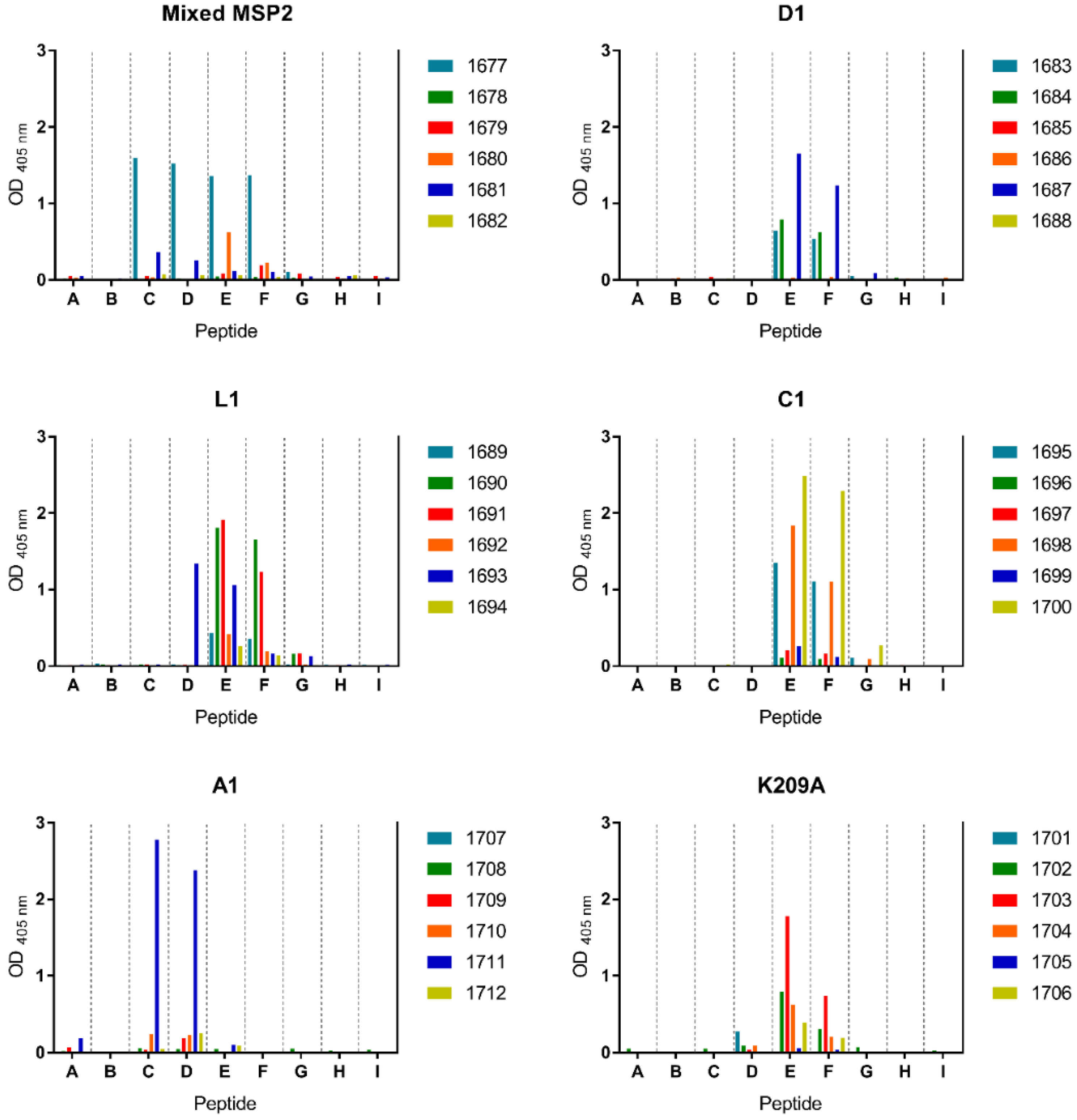
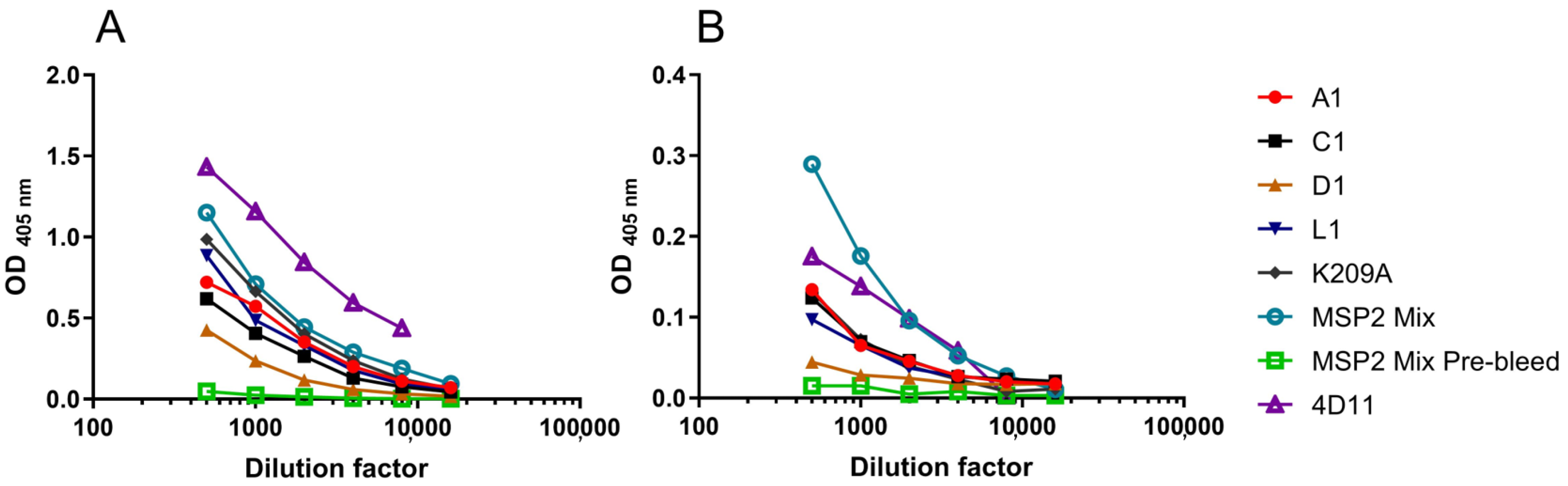
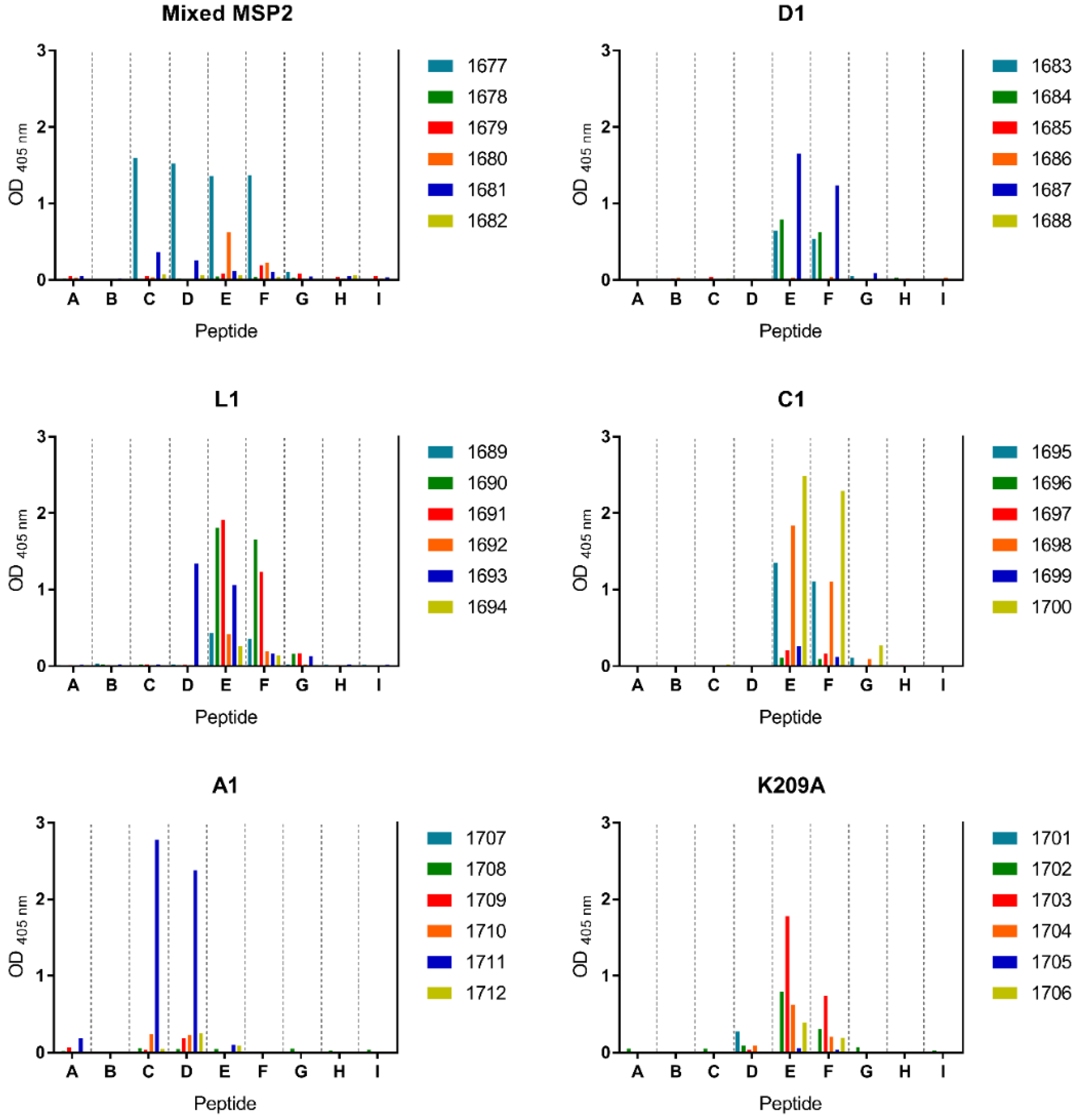
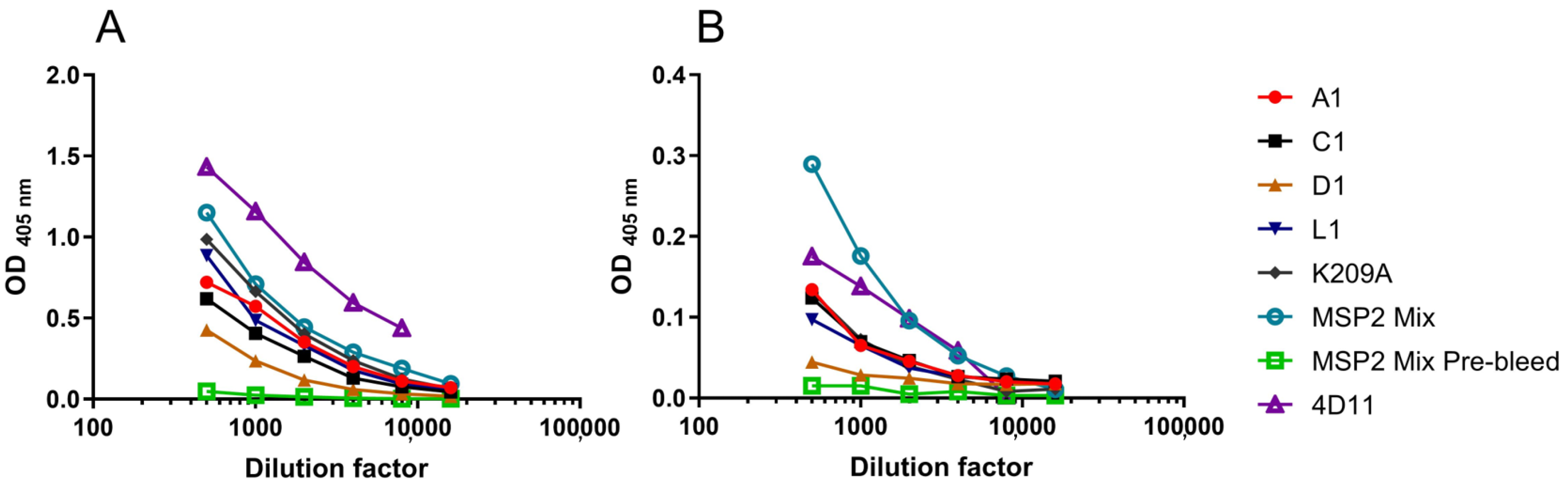
3.4. Peptide-KLH Conjugates Were Able to Induce Epitope-Specific Antibody Responses
3.5. Peptide-KLH Conjugates Induced Antibodies Recognising the 4D11-Specific Epitope
3.6. The K209A Mutation Biased the Antibody Response towards the 4D11-Specific Epitope
3.7. Peptide-Induced Antibody Responses Recognised Native MSP2 on the Merozoite Surface
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organisation. World Malaria Report 2020; WHO: Geneva, Switzerland, 2020. [Google Scholar]
- Greenwood, B.M. Efficacy and safety of RTS,S/AS01 malaria vaccine with or without a booster dose in infants and children in Africa: Final results of a phase 3, individually randomised, controlled trial. Lancet 2015, 386, 31–45. [Google Scholar] [CrossRef] [Green Version]
- Vekemans, J.; Schellenberg, D.; Benns, S.; O’Brien, K.; Alonso, P. Meeting report: WHO consultation on malaria vaccine development, Geneva, 15–16 July 2019. Vaccine 2021, 39, 2907–2916. [Google Scholar] [CrossRef]
- Datoo, M.S.; Natama, M.H.; Somé, A.; Traoré, O.; Rouamba, T.; Bellamy, D.; Yameogo, P.; Valia, D.; Tegneri, M.; Ouedraogo, F.; et al. Efficacy of a low-dose candidate malaria vaccine, R21 in adjuvant Matrix-M, with seasonal administration to children in Burkina Faso: A randomised controlled trial. Lancet 2021, 397, 1809–1818. [Google Scholar] [CrossRef]
- Smythe, J.A.; Coppel, R.; Day, K.; Martin, R.K.; Oduola, A.M.; Kemp, D.J.; Anders, R.F. Structural diversity in the Plasmodium falciparum merozoite surface antigen 2. Proc. Natl. Acad. Sci. USA 1991, 88, 1751–1755. [Google Scholar] [CrossRef] [Green Version]
- Fenton, B.; Clark, J.T.; Khan, C.M.; Robinson, J.V.; Walliker, D.; Ridley, R.; Scaife, J.G.; McBride, J.S. Structural and antigenic polymorphism of the 35- to 48-kilodalton merozoite surface antigen (MSA-2) of the malaria parasite Plasmodium falciparum. Mol. Cell. Biol. 1991, 11, 963–971. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Perugini, M.; Yao, S.; Adda, C.; Murphy, V.J.; Low, A.; Anders, R.F.; Norton, R.S. Solution conformation, backbone dynamics and lipid interactions of the intrinsically unstructured malaria surface protein MSP2. J. Mol. Biol. 2008, 379, 105–121. [Google Scholar] [CrossRef] [Green Version]
