
New Data on Vaccine Antigen Deficient Bordetella pertussis Isolates

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bordetella pertussis Isolates and Growth Conditions

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Year of Collection | ptxP | ptxA | prn | fim2; fim3 | Sero typing | Western Blot | Reference |
|---|---|---|---|---|---|---|---|---|
| Tohama | 1950 | 1 | 2 | 1 | 2-1; 3-1 | 2 | + | [8] |
| CIP1672 | 1954 | 1 | 1 | 1 | 2-1; 3-1 | 2 | FHA− | [8] |
| FR4624 | 2009 | 3 | 1 | 2 | 2-1; 3-2 | 3 | PRN−/FHA− | [8] |
| FR4684 | 2010 | 1 | 2 | 1 | 2-1; 3-1 | 3 | PRN− | [19] |
| FR4929 | 2011 | 21 | 1 | 2 | 2-1; 3-2 | 3 | + | [19] |
| FR5133 | 2012 | 3 | 1 | 2 | 2-1; 3-2 | 3 | + | [19] |
| FR5187 | 2012 | 3 | 1 | 2 | 2-1; 3-1 | 3 | PRN− | [19] |
2.2. DNA Preparation
2.3. Whole Genome Sequencing (WGS)/Single Nucleotide Polymorphism (SNP) Analysis
