
Targeting Transcriptional Regulators of CD8+ T Cell Dysfunction to Boost Anti-Tumor Immunity

Abstract
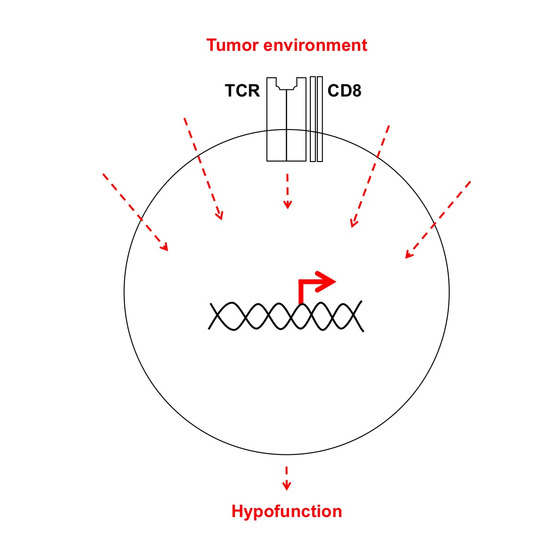
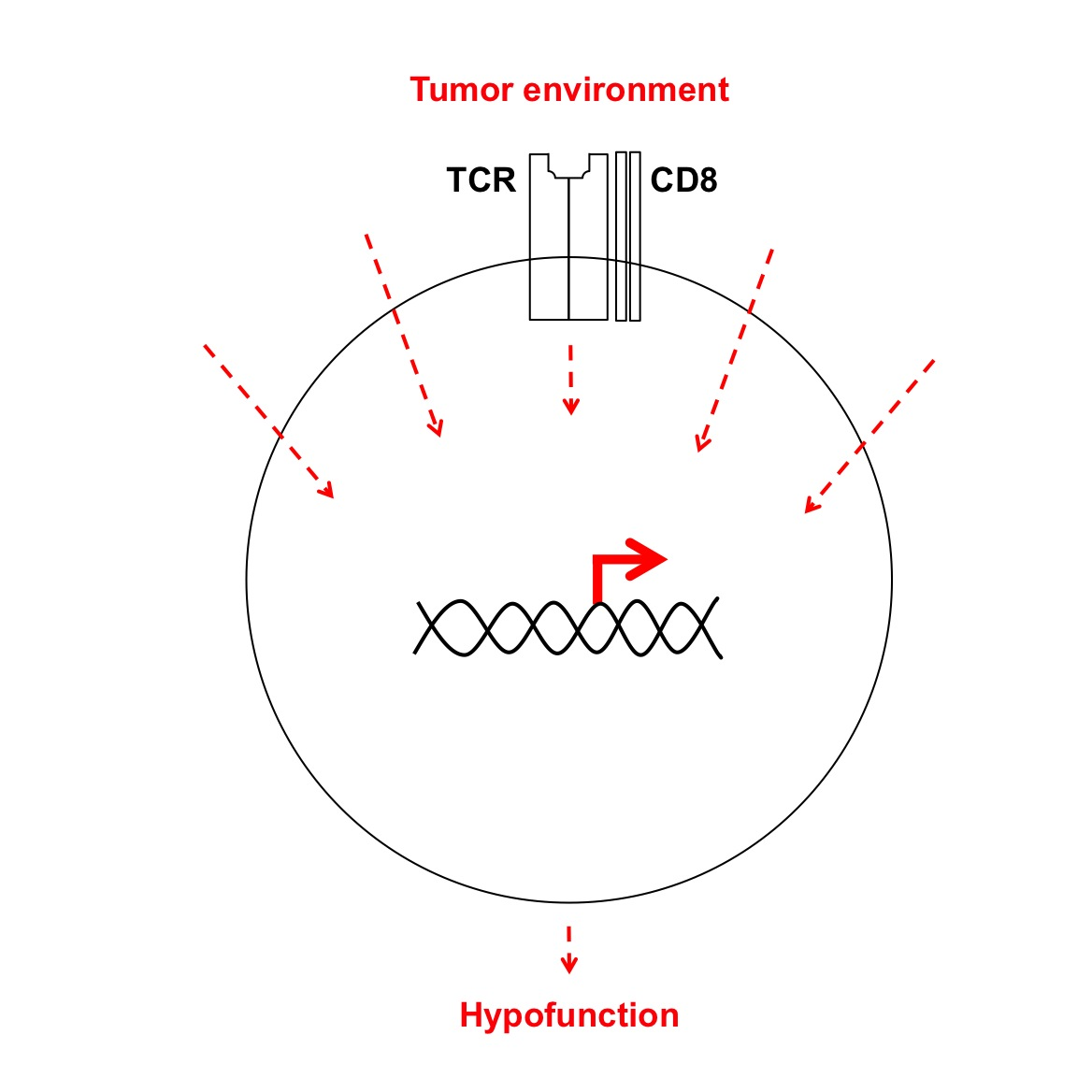
:
1. Brief History of Cancer Immunotherapy
2. Rationale to Pursue Transcriptional Regulation of TIL Dysfunction

3. Transcriptional Regulators of Anergy and Tolerance in TIL
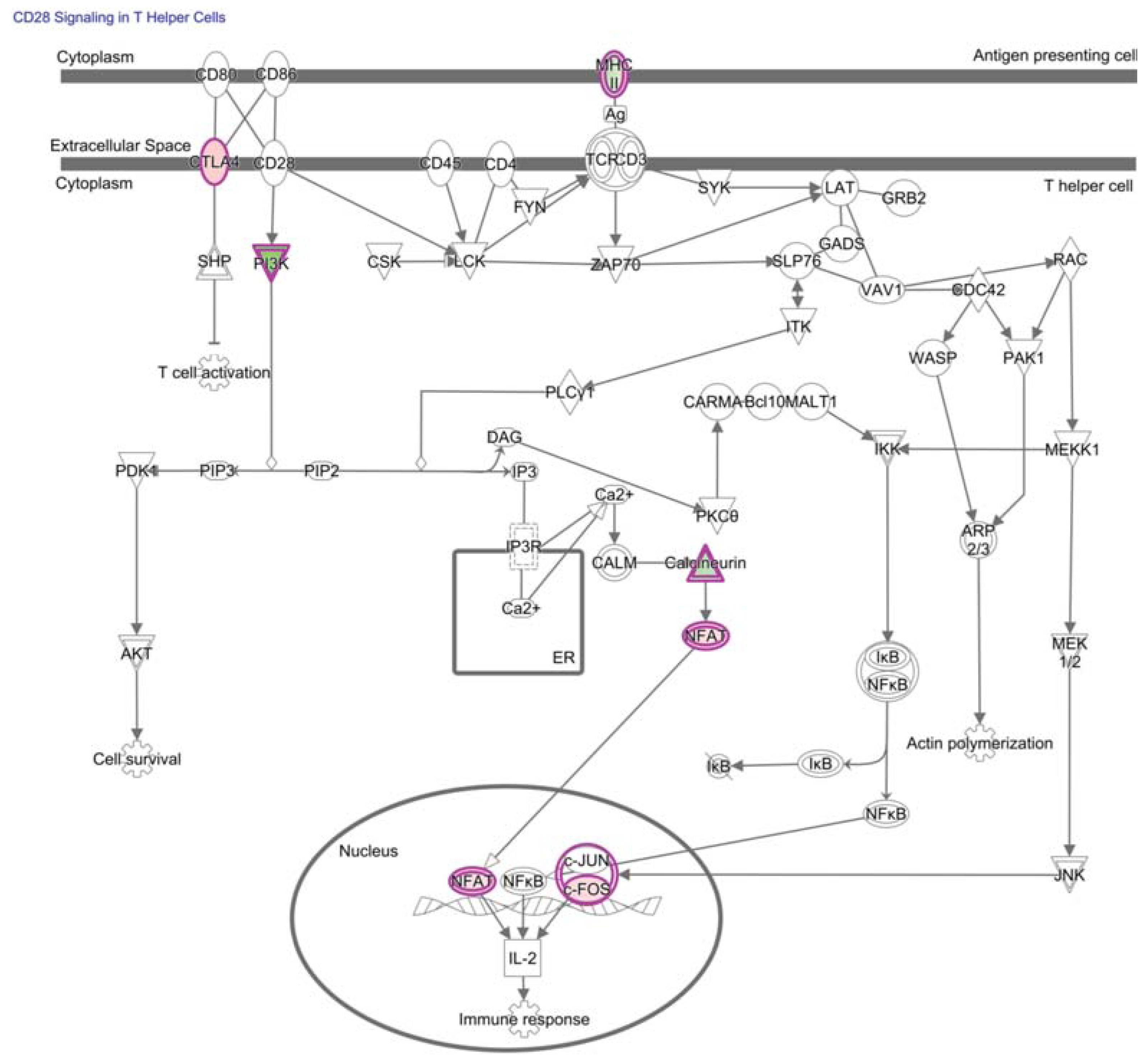
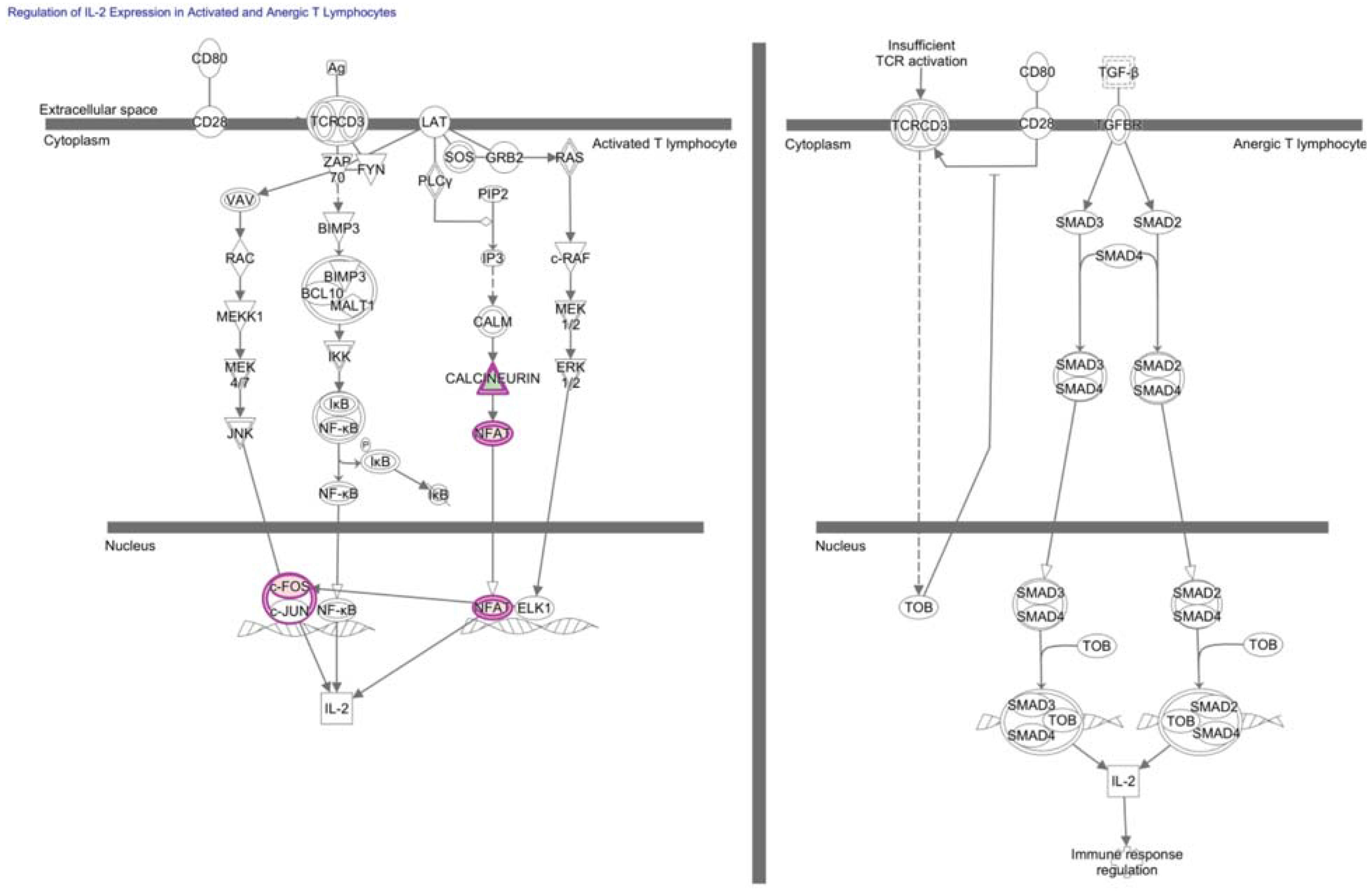
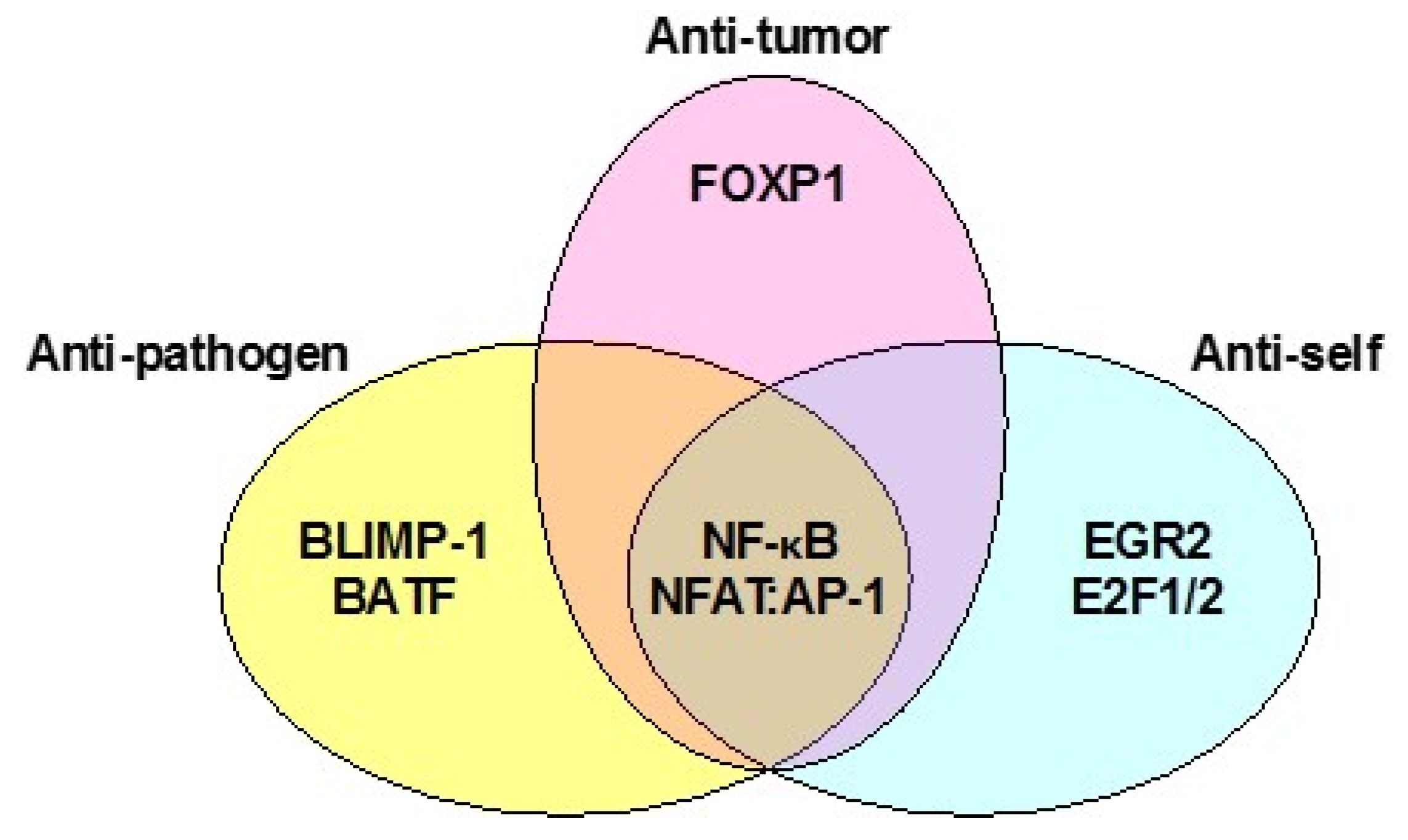
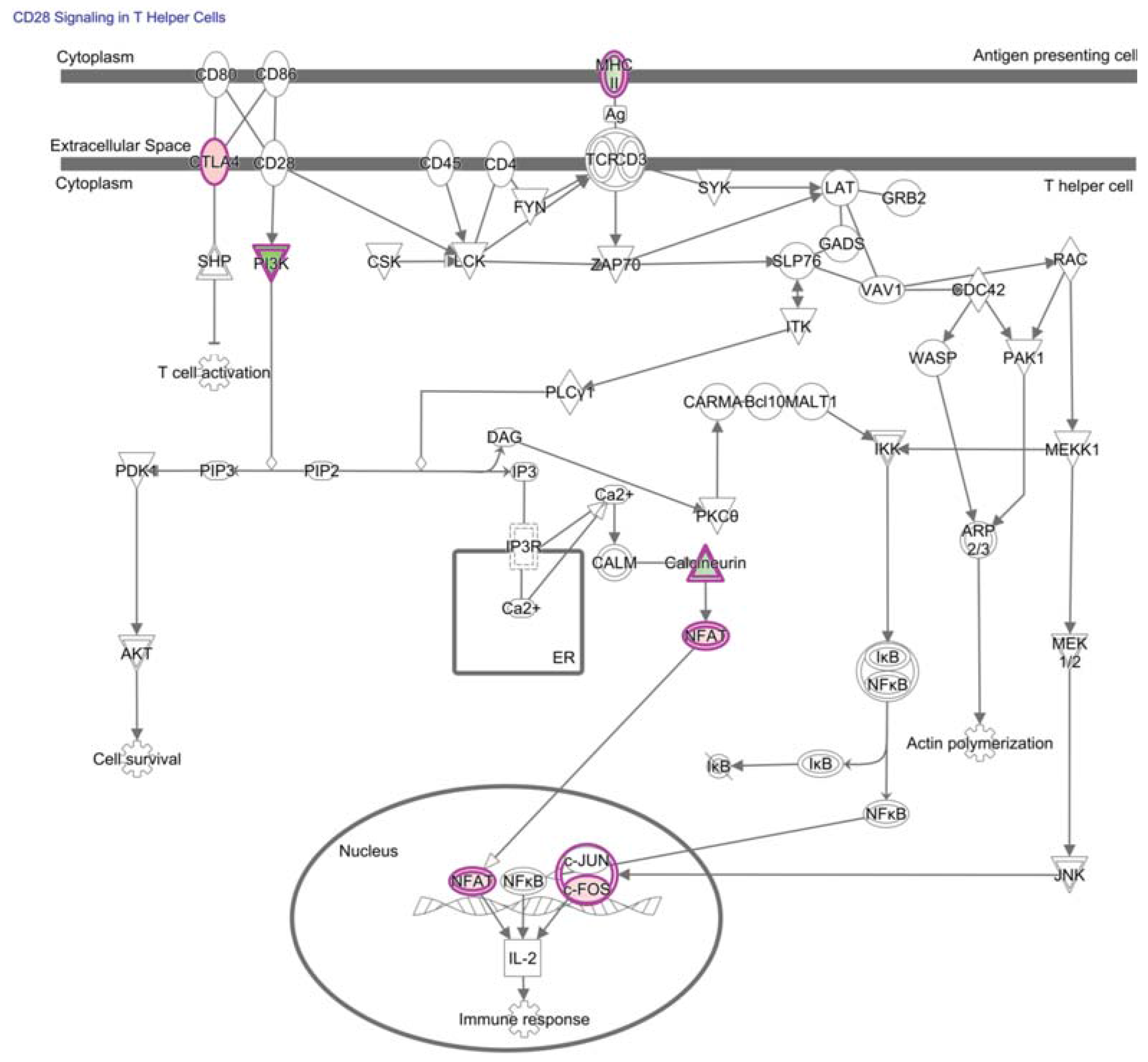
4. NF-κB in Hypofunctional Anti-Self and Tumor Infiltrating CD8+ T Cells
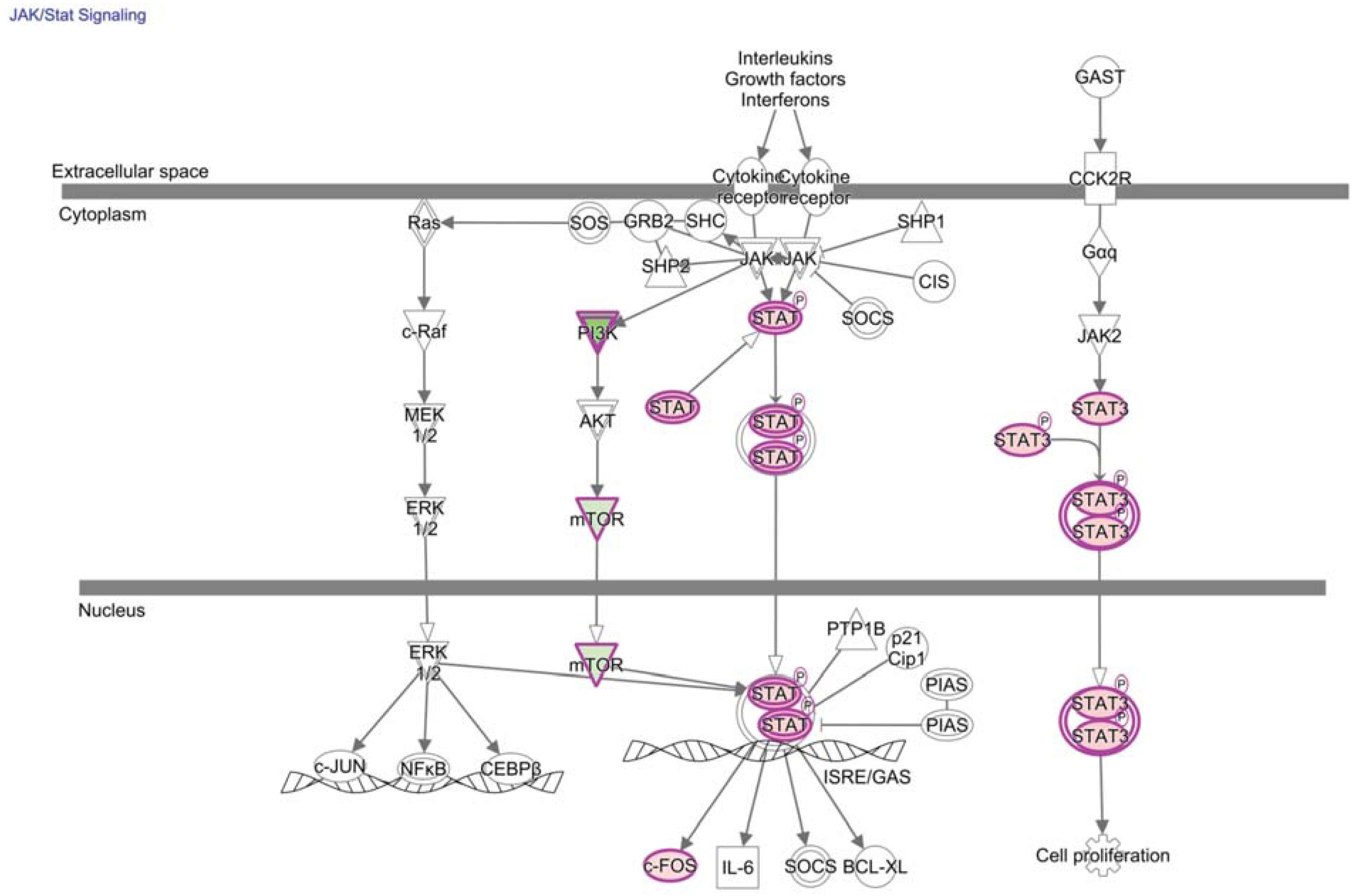
5. TIL and JAK/STAT Relay of Extracellular Signals for T Cell Programming

6. Transcriptional Regulators Underlying PD-1 Signaling and Expression
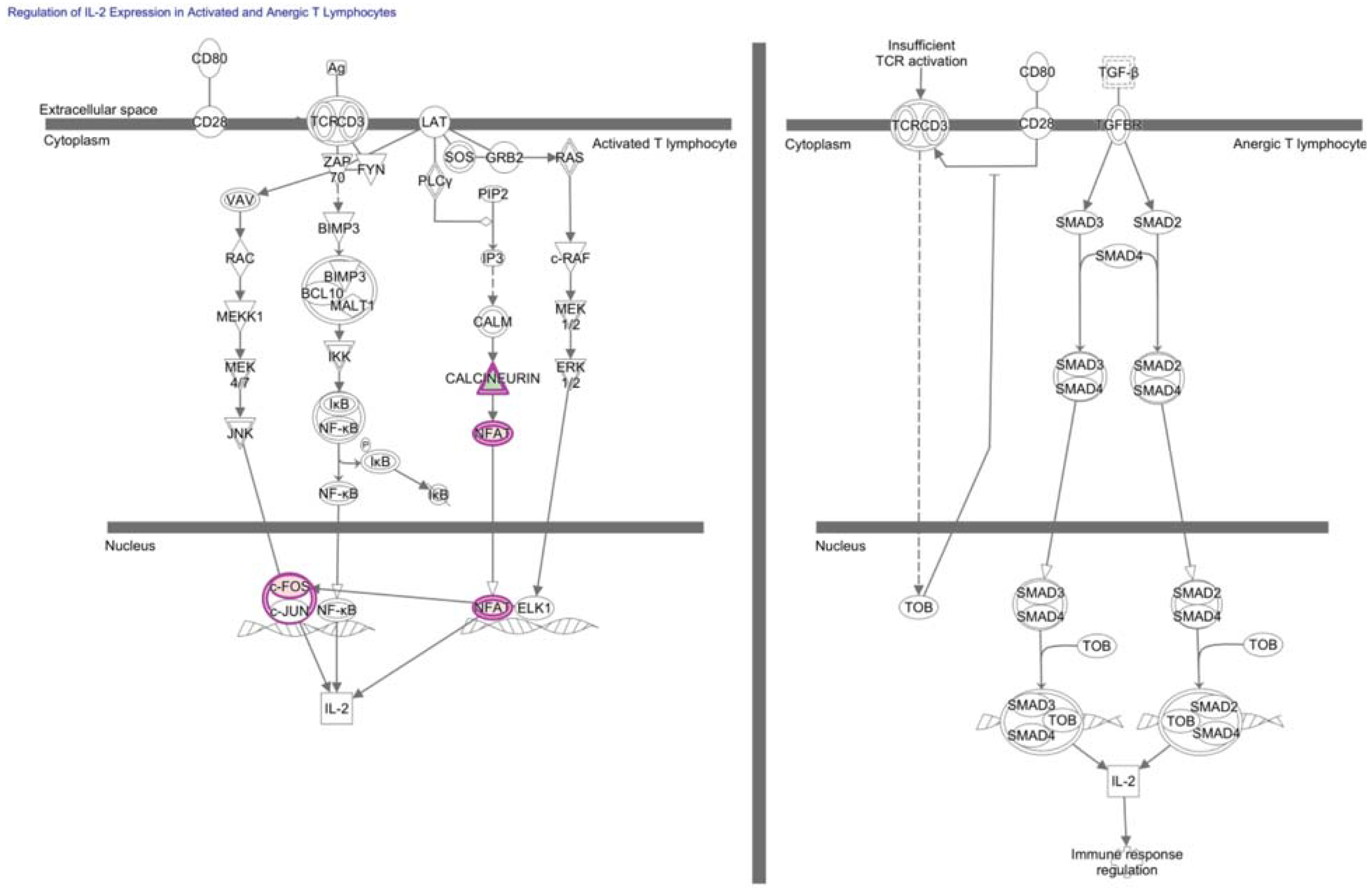
7. NFAT in Hypofunctional Anti-Self and Tumor Infiltrating CD8+ T Cells
8. Transcriptional Regulators of Exhaustion in TIL
9. Transcriptome of TAA-Specific CD8+ T Cells Reanalyzed to Identify Transcriptional Networks of TIL Hypofunction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Probe | Fold change (TILN/PBMC) | Gene Probe | Fold change (TILN/PBMC) |
|---|---|---|---|
| ERF | 16.8 | ZFP36L1 | 4.0 |
| HIP2 | 7.5 | ATF4 | 3.8 |
| CD619445 | 7.5 | ZFP36L1 | 3.3 |
| AI718865 | 7.4 | IRF4 | 3.2 |
| ILF2 | 7.1 | E2F1 | -3.0 |
| STAT3 | 7.1 | EIF4G3 | -4.9 |
| ATF3 | 6.4 | SSBP4 | -5.4 |
| BE839843 | 5.7 | SSBP3 | -5.5 |
| FOS | 5.7 | EIF3S9 | -5.9 |
| NFAT5 | 5.3 |
| Upstream Regulator | Activation z-score | p-value of overlap | Target molecules in dataset |
|---|---|---|---|
| STAT5A | 1.342 | 4.89E-06 | CASP8, DUSP5, FASLG, FOS, IFNG, MCL1, S1PR5, TNFRSF25, TNFRSF9, TRAF3 |
| ID3 | 0 | 1.05E-04 | DUSP1, DUSP4, IFNG, IRF4, NFAT5, PIK3IP1, PIK3R1, TNFRSF25, TRAF3, TRAF5 |
| ID2 | 0 | 1.12E-04 | DUSP1, DUSP4, IFNG, IRF4, NFAT5, PIK3IP1, PIK3R1, TNFRSF25, TRAF3, TRAF5 |
| FOXP3 | -0.555 | 1.22E-04 | CTLA4, DUSP4, ICOS, IFNG, IRF4, RGS1 |
| CYLD | 7.31E-04 | CTLA4, ICOS, IFNG | |
| STAT5B | 1.342 | 1.07E-03 | CASP8, IFNG, MCL1, TNFRSF25, TRAF3 |
| ELF4 | 1.70E-03 | DUSP1, DUSP5 | |
| SATB1 | -1.741 | 2.28E-03 | DUSP4, PIK3IP1, RGS1, S1PR1, TUBA4A, VTA1 |
| IRF1 | 2.80E-03 | FASLG, IFNG | |
| EGR3 | 4.16E-03 | CBLB, FASLG | |
| JUND | 4.16E-03 | CTLA4, IFNG | |
| ATF2 | 4.16E-03 | DUSP1, IFNG | |
| NFKB1 | 5.75E-03 | FASLG, IFNG | |
| GATA3 | 7.12E-03 | CTLA4, FOS, ICOS, IFNG | |
| CREB1 | 7.59E-03 | FOS, IFNG | |
| STAT3 | 7.62E-03 | CTLA4, IFNG, IRF4 | |
| PRDM1 | 9.64E-03 | FOS, IFNG | |
| HDAC2 | 1.29E-02 | CD27, DCLRE1C, MYO1F | |
| BACH2 | 1.42E-02 | IFNG, IRF4, MCL1 | |
| NCOR2 | 1.71E-02 | FOS | |
| IRF2 | 1.71E-02 | FASLG | |
| STAT2 | 1.71E-02 | IFNG | |
| MYBL2 | 1.71E-02 | FASLG | |
| ATF1 | 1.71E-02 | IFNG | |
| NFATC1 | 1.71E-02 | FASLG, IFNG | |
| BCL6 | 1.99E-02 | CTLA4, IFNG, IRF4 | |
| HDAC1 | 1.99E-02 | CD27, DCLRE1C, MYO1F | |
| NFATC2 | 2.97E-02 | ICOS, IFNG | |
| NFKBID | 3.38E-02 | IFNG | |
| TRIM27 | 3.38E-02 | IFNG | |
| CALR | 3.38E-02 | IFNG | |
| CREBBP | 3.85E-02 | DGKE, DUSP4, FASLG, IFNG, MYO1F, NR3C1, ST6GAL1 | |
| STAT6 | 4.59E-02 | HIPK2, IFNG, IRF4 |
10. NF-κB and NFAT in Exhausted Anti-Pathogen and Anti-Tumor CD8+ T Cells
11. T-Bet and Eomes in Exhausted Anti-Pathogen and Tumor Infiltrating T Cells
12. Transcriptional Regulators Underlying TGF-β Inhibition of TIL Function
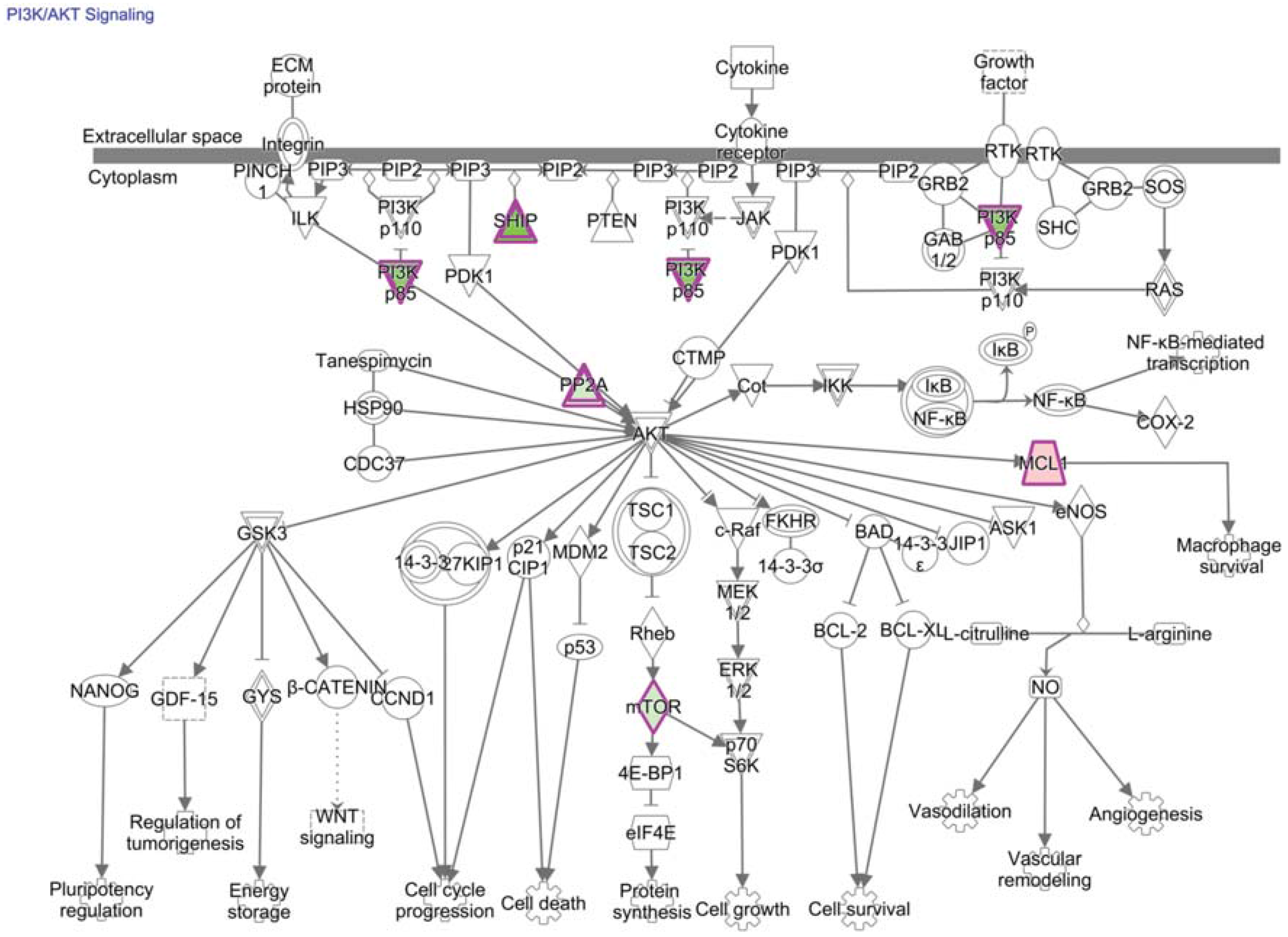
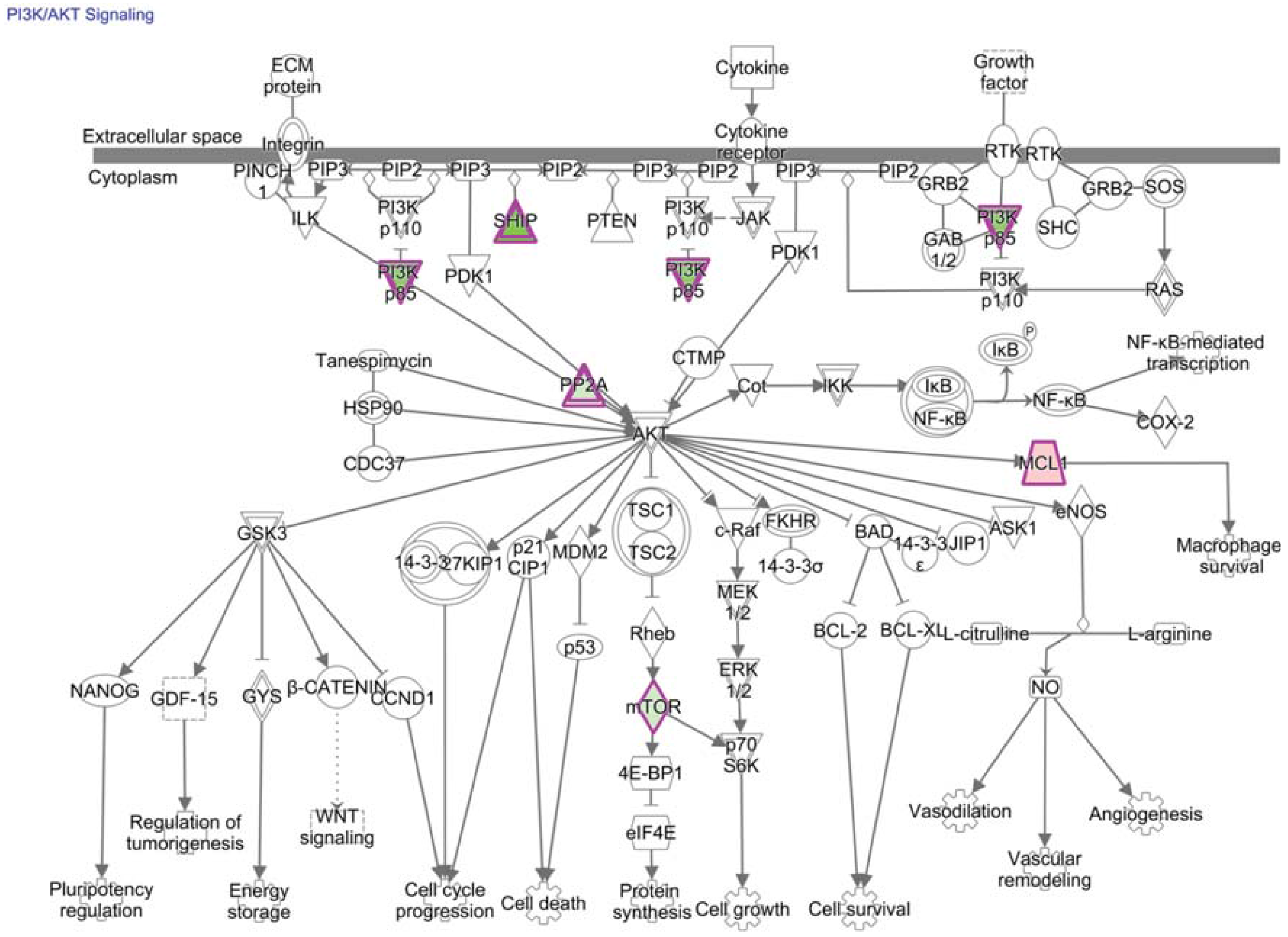
13. PI3K/AKT/mTOR Signaling and Transcriptional Consequences in TIL
14. Technological Advances to Utilize Transcriptional Regulators to Increase the Persistence of Functional TIL
15. Technological Advances to Identify Novel Transcriptional Regulators of TIL
16. Discussion and Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Coley, W.B., II. Contribution to the knowledge of sarcoma. Ann. Surg. 1891, 14, 199–220. [Google Scholar] [CrossRef] [PubMed]
- Richardson, M.A.; Ramirez, T.; Russell, N.C.; Moye, L.A. Coley toxins immunotherapy: A retrospective review. Altern. Ther. Health Med. 1999, 5, 42–47. [Google Scholar] [PubMed]
- Strebhardt, K.; Ullrich, A. Paul ehrlich’s magic bullet concept: 100 Years of progress. Nat. Rev. Cancer 2008, 8, 473–480. [Google Scholar] [CrossRef] [PubMed]
- Foley, E.J. Antigenic properties of methylcholanthrene-induced tumors in mice of the strain of origin. Cancer Res. 1953, 13, 835–837. [Google Scholar] [PubMed]
- Klein, G. Tumor antigens. Annu. Rev. Microbiol. 1966, 20, 223–252. [Google Scholar] [CrossRef] [PubMed]
- Old, L.J.; Boyse, E.A. Specific antigens of tumors and leukemias of experimental animals. Med. Clin. North Am. 1966, 50, 901–912. [Google Scholar] [PubMed]
- Old, L.J.; Boyse, E.A. Immunology of experimental tumors. Annu. Rev. Med. 1964, 15, 167–186. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.P.; Old, L.J.; Schreiber, R.D. The three es of cancer immunoediting. Annu. Rev. Immunol. 2004, 22, 329–360. [Google Scholar] [CrossRef] [PubMed]
- Thomas, L. Discussion. In Cellular and Humoral Aspects of the Hypersensitive States; Lawrence, H.S., Ed.; Hoeber-Harper: New York, NY, USA, 1959; pp. 529–533. [Google Scholar]
- Burnet, F.M. The concept of immunological surveillance. Prog. Exp. Tumor Res. 1970, 13, 1–27. [Google Scholar] [PubMed]
- Stutman, O. Tumor development after 3-methylcholanthrene in immunologically deficient athymic-nude mice. Science 1974, 183, 534–536. [Google Scholar] [CrossRef] [PubMed]
- Van der Bruggen, P.; Traversari, C.; Chomez, P.; Lurquin, C.; de Plaen, E.; van den Eynde, B.; Knuth, A.; Boon, T. A gene encoding an antigen recognized by cytolytic t lymphocytes on a human melanoma. Science 1991, 254, 1643–1647. [Google Scholar] [CrossRef] [PubMed]
- Coulie, P.G.; van den Eynde, B.J.; van der Bruggen, P.; Boon, T. Tumour antigens recognized by T lymphocytes: At the core of cancer immunotherapy. Nat. Rev. Cancer 2014, 14, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Galon, J.; Costes, A.; Sanchez-Cabo, F.; Kirilovsky, A.; Mlecnik, B.; Lagorce-Pages, C.; Tosolini, M.; Camus, M.; Berger, A.; Wind, P.; et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006, 313, 1960–1964. [Google Scholar] [CrossRef] [PubMed]