- Adda, C.; Murphy, V.J.; Sunde, M.; Waddington, L.J.; Schloegel, J.; Talbo, G.H.; Vingas, K.; Kienzle, V.; Masciantonio, R.; Howlett, G.J.; et al. Plasmodium falciparum merozoite surface protein 2 is unstructured and forms amyloid-like fibrils. Mol. Biochem. Parasitol. 2009, 166, 159–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genton, B.; Betuela, I.; Felger, I.; Al-Yaman, F.; Anders, R.F.; Saul, A.; Rare, L.; Baisor, M.; Lorry, K.; Brown, G.V.; et al. A Recombinant blood-stage malaria vaccine reduces Plasmodium falciparum density and exerts selective pressure on parasite populations in a Phase 1–2b Trial in Papua New Guinea. J. Infect. Dis. 2002, 185, 820–827. [Google Scholar] [CrossRef] [Green Version]
- McCarthy, J.S.; Marjason, J.; Elliott, S.; Fahey, P.; Bang, G.; Malkin, E.; Tierney, E.; Aked-Hurditch, H.; Adda, C.; Cross, N.; et al. A Phase 1 trial of MSP2-C1, a blood-stage malaria vaccine containing 2 isoforms of MSP2 formulated with Montanide® ISA 720. PLoS ONE 2011, 6, e24413. [Google Scholar] [CrossRef]
- Krishnarjuna, B.; Andrew, D.; MacRaild, C.; Morales, R.; Beeson, J.G.; Anders, R.F.; Richards, J.S.; Norton, R.S. Strain-transcending immune response generated by chimeras of the malaria vaccine candidate merozoite surface protein 2. Sci. Rep. 2016, 6, 20613. [Google Scholar] [CrossRef]
- Eacret, J.S.; Gonzales, D.M.; Franks, R.G.; Burns, J.M., Jr. Immunization with merozoite surface protein 2 fused to a Plasmodium-specific carrier protein elicits strain-specific and strain-transcending, opsonizing antibody. Sci. Rep. 2019, 9, 9022. [Google Scholar] [CrossRef]
- Adda, C.; MacRaild, C.; Reiling, L.; Wycherley, K.; Boyle, M.; Kienzle, V.; Masendycz, P.; Foley, M.; Beeson, J.G.; Norton, R.S.; et al. Antigenic characterization of an intrinsically unstructured protein, Plasmodium falciparum merozoite surface protein 2. Infect. Immun. 2012, 80, 4177–4185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skwarczynski, M.; Toth, I. Peptide-based synthetic vaccines. Chem. Sci. 2015, 7, 842–854. [Google Scholar] [CrossRef] [Green Version]
