Investigating Prime-Pull Vaccination through a Combination of Parenteral Vaccination and Intranasal Boosting



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of Freeze-Dried PLGA:H56 Nanoparticles and PLGA:H56 Microparticles
2.3. Particle Size and Zeta Potential of PLGA Nano- and Microparticles
2.4. Pharmacopoeia Airway Simulator: Next Generation Impactor (NGI)
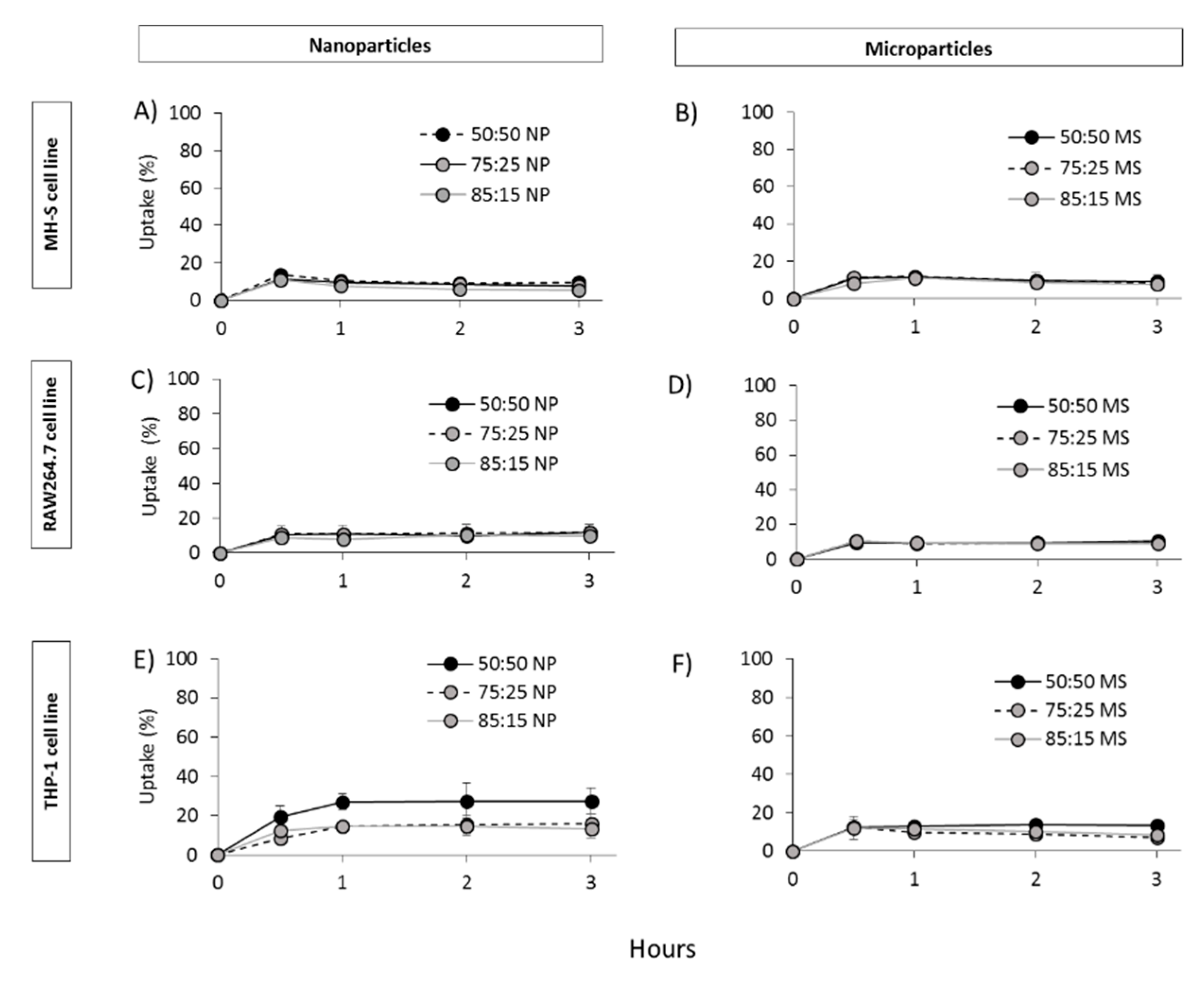
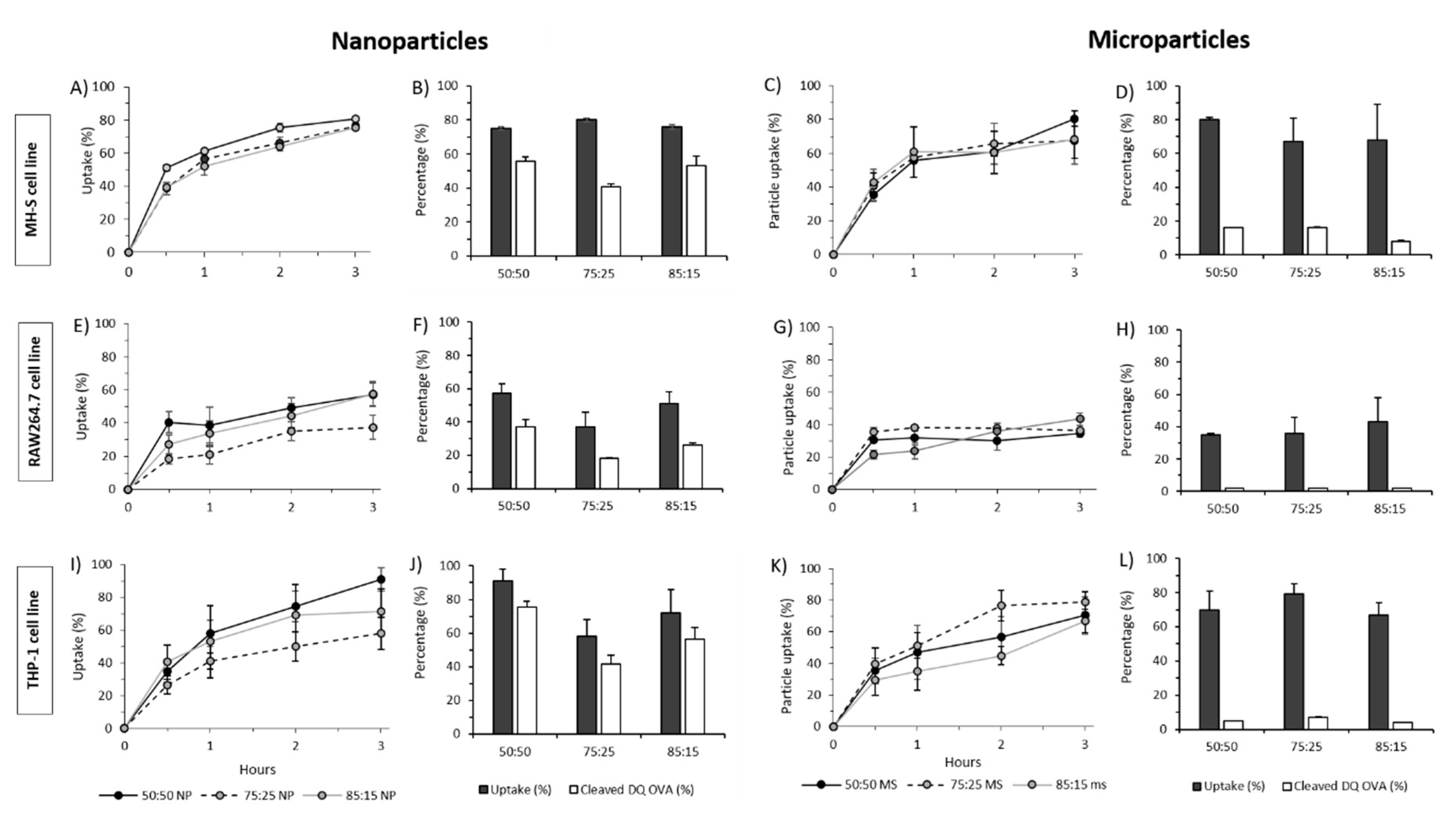
2.5. In Vitro Cell Viability, Particle Uptake and Antigen Processing in Three Macrophages Cell Lines: THP-1, MH-S and RAW264.7
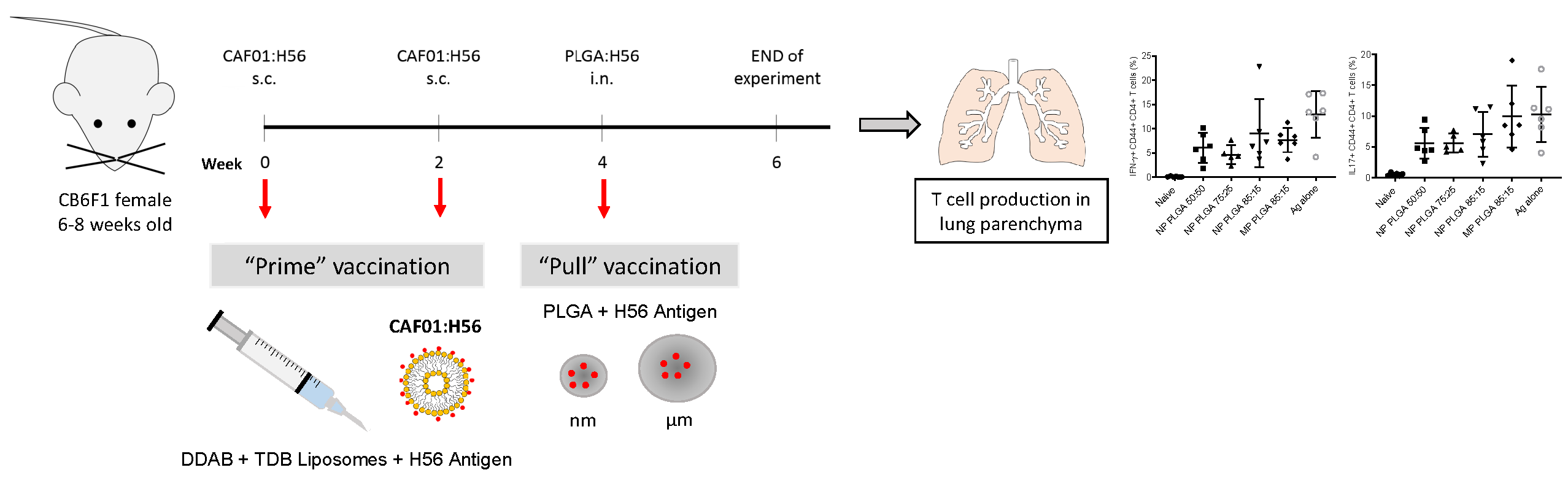
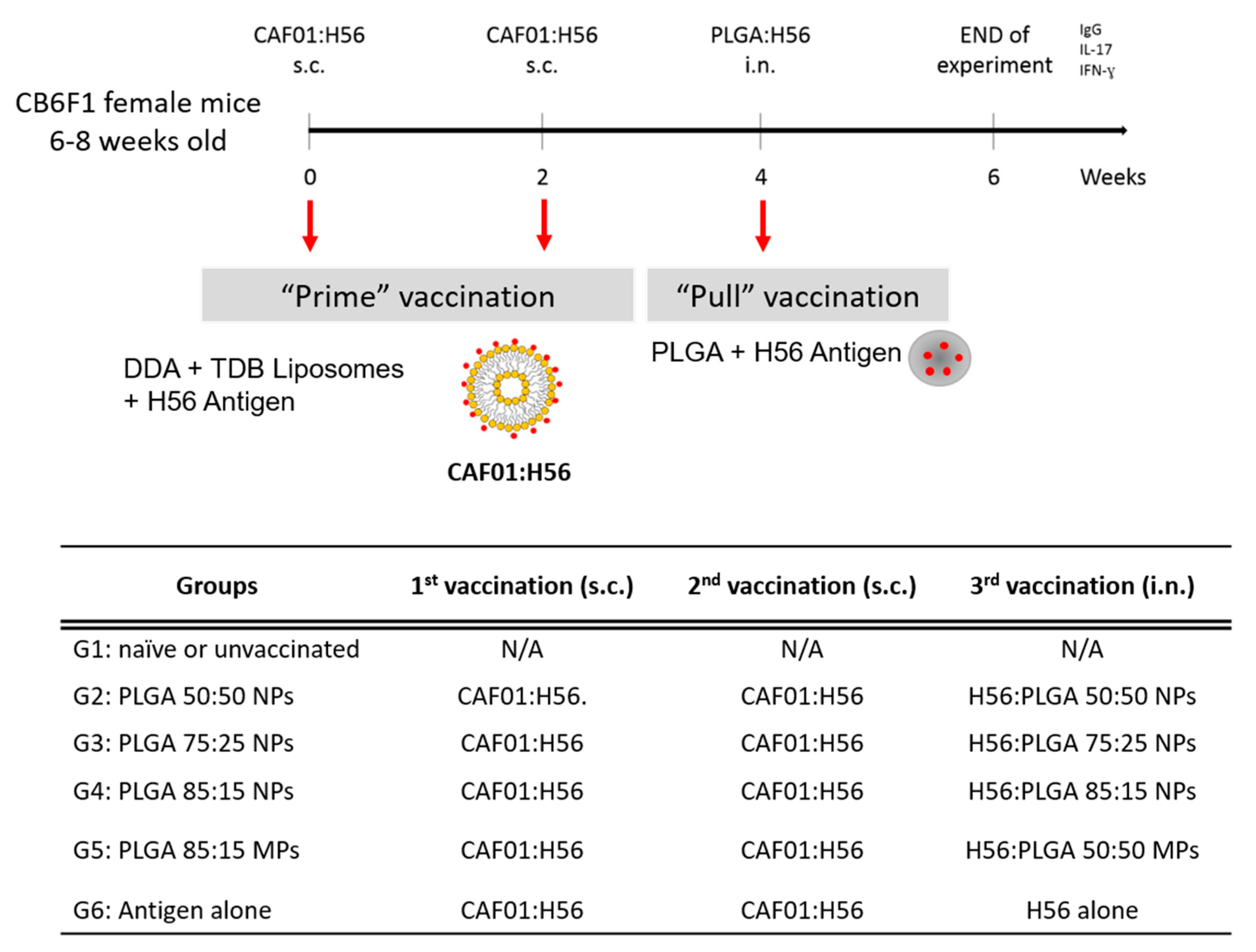
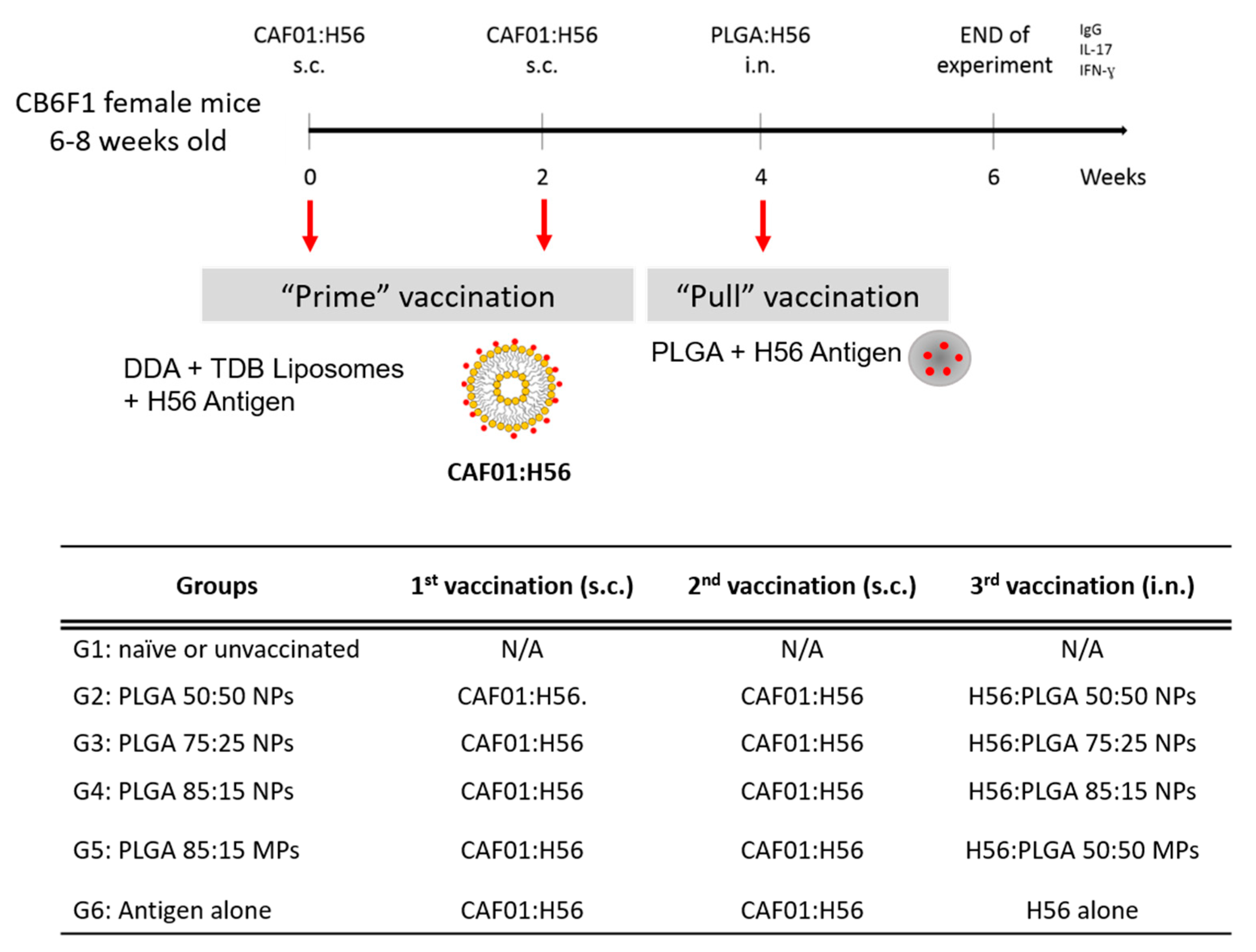
2.6. Prime-Pull Immunisation Protocol
2.6.1. Organ Processing
2.6.2. Intracellular Fluorescence Activated Cell Sorting Staining (icFACS)
2.7. Statistical Analysis
3. Results
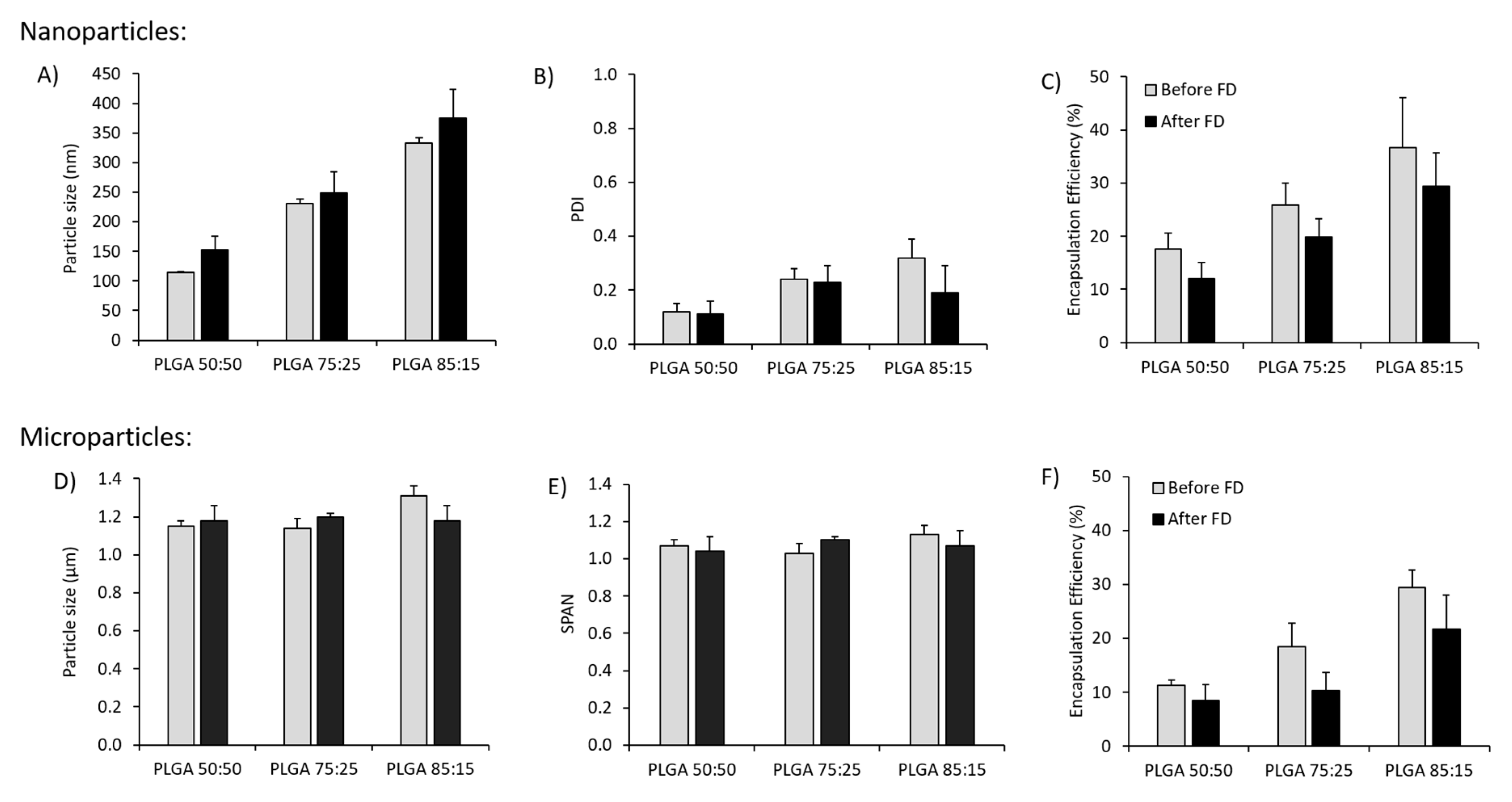
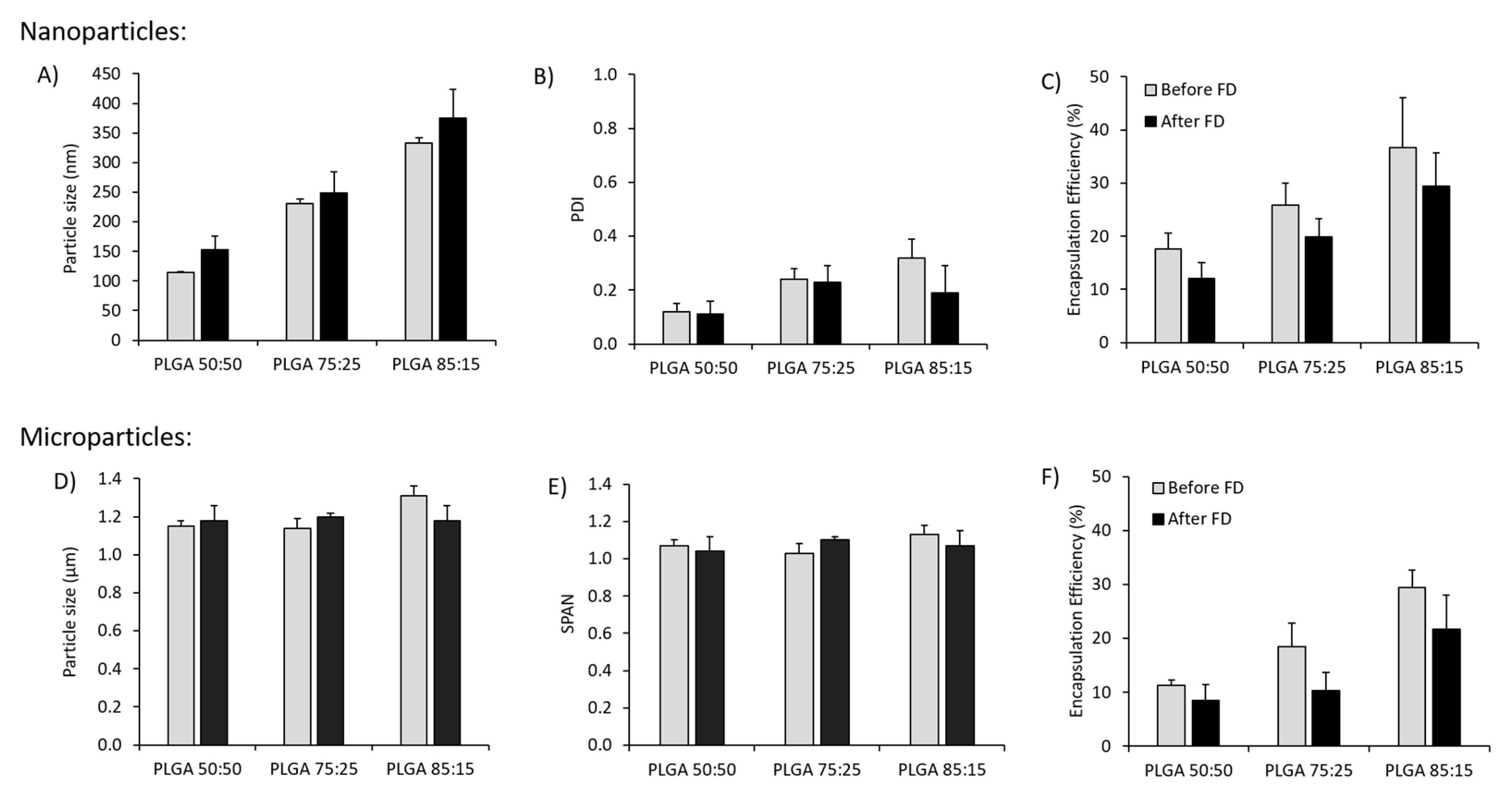
3.1. Effect of Freeze-Drying on the Physicochemical Characteristics of Antigen Loaded PLGA Nano- and Microparticles
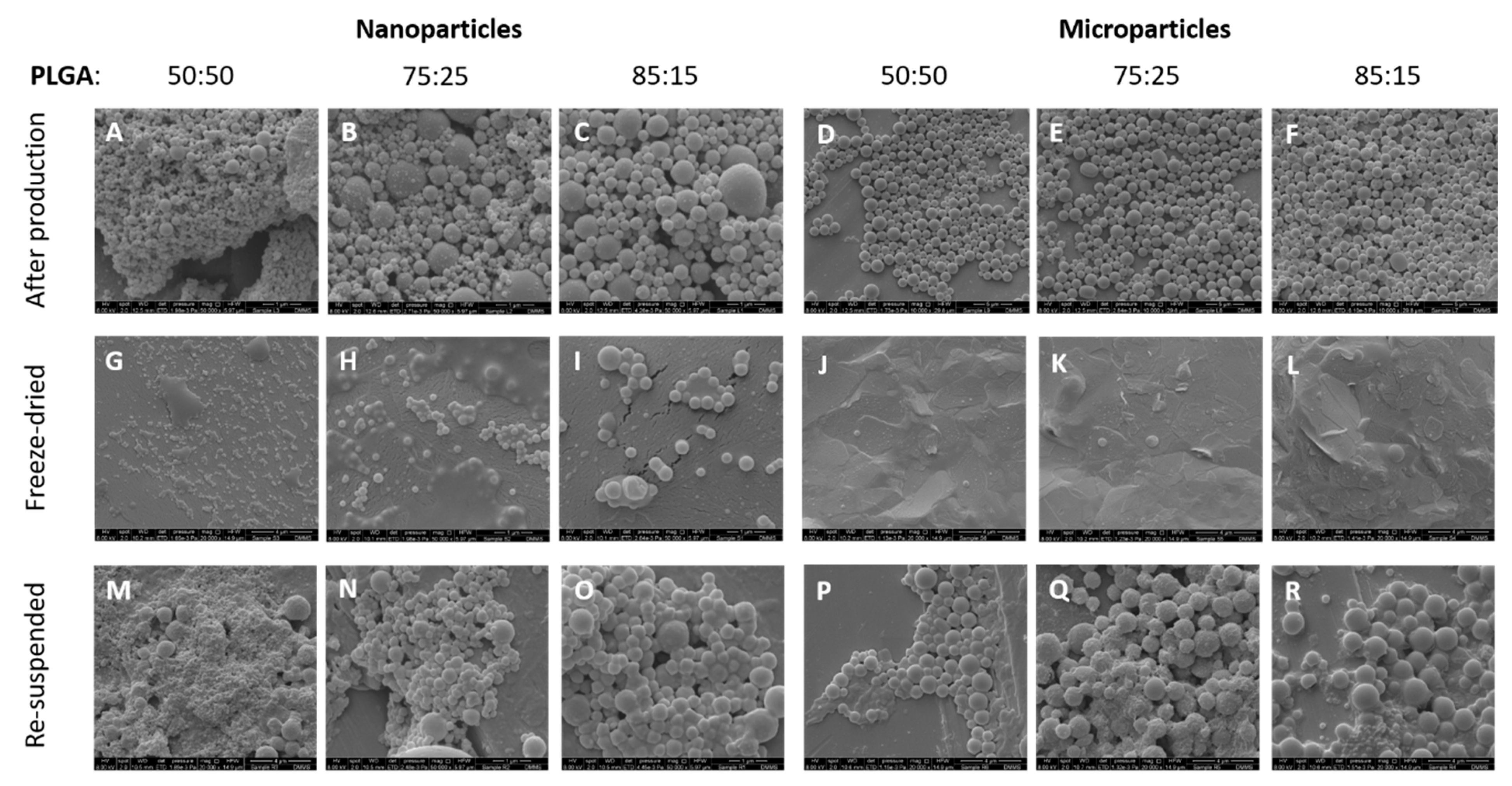
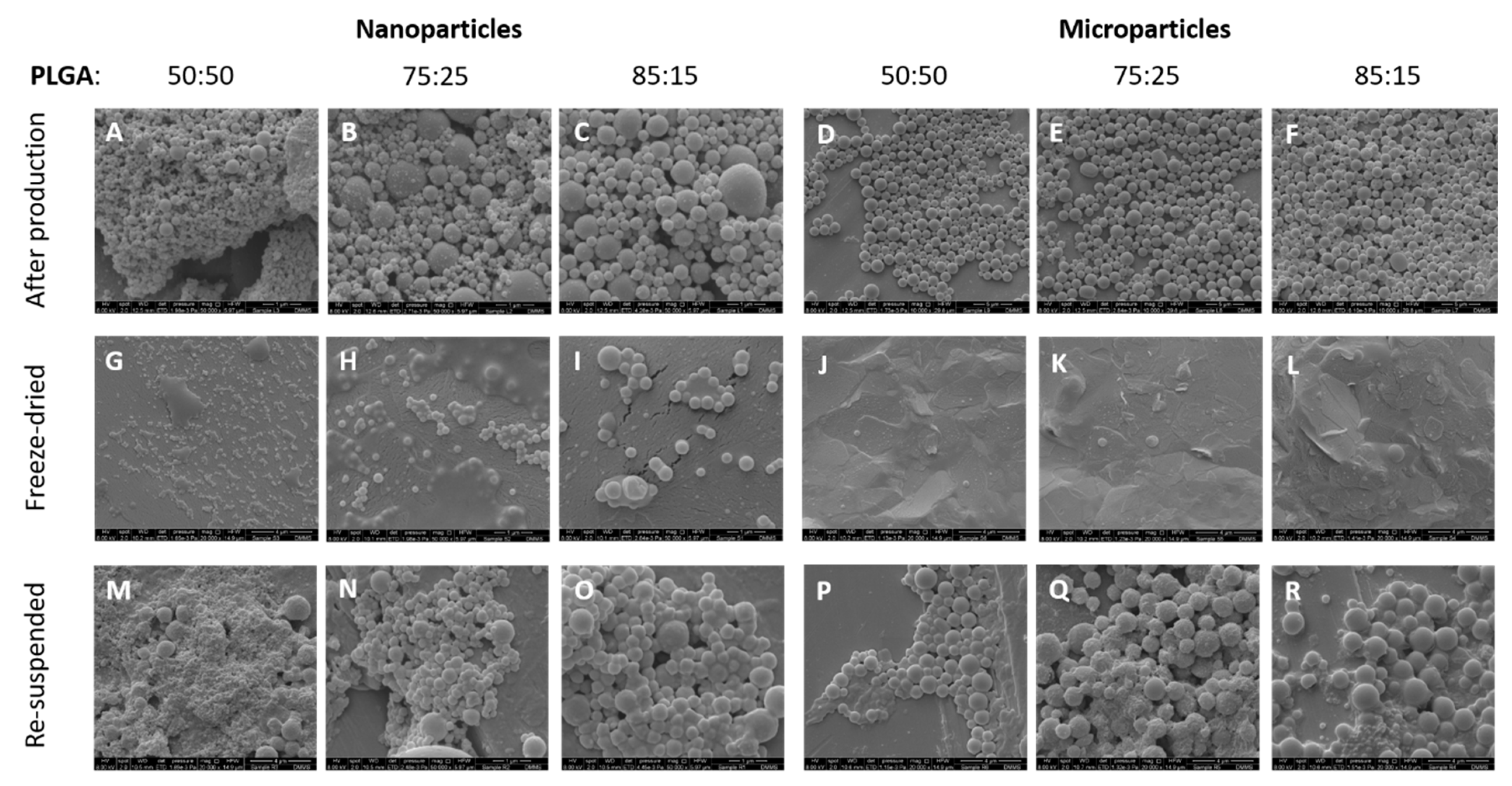
3.2. Morphological Characterisation of the Antigen Loaded PLGA Nano- and Micro-Particles
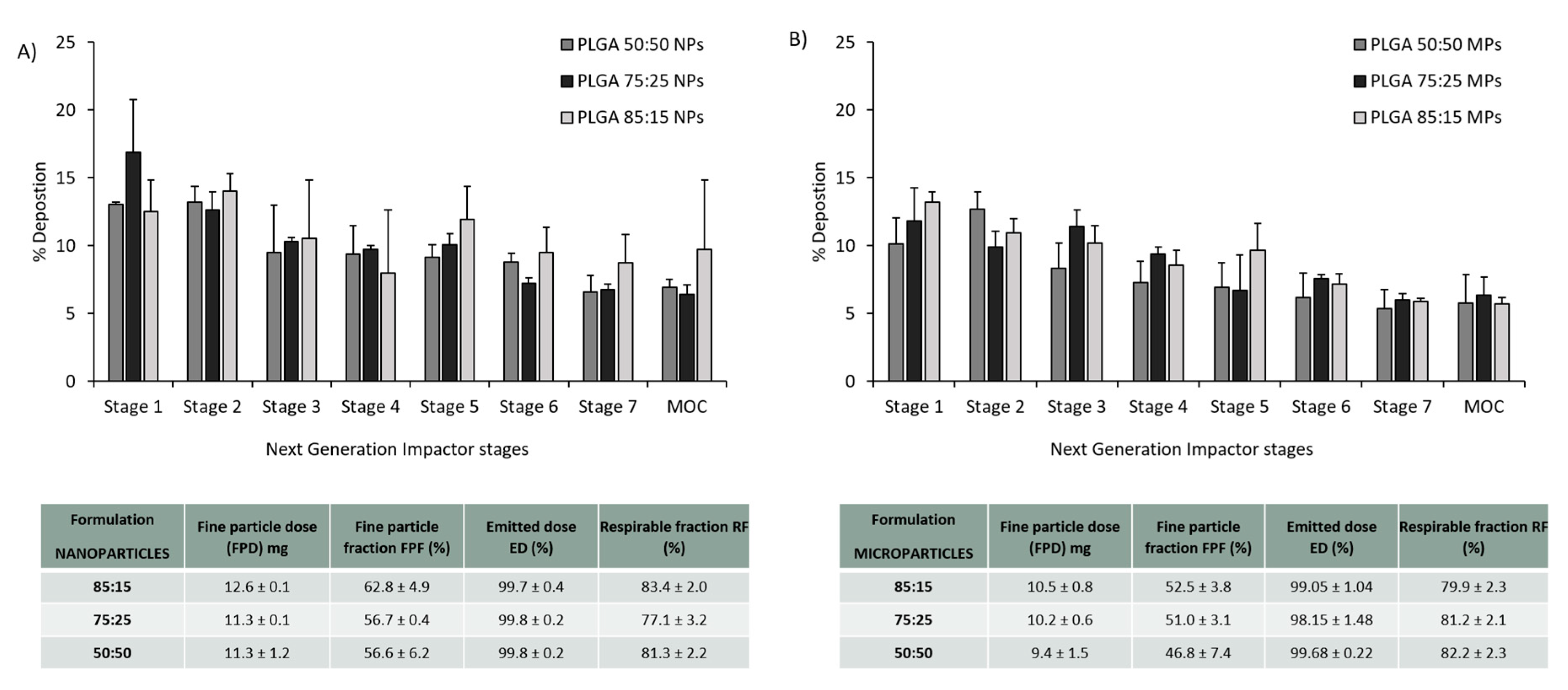
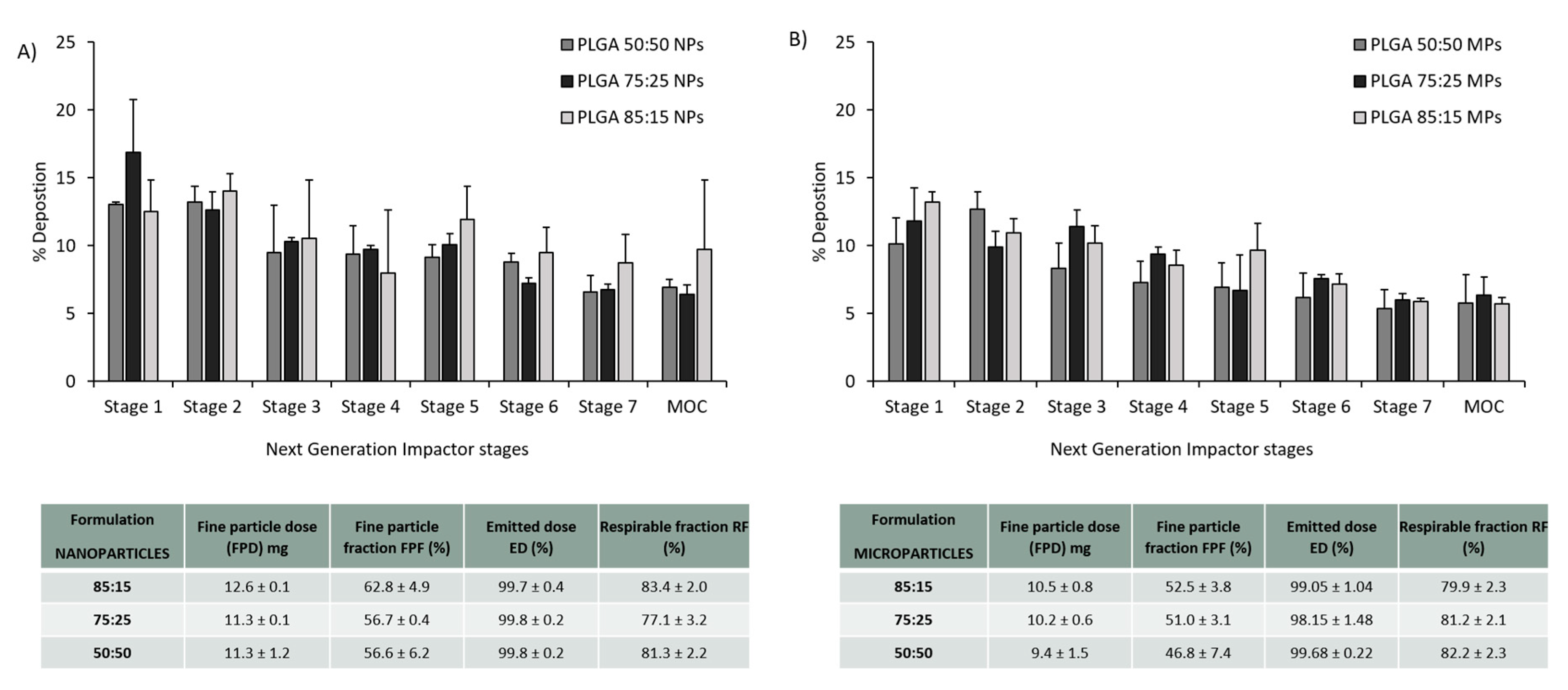
3.3. PLGA Nano- and Microparticles Are Deposited in the Deep Lungs
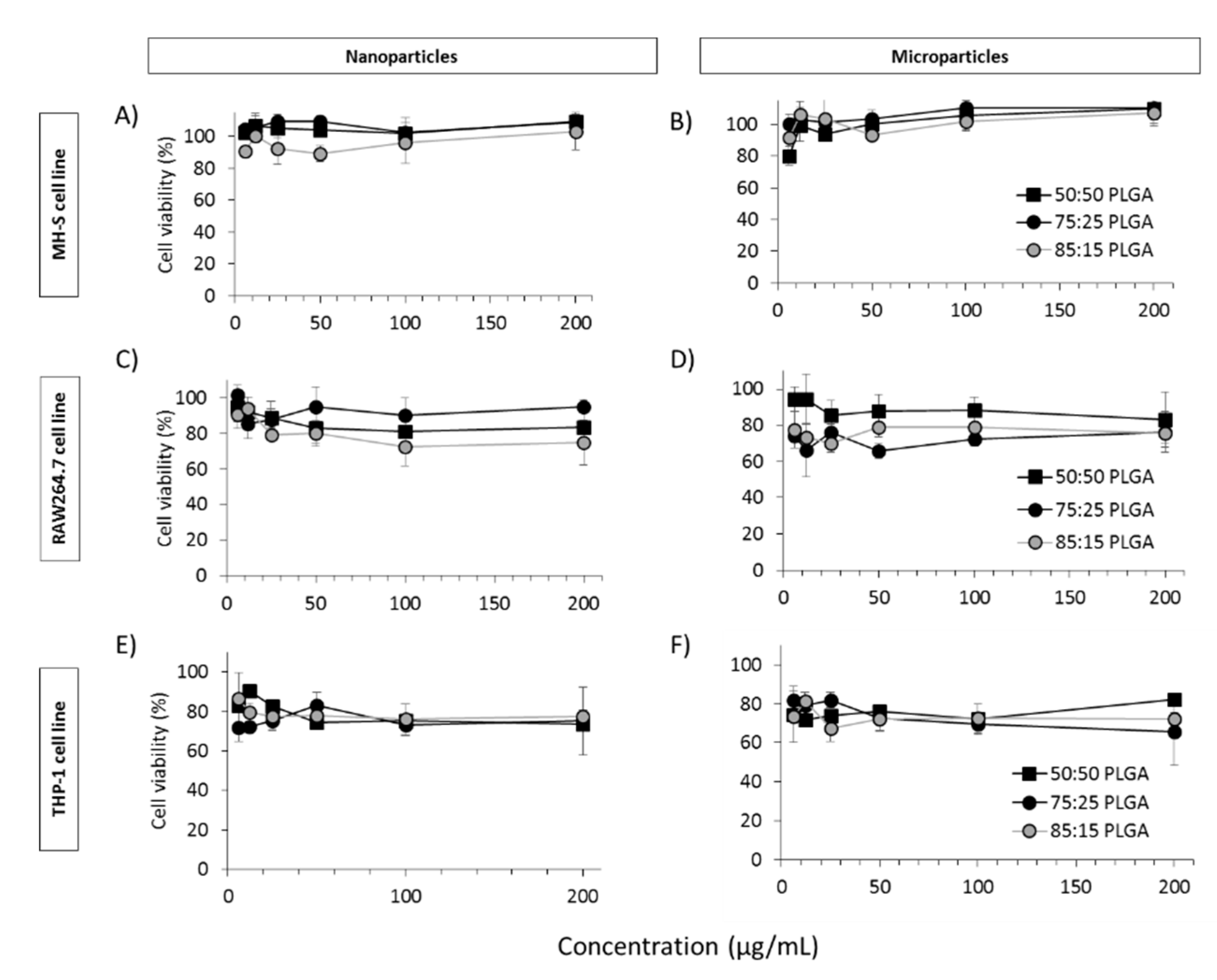
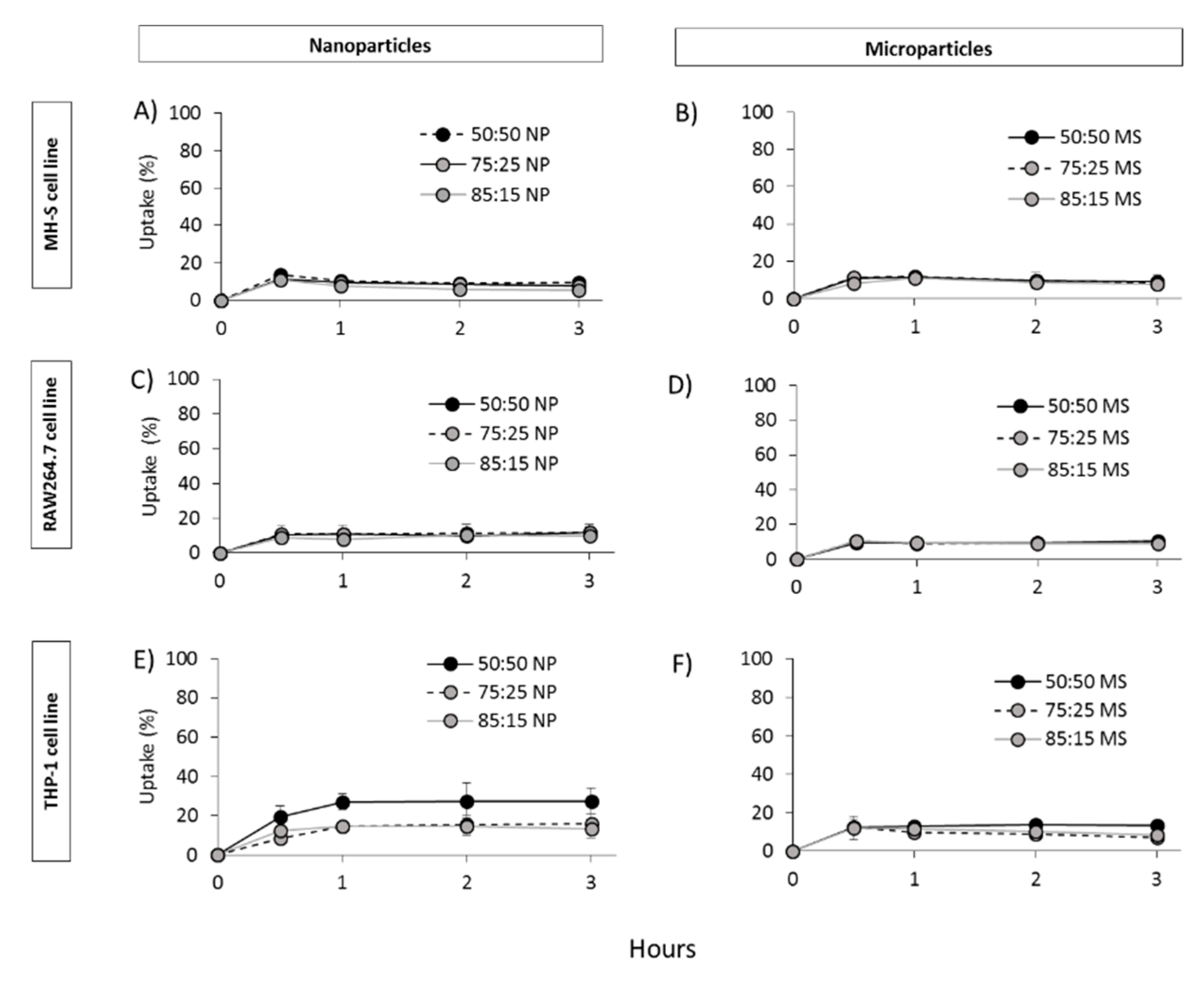
3.4. In Vitro Studies in Murine and Human Origin Cell Lines: Cell Viability, Uptake and Antigen Processing
3.5. Prime-Pull Vaccination Using CAF01:H56 as Primer and PLGA:H56 as Mucosal Booster
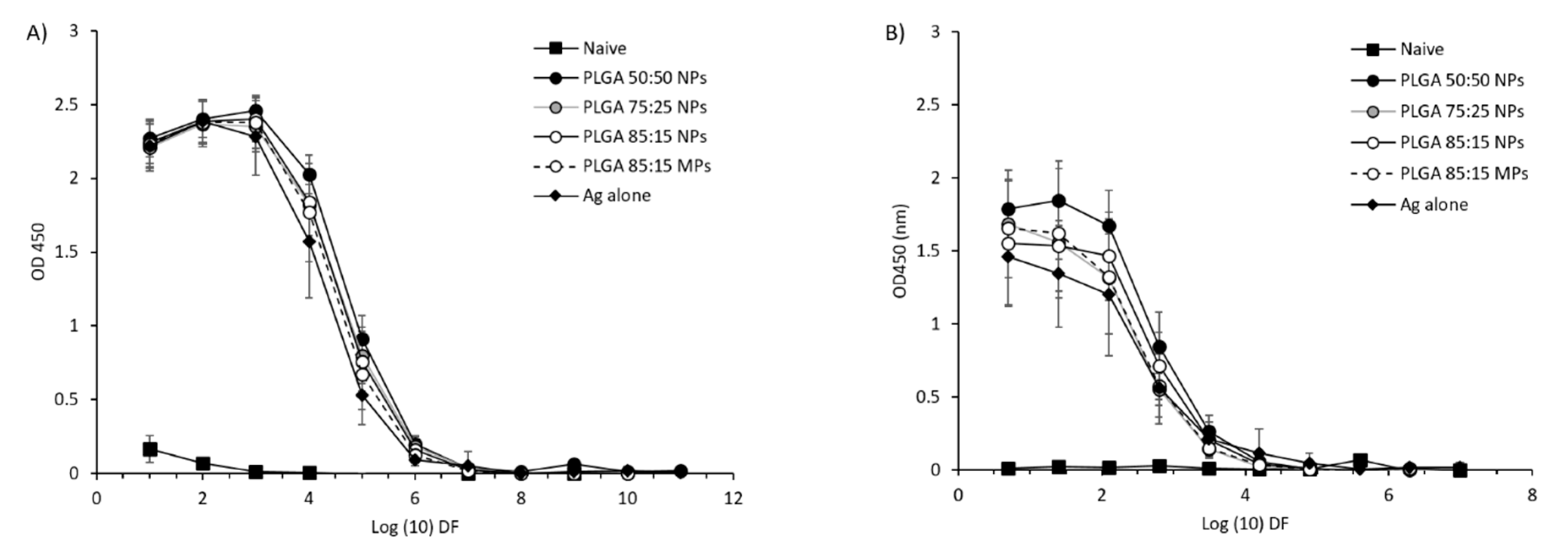
3.5.1. Mucosal Boosting with PLGA:H56 Nano- or Microparticles Does not Enhance the Humoral and Cellular Immune Responses
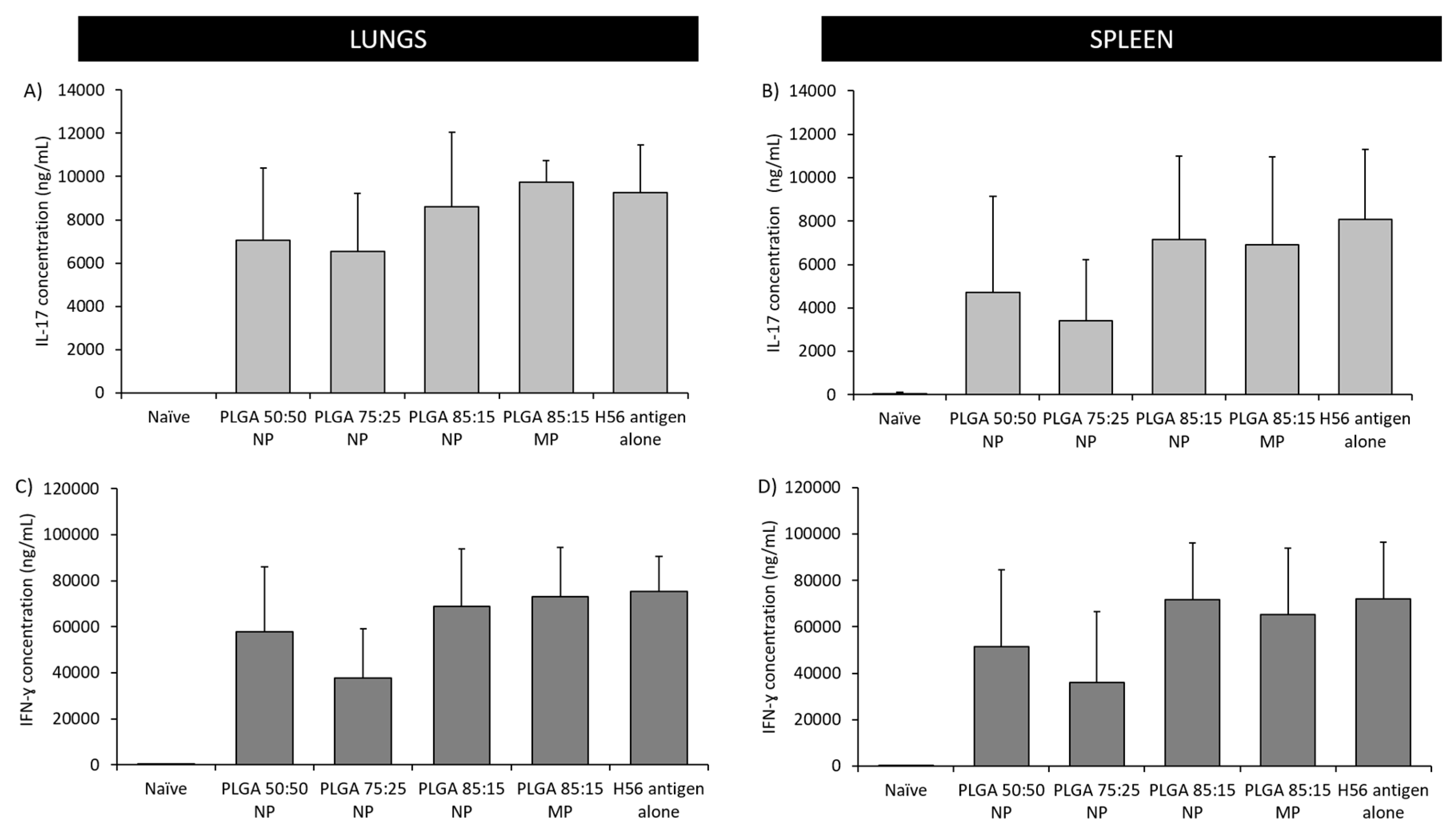
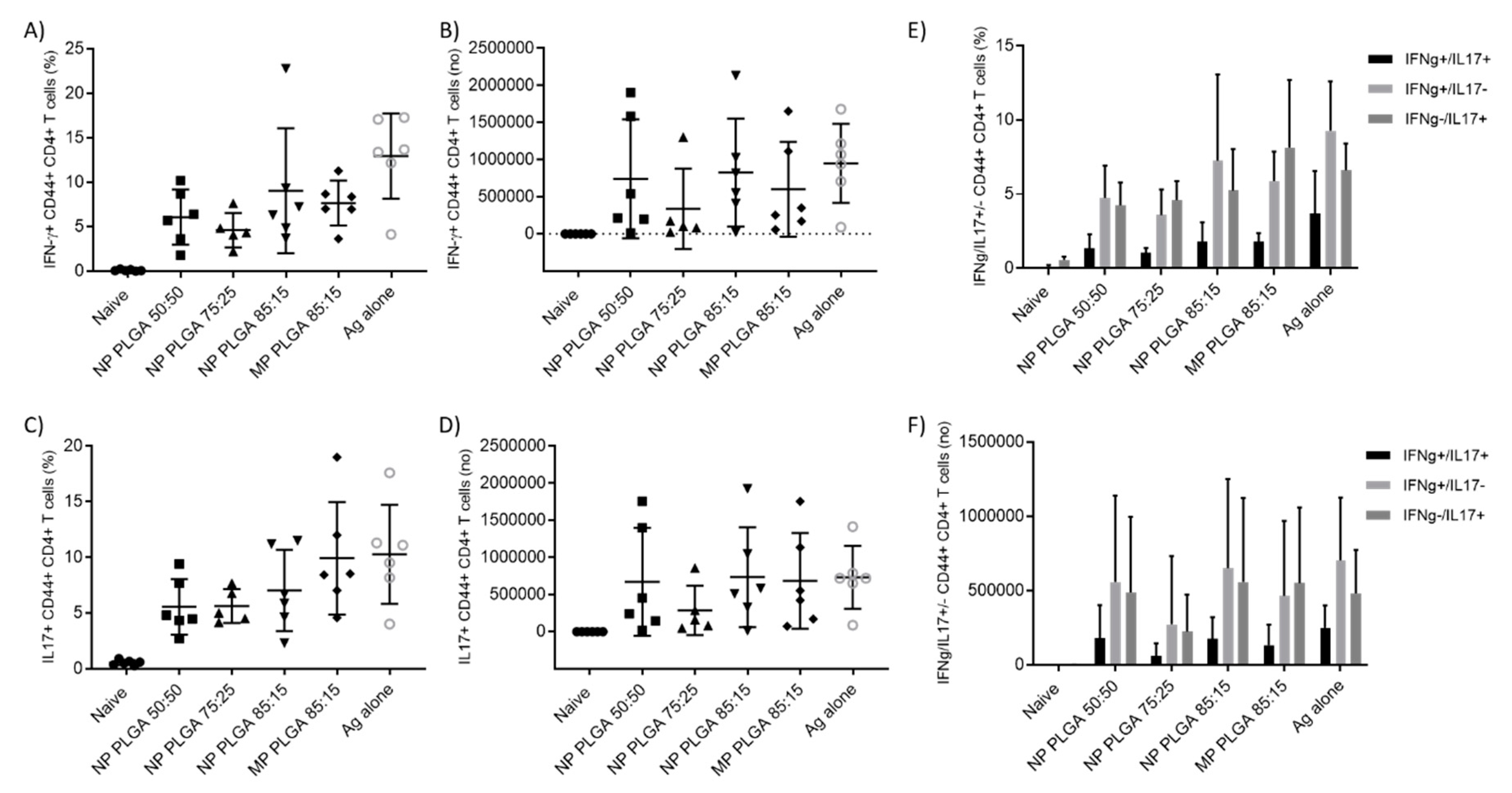
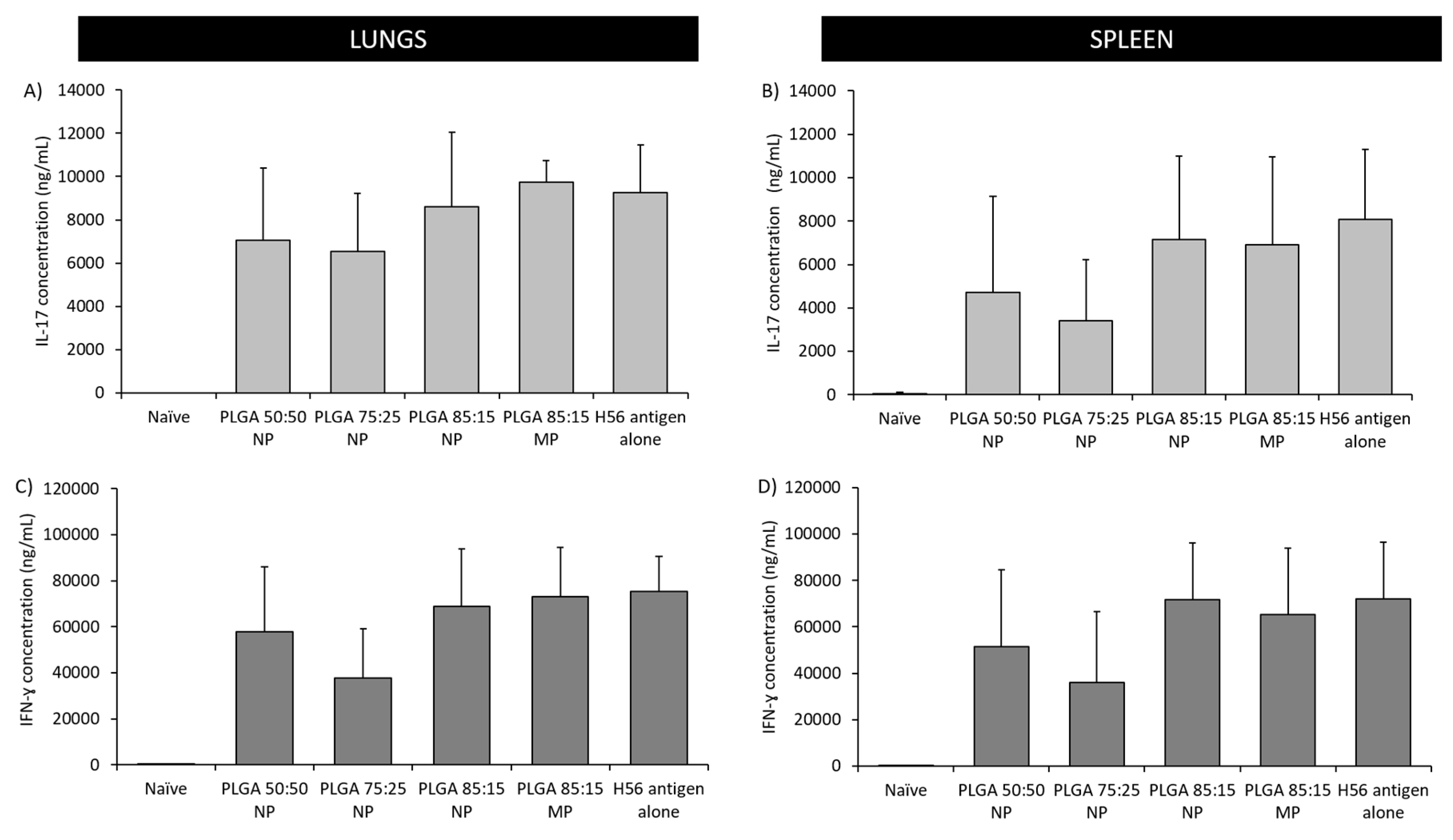
3.5.2. The Location of T-Cell Responses is Important for Protection against TB: Prime-Pull Immunisation Stimulates Cytokine Production in the Lung Parenchyma
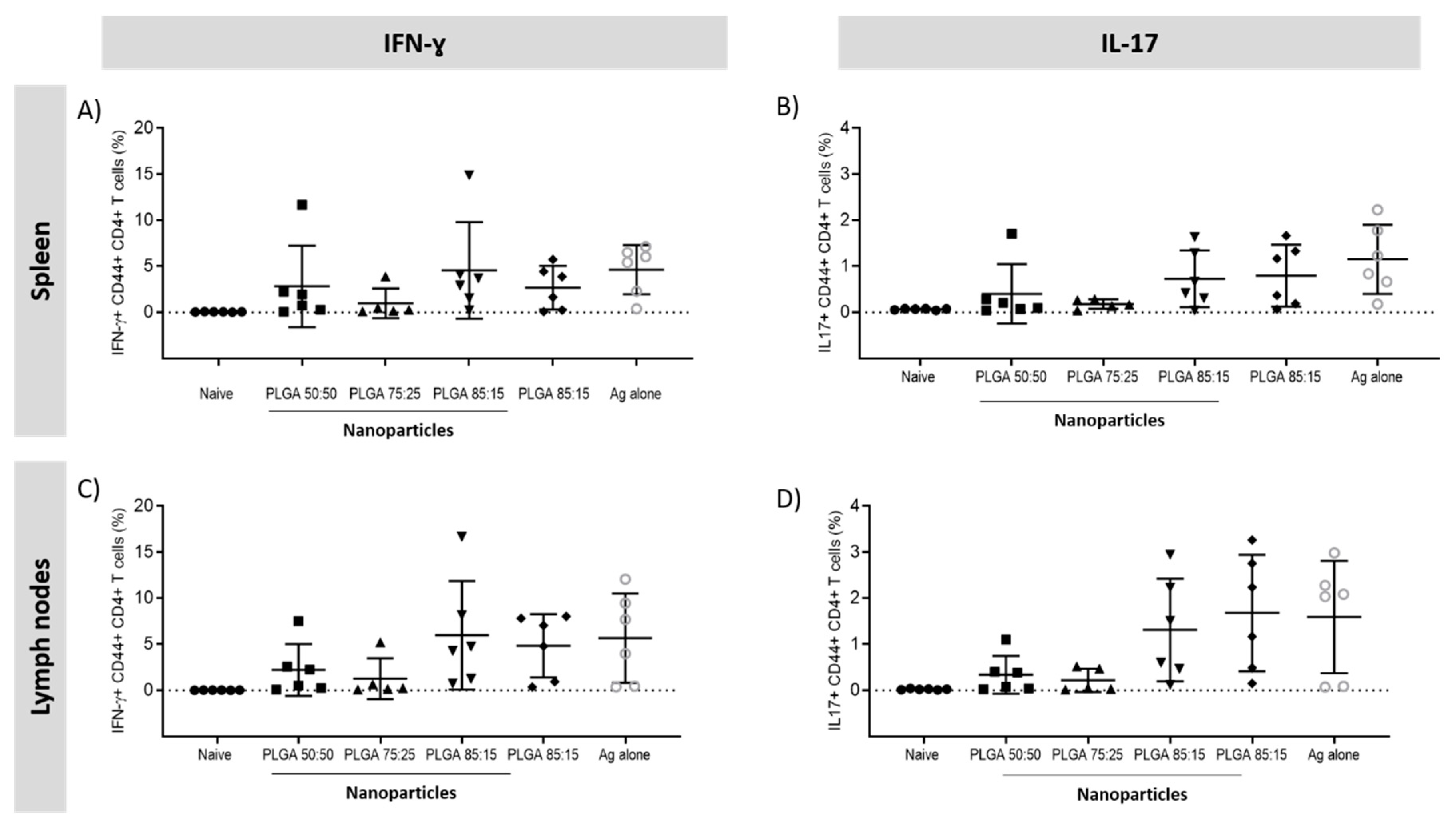
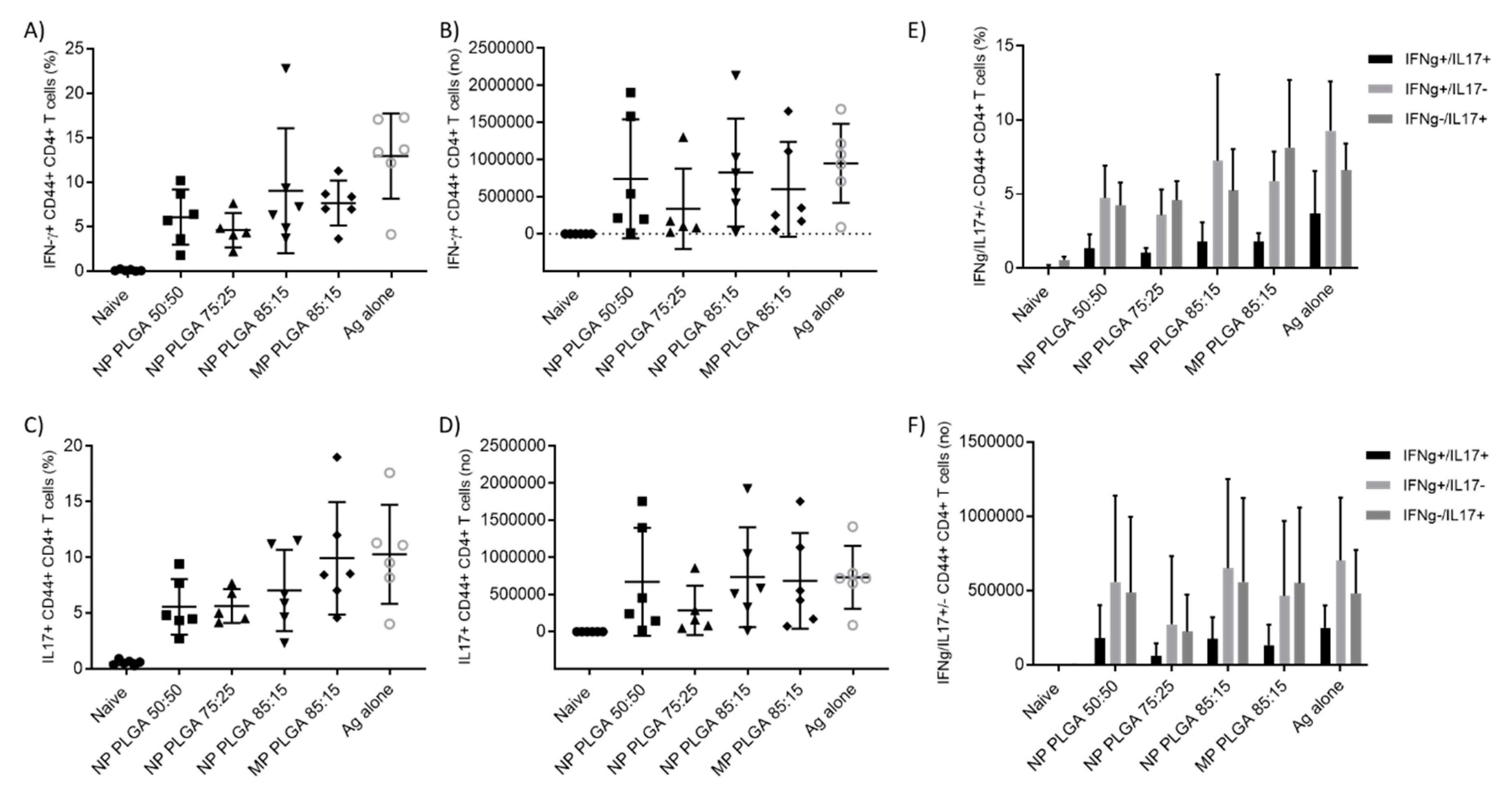
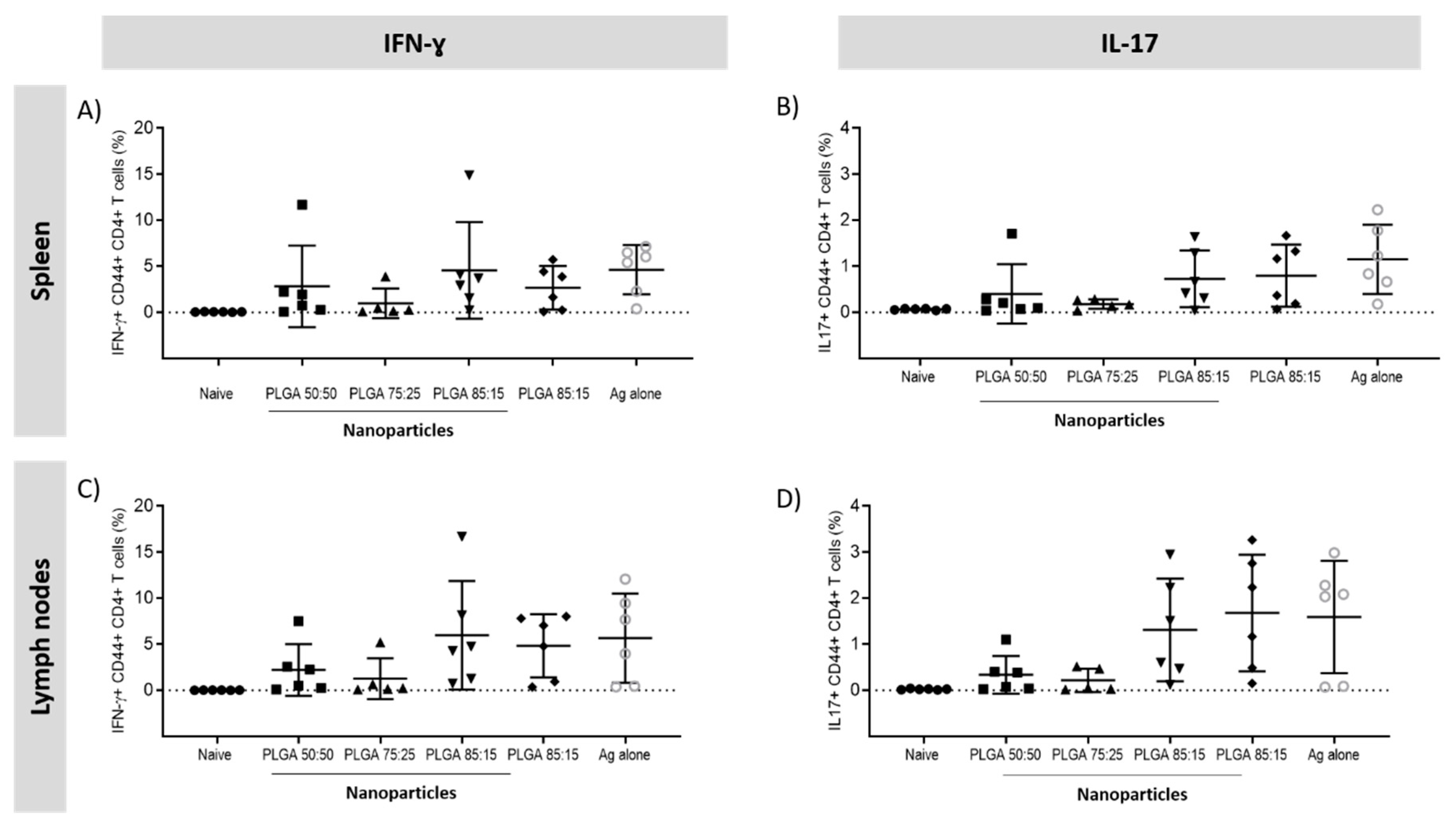