- Fridman, W.H.; Pages, F.; Sautes-Fridman, C.; Galon, J. The immune contexture in human tumours: Impact on clinical outcome. Nat. Rev. Cancer 2012, 12, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Bindea, G.; Mlecnik, B.; Tosolini, M.; Kirilovsky, A.; Waldner, M.; Obenauf, A.C.; Angell, H.; Fredriksen, T.; Lafontaine, L.; Berger, A.; et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity 2013, 39, 782–795. [Google Scholar] [CrossRef] [PubMed]
- Reichert, T.E.; Day, R.; Wagner, E.M.; Whiteside, T.L. Absent or low expression of the zeta chain in T cells at the tumor site correlates with poor survival in patients with oral carcinoma. Cancer Res. 1998, 58, 5344–5347. [Google Scholar] [PubMed]
- Naito, Y.; Saito, K.; Shiiba, K.; Ohuchi, A.; Saigenji, K.; Nagura, H.; Ohtani, H. CD8+ T cells infiltrated within cancer cell nests as a prognostic factor in human colorectal cancer. Cancer Res. 1998, 58, 3491–3494. [Google Scholar] [PubMed]
- Schumacher, K.; Haensch, W.; Roefzaad, C.; Schlag, P.M. Prognostic significance of activated CD8(+) T cell infiltrations within esophageal carcinomas. Cancer Res. 2001, 61, 3932–3936. [Google Scholar] [PubMed]
- Zhou, G.; Lu, Z.; McCadden, J.D.; Levitsky, H.I.; Marson, A.L. Reciprocal changes in tumor antigenicity and antigen-specific T cell function during tumor progression. J. Exp. Med. 2004, 200, 1581–1592. [Google Scholar] [CrossRef] [PubMed]
- Baitsch, L.; Fuertes-Marraco, S.A.; Legat, A.; Meyer, C.; Speiser, D.E. The three main stumbling blocks for anticancer T cells. Trends Immunol. 2012, 33, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Couzin-Frankel, J. Breakthrough of the year 2013. Cancer immunotherapy. Science 2013, 342, 1432–1433. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vacchelli, E.; Bravo-San Pedro, J.M.; Buque, A.; Senovilla, L.; Baracco, E.E.; Bloy, N.; Castoldi, F.; Abastado, J.P.; Agostinis, P.; et al. Classification of current anticancer immunotherapies. Oncotarget 2014, 5, 12472–12508. [Google Scholar] [CrossRef] [PubMed]
- Zavala, V.A.; Kalergis, A.M. New clinical advances in immunotherapy for the treatment of solid tumours. Immunology 2015, 145, 182–201. [Google Scholar] [CrossRef] [PubMed]
- Wolchok, J.D.; Kluger, H.; Callahan, M.K.; Postow, M.A.; Rizvi, N.A.; Lesokhin, A.M.; Segal, N.H.; Ariyan, C.E.; Gordon, R.A.; Reed, K.; et al. Nivolumab plus ipilimumab in advanced melanoma. N. Engl. J. Med. 2013, 369, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Zamarin, D.; Postow, M.A. Immune checkpoint modulation: Rational design of combination strategies. Pharmacol. Ther. 2015, 150, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Ascierto, P.A.; Addeo, R.; Carteni, G.; Daniele, B.; de Laurentis, M.; Ianniello, G.P.; Morabito, A.; Palmieri, G.; Pepe, S.; Perrone, F.; et al. The role of immunotherapy in solid tumors: Report from the campania society of oncology immunotherapy (SCITO) meeting, Naples 2014. J. Transl. Med. 2014. [Google Scholar] [CrossRef] [PubMed]
- Barbee, M.S.; Ogunniyi, A.; Horvat, T.Z.; Dang, T.O. Current status and future directions of the immune checkpoint inhibitors ipilimumab, pembrolizumab, and nivolumab in oncology. Ann. Pharmacother. 2015, 49, 907–937. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Allison, J.P. The future of immune checkpoint therapy. Science 2015, 348, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Whiteside, T.L. The tumor microenvironment and its role in promoting tumor growth. Oncogene 2008, 27, 5904–5912. [Google Scholar] [CrossRef] [PubMed]
- Bucks, C.M.; Norton, J.A.; Boesteanu, A.C.; Mueller, Y.M.; Katsikis, P.D. Chronic antigen stimulation alone is sufficient to drive CD8+ T cell exhaustion. J. Immunol. 2009, 182, 6697–6708. [Google Scholar] [CrossRef]
- Ledford, H. Immunotherapy’s cancer remit widens. Nature 2013. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Macian, F.; Garcia-Cozar, F.; Im, S.H.; Horton, H.F.; Byrne, M.C.; Rao, A. Transcriptional mechanisms underlying lymphocyte tolerance. Cell 2002, 109, 719–731. [Google Scholar] [CrossRef]
- Wherry, E.J.; Ha, S.J.; Kaech, S.M.; Haining, W.N.; Sarkar, S.; Kalia, V.; Subramaniam, S.; Blattman, J.N.; Barber, D.L.; Ahmed, R. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity 2007, 27, 670–684. [Google Scholar] [CrossRef] [PubMed]
- Baitsch, L.; Baumgaertner, P.; Devevre, E.; Raghav, S.K.; Legat, A.; Barba, L.; Wieckowski, S.; Bouzourene, H.; Deplancke, B.; Romero, P.; et al. Exhaustion of tumor-specific CD8(+) T cells in metastases from melanoma patients. J. Clin. Investig. 2011, 121, 2350–2360. [Google Scholar] [PubMed]
- Schietinger, A.; Delrow, J.J.; Basom, R.S.; Blattman, J.N.; Greenberg, P.D. Rescued tolerant CD8 T cells are preprogrammed to reestablish the tolerant state. Science 2012, 335, 723–727. [Google Scholar] [CrossRef] [PubMed]
- Schietinger, A.; Greenberg, P.D. Tolerance and exhaustion: Defining mechanisms of T cell dysfunction. Trends Immunol. 2014, 35, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Vigneron, N.; Stroobant, V.; van den Eynde, B.J.; van der Bruggen, P. Database of T cell-defined human tumor antigens: The 2013 update. Cancer Immun. 2013, 13, PMC3718731. [Google Scholar]
- Hogquist, K.A.; Baldwin, T.A.; Jameson, S.C. Central tolerance: Learning self-control in the thymus. Nat. Rev. Immunol. 2005, 5, 772–782. [Google Scholar] [CrossRef] [PubMed]
- Mueller, D.L. Mechanisms maintaining peripheral tolerance. Nat. Immunol. 2010, 11, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Buhrman, J.D.; Slansky, J.E. Improving T cell responses to modified peptides in tumor vaccines. Immunol. Res. 2013, 55, 34–47. [Google Scholar] [CrossRef] [PubMed]
- Ochsenbein, A.F.; Sierro, S.; Odermatt, B.; Pericin, M.; Karrer, U.; Hermans, J.; Hemmi, S.; Hengartner, H.; Zinkernagel, R.M. Roles of tumour localization, second signals and cross priming in cytotoxic T cell induction. Nature 2001, 411, 1058–1064. [Google Scholar] [CrossRef] [PubMed]
- Spiotto, M.T.; Yu, P.; Rowley, D.A.; Nishimura, M.I.; Meredith, S.C.; Gajewski, T.F.; Fu, Y.X.; Schreiber, H. Increasing tumor antigen expression overcomes “ignorance” to solid tumors via crosspresentation by bone marrow-derived stromal cells. Immunity 2002, 17, 737–747. [Google Scholar] [CrossRef]
- Bandyopadhyay, S.; Soto-Nieves, N.; Macian, F. Transcriptional regulation of T cell tolerance. Semin. Immunol. 2007, 19, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef] [PubMed]