- MacRaild, C.A.; Seow, J.; Das, S.; Norton, R.S. Disordered epitopes as peptide vaccines. Pept. Sci. 2018, 110, e24067. [Google Scholar] [CrossRef] [Green Version]
- Skwarczynski, M.; Toth, I. Peptide-based subunit nanovaccines. Curr. Drug Deliv. 2011, 8, 282–289. [Google Scholar] [CrossRef]
- Stephens, A.J.; Burgess-Brown, N.A.; Jiang, S. Beyond just peptide antigens: The complex world of peptide-based cancer vaccines. Front. Immunol. 2021, 12, 696–791. [Google Scholar] [CrossRef]
- Skwarczynski, M.; Zhao, G.; Boer, J.C.; Ozberk, V.; Azuar, A.; Cruz, J.G.; Giddam, A.K.; Khalil, Z.G.; Pandey, M.; Shibu, M.A.; et al. Poly(amino acids) as a potent self-adjuvanting delivery system for peptide-based nanovaccines. Sci. Adv. 2020, 6, eaax2285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patarroyo, M.E.; Patarroyo, M.-A. Emerging rules for subunit-based, multiantigenic, multistage chemically synthesized vaccines. Acc. Chem. Res. 2008, 41, 377–386. [Google Scholar] [CrossRef]
- Cozzi, R.; Scarselli, M. Structural vaccinology: A three-dimensional view for vaccine development. Curr. Top. Med. Chem. 2013, 13, 2629–2637. [Google Scholar] [CrossRef]
- Dormitzer, P.R.; Grandi, G.; Rappuoli, R. Structural vaccinology starts to deliver. Nat. Rev. Genet. 2012, 10, 807–813. [Google Scholar] [CrossRef] [PubMed]
- Kwong, P.D.; DeKosky, B.J.; Ulmer, J.B. Antibody-guided structure-based vaccines. Semin. Immunol. 2020, 50, 101428. [Google Scholar] [CrossRef] [PubMed]
- Kulp, D.W.; Schief, W.R. Advances in structure-based vaccine design. Curr. Opin. Virol. 2013, 3, 322–331. [Google Scholar] [CrossRef] [Green Version]
- Scarselli, M.; Aricò, B.; Brunelli, B.; Savino, S.; Di Marcello, F.; Palumbo, E.; Veggi, D.; Ciucchi, L.; Cartocci, E.; Bottomley, M.J.; et al. Rational design of a meningococcal antigen inducing broad protective immunity. Sci. Transl. Med. 2011, 3, 91ra62. [Google Scholar] [CrossRef] [PubMed]