3.5.3. Antigen Specific CD4+ T Cells Responses in the Spleen and Lymph Nodes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Roy, A.; Eisenhut, M.; Harris, R.J.; Rodrigues, L.C.; Sridhar, S.; Habermann, S.; Snell, L.; Mangtani, P.; Adetifa, I.; Lalvani, A.; et al. Effect of BCG vaccination against Mycobacterium tuberculosis infection in children: Systematic review and meta-analysis. BMJ Br. Med. J. 2014, 349, g4643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horvath, C.N.; Shaler, C.R.; Jeyanathan, M.; Zganiacz, A.; Xing, Z. Mechanisms of delayed anti-tuberculosis protection in the lung of parenteral BCG-vaccinated hosts: A critical role of airway luminal T cells. Mucosal Immunol. 2012, 5, 420–431. Available online: https://www.nature.com/articles/mi201219#supplementary-information (accessed on 25 November 2019). [CrossRef] [PubMed]
- Mogues, T.; Goodrich, M.E.; Ryan, L.; LaCourse, R.; North, R.J. The Relative Importance of T Cell Subsets in Immunity and Immunopathology of Airborne Mycobacterium tuberculosis Infection in Mice. J. Exp. Med. 2001, 193, 271–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakai, S.; Kauffman, K.D.; Schenkel, J.M.; McBerry, C.C.; Mayer-Barber, K.D.; Masopust, D.; Barber, D.L. Cutting Edge: Control of Mycobacterium tuberculosis Infection by a Subset of Lung Parenchyma–Homing CD4 T Cells. J. Immunol. 2014, 192, 2965–2969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woodworth, J.S.; Cohen, S.B.; Moguche, A.O.; Plumlee, C.R.; Agger, E.M.; Urdahl, K.B.; Andersen, P. Subunit vaccine H56/CAF01 induces a population of circulating CD4 T cells that traffic into the Mycobacterium tuberculosis-infected lung. Mucosal Immunol. 2017, 10, 555–564. [Google Scholar] [CrossRef] [Green Version]
- Jeyanathan, M.; Yao, Y.; Afkhami, S.; Smaill, F.; Xing, Z. New tuberculosis vaccine strategies: Taking aim at un-natural immunity. Trends Immunol. 2018, 39, 419–433. [Google Scholar] [CrossRef]
- Christensen, D.; Mortensen, R.; Rosenkrands, I.; Dietrich, J.; Andersen, P. Vaccine-induced Th17 cells are established as resident memory cells in the lung and promote local IgA responses. Mucosal Immunol. 2017, 10, 260–270. [Google Scholar] [CrossRef] [Green Version]
- Ungaro, F.; d’Angelo, I.; Miro, A.; La Rotonda, M.I.; Quaglia, F. Engineered PLGA nano- and micro-carriers for pulmonary delivery: Challenges and promises. J. Pharm. Pharmacol. 2012, 64, 1217–1235. [Google Scholar] [CrossRef]
- Mohammed, A.R.; Bramwell, V.W.; Coombes, A.G.A.; Perrie, Y. Lyophilisation and sterilisation of liposomal vaccines to produce stable and sterile products. Methods 2006, 40, 30–38. [Google Scholar] [CrossRef]