- Kantoff, P.W.; Higano, C.S.; Shore, N.D.; Berger, E.R.; Small, E.J.; Penson, D.F.; Redfern, C.H.; Ferrari, A.C.; Dreicer, R.; Sims, R.B.; et al. Sipuleucel-t immunotherapy for castration-resistant prostate cancer. N. Engl. J. Med. 2010, 363, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Robbins, P.F.; Morgan, R.A.; Feldman, S.A.; Yang, J.C.; Sherry, R.M.; Dudley, M.E.; Wunderlich, J.R.; Nahvi, A.V.; Helman, L.J.; Mackall, C.L.; et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J. Clin. Oncol. 2011, 29, 917–924. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.E.; Bishop, K.D.; Phillips, N.E.; Mordes, J.P.; Greiner, D.L.; Rossini, A.A.; Czech, M.P. Early growth response gene-2, a zinc-finger transcription factor, is required for full induction of clonal anergy in CD4+ T cells. J. Immunol. 2004, 173, 7331–7338. [Google Scholar] [CrossRef] [PubMed]
- Safford, M.; Collins, S.; Lutz, M.A.; Allen, A.; Huang, C.T.; Kowalski, J.; Blackford, A.; Horton, M.R.; Drake, C.; Schwartz, R.H.; et al. Egr-2 and egr-3 are negative regulators of T cell activation. Nat. Immunol. 2005, 6, 472–480. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Zha, Y.; Driessens, G.; Locke, F.; Gajewski, T.F. Transcriptional regulator early growth response gene 2 (Egr2) is required for T cell anergy in vitro and in vivo. J. Exp. Med. 2012, 209, 2157–2163. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Zha, Y.; Spaapen, R.M.; Mathew, R.; Barr, K.; Bendelac, A.; Gajewski, T.F. Egr2-dependent gene expression profiling and CHiP-Seq reveal novel biologic targets in T cell anergy. Mol. Immunol. 2013, 55, 283–291. [Google Scholar] [CrossRef] [PubMed]
- DeGregori, J.; Johnson, D.G. Distinct and overlapping roles for e2f family members in transcription, proliferation and apoptosis. Curr. Mol. Med. 2006, 6, 739–748. [Google Scholar] [PubMed]
- Murga, M.; Fernandez-Capetillo, O.; Field, S.J.; Moreno, B.; Borlado, L.R.; Fujiwara, Y.; Balomenos, D.; Vicario, A.; Carrera, A.C.; Orkin, S.H.; et al. Mutation of E2F2 in mice causes enhanced T lymphocyte proliferation, leading to the development of autoimmunity. Immunity 2001, 15, 959–970. [Google Scholar] [CrossRef]
- Zhu, J.W.; Field, S.J.; Gore, L.; Thompson, M.; Yang, H.; Fujiwara, Y.; Cardiff, R.D.; Greenberg, M.; Orkin, S.H.; DeGregori, J. E2F1 and E2F2 determine thresholds for antigen-induced T cell proliferation and suppress tumorigenesis. Mol. Cell. Biol. 2001, 21, 8547–8564. [Google Scholar] [CrossRef] [PubMed]
- DeRyckere, D.; DeGregori, J. E2F1 and E2F2 are differentially required for homeostasis-driven and antigen-induced T cell proliferation in vivo. J. Immunol. 2005, 175, 647–655. [Google Scholar] [CrossRef] [PubMed]
- Li, F.X.; Zhu, J.W.; Tessem, J.S.; Beilke, J.; Varella-Garcia, M.; Jensen, J.; Hogan, C.J.; DeGregori, J. The development of diabetes in e2f1/e2f2 mutant mice reveals important roles for bone marrow-derived cells in preventing islet cell loss. Proc. Natl. Acad. Sci. USA 2003, 100, 12935–12940. [Google Scholar] [CrossRef] [PubMed]
- Li, F.X.; Zhu, J.W.; Hogan, C.J.; DeGregori, J. Defective gene expression, s phase progression, and maturation during hematopoiesis in e2f1/e2f2 mutant mice. Mol. Cell. Biol. 2003, 23, 3607–3622. [Google Scholar] [CrossRef] [PubMed]
- Field, S.J.; Tsai, F.Y.; Kuo, F.; Zubiaga, A.M.; Kaelin, W.G., Jr.; Livingston, D.M.; Orkin, S.H.; Greenberg, M.E. E2f-1 functions in mice to promote apoptosis and suppress proliferation. Cell 1996, 85, 549–561. [Google Scholar] [CrossRef]
- Zhu, J.W.; DeRyckere, D.; Li, F.X.; Wan, Y.Y.; DeGregori, J. A role for E2F1 in the induction of ARF, p53, and apoptosis during thymic negative selection. Cell Growth Differ. 1999, 10, 829–838. [Google Scholar] [PubMed]
- Lissy, N.A.; Davis, P.K.; Irwin, M.; Kaelin, W.G.; Dowdy, S.F. A common E2F-1 and p73 pathway mediates cell death induced by tcr activation. Nature 2000, 407, 642–645. [Google Scholar] [PubMed]
- Intlekofer, A.M.; Takemoto, N.; Wherry, E.J.; Longworth, S.A.; Northrup, J.T.; Palanivel, V.R.; Mullen, A.C.; Gasink, C.R.; Kaech, S.M.; Miller, J.D.; et al. Effector and memory CD8+ T cell fate coupled by T-bet and eomesodermin. Nat. Immunol. 2005, 6, 1236–1244. [Google Scholar] [CrossRef] [PubMed]
- Pearce, E.L.; Mullen, A.C.; Martins, G.A.; Krawczyk, C.M.; Hutchins, A.S.; Zediak, V.P.; Banica, M.; DiCioccio, C.B.; Gross, D.A.; Mao, C.A.; et al. Control of effector CD8+ T cell function by the transcription factor eomesodermin. Science 2003, 302, 1041–1043. [Google Scholar] [CrossRef] [PubMed]
- Thierfelder, W.E.; van Deursen, J.M.; Yamamoto, K.; Tripp, R.A.; Sarawar, S.R.; Carson, R.T.; Sangster, M.Y.; Vignali, D.A.; Doherty, P.C.; Grosveld, G.C.; et al. Requirement for STAT4 in interleukin-12-mediated responses of natural killer and T cells. Nature 1996, 382, 171–174. [Google Scholar] [CrossRef]
- Ebert, P.J.; Jiang, S.; Xie, J.; Li, Q.J.; Davis, M.M. An endogenous positively selecting peptide enhances mature T cell responses and becomes an autoantigen in the absence of microrna miR-181a. Nat. Immunol. 2009, 10, 1162–1169. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.J.; Chau, J.; Ebert, P.J.; Sylvester, G.; Min, H.; Liu, G.; Braich, R.; Manoharan, M.; Soutschek, J.; Skare, P.; et al. miR-181a is an intrinsic modulator of T cell sensitivity and selection. Cell 2007, 129, 147–161. [Google Scholar] [CrossRef] [PubMed]
- Grosso, J.F.; Kelleher, C.C.; Harris, T.J.; Maris, C.H.; Hipkiss, E.L.; de Marzo, A.; Anders, R.; Netto, G.; Getnet, D.; Bruno, T.C.; et al. Lag-3 regulates CD8+ T cell accumulation and effector function in murine self- and tumor-tolerance systems. J. Clin. Investig. 2007, 117, 3383–3392. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.C.; Chang, J.H.; Jin, J. Regulation of nuclear factor-kappab in autoimmunity. Trends Immunol. 2013, 34, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Krishna, S.; Xie, D.; Gorentla, B.; Shin, J.; Gao, J.; Zhong, X.P. Chronic activation of the kinase IKKbeta impairs T cell function and survival. J. Immunol. 2012, 189, 1209–1219. [Google Scholar] [CrossRef] [PubMed]