- Swanson, K.A.; Settembre, E.C.; Shaw, C.A.; Dey, A.K.; Rappuoli, R.; Mandl, C.W.; Dormitzer, P.R.; Carfi, A. Structural basis for immunization with postfusion respiratory syncytial virus fusion F glycoprotein (RSV F) to elicit high neutralizing antibody titers. Proc. Natl. Acad. Sci. USA 2011, 108, 9619–9624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLellan, J.; Chen, M.; Joyce, M.G.; Sastry, M.; Stewart-Jones, G.B.E.; Yang, Y.; Zhang, B.; Chen, L.; Srivatsan, S.; Zheng, A.; et al. Structure-based design of a fusion glycoprotein vaccine for respiratory syncytial virus. Science 2013, 342, 592–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crank, M.C.; Ruckwardt, T.J.; Chen, M.; Morabito, K.M.; Phung, E.; Costner, P.J.; Holman, L.A.; Hickman, S.P.; Berkowitz, N.M.; Gordon, I.J.; et al. A proof of concept for structure-based vaccine design targeting RSV in humans. Science 2019, 365, 505–509. [Google Scholar] [CrossRef] [PubMed]
- Nuccitelli, A.; Cozzi, R.; Gourlay, L.J.; Donnarumma, D.; Necchi, F.; Norais, N.; Telford, J.L.; Rappuoli, R.; Bolognesi, M.; Maione, D.; et al. Structure-based approach to rationally design a chimeric protein for an effective vaccine against Group B Streptococcus infections. Proc. Natl. Acad. Sci. USA 2011, 108, 10278–10283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jardine, J.; Julien, J.-P.; Menis, S.; Ota, T.; Kalyuzhniy, O.; McGuire, A.; Sok, D.; Huang, P.-S.; MacPherson, S.; Jones, M.; et al. Rational HIV immunogen design to target specific germline B cell receptors. Science 2013, 340, 711–716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ofek, G.; Guenaga, F.J.; Schief, W.R.; Skinner, J.; Baker, D.; Wyatt, R.; Kwong, P.D. Elicitation of structure-specific antibodies by epitope scaffolds. Proc. Natl. Acad. Sci. USA 2010, 107, 17880–17887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azoitei, M.L.; Ban, Y.-E.A.; Julien, J.-P.; Bryson, S.; Schroeter, A.; Kalyuzhniy, O.; Porter, J.; Adachi, Y.; Baker, D.; Pai, E.F.; et al. Computational design of high-affinity epitope scaffolds by backbone grafting of a linear epitope. J. Mol. Biol. 2012, 415, 175–192. [Google Scholar] [CrossRef]
- Seow, J.; Morales, R.; MacRaild, C.; Krishnarjuna, B.; McGowan, S.; Dingjan, T.; Jaipuria, G.; Rouet, R.; Wilde, K.; Atreya, H.S.; et al. Structure and characterisation of a key epitope in the conserved C-terminal domain of the malaria vaccine candidate MSP2. J. Mol. Biol. 2017, 429, 836–846. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Schmid, N.; Eichenberger, A.P.; Choutko, A.; Riniker, S.; Winger, M.; Mark, A.; Van Gunsteren, W.F. Definition and testing of the GROMOS force-field versions 54A7 and 54B7. Eur. Biophys. J. 2011, 40, 843–856. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [Green Version]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Postma, T.M.; Albericio, F. N-Chlorosuccinimide, an efficient reagent for on-resin disulfide formation in solid-phase peptide synthesis. Org. Lett. 2013, 15, 616–619. [Google Scholar] [CrossRef] [PubMed]
- Postma, T.M.; Albericio, F. Disulfide formation strategies in peptide synthesis. Eur. J. Org. Chem. 2014, 2014, 3519–3530. [Google Scholar] [CrossRef]
- Boyle, M.; Wilson, D.; Richards, J.; Riglar, D.; Tetteh, K.K.A.; Conway, D.; Ralph, S.; Baum, J.; Beeson, J.G. Isolation of viable Plasmodium falciparum merozoites to define erythrocyte invasion events and advance vaccine and drug development. Proc. Natl. Acad. Sci. USA 2010, 107, 14378–14383. [Google Scholar] [CrossRef] [Green Version]
- Salinas, N.; Tang, W.K.; Tolia, N.H.; Salina, N.D. Blood-stage malaria parasite antigens: Structure, function, and vaccine potential. J. Mol. Biol. 2019, 431, 4259–4280. [Google Scholar] [CrossRef]
- Rappuoli, R.; Bottomley, M.J.; D’Oro, U.; Finco, O.; De Gregorio, E. Reverse vaccinology 2.0: Human immunology instructs vaccine antigen design. J. Exp. Med. 2016, 213, 469–481. [Google Scholar] [CrossRef]
- Kaufmann, S.H.; McElrath, M.J.; Lewis, D.J.; Del Giudice, G. Challenges and responses in human vaccine development. Curr. Opin. Immunol. 2014, 28, 18–26. [Google Scholar] [CrossRef] [Green Version]
- Cui, J.J.; Tran-Dubé, M.; Shen, H.; Nambu, M.; Kung, P.-P.; Pairish, M.; Jia, L.; Meng, J.; Funk, L.; Botrous, I.; et al. Structure based drug design of crizotinib (PF-02341066), a potent and selective dual inhibitor of mesenchymal–epithelial transition factor (c-MET) kinase and anaplastic lymphoma kinase (ALK). J. Med. Chem. 2011, 54, 6342–6363. [Google Scholar] [CrossRef] [PubMed]