- Mohammed, A.R.; Coombes, A.G.A.; Perrie, Y. Amino acids as cryoprotectants for liposomal delivery systems. Eur. J. Pharm. Sci. 2007, 30, 406–413. [Google Scholar] [CrossRef]
- Anderson, K.G.; Mayer-Barber, K.; Sung, H.; Beura, L.; James, B.R.; Taylor, J.J.; Qunaj, L.; Griffith, T.S.; Vezys, V.; Barber, D.L.; et al. Intravascular staining for discrimination of vascular and tissue leukocytes. Nat. Protoc. 2014, 9, 209–222. Available online: https://www.nature.com/articles/nprot.2014.005#supplementary-information (accessed on 25 November 2019). [CrossRef] [PubMed] [Green Version]
- He, C.; Hu, Y.; Yin, L.; Tang, C.; Yin, C. Effects of particle size and surface charge on cellular uptake and biodistribution of polymeric nanoparticles. Biomaterials 2010, 31, 3657–3666. [Google Scholar] [CrossRef] [PubMed]
- Hirota, K.; Hasegawa, T.; Nakajima, T.; Inagawa, H.; Kohchi, C.; Soma, G.-I.; Makino, K.; Terada, H. Delivery of rifampicin–PLGA microspheres into alveolar macrophages is promising for treatment of tuberculosis. J. Control. Release 2010, 142, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, T.C.; Peters, J.I.; Williams, R.O., III. Influence of particle size on regional lung deposition–what evidence is there? Int. J. Pharm. 2011, 406, 1–10. [Google Scholar] [CrossRef]
- Pilcer, G.; Amighi, K. Formulation strategy and use of excipients in pulmonary drug delivery. Int. J. Pharm. 2010, 392, 1–19. [Google Scholar] [CrossRef]
- Hinds, W.C. Aerosol Technology: Properties, Behavior, and Measurement of Airborne Particles; John Wiley & Sons: Hoboken, NJ, USA, 2012. [Google Scholar]
- Seville, P.; Learoyd, T.P.; Li, H.; Williamson, I.J.; Birchall, J. Amino acid-modified spray-dried powders with enhanced aerosolisation properties for pulmonary drug delivery. Powder Technol. 2007, 178, 40–50. [Google Scholar] [CrossRef]
- Adler, M.; Unger, M.; Lee, G. Surface Composition of Spray-Dried Particles of Bovine Serum Albumin/Trehalose/Surfactant. Pharm. Res. 2000, 17, 863–870. [Google Scholar] [CrossRef]
- Fonte, P.; Araújo, F.; Seabra, V.; Reis, S.; van de Weert, M.; Sarmento, B. Co-encapsulation of lyoprotectants improves the stability of protein-loaded PLGA nanoparticles upon lyophilization. Int. J. Pharm. 2015, 496, 850–862. [Google Scholar] [CrossRef]