2.4. RNA Preparation
2.5. Transcriptomic Analyses of Targeted Virulence Factors
| Primers Names | Primers Sequences 5' ≥ 3' |
|---|---|
| recA-F | TGGACGTGCAATACGC |
| recA-R | GACCATGCAGTTGGTG |
| ptxA-F | CCTACCAGAGCGAATATCTGGCAC |
| ptxA-R | GATTGGCGCGAGTCTGCT |
| fhaB-F | TCGGAGAGCCACAACT |
| fhaB-R | GTTCCGTATTGAAATTGAAGCC |
| Prn-F | CGGCGACCTTTACCCTTG |
| Prn-R | GGCTCCACTGCCCATTG |
| cyaA-F | GGTCAGCTATGCCGCCCT |
| cyaA-R | TTCTCCGTGCGCTTGCCGTA |
2.6. Interaction with Human Tracheal Epithelial Cells
3. Results
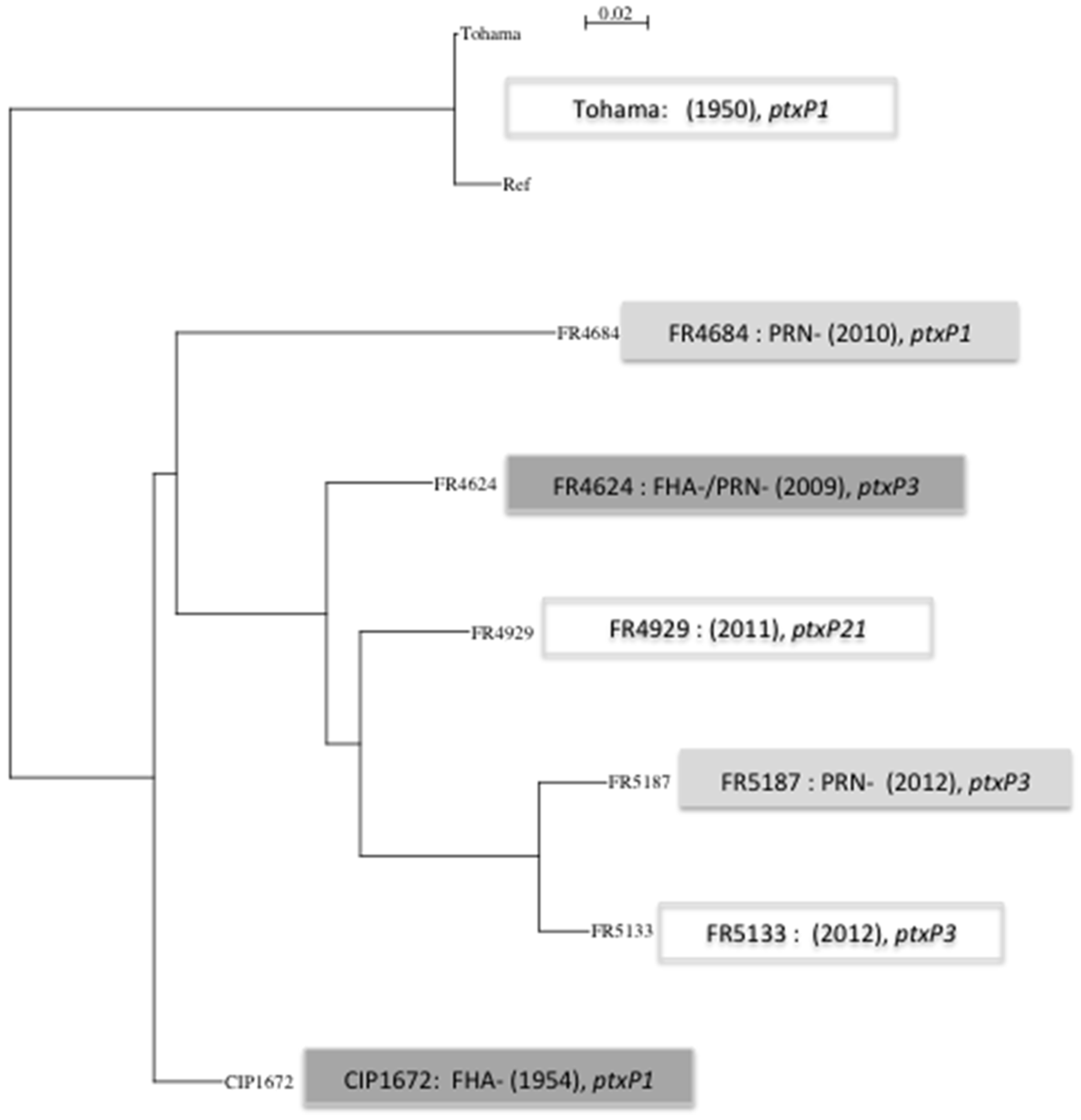
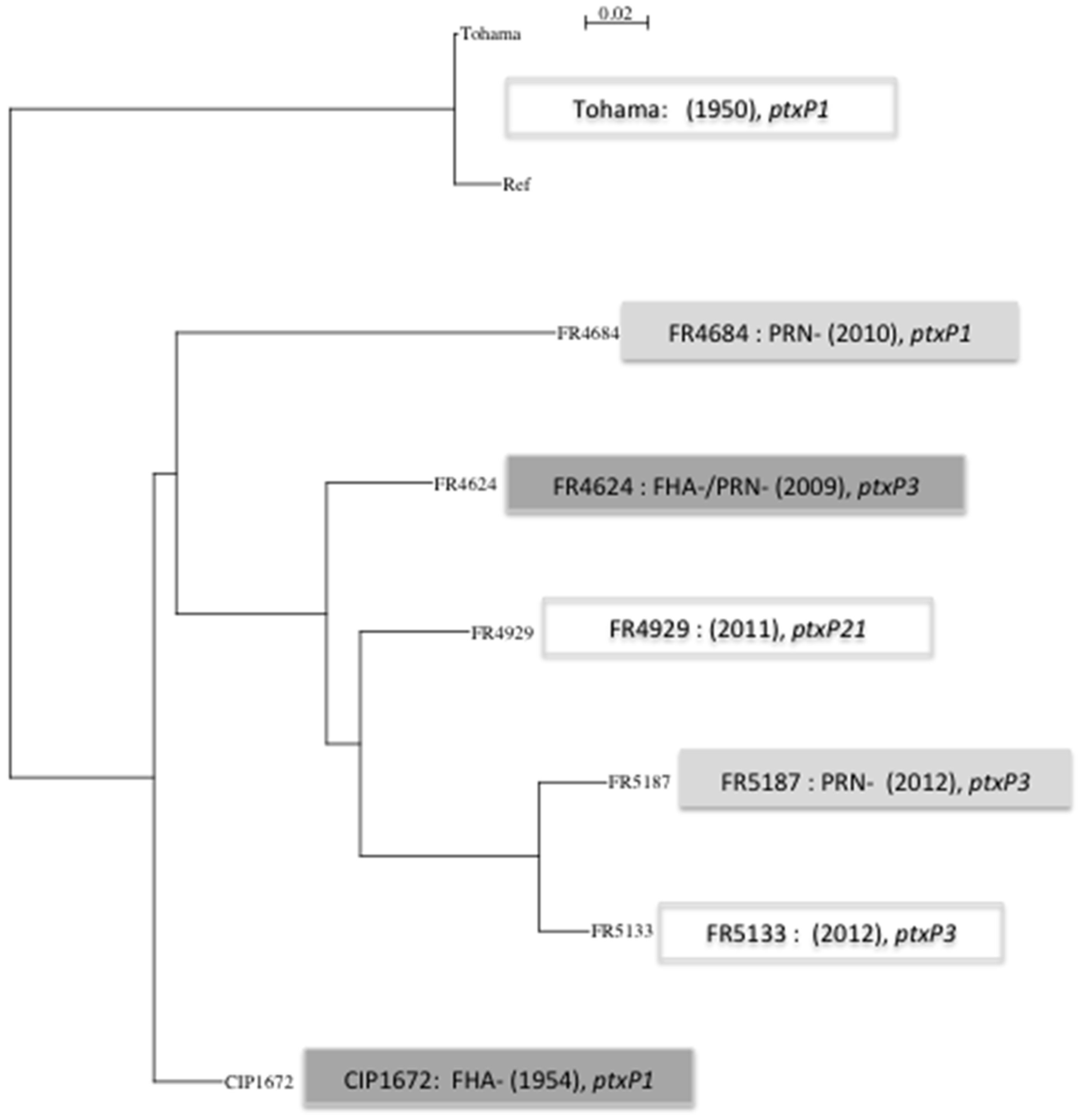
3.1. Whole Genome Data Analysis
| SNP details | Tohama (+) | CIP1672 (FHA−) | FR4624 (PRN−/FHA−) | FR4684 (PRN−) | FR4929 (+) | FR5133 (+) | FR5187 (PRN−) |
|---|---|---|---|---|---|---|---|