- Clavijo, P.E.; Frauwirth, K.A. Anergic CD8+ T lymphocytes have impaired NF-kappab activation with defects in p65 phosphorylation and acetylation. J. Immunol. 2012, 188, 1213–1221. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, P.; Sica, A.; Young, H.A.; Ye, J.; Franco, J.L.; Wiltrout, R.H.; Longo, D.L.; Rice, N.R.; Komschlies, K.L. Alterations in NF kappa B/Rel family proteins in splenic T cells from tumor-bearing mice and reversal following therapy. Cancer Res. 1994, 54, 2969–2972. [Google Scholar] [PubMed]
- Girolomoni, G.; Ricciardi-Castagnoli, P. Dendritic cells hold promise for immunotherapy. Immunol. Today 1997, 18, 102–104. [Google Scholar] [CrossRef]
- Schier, W.W. Cutaneous anergy and hodgkin’s disease. N. Engl. J. Med. 1954, 250, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Ashikawa, K.; Motoya, K.; Sekiguchi, M.; Ishibashi, Y. Immune response in tumor-bearing patients and animals. II. Incidence of tuberculin anergy in cancer patients. Gan 1967, 58, 565–573. [Google Scholar] [PubMed]
- Barnes, S.E.; Wang, Y.; Chen, L.; Molinero, L.L.; Gajewski, T.F.; Evaristo, C.; Alegre, M.L. T cell-NF-kappab activation is required for tumor control in vivo. J. Immunother. Cancer 2015. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, J.J.; Lahesmaa, R.; Vahedi, G.; Laurence, A.; Kanno, Y. Genomic views of STAT function in CD4+ T helper cell differentiation. Nat. Rev. Immunol. 2011, 11, 239–250. [Google Scholar] [CrossRef] [PubMed]
- Hirahara, K.; Poholek, A.; Vahedi, G.; Laurence, A.; Kanno, Y.; Milner, J.D.; O’Shea, J.J. Mechanisms underlying helper T cell plasticity: Implications for immune-mediated disease. J. Allergy Clin. Immunol. 2013, 131, 1276–1287. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Pardoll, D.; Jove, R. STATs in cancer inflammation and immunity: A leading role for STAT3. Nat. Rev. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef] [PubMed]
- Triplett, T.A.; Tucker, C.G.; Triplett, K.C.; Alderman, Z.; Sun, L.; Ling, L.E.; Akporiaye, E.T.; Weinberg, A.D. STAT3 signaling is required for optimal regression of large established tumors in mice treated with anti-OX40 and TGFbeta receptor blockade. Cancer Immunol. Res. 2015, 3, 526–535. [Google Scholar] [CrossRef] [PubMed]
- Pauken, K.E.; Wherry, E.J. Overcoming T cell exhaustion in infection and cancer. Trends Immunol. 2015, 36, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Patsoukis, N.; Brown, J.; Petkova, V.; Liu, F.; Li, L.; Boussiotis, V.A. Selective effects of PD-1 on Akt and Ras pathways regulate molecular components of the cell cycle and inhibit T cell proliferation. Sci. Signal. 2012. [Google Scholar] [CrossRef] [PubMed]
- Patsoukis, N.; Sari, D.; Boussiotis, V.A. PD-1 inhibits T cell proliferation by upregulating p27 and p15 and suppressing CDC25a. Cell Cycle 2012, 11, 4305–4309. [Google Scholar] [CrossRef] [PubMed]
- Boussiotis, V.A.; Freeman, G.J.; Taylor, P.A.; Berezovskaya, A.; Grass, I.; Blazar, B.R.; Nadler, L.M. P27Kip1 functions as an anergy factor inhibiting interleukin 2 transcription and clonal expansion of alloreactive human and mouse helper T lymphocytes. Nat. Med. 2000, 6, 290–297. [Google Scholar] [CrossRef] [PubMed]
- Kudo, H.; Matsuoka, T.; Mitsuya, H.; Nishimura, Y.; Matsushita, S. Cross-linking HLA-DR molecules on Th1 cells induces anergy in association with increased level of cyclin-dependent kinase inhibitor p27kip1. Immunol. Lett. 2002, 81, 149–155. [Google Scholar] [CrossRef]
- Asai, K.; Hachimura, S.; Kimura, M.; Toraya, T.; Yamashita, M.; Nakayama, T.; Kaminogawa, S. T cell hyporesponsiveness induced by oral administration of ovalbumin is associated with impaired nfat nuclear translocation and p27Kip1 degradation. J. Immunol. 2002, 169, 4723–4731. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.K.; DeLoose, A.; Gilbert, K.M. Induction of anergy in th1 cells associated with increased levels of cyclin-dependent kinase inhibitors p21Cip1 and p27Kip1. J. Immunol. 2001, 166, 952–958. [Google Scholar] [CrossRef] [PubMed]
- Macian, F.; Im, S.H.; Garcia-Cozar, F.J.; Rao, A. T cell anergy. Curr. Opin. Immunol. 2004, 16, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Appleman, L.J.; van Puijenbroek, A.A.; Shu, K.M.; Nadler, L.M.; Boussiotis, V.A. CD28 costimulation mediates down-regulation of p27Kip1 and cell cycle progression by activation of the PI3K/PKB signaling pathway in primary human T cells. J. Immunol. 2002, 168, 2729–2736. [Google Scholar] [CrossRef] [PubMed]
- Keir, M.E.; Butte, M.J.; Freeman, G.J.; Sharpe, A.H. PD-1 and its ligands in tolerance and immunity. Annu. Rev. Immunol. 2008, 26, 677–704. [Google Scholar] [CrossRef] [PubMed]
- Fife, B.T.; Pauken, K.E. The role of the PD-1 pathway in autoimmunity and peripheral tolerance. Ann. NY Acad. Sci. 2011, 1217, 45–59. [Google Scholar] [CrossRef] [PubMed]
- Quigley, M.; Pereyra, F.; Nilsson, B.; Porichis, F.; Fonseca, C.; Eichbaum, Q.; Julg, B.; Jesneck, J.L.; Brosnahan, K.; Imam, S.; et al. Transcriptional analysis of HIV-specific CD8+ T cells shows that PD-1 inhibits T cell function by upregulating BATF. Nat. Med. 2010, 16, 1147–1151. [Google Scholar] [CrossRef] [PubMed]
- Murphy, T.L.; Tussiwand, R.; Murphy, K.M. Specificity through cooperation: BATF-IRF interactions control immune-regulatory networks. Nat. Rev. Immunol. 2013, 13, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Levy, D.E.; Kessler, D.S.; Pine, R.; Darnell, J.E., Jr. Cytoplasmic activation of ISGF3, the positive regulator of interferon-alpha-stimulated transcription, reconstituted in vitro. Genes Dev. 1989, 3, 1362–1371. [Google Scholar] [CrossRef] [PubMed]
- Kao, C.; Oestreich, K.J.; Paley, M.A.; Crawford, A.; Angelosanto, J.M.; Ali, M.A.; Intlekofer, A.M.; Boss, J.M.; Reiner, S.L.; Weinmann, A.S.; et al. Transcription factor t-bet represses expression of the inhibitory receptor PD-1 and sustains virus-specific CD8+ T cell responses during chronic infection. Nat. Immunol. 2011, 12, 663–671. [Google Scholar] [CrossRef] [PubMed]
- Staron, M.M.; Gray, S.M.; Marshall, H.D.; Parish, I.A.; Chen, J.H.; Perry, C.J.; Cui, G.; Li, M.O.; Kaech, S.M. The transcription factor FoxO1 sustains expression of the inhibitory receptor PD-1 and survival of antiviral CD8(+) T cells during chronic infection. Immunity 2014, 41, 802–814. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.; Blackburn, S.D.; Intlekofer, A.M.; Kao, C.; Angelosanto, J.M.; Reiner, S.L.; Wherry, E.J. A role for the transcriptional repressor BLIMP-1 in CD8(+) T cell exhaustion during chronic viral infection. Immunity 2009, 31, 309–320. [Google Scholar] [CrossRef] [PubMed]