- Abdelwahed, W.; Degobert, G.; Stainmesse, S.; Fessi, H. Freeze-drying of nanoparticles: Formulation, process and storage considerations. Adv. Drug Deliv. Rev. 2006, 58, 1688–1713. [Google Scholar] [CrossRef]
- Fonte, P.; Soares, S.; Costa, A.; Andrade, J.C.; Seabra, V.; Reis, S.; Sarmento, B. Effect of cryoprotectants on the porosity and stability of insulin-loaded PLGA nanoparticles after freeze-drying. Biomatter 2012, 2, 329–339. [Google Scholar] [CrossRef] [Green Version]
- Woodworth, J.S.; Christensen, D.; Cassidy, J.P.; Agger, E.M.; Mortensen, R.; Andersen, P. Mucosal boosting of H56:CAF01 immunization promotes lung-localized T cells and an accelerated pulmonary response to Mycobacterium tuberculosis infection without enhancing vaccine protection. Mucosal Immunol. 2019, 12, 816–826. [Google Scholar] [CrossRef] [PubMed]
- Lorenzen, E.; Follmann, F.; Bøje, S.; Erneholm, K.; Olsen, A.W.; Agerholm, J.S.; Jungersen, G.; Andersen, P. Intramuscular Priming and Intranasal Boosting Induce Strong Genital Immunity Through Secretory IgA in Minipigs Infected with Chlamydia trachomatis. Front. Immunol. 2015, 6, 628. [Google Scholar] [CrossRef] [PubMed]
- Desvignes, L.; Ernst, J.D. Interferon-γ-responsive nonhematopoietic cells regulate the immune response to Mycobacterium tuberculosis. Immunity 2009, 31, 974–985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baaten, B.J.G.; Li, C.-R.; Deiro, M.F.; Lin, M.M.; Linton, P.J.; Bradley, L.M. CD44 Regulates Survival and Memory Development in Th1 Cells. Immunity 2010, 32, 104–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konan, Y.N.; Gurny, R.; Allémann, E. Preparation and characterization of sterile and freeze-dried sub-200 nm nanoparticles. Int. J. Pharm. 2002, 233, 239–252. [Google Scholar] [CrossRef]
- Bozdag, S.; Dillen, K.; Vandervoort, J.; Ludwig, A. The effect of freeze-drying with different cryoprotectants and gamma-irradiation sterilization on the characteristics of ciprofloxacin HCl-loaded poly (D, L-lactide-glycolide) nanoparticles. J. Pharm. Pharmacol. 2005, 57, 699–708. [Google Scholar] [CrossRef]
- Allison, S.; Molina, M.D.; Anchordoquy, T.J. Stabilization of lipid/DNA complexes during the freezing step of the lyophilization process: The particle isolation hypothesis. Biochim. Biophys. Acta (BBA)-Biomembr. 2000, 1468, 127–138. [Google Scholar] [CrossRef] [Green Version]