- Lounnas, V.; Ritschel, T.; Kelder, J.; McGuire, R.; Bywater, R.P.; Foloppe, N. Current progress in structure-based rational drug design marks a new mindset in drug discovery. Comput. Struct. Biotechnol. J. 2013, 5, e201302011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oyen, D.; Torres, J.L.; Wille-Reece, U.; Ockenhouse, C.F.; Emerling, D.; Glanville, J.; Volkmuth, W.; Flores-Garcia, Y.; Zavala, F.; Ward, A.B.; et al. Structural basis for antibody recognition of the NANP repeats in Plasmodium falciparum circumsporozoite protein. Proc. Natl. Acad. Sci. USA 2017, 114, E10438–E10445. [Google Scholar] [CrossRef] [Green Version]
- Frimpong, A.; Kusi, K.; Ofori, M.F.; Ndifon, W. Novel strategies for malaria vaccine design. Front. Immunol. 2018, 9, 2769. [Google Scholar] [CrossRef] [PubMed]
- Burton, D.R. Scaffolding to build a rational vaccine design strategy. Proc. Natl. Acad. Sci. USA 2010, 107, 17859–17860. [Google Scholar] [CrossRef] [Green Version]
- Guenaga, J.; Dosenovic, P.; Ofek, G.; Baker, D.; Schief, W.R.; Kwong, P.D.; Hedestam, G.B.K.; Wyatt, R.T. Heterologous epitope-scaffold prime∶ boosting immuno-focuses B cell responses to the HIV-1 gp41 2F5 neutralization determinant. PLoS ONE 2011, 6, e16074. [Google Scholar] [CrossRef] [Green Version]
- MacRaild, C.A.; Richards, J.S.; Anders, R.F.; Norton, R.S. Antibody recognition of disordered antigens. Structure 2016, 24, 148–157. [Google Scholar] [CrossRef] [Green Version]
- Triller, G.; Scally, S.W.; Costa, G.; Pissarev, M.; Kreschel, C.; Bosch, A.; Marois, E.; Sack, B.K.; Murugan, R.; Salman, A.; et al. Natural parasite exposure induces protective human anti-malarial antibodies. Immunology 2017, 47, 1197–1209.e10. [Google Scholar] [CrossRef] [Green Version]
- Meola, A.; Tarr, A.; England, P.; Meredith, L.; McClure, P.; Foung, S.K.H.; McKeating, J.; Ball, J.; Rey, F.; Krey, T. Structural flexibility of a conserved antigenic region in hepatitis C virus glycoprotein E2 recognized by broadly neutralizing antibodies. J. Virol. 2014, 89, 2170–2181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabatino, D. Medicinal chemistry and methodological advances in the development of peptide-based vaccines. J. Med. Chem. 2020, 63, 14184–14196. [Google Scholar] [CrossRef]