- Oestreich, K.J.; Yoon, H.; Ahmed, R.; Boss, J.M. Nfatc1 regulates PD-1 expression upon T cell activation. J. Immunol. 2008, 181, 4832–4839. [Google Scholar] [CrossRef] [PubMed]
- Youngblood, B.; Oestreich, K.J.; Ha, S.J.; Duraiswamy, J.; Akondy, R.S.; West, E.E.; Wei, Z.; Lu, P.; Austin, J.W.; Riley, J.L.; et al. Chronic virus infection enforces demethylation of the locus that encodes PD-1 in antigen-specific CD8(+) T cells. Immunity 2011, 35, 400–412. [Google Scholar] [CrossRef] [PubMed]
- Fehr, T.; Lucas, C.L.; Kurtz, J.; Onoe, T.; Zhao, G.; Hogan, T.; Vallot, C.; Rao, A.; Sykes, M. A CD8 T cell-intrinsic role for the calcineurin-NFAT pathway for tolerance induction in vivo. Blood 2010, 115, 1280–1287. [Google Scholar] [CrossRef] [PubMed]
- Heissmeyer, V.; Macian, F.; Im, S.H.; Varma, R.; Feske, S.; Venuprasad, K.; Gu, H.; Liu, Y.C.; Dustin, M.L.; Rao, A. Calcineurin imposes T cell unresponsiveness through targeted proteolysis of signaling proteins. Nat. Immunol. 2004, 5, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Macian, F. NFAT proteins: Key regulators of T cell development and function. Nat. Rev. Immunol. 2005, 5, 472–484. [Google Scholar] [CrossRef] [PubMed]
- Thomas, R.M.; Chunder, N.; Chen, C.; Umetsu, S.E.; Winandy, S.; Wells, A.D. Ikaros enforces the costimulatory requirement for IL2 gene expression and is required for anergy induction in CD4+ T lymphocytes. J. Immunol. 2007, 179, 7305–7315. [Google Scholar] [CrossRef] [PubMed]
- Baine, I.; Basu, S.; Ames, R.; Sellers, R.S.; Macian, F. Helios induces epigenetic silencing of IL2 gene expression in regulatory T cells. J. Immunol. 2013, 190, 1008–1016. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhyay, S.; Dure, M.; Paroder, M.; Soto-Nieves, N.; Puga, I.; Macian, F. Interleukin 2 gene transcription is regulated by ikaros-induced changes in histone acetylation in anergic T cells. Blood 2007, 109, 2878–2886. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, S.; Thomas, R.M.; Wertheim, G.B.; Zhang, F.; Shen, H.; Wells, A.D. Ikaros imposes a barrier to CD8+ T cell differentiation by restricting autocrine IL-2 production. J. Immunol. 2014, 192, 5118–5129. [Google Scholar] [CrossRef] [PubMed]
- Paley, M.A.; Kroy, D.C.; Odorizzi, P.M.; Johnnidis, J.B.; Dolfi, D.V.; Barnett, B.E.; Bikoff, E.K.; Robertson, E.J.; Lauer, G.M.; Reiner, S.L.; et al. Progenitor and terminal subsets of CD8+ T cells cooperate to contain chronic viral infection. Science 2012, 338, 1220–1225. [Google Scholar] [CrossRef] [PubMed]
- Wherry, E.J. T cell exhaustion. Nat. Immunol. 2011, 12, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Zajac, A.J.; Blattman, J.N.; Murali-Krishna, K.; Sourdive, D.J.; Suresh, M.; Altman, J.D.; Ahmed, R. Viral immune evasion due to persistence of activated T cells without effector function. J. Exp. Med. 1998, 188, 2205–2213. [Google Scholar] [CrossRef] [PubMed]
- Gallimore, A.; Glithero, A.; Godkin, A.; Tissot, A.C.; Pluckthun, A.; Elliott, T.; Hengartner, H.; Zinkernagel, R. Induction and exhaustion of lymphocytic choriomeningitis virus-specific cytotoxic T lymphocytes visualized using soluble tetrameric major histocompatibility complex class I-peptide complexes. J. Exp. Med. 1998, 187, 1383–1393. [Google Scholar] [CrossRef] [PubMed]
- Doering, T.A.; Crawford, A.; Angelosanto, J.M.; Paley, M.A.; Ziegler, C.G.; Wherry, E.J. Network analysis reveals centrally connected genes and pathways involved in CD8+ T cell exhaustion versus memory. Immunity 2012, 37, 1130–1144. [Google Scholar] [CrossRef] [PubMed]
- Rangachari, M.; Zhu, C.; Sakuishi, K.; Xiao, S.; Karman, J.; Chen, A.; Angin, M.; Wakeham, A.; Greenfield, E.A.; Sobel, R.A.; et al. Bat3 promotes T cell responses and autoimmunity by repressing Tim-3-mediated cell death and exhaustion. Nat. Med. 2012, 18, 1394–1400. [Google Scholar] [CrossRef] [PubMed]
- Larsson, M.; Shankar, E.M.; Che, K.F.; Saeidi, A.; Ellegard, R.; Barathan, M.; Velu, V.; Kamarulzaman, A. Molecular signatures of T cell inhibition in HIV-1 infection. Retrovirology 2013. [Google Scholar] [CrossRef] [PubMed]
- Angelosanto, J.M.; Wherry, E.J. Transcription factor regulation of CD8+ T cell memory and exhaustion. Immunol. Rev. 2010, 236, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Hess Michelini, R.; Doedens, A.L.; Goldrath, A.W.; Hedrick, S.M. Differentiation of CD8 memory T cells depends on FoxO1. J. Exp. Med. 2013, 210, 1189–1200. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.V.; Ouyang, W.; Liao, W.; Zhang, M.Q.; Li, M.O. The transcription factor FoxO1 controls central-memory CD8+ T cell responses to infection. Immunity 2013, 39, 286–297. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, J.A.; Kim, E.H.; Plisch, E.H.; Suresh, M. FoxO3 regulates the CD8 T cell response to a chronic viral infection. J. Virol. 2012, 86, 9025–9034. [Google Scholar] [CrossRef] [PubMed]
- Tzelepis, F.; Joseph, J.; Haddad, E.K.; Maclean, S.; Dudani, R.; Agenes, F.; Peng, S.L.; Sekaly, R.P.; Sad, S. Intrinsic role of foxO3a in the development of CD8+ T cell memory. J. Immunol. 2013, 190, 1066–1075. [Google Scholar] [CrossRef] [PubMed]
- Togher, S.; Larange, A.; Schoenberger, S.P.; Feau, S. Foxo3 is a negative regulator of primary CD8+ T cell expansion but not of memory formation. Immunol. Cell Biol. 2015, 93, 120–125. [Google Scholar] [CrossRef] [PubMed]
- Crespo, J.; Sun, H.; Welling, T.H.; Tian, Z.; Zou, W. T cell anergy, exhaustion, senescence, and stemness in the tumor microenvironment. Curr. Opin. Immunol. 2013, 25, 214–221. [Google Scholar] [CrossRef] [PubMed]
- Klebanoff, C.A.; Gattinoni, L.; Restifo, N.P. CD8+ T cell memory in tumor immunology and immunotherapy. Immunol. Rev. 2006, 211, 214–224. [Google Scholar] [CrossRef] [PubMed]
- John, S.A.; Garrett-Sinha, L.A. Blimp1: A conserved transcriptional repressor critical for differentiation of many tissues. Exp. Cell Res. 2009, 315, 1077–1084. [Google Scholar] [CrossRef] [PubMed]