- Raula, J.; Thielmann, F.; Naderi, M.; Lehto, V.-P.; Kauppinen, E.I. Investigations on particle surface characteristics vs. dispersion behaviour of l-leucine coated carrier-free inhalable powders. Int. J. Pharm. 2010, 385, 79–85. [Google Scholar] [CrossRef]
- Saez, A.; Guzman, M.; Molpeceres, J.; Aberturas, M. Freeze-drying of polycaprolactone and poly (D, L-lactic-glycolic) nanoparticles induce minor particle size changes affecting the oral pharmacokinetics of loaded drugs. Eur. J. Pharm. Biopharm. 2000, 50, 379–387. [Google Scholar] [CrossRef]
- Chacon, M.; Molpeceres, J.; Berges, L.; Guzman, M.; Aberturas, M. Stability and freeze-drying of cyclosporine loaded poly (D, L lactide–glycolide) carriers. Eur. J. Pharm. Sci. 1999, 8, 99–107. [Google Scholar] [CrossRef]
- Najafabadi, A.; Gilani, K.; Barghi, M.; Rafiee-Tehrani, M. The effect of vehicle on physical properties and aerosolisation behavior of disodium cromoglycate microparticles spray dried alone or with L-Leucine. Int. J. Pharm. 2004, 285, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Chew, N.Y.K.; Tang, P.; Chan, H.-K.; Raper, J.A. How Much Particle Surface Corrugation Is Sufficient to Improve Aerosol Performance of Powders? Pharm. Res. 2005, 22, 148–152. [Google Scholar] [CrossRef] [PubMed]
- Chow, M.Y.T.; Qiu, Y.; Lo, F.F.K.; Lin, H.H.S.; Chan, H.-K.; Kwok, P.C.L.; Lam, J.K.W. Inhaled powder formulation of naked siRNA using spray drying technology with l-leucine as dispersion enhancer. Int. J. Pharm. 2017, 530, 40–52. [Google Scholar] [CrossRef] [PubMed]
- Sou, T.; Kaminskas, L.M.; Nguyen, T.-H.; Carlberg, R.; McIntosh, M.P.; Morton, D.A.V. The effect of amino acid excipients on morphology and solid-state properties of multi-component spray-dried formulations for pulmonary delivery of biomacromolecules. Eur. J. Pharm. Biopharm. 2013, 83, 234–243. [Google Scholar] [CrossRef]
- Eedara, B.B.; Rangnekar, B.; Sinha, S.; Doyle, C.; Cavallaro, A.; Das, S.C. Development and characterization of high payload combination dry powders of anti-tubercular drugs for treating pulmonary tuberculosis. Eur. J. Pharm. Sci. 2018, 118, 216–226. [Google Scholar] [CrossRef]
- Wolfenden, R.; Andersson, L.; Cullis, P.M.; Southgate, C.C.B. Affinities of Amino Acid Side Chains for Solvent Water. Biochemistry 1981, 20, 849–855. [Google Scholar] [CrossRef]
- Xie, X.; Lin, W.; Xing, C.; Yang, Y.; Chi, Q.; Zhang, H.; Li, Y.; Li, Z.; Yang, Y.; Yang, Z.; et al. In Vitro and In Vivo Evaluations of PLGA Microspheres Containing Nalmefene. PLoS ONE 2015, 10, e0125953. [Google Scholar] [CrossRef]