| SNP | 22 | 205 | 252 | 268 | 266 | 279 | 286 |
| Synonymous | 4 | 54 | 67 | 69 | 70 | 75 | 78 |
| Non synonymous | 8 | 82 | 100 | 98 | 105 | 100 | 105 |
| Intragenic | 4 | 28 | 36 | 35 | 34 | 29 | 36 |
| Stop | 0 | 0 | 2 | 0 | 1 | 2 | 3 |
| Intergenic | 6 | 27 | 30 | 44 | 40 | 44 | 43 |
| Frame shift | 0 | 12 | 15 | 20 | 14 | 27 | 19 |
| Insertion codon | 0 | 2 | 2 | 2 | 2 | 2 | 2 |
| Tohama Reference Genome NC_02929 Position | AN in theRef. | Tohama ptxP1 | CIP1672 (FHA-) ptxP1 | FR4624 (FHA-/PRN-) ptxP3 | FR4684 (PRN-) ptxP1 | FR4929 ptxP21 | FR5133 ptxP3 | FR518 (PRN-) ptxP3 | SNP Observed | SNP Type | Gene or Promoter Localisation | Nucleic Acid Change within Codon | AminoAcid Change |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 3988168 | G | G | G | A | G | A | A | A | A | intergenic | ptxp | ||
| 3988244 | G | G | G | G | G | A | G | G | A | intergenic | ptxp | ||
| 3988941 | G | G | A | A | G | A | A | A | A | non synonymous | ptxA | atG/atA | M/I |
| 3989239 | G | G | A | A | A | A | A | A | A | non synonymous | ptxB | Ggc/Agc | G/S |
| 3991376 | C | C | C | T | C | T | T | T | T | synonymous | ptxC | tgC/tgT | |
| 1098920 | * | * | * | * | * | C | C | C | INS (C) | frameshift | prn | ggt/gCgt | G/A |
| 1098922 | G | G | G | G | G | T | T | T | T | non synonymous | prn | Gcg/Tcg | A/S |
| 1098926 | T | T | T | T | T | * | * | * | DEL (T) | frameshift | prn | ||
| 1098918 | T | T | T | T | T | C | C | C | C | synonymous | prn | ggT/ggC | |
| 1176534 | * | G | G | G | * | G | G | G | INS (G) | intergenic | pfim2 | ||
| 1176535 | * | G | G | G | * | G | G | G | INS (G) | intergenic | pfim2 | ||
| 1176536 | * | G | G | G | * | G | G | G | INS (G) | intergenic | pfim2 | ||
| 1176541 | * | G | G | G | G | * | G | * | INS (G) | intergenic | pfim2 | ||
| 1176542 | * | G | G | G | G | * | * | * | INS (G) | intergenic | pfim2 | ||
| 1176543 | * | G | G | G | G | * | * | * | INS (G) | intergenic | pfim2 | ||
| 1176544 | * | G | G | G | G | * | * | * | INS (G) | intergenic | pfim2 | ||
| 1176545 | * | G | G | G | G | * | * | * | INS (G) | intergenic | pfim2 | ||
| 1176546 | * | A | A | A | A | * | * | * | INS (A) | intergenic | pfim2 | ||
| 1626880 | G | G | G | G | G | C | C | C | C | intergenic | pfim2 | ||
| 1637246 | C | C | C | C | T | C | C | C | T | intergenic | pfim2 | ||
| 1647861 | C | C | C | A | C | A | A | C | A | non synonymous | fim3 | gCg/gAg | A/E |
| 1984103 | T | T | C | C | C | C | C | C | C | non synonymous | fimD | tTc/tCc | F/S |
| 1965604 | T | T | C | C | C | C | C | C | C | non synonymous | bvgS | Aag/Gag | K/E |
| 1968699 | G | G | G | A | G | G | G | G | A | intergenic | pfhaB | ||
| 2826237 | G | G | G | G | G | A | A | G | A | non synonymous | fhaS | Cac/Tac | H/Y |
| 223961 | G | G | G | G | G | G | G | A | A | non synonymous | sphB1 | Gta/Ata | V/I |
| 224066 | G | G | G | G | G | A | A | G | A | non synonymous | sphB1 | Gcc/Acc | A/T |
| 511992 | A | A | A | G | A | G | G | G | G | intergenic | pbteA | ||
| 514171 | G | G | G | A | G | A | A | A | A | intergenic | pbteA | ||
| 2374322 | T | T | T | C | T | C | C | C | C | non synonymous | bscI | tAc/tGc | Y/C |
| 2376650 | G | G | G | A | G | A | A | A | A | non synonymous | bopB | Ccc/Tcc | P/S |
| 2363842 | C | C | C | C | T | C | C | C | T | synonymous | bscC | ctG/ctA |
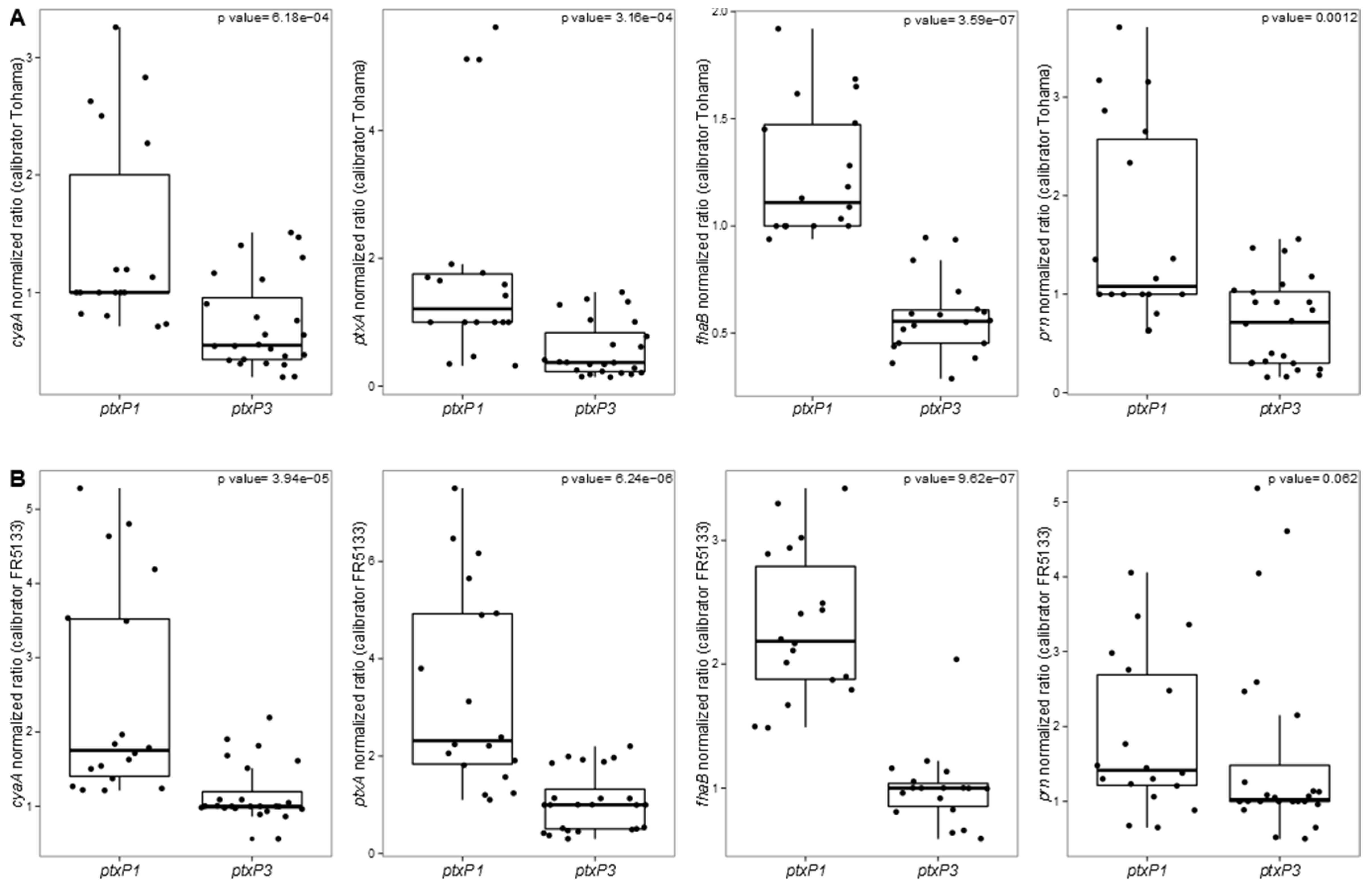
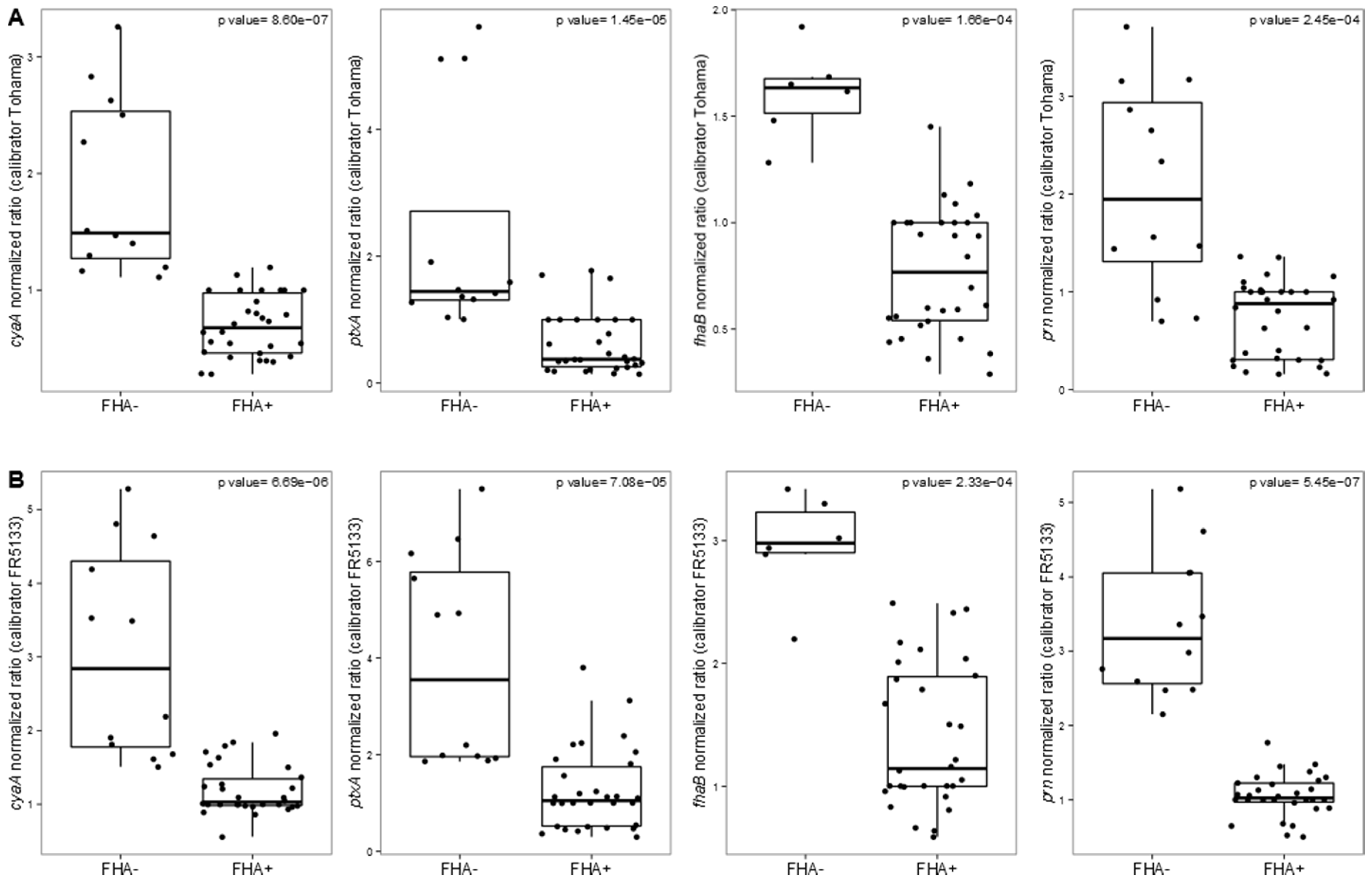
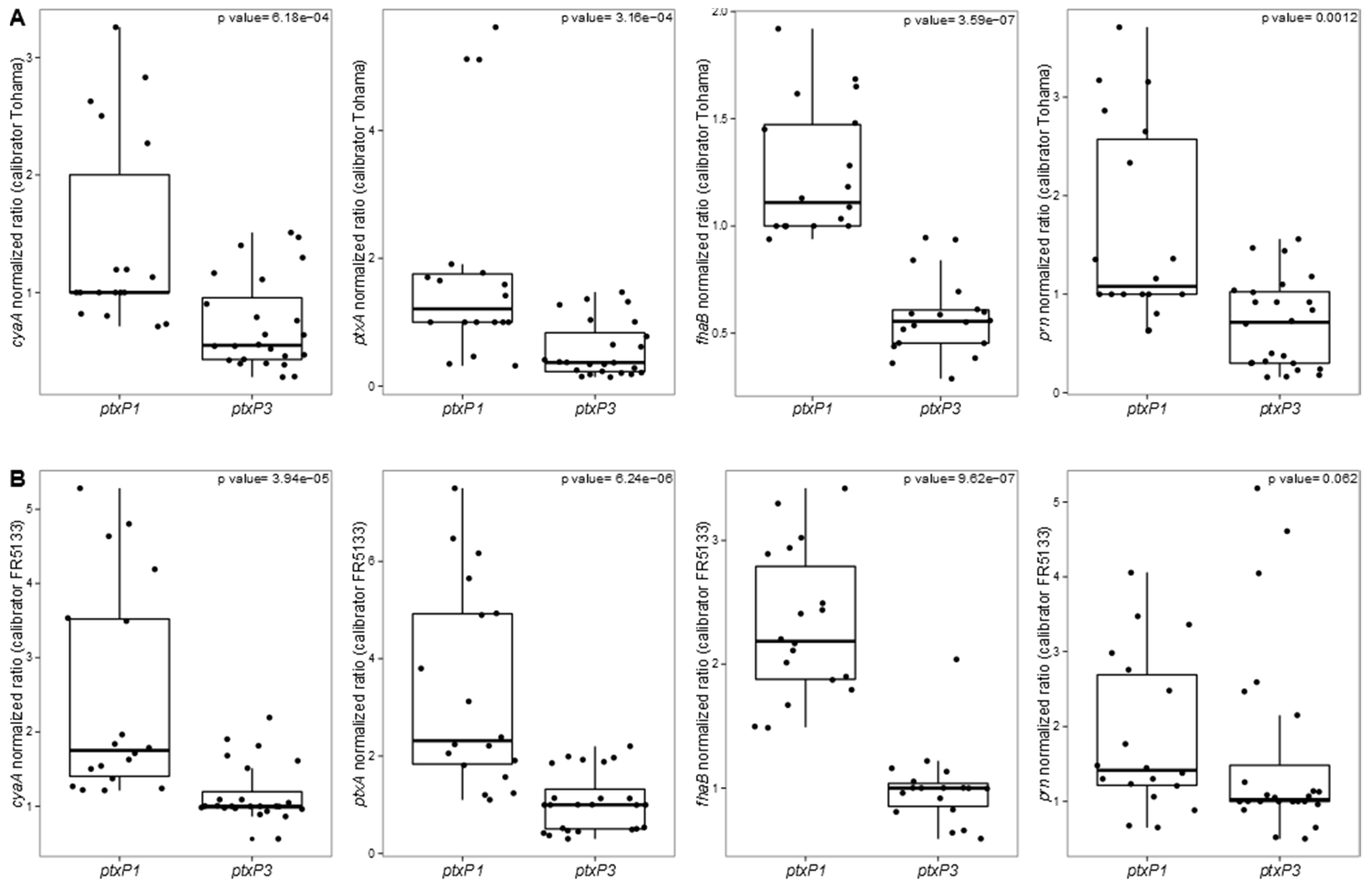
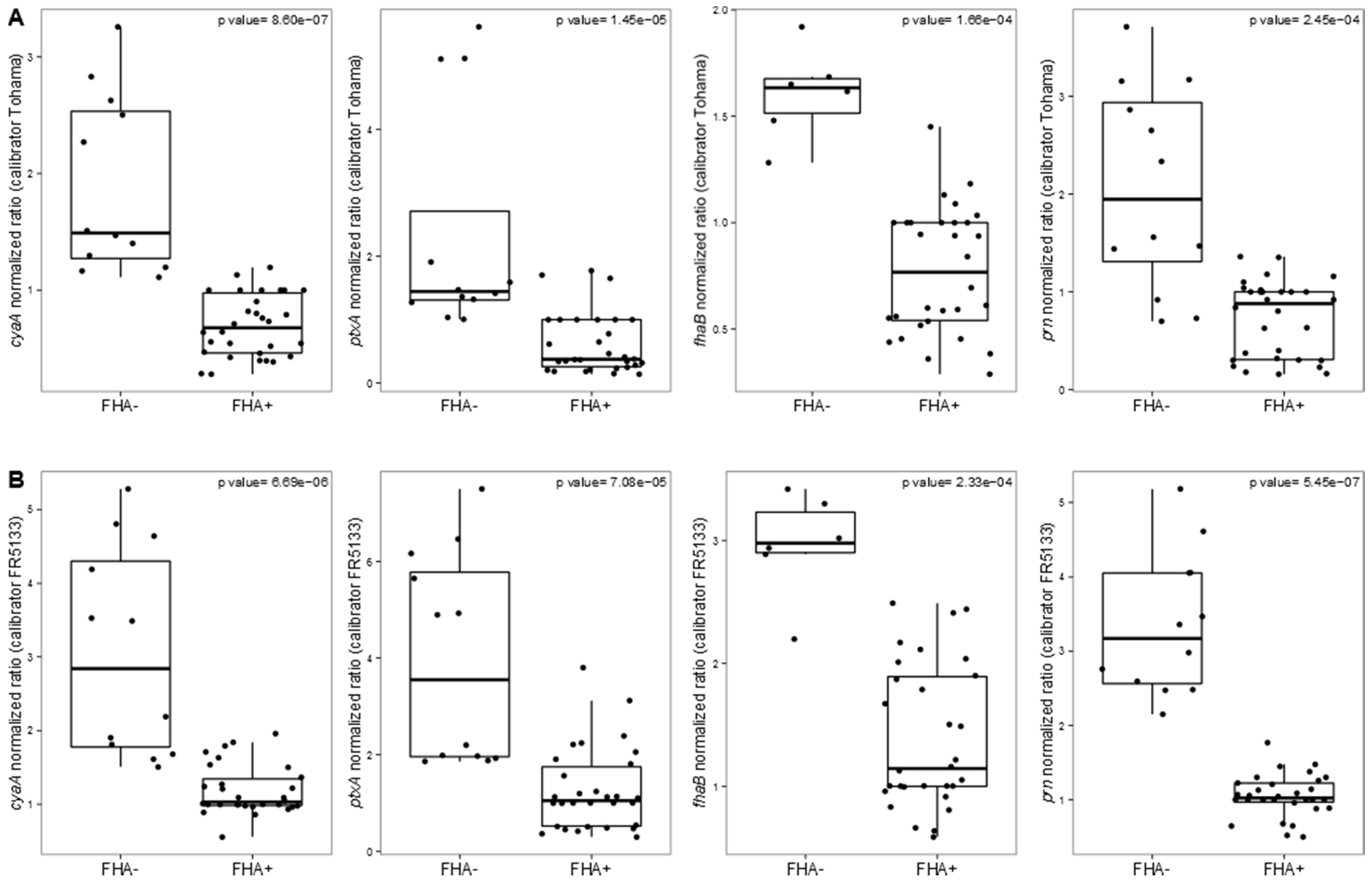
3.2. Targeted Transcriptomic Analysis of cyaA, ptxA, fhaB and prn Expression
3.3. Properties toward Human Tracheal Epithelial Cells
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hegerle, N.; Guiso, N. Epidemiology of whooping cough & typing of Bordetella pertussis. Future Microbiol. 2013, 8, 1391–1403. [Google Scholar] [PubMed]
- Sheridan, S.L.; Frith, K.; Snelling, T.L.; Grimwood, K.; McIntyre, P.B.; Lambert, S.B. Waning vaccine immunity in teenagers primed with whole cell and acellular pertussis vaccine: Recent epidemiology. Expert Rev. Vaccines 2014, 13, 1081–1106. [Google Scholar] [CrossRef] [PubMed]
- Sheridan, S.L.; Ware, R.S.; Grimwood, K.; Lambert, S.B. Number and order of whole cell pertussis vaccines in infancy and disease protection. JAMA 2012, 308, 454–456. [Google Scholar] [CrossRef] [PubMed]
- Gambhir, M.; Clark, T.A.; Cauchemez, S.; Tartof, S.Y.; Swerdlow, D.L.; Ferguson, N.M. A change in vaccine efficacy and duration of protection explains recent rises in pertussis incidence in the United States. PLoS Comput. Biol. 2015, 11, e1004138. [Google Scholar] [CrossRef] [PubMed]
- Parkhill, J.; Sebaihia, M.; Preston, A.; Murphy, L.D.; Thomson, N.; Harris, D.E.; Holden, M.T.; Churcher, C.M.; Bentley, S.D.; Mungall, K.L.; et al. Comparative analysis of the genome sequences of Bordetella pertussis, Bordetella parapertussis and Bordetella bronchiseptica. Nat. Genet. 2003, 35, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Diavatopoulos, D.A.; Cummings, C.A.; Schouls, L.M