- Martins, G.A.; Cimmino, L.; Shapiro-Shelef, M.; Szabolcs, M.; Herron, A.; Magnusdottir, E.; Calame, K. Transcriptional repressor BLIMP-1 regulates T cell homeostasis and function. Nat. Immunol. 2006, 7, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Kallies, A.; Hawkins, E.D.; Belz, G.T.; Metcalf, D.; Hommel, M.; Corcoran, L.M.; Hodgkin, P.D.; Nutt, S.L. Transcriptional repressor BLIMP-1 is essential for T cell homeostasis and self-tolerance. Nat. Immunol. 2006, 7, 466–474. [Google Scholar] [CrossRef] [PubMed]
- Rutishauser, R.L.; Martins, G.A.; Kalachikov, S.; Chandele, A.; Parish, I.A.; Meffre, E.; Jacob, J.; Calame, K.; Kaech, S.M. Transcriptional repressor BLIMP-1 promotes CD8+ T cell terminal differentiation and represses the acquisition of central memory T cell properties. Immunity 2009, 31, 296–308. [Google Scholar] [CrossRef] [PubMed]
- Kallies, A.; Xin, A.; Belz, G.T.; Nutt, S.L. BLIMP-1 transcription factor is required for the differentiation of effector CD8(+) T cells and memory responses. Immunity 2009, 31, 283–295. [Google Scholar] [CrossRef] [PubMed]
- Cimmino, L.; Martins, G.A.; Liao, J.; Magnusdottir, E.; Grunig, G.; Perez, R.K.; Calame, K.L. BLIMP-1 attenuates Th1 differentiation by repression of Ifng, tbx21, and bcl6 gene expression. J. Immunol. 2008, 181, 2338–2347. [Google Scholar] [CrossRef] [PubMed]
- Martins, G.A.; Cimmino, L.; Liao, J.; Magnusdottir, E.; Calame, K. Blimp-1 directly represses IL2 and the IL2 activator Fos, attenuating T cell proliferation and survival. J. Exp. Med. 2008, 205, 1959–1965. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Pos, Z.; Rao, M.; Klebanoff, C.A.; Yu, Z.; Sukumar, M.; Reger, R.N.; Palmer, D.C.; Borman, Z.A.; Muranski, P.; et al. Repression of the DNA-binding inhibitor Id3 by Blimp-1 limits the formation of memory CD8+ T cells. Nat. Immunol. 2011, 12, 1230–1237. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Youngblood, B.A.; Austin, J.W.; Mohammed, A.U.; Butler, R.; Ahmed, R.; Boss, J.M. Blimp-1 represses CD8 T cell expression of PD-1 using a feed-forward transcriptional circuit during acute viral infection. J. Exp. Med. 2014, 211, 515–527. [Google Scholar] [CrossRef] [PubMed]
- Crotty, S.; Johnston, R.J.; Schoenberger, S.P. Effectors and memories: Bcl-6 and Blimp-1 in T and B lymphocyte differentiation. Nat. Immunol. 2010, 11, 114–120. [Google Scholar] [CrossRef] [PubMed]
- Ancelin, K.; Lange, U.C.; Hajkova, P.; Schneider, R.; Bannister, A.J.; Kouzarides, T.; Surani, M.A. Blimp1 associates with prmt5 and directs histone arginine methylation in mouse germ cells. Nat. Cell Biol. 2006, 8, 623–630. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Angelin-Duclos, C.; Greenwood, J.; Liao, J.; Calame, K. Transcriptional repression by Blimp-1 (PRDI-BF1) involves recruitment of histone deacetylase. Mol. Cell. Biol. 2000, 20, 2592–2603. [Google Scholar] [CrossRef] [PubMed]
- Gyory, I.; Wu, J.; Fejer, G.; Seto, E.; Wright, K.L. PRDI-BF1 recruits the histone H3 methyltransferase G9a in transcriptional silencing. Nat. Immunol. 2004, 5, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Su, S.T.; Ying, H.Y.; Chiu, Y.K.; Lin, F.R.; Chen, M.Y.; Lin, K.I. Involvement of histone demethylase LSD1 in Blimp-1-mediated gene repression during plasma cell differentiation. Mol. Cell. Biol. 2009, 29, 1421–1431. [Google Scholar] [CrossRef] [PubMed]
- Heng, T.S.; Painter, M.W.; Immunological Genome Project, C. The immunological genome project: Networks of gene expression in immune cells. Nat. Immunol. 2008, 9, 1091–1094. [Google Scholar] [CrossRef] [PubMed]
- Schoenborn, J.R.; Dorschner, M.O.; Sekimata, M.; Santer, D.M.; Shnyreva, M.; Fitzpatrick, D.R.; Stamatoyannopoulos, J.A.; Wilson, C.B. Comprehensive epigenetic profiling identifies multiple distal regulatory elements directing transcription of the gene encoding interferon-gamma. Nat. Immunol. 2007, 8, 732–742. [Google Scholar] [CrossRef] [PubMed]
- Creyghton, M.P.; Cheng, A.W.; Welstead, G.G.; Kooistra, T.; Carey, B.W.; Steine, E.J.; Hanna, J.; Lodato, M.A.; Frampton, G.M.; Sharp, P.A.; et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. USA 2010, 107, 21931–21936. [Google Scholar] [CrossRef] [PubMed]
- Rada-Iglesias, A.; Bajpai, R.; Swigut, T.; Brugmann, S.A.; Flynn, R.A.; Wysocka, J. A unique chromatin signature uncovers early developmental enhancers in humans. Nature 2011, 470, 279–283. [Google Scholar] [CrossRef] [PubMed]
- Ooi, S.K.; Qiu, C.; Bernstein, E.; Li, K.; Jia, D.; Yang, Z.; Erdjument-Bromage, H.; Tempst, P.; Lin, S.P.; Allis, C.D.; et al. DNMT3l connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature 2007, 448, 714–717. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Thieu, V.T.; Kaplan, M.H. Stat4 limits DNA methyltransferase recruitment and DNA methylation of the IL-18ralpha gene during Th1 differentiation. EMBO J. 2007, 26, 2052–2060. [Google Scholar] [CrossRef] [PubMed]
- Parish, I.A.; Rao, S.; Smyth, G.K.; Juelich, T.; Denyer, G.S.; Davey, G.M.; Strasser, A.; Heath, W.R. The molecular signature of CD8+ T cells undergoing deletional tolerance. Blood 2009, 113, 4575–4585. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstrale, M.; Laurila, E.; et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Huang da, W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using david bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Huang da, W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucl. Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.T.; Nevins, J.R. Gather: A systems approach to interpreting genomic signatures. Bioinformatics 2006, 22, 2926–2933. [Google Scholar] [CrossRef] [PubMed]
- Sakuishi, K.; Apetoh, L.; Sullivan, J.M.; Blazar, B.R.; Kuchroo, V.K.; Anderson, A.C. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J. Exp. Med. 2010, 207, 2187–2194. [Google Scholar] [CrossRef] [PubMed]
- Chemnitz, J.M.; Parry, R.V.; Nichols, K.E.; June, C.H.; Riley, J.L. SHP-1 AND SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J. Immunol. 2004, 173, 945–954. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Su, E.W.; Zhu, C.; Hainline, S.; Phuah, J.; Moroco, J.A.; Smithgall, T.E.; Kuchroo, V.K.; Kane, L.P. Phosphotyrosine-dependent coupling of Tim-3 to T cell receptor signaling pathways. Mol. Cell. Biol. 2011, 31, 3963–3974. [Google Scholar] [CrossRef] [PubMed]
- Ferris, R.L.; Lu, B.; Kane, L.P. Too much of a good thing? Tim-3 and TCR signaling in T cell exhaustion. J. Immunol. 2014, 193, 1525–1530. [Google Scholar] [CrossRef] [PubMed]