- Liu, C.W.; Lin, W.J. Polymeric nanoparticles conjugate a novel heptapeptide as an epidermal growth factor receptor-active targeting ligand for doxorubicin. Int. J. Nanomed. 2012, 7, 4749–4767. [Google Scholar] [CrossRef] [Green Version]
- Akagi, T.; Shima, F.; Akashi, M. Intracellular degradation and distribution of protein-encapsulated amphiphilic poly (amino acid) nanoparticles. Biomaterials 2011, 32, 4959–4967. [Google Scholar] [CrossRef]
- Andersen, P.; Urdahl, K.B. TB vaccines; promoting rapid and durable protection in the lung. Curr. Opin. Immunol. 2015, 35, 55–62. [Google Scholar] [CrossRef] [Green Version]
- Aagaard, C.; Hoang, T.; Dietrich, J.; Cardona, P.-J.; Izzo, A.; Dolganov, G.; Schoolnik, G.K.; Cassidy, J.P.; Billeskov, R.; Andersen, P. A multistage tuberculosis vaccine that confers efficient protection before and after exposure. Nat. Med. 2011, 17, 189–194. Available online: https://www.nature.com/articles/nm.2285#supplementary-information (accessed on 25 November 2019). [CrossRef] [PubMed]
- Woodworth, J.S.; Christensen, D.; Andersen, P. Parenteral-Mucosal prime-boost with a TB subunit vaccine enhances early lung T cells against aerosol M. tuberculosis infection with transient improved protection. J. Immunol. 2017, 198, 73–75. [Google Scholar]
- Gutierro, I.; Hernandez, R.; Igartua, M.; Gascon, A.; Pedraz, J. Size dependent immune response after subcutaneous, oral and intranasal administration of BSA loaded nanospheres. Vaccine 2002, 21, 67–77. [Google Scholar] [CrossRef]
- Carcaboso, A.; Hernandez, R.; Igartua, M.; Rosas, J.; Patarroyo, M.; Pedraz, J. Potent, long lasting systemic antibody levels and mixed Th1/Th2 immune response after nasal immunization with malaria antigen loaded PLGA microparticles. Vaccine 2004, 22, 1423–1432. [Google Scholar] [CrossRef]
- Wendorf, J.; Chesko, J.; Kazzaz, J.; Ugozzoli, M.; Vajdy, M.; O’hagan, D.; Singh, M. A comparison of anionic nanoparticles and microparticles as vaccine delivery systems. Hum. Vaccines 2008, 4, 44–49. [Google Scholar] [CrossRef] [Green Version]
- Román, B.S.; Irache, J.M.; Gómez, S.; Tsapis, N.; Gamazo, C.; Espuelas, M.S. Co-encapsulation of an antigen and CpG oligonucleotides into PLGA microparticles by TROMS technology. Eur. J. Pharm. Biopharm. 2008, 70, 98–108. [Google Scholar] [CrossRef] [Green Version]