- Boyle, M.; Reiling, L.; Feng, G.; Langer, C.; Osier, F.; Aspeling-Jones, H.; Cheng, Y.S.; Stubbs, J.; Tetteh, K.K.; Conway, D.; et al. Human antibodies fix complement to inhibit Plasmodium falciparum invasion of erythrocytes and are associated with protection against malaria. Immunology 2015, 42, 580–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duncan, C.J.; Hill, A.V.; Ellis, R.D. Can growth inhibition assays (GIA) predict blood-stage malaria vaccine efficacy? Hum. Vaccines Immunother. 2012, 8, 706–714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osier, F.H.; Feng, G.; Boyle, M.J.; Langer, C.; Zhou, J.; Richards, J.S.; McCallum, F.J.; Reiling, L.; Jaworowski, A.; Anders, R.F.; et al. Opsonic phagocytosis of Plasmodium falciparum merozoites: Mechanism in human immunity and a correlate of protection against malaria. BMC Med. 2014, 12, 108. [Google Scholar] [CrossRef] [Green Version]
- Menezes-Souza, D.; Mendes, T.A.D.O.; Nagem, R.A.P.; Santos, T.T.D.O.; Silva, A.L.T.; Santoro, M.M.; De Carvalho, S.F.G.; Coelho, E.A.F.; Bartholomeu, D.C.; Fujiwara, R.T. Mapping B-cell epitopes for the peroxidoxin of Leishmania (Viannia) braziliensis and its potential for the clinical diagnosis of tegumentary and visceral leishmaniasis. PLoS ONE 2014, 9, e99216. [Google Scholar] [CrossRef]
- Ramamurthy, M.; Sankar, S.; Abraham, A.M.; Nandagopal, B.; Sridharan, G. B cell epitopes in the intrinsically disordered regions of neuraminidase and hemagglutinin proteins of H5N1 and H9N2 avian influenza viruses for peptide-based vaccine development. J. Cell. Biochem. 2019, 120, 17534–17544. [Google Scholar] [CrossRef]
- Feng, Z.-P.; Zhang, X.; Han, P.; Arora, N.; Anders, R.F.; Norton, R.S. Abundance of intrinsically unstructured proteins in P. falciparum and other apicomplexan parasite proteomes. Mol. Biochem. Parasitol. 2006, 150, 256–267. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | Sequence | Kd against 9H4 IgG (µM) | Kd against 4D11 IgG (µM) |
|---|---|---|---|
| 3D7-MSP2207–222 (WT) | SQKECTDGNKENCGAA | 1.5 | 0.9 |
| S207A | AQKECTDGNKENCGAA | 2.9 | 0.5 |
| Q208A | SAKECTDGNKENCGAA | 2.9 | 0.4 |
| K209A | SQAECTDGNKENCGAA | 7.3 | 0.3 |
| E210A | SQKACTDGNKENCGAA | 1.4 | 0.5 |
| T212A | SQKECADGNKENCGAA | 4.8 | 0.5 |
| D213A | SQKECTAGNKENCGAA | 0.6 | 0.5 |
| G214A | SQKECTDANKENCGAA | 0.07 | 0.4 |
| N215A | SQKECTDGAKENCGAA | 0.1 | 0.5 |
| K216A | SQKECTDGNAENCGAA | 0.09 | 21.0 |