- Agnellini, P.; Wolint, P.; Rehr, M.; Cahenzli, J.; Karrer, U.; Oxenius, A. Impaired NFAT nuclear translocation results in split exhaustion of virus-specific CD8+ T cell functions during chronic viral infection. Proc. Natl. Acad. Sci. USA 2007, 104, 4565–4570. [Google Scholar] [CrossRef] [PubMed]
- Martinez, G.J.; Pereira, R.M.; Aijo, T.; Kim, E.Y.; Marangoni, F.; Pipkin, M.E.; Togher, S.; Heissmeyer, V.; Zhang, Y.C.; Crotty, S.; et al. The transcription factor NFAT promotes exhaustion of activated CD8(+) T cells. Immunity 2015, 42, 265–278. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, S.D.; Shin, H.; Freeman, G.J.; Wherry, E.J. Selective expansion of a subset of exhausted CD8 T cells by alphapd-l1 blockade. Proc. Natl. Acad. Sci. USA 2008, 105, 15016–15021. [Google Scholar] [CrossRef] [PubMed]
- Buggert, M.; Tauriainen, J.; Yamamoto, T.; Frederiksen, J.; Ivarsson, M.A.; Michaelsson, J.; Lund, O.; Hejdeman, B.; Jansson, M.; Sonnerborg, A.; et al. T-bet and eomes are differentially linked to the exhausted phenotype of CD8+ T cells in hiv infection. PLoS Pathog. 2014, 10, e1004251. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Ju, S.; Chen, E.; Dai, S.; Li, C.; Morel, P.; Liu, L.; Zhang, X.; Lu, B. T-bet and eomesodermin are required for T cell-mediated antitumor immune responses. J. Immunol. 2010, 185, 3174–3183. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Sadashivaiah, K.; Furusawa, A.; Davila, E.; Tamada, K.; Banerjee, A. Eomesodermin is required for antitumor immunity mediated by 4-1BB-agonist immunotherapy. Oncoimmunology 2014, 3, e27680. [Google Scholar] [CrossRef] [PubMed]
- Berrien-Elliott, M.M.; Yuan, J.; Swier, L.E.; Jackson, S.R.; Chen, C.L.; Donlin, M.J.; Teague, R.M. Checkpoint blockade immunotherapy relies on T-bet but not eomes to induce effector function in tumor-infiltrating CD8+ T cells. Cancer Immunol. Res. 2015, 3, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Zitvogel, L.; Kroemer, G. Targeting foxp1 for reinstating anticancer immunosurveillance. Immunity 2014, 41, 345–347. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Bevan, M.J. Tgf-beta signaling to T cells inhibits autoimmunity during lymphopenia-driven proliferation. Nat. Immunol. 2012, 13, 667–673. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, N.; Kang, J. Smad regulatory networks construct a balanced immune system. Immunology 2013, 139, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Tinoco, R.; Alcalde, V.; Yang, Y.; Sauer, K.; Zuniga, E.I. Cell-intrinsic transforming growth factor-beta signaling mediates virus-specific CD8+ T cell deletion and viral persistence in vivo. Immunity 2009, 31, 145–157. [Google Scholar] [CrossRef] [PubMed]
- Garidou, L.; Heydari, S.; Gossa, S.; McGavern, D.B. Therapeutic blockade of transforming growth factor beta fails to promote clearance of a persistent viral infection. J. Virol. 2012, 86, 7060–7071. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Massague, J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell 2003, 113, 685–700. [Google Scholar] [CrossRef]
- Mokrani, M.; Klibi, J.; Bluteau, D.; Bismuth, G.; Mami-Chouaib, F. Smad and nfat pathways cooperate to induce CD103 expression in human CD8 T lymphocytes. J. Immunol. 2014, 192, 2471–2479. [Google Scholar] [CrossRef] [PubMed]
- Stephen, T.L.; Rutkowski, M.R.; Allegrezza, M.J.; Perales-Puchalt, A.; Tesone, A.J.; Svoronos, N.; Nguyen, J.M.; Sarmin, F.; Borowsky, M.E.; Tchou, J.; et al. Transforming growth factor beta-mediated suppression of antitumor T cells requires FoxP1 transcription factor expression. Immunity 2014, 41, 427–439. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Wang, H.; Takata, H.; Day, T.J.; Willen, J.; Hu, H. Transcription factor FoxP1 exerts essential cell-intrinsic regulation of the quiescence of naive T cells. Nat. Immunol. 2011, 12, 544–550. [Google Scholar] [CrossRef] [PubMed]
- Skon, C.N.; Jameson, S.C. Fox factors fight over T cell quiescence. Nat. Immunol. 2011, 12, 522–524. [Google Scholar] [CrossRef] [PubMed]
- Chapman, N.M.; Chi, H. mTOR links environmental signals to T cell fate decisions. Front. Immunol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Sinclair, L.V.; Finlay, D.; Feijoo, C.; Cornish, G.H.; Gray, A.; Ager, A.; Okkenhaug, K.; Hagenbeek, T.J.; Spits, H.; Cantrell, D.A. Phosphatidylinositol-3-oh kinase and nutrient-sensing mtor pathways control t lymphocyte trafficking. Nat. Immunol. 2008, 9, 513–521. [Google Scholar] [CrossRef] [PubMed]
- Sukumar, M.; Liu, J.; Ji, Y.; Subramanian, M.; Crompton, J.G.; Yu, Z.; Roychoudhuri, R.; Palmer, D.C.; Muranski, P.; Karoly, E.D.; et al. Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. J. Clin. Investig. 2013, 123, 4479–4488. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Gabrilovich, D.I. Hypoxia-inducible factors in regulation of immune responses in tumour microenvironment. Immunology 2014, 143, 512–519. [Google Scholar] [CrossRef] [PubMed]
- Palazon, A.; Martinez-Forero, I.; Teijeira, A.; Morales-Kastresana, A.; Alfaro, C.; Sanmamed, M.F.; Perez-Gracia, J.L.; Penuelas, I.; Hervas-Stubbs, S.; Rouzaut, A.; et al. The HIF-1alpha hypoxia response in tumor-infiltrating T lymphocytes induces functional CD137 (4-1bb) for immunotherapy. Cancer Discov. 2012, 2, 608–623. [Google Scholar] [CrossRef] [PubMed]
- Yonezawa, A.; Dutt, S.; Chester, C.; Kim, J.; Kohrt, H.E. Boosting cancer immunotherapy with anti-CD137 antibody therapy. Clin. Cancer Res. 2015. [Google Scholar] [CrossRef] [PubMed]
- Doedens, A.L.; Phan, A.T.; Stradner, M.H.; Fujimoto, J.K.; Nguyen, J.V.; Yang, E.; Johnson, R.S.; Goldrath, A.W. Hypoxia-inducible factors enhance the effector responses of CD8(+) T cells to persistent antigen. Nat. Immunol. 2013, 14, 1173–1182. [Google Scholar] [CrossRef] [PubMed]
- Berezhnoy, A.; Castro, I.; Levay, A.; Malek, T.R.; Gilboa, E. Aptamer-targeted inhibition of mtor in T cells enhances antitumor immunity. J. Clin. Investig. 2014, 124, 188–197. [Google Scholar] [CrossRef] [PubMed]