- Kang, P.B.; Azad, A.K.; Torrelles, J.B.; Kaufman, T.M.; Beharka, A.; Tibesar, E.; DesJardin, L.E.; Schlesinger, L.S. The human macrophage mannose receptor directs Mycobacterium tuberculosis lipoarabinomannan-mediated phagosome biogenesis. J. Exp. Med. 2005, 202, 987–999. [Google Scholar] [CrossRef]
- Kohama, H.; Umemura, M.; Okamoto, Y.; Yahagi, A.; Goga, H.; Harakuni, T.; Matsuzaki, G.; Arakawa, T. Mucosal immunization with recombinant heparin-binding haemagglutinin adhesin suppresses extrapulmonary dissemination of Mycobacterium bovis bacillus Calmette-Guérin (BCG) in infected mice. Vaccine 2008, 26, 924–932. [Google Scholar] [CrossRef]
- Chong, C.S.W.; Cao, M.; Wong, W.W.; Fischer, K.P.; Addison, W.R.; Kwon, G.S.; Tyrrell, D.L.; Samuel, J. Enhancement of T helper type 1 immune responses against hepatitis B virus core antigen by PLGA nanoparticle vaccine delivery. J. Control. Release 2005, 102, 85–99. [Google Scholar] [CrossRef]
- Thomas, C.; Rawat, A.; Hope-Weeks, L.; Ahsan, F. Aerosolized PLA and PLGA nanoparticles enhance humoral, mucosal and cytokine responses to hepatitis B vaccine. Mol. Pharm. 2011, 8, 405–415. [Google Scholar] [CrossRef]
- Saini, V.; Jain, V.; Sudheesh, M.S.; Jaganathan, K.S.; Murthy, P.K.; Kohli, D.V. Comparison of humoral and cell-mediated immune responses to cationic PLGA microspheres containing recombinant hepatitis B antigen. Int. J. Pharm. 2011, 408, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Shafiani, S.; Dinh, C.; Ertelt, J.M.; Moguche, A.O.; Siddiqui, I.; Smigiel, K.S.; Sharma, P.; Campbell, D.J.; Way, S.S.; Urdahl, K.B. Pathogen-Specific Treg Cells Expand Early during Mycobacterium tuberculosis Infection but Are Later Eliminated in Response to Interleukin-12. Immunity 2013, 38, 1261–1270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakai, S.; Mayer-Barber, K.D.; Barber, D.L. Defining features of protective CD4 T cell responses to Mycobacterium tuberculosis. Curr. Opin. Immunol. 2014, 29, 137–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gopal, R.; Rangel-Moreno, J.; Slight, S.; Lin, Y.; Nawar, H.F.; Fallert Junecko, B.A.; Reinhart, T.A.; Kolls, J.; Randall, T.D.; Connell, T.D.; et al. Interleukin-17-dependent CXCL13 mediates mucosal vaccine–induced immunity against tuberculosis. Mucosal Immunol. 2013, 6, 972–984. Available online: https://www.nature.com/articles/mi2012135#supplementary-information (accessed on 25 November 2019). [CrossRef] [PubMed]