| E217A | SQKECTDGNKANCGAA | 0.09 | 219.4 |
| N218A | SQKECTDGNKEACGAA | 2.7 | 6.0 |
| G220A | SQKECTDGNKENCAAA | 2.9 | 42.2 |
| Name | Sequence | Kd against 4D11 IgG (uM) |
|---|---|---|
| 8mer | NKENCGAA | 0.24 |
| D1 | NKENCGAANKENCGAAC | 2.38 |
| L1 | NKENCGAAGNKENCGAAC | 0.47 |
| L2 | NKENCGAAGGNKENCGAAC | 0.67 |
| L3 | NKENCGAAGGGNKENCGAAC | 0.58 |
| C1 | c[NKENCGAAGNKENCGAAC] | 0.44 |
| C2 | c[NKENCGAAGGNKENCGAACG] | 0.68 |
| C3 | c[NKENCGAAGGGNKENCGAAGCG] | 1.04 |
| A1 | SQKECTDGNKENCGAAC | 0.20 |
| K209A | SQAECTDGNKENCGAAC | 0.20 |
| Peptide Name | Sequence |
|---|---|
| A | HPQNTSDSQKECT |
| B | QNTSDSQKECTDG |
| C | SDSQKESTDGNKE |
| D | SQKECTDGNKENC |
| E | ECTDGNKENCGAA |
| F | TDGNKENCGAATS |
| G | NKENCGAATSLLN |
| H | ENCGAATSLLNNS |
| I | CGAATSLLNNSSN |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seow, J.; Das, S.C.; Morales, R.A.V.; Ataide, R.; Krishnarjuna, B.; Silk, M.; Chalmers, D.K.; Richards, J.; Anders, R.F.; MacRaild, C.A.; et al. Guiding the Immune Response to a Conserved Epitope in MSP2, an Intrinsically Disordered Malaria Vaccine Candidate. Vaccines 2021, 9, 855. https://doi.org/10.3390/vaccines9080855
Seow J, Das SC, Morales RAV, Ataide R, Krishnarjuna B, Silk M, Chalmers DK, Richards J, Anders RF, MacRaild CA, et al. Guiding the Immune Response to a Conserved Epitope in MSP2, an Intrinsically Disordered Malaria Vaccine Candidate. Vaccines. 2021; 9(8):855. https://doi.org/10.3390/vaccines9080855
Chicago/Turabian StyleSeow, Jeffrey, Sreedam C. Das, Rodrigo A. V. Morales, Ricardo Ataide, Bankala Krishnarjuna, Mitchell Silk, David K. Chalmers, Jack Richards, Robin F. Anders, Christopher A. MacRaild, and et al. 2021. "Guiding the Immune Response to a Conserved Epitope in MSP2, an Intrinsically Disordered Malaria Vaccine Candidate" Vaccines 9, no. 8: 855. https://doi.org/10.3390/vaccines9080855
APA StyleSeow, J., Das, S. C., Morales, R. A. V., Ataide, R., Krishnarjuna, B., Silk, M., Chalmers, D. K., Richards, J., Anders, R. F., MacRaild, C. A., & Norton, R. S. (2021). Guiding the Immune Response to a Conserved Epitope in MSP2, an Intrinsically Disordered Malaria Vaccine Candidate. Vaccines, 9(8), 855. https://doi.org/10.3390/vaccines9080855