- Araki, K.; Turner, A.P.; Shaffer, V.O.; Gangappa, S.; Keller, S.A.; Bachmann, M.F.; Larsen, C.P.; Ahmed, R. Mtor regulates memory CD8 T cell differentiation. Nature 2009, 460, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Joshi, N.S.; Cui, W.; Chandele, A.; Lee, H.K.; Urso, D.R.; Hagman, J.; Gapin, L.; Kaech, S.M. Inflammation directs memory precursor and short-lived effector CD8(+) T cell fates via the graded expression of t-bet transcription factor. Immunity 2007, 27, 281–295. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Yu, S.; Zhao, D.M.; Harty, J.T.; Badovinac, V.P.; Xue, H.H. Differentiation and persistence of memory CD8(+) T cells depend on T cell factor 1. Immunity 2010, 33, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Pearce, E.L.; Walsh, M.C.; Cejas, P.J.; Harms, G.M.; Shen, H.; Wang, L.S.; Jones, R.G.; Choi, Y. Enhancing CD8 T cell memory by modulating fatty acid metabolism. Nature 2009, 460, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Jeannet, G.; Boudousquie, C.; Gardiol, N.; Kang, J.; Huelsken, J.; Held, W. Essential role of the Wnt pathway effector TCF-1 for the establishment of functional CD8 T cell memory. Proc. Natl. Acad. Sci. USA 2010, 107, 9777–9782. [Google Scholar] [CrossRef] [PubMed]
- Keefe, A.D.; Pai, S.; Ellington, A. Aptamers as therapeutics. Nat. Rev. Drug Discov. 2010, 9, 537–550. [Google Scholar] [CrossRef] [PubMed]
- Delgoffe, G.M.; Kole, T.P.; Zheng, Y.; Zarek, P.E.; Matthews, K.L.; Xiao, B.; Worley, P.F.; Kozma, S.C.; Powell, J.D. The mtor kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity 2009, 30, 832–844. [Google Scholar] [CrossRef] [PubMed]
- Dassie, J.P.; Liu, X.Y.; Thomas, G.S.; Whitaker, R.M.; Thiel, K.W.; Stockdale, K.R.; Meyerholz, D.K.; McCaffrey, A.P.; McNamara, J.O., II; Giangrande, P.H. Systemic administration of optimized aptamer-sirna chimeras promotes regression of psma-expressing tumors. Nat. Biotechnol. 2009, 27, 839–849. [Google Scholar] [CrossRef] [PubMed]
- Ni, X.; Zhang, Y.; Ribas, J.; Chowdhury, W.H.; Castanares, M.; Zhang, Z.; Laiho, M.; DeWeese, T.L.; Lupold, S.E. Prostate-targeted radiosensitization via aptamer-shrna chimeras in human tumor xenografts. J. Clin. Investig. 2011, 121, 2383–2390. [Google Scholar] [CrossRef] [PubMed]
- Pastor, F.; Kolonias, D.; Giangrande, P.H.; Gilboa, E. Induction of tumour immunity by targeted inhibition of nonsense-mediated mrna decay. Nature 2010, 465, 227–230. [Google Scholar] [CrossRef] [PubMed]
- Pastor, F.; Kolonias, D.; McNamara, J.O., 2nd; Gilboa, E. Targeting 4-1BB costimulation to disseminated tumor lesions with bi-specific oligonucleotide aptamers. Mol. Ther. 2011, 19, 1878–1886. [Google Scholar] [CrossRef] [PubMed]
- Gattinoni, L.; Klebanoff, C.A.; Restifo, N.P. Paths to stemness: Building the ultimate antitumour T cell. Nat. Rev. Cancer 2012, 12, 671–684. [Google Scholar] [CrossRef] [PubMed]
- Gattinoni, L.; Lugli, E.; Ji, Y.; Pos, Z.; Paulos, C.M.; Quigley, M.F.; Almeida, J.R.; Gostick, E.; Yu, Z.; Carpenito, C.; et al. A human memory T cell subset with stem cell-like properties. Nat. Med. 2011, 17, 1290–1297. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.M.; Yu, S.; Zhou, X.; Haring, J.S.; Held, W.; Badovinac, V.P.; Harty, J.T.; Xue, H.H. Constitutive activation of Wnt signaling favors generation of memory CD8 T cells. J. Immunol. 2010, 184, 1191–1199. [Google Scholar] [CrossRef] [PubMed]
- Kalos, M.; Levine, B.L.; Porter, D.L.; Katz, S.; Grupp, S.A.; Bagg, A.; June, C.H. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci. Transl. Med. 2011. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A.; Yang, J.C.; Sherry, R.M.; Kammula, U.S.; Hughes, M.S.; Phan, G.Q.; Citrin, D.E.; Restifo, N.P.; Robbins, P.F.; Wunderlich, J.R.; et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T cell transfer immunotherapy. Clin. Cancer Res. 2011, 17, 4550–4557. [Google Scholar] [CrossRef] [PubMed]
- Robbins, P.F.; Dudley, M.E.; Wunderlich, J.; El-Gamil, M.; Li, Y.F.; Zhou, J.; Huang, J.; Powell, D.J., Jr.; Rosenberg, S.A. Cutting edge: Persistence of transferred lymphocyte clonotypes correlates with cancer regression in patients receiving cell transfer therapy. J. Immunol. 2004, 173, 7125–7130. [Google Scholar] [CrossRef]
- Pule, M.A.; Savoldo, B.; Myers, G.D.; Rossig, C.; Russell, H.V.; Dotti, G.; Huls, M.H.; Liu, E.; Gee, A.P.; Mei, Z.; et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: Persistence and antitumor activity in individuals with neuroblastoma. Nat. Med. 2008, 14, 1264–1270. [Google Scholar] [CrossRef] [PubMed]
- Gattinoni, L.; Klebanoff, C.A.; Restifo, N.P. Pharmacologic induction of CD8+ T cell memory: Better living through chemistry. Sci. Transl. Med. 2009. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Shaffer, D.R.; Alvarez Arias, D.A.; Nakazaki, Y.; Pos, W.; Torres, A.J.; Cremasco, V.; Dougan, S.K.; Cowley, G.S.; Elpek, K.; et al. In vivo discovery of immunotherapy targets in the tumour microenvironment. Nature 2014, 506, 52–57. [Google Scholar] [CrossRef] [PubMed]
- Barrangou, R.; Birmingham, A.; Wiemann, S.; Beijersbergen, R.L.; Hornung, V.; Smith, A. Advances in CRISPR-Cas9 genome engineering: Lessons learned from RNA interference. Nucl. Acids Res. 2015, 43, 3407–3419. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.; Moehlenbrink, J.; Lu, Y.C.; Zalmas, L.P.; Sagum, C.A.; Carr, S.; McGouran, J.F.; Alexander, L.; Fedorov, O.; Munro, S.; et al. Arginine methylation-dependent reader-writer interplay governs growth control by e2f-1. Mol. Cell 2013, 52, 37–51. [Google Scholar] [CrossRef] [PubMed]
- Dey, S.S.; Kester, L.; Spanjaard, B.; Bienko, M.; van Oudenaarden, A. Integrated genome and transcriptome sequencing of the same cell. Nat. Biotechnol. 2015, 33, 285–289. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Waugh, K.A.; Leach, S.M.; Slansky, J.E. Targeting Transcriptional Regulators of CD8+ T Cell Dysfunction to Boost Anti-Tumor Immunity. Vaccines 2015, 3, 771-802. https://doi.org/10.3390/vaccines3030771
Waugh KA, Leach SM, Slansky JE. Targeting Transcriptional Regulators of CD8+ T Cell Dysfunction to Boost Anti-Tumor Immunity. Vaccines. 2015; 3(3):771-802. https://doi.org/10.3390/vaccines3030771
Chicago/Turabian StyleWaugh, Katherine A., Sonia M. Leach, and Jill E. Slansky. 2015. "Targeting Transcriptional Regulators of CD8+ T Cell Dysfunction to Boost Anti-Tumor Immunity" Vaccines 3, no. 3: 771-802. https://doi.org/10.3390/vaccines3030771
APA StyleWaugh, K. A., Leach, S. M., & Slansky, J. E. (2015). Targeting Transcriptional Regulators of CD8+ T Cell Dysfunction to Boost Anti-Tumor Immunity. Vaccines, 3(3), 771-802. https://doi.org/10.3390/vaccines3030771