- Kaushal, D.; Foreman, T.W.; Gautam, U.S.; Alvarez, X.; Adekambi, T.; Rangel-Moreno, J.; Golden, N.A.; Johnson, A.-M.F.; Phillips, B.L.; Ahsan, M.H.; et al. Mucosal vaccination with attenuated Mycobacterium tuberculosis induces strong central memory responses and protects against tuberculosis. Nat. Commun. 2015, 6, 8533. Available online: https://www.nature.com/articles/ncomms9533#supplementary-information (accessed on 25 November 2019). [CrossRef] [PubMed] [Green Version]
- Garcia-Contreras, L.; Wong, Y.-L.; Muttil, P.; Padilla, D.; Sadoff, J.; DeRousse, J.; Germishuizen, W.A.; Goonesekera, S.; Elbert, K.; Bloom, B.R.; et al. Immunization by a bacterial aerosol. Proc. Natl. Acad. Sci. USA 2008, 105, 4656–4660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeyanathan, M.; Heriazon, A.; Xing, Z. Airway luminal T cells: A newcomer on the stage of TB vaccination strategies. Trends Immunol. 2010, 31, 247–252. [Google Scholar] [CrossRef]
- Ashhurst, A.S.; Parumasivam, T.; Chan, J.G.Y.; Lin, L.C.W.; Flórido, M.; West, N.P.; Chan, H.-K.; Britton, W.J. PLGA particulate subunit tuberculosis vaccines promote humoral and Th17 responses but do not enhance control of Mycobacterium tuberculosis infection. PLoS ONE 2018, 13, e0194620. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roces, C.B.; Hussain, M.T.; Schmidt, S.T.; Christensen, D.; Perrie, Y. Investigating Prime-Pull Vaccination through a Combination of Parenteral Vaccination and Intranasal Boosting. Vaccines 2020, 8, 10. https://doi.org/10.3390/vaccines8010010
Roces CB, Hussain MT, Schmidt ST, Christensen D, Perrie Y. Investigating Prime-Pull Vaccination through a Combination of Parenteral Vaccination and Intranasal Boosting. Vaccines. 2020; 8(1):10. https://doi.org/10.3390/vaccines8010010
Chicago/Turabian StyleRoces, Carla B., Maryam T. Hussain, Signe T. Schmidt, Dennis Christensen, and Yvonne Perrie. 2020. "Investigating Prime-Pull Vaccination through a Combination of Parenteral Vaccination and Intranasal Boosting" Vaccines 8, no. 1: 10. https://doi.org/10.3390/vaccines8010010
APA StyleRoces, C. B., Hussain, M. T., Schmidt, S. T., Christensen, D., & Perrie, Y. (2020). Investigating Prime-Pull Vaccination through a Combination of Parenteral Vaccination and Intranasal Boosting. Vaccines, 8(1), 10. https://doi.org/10.3390/vaccines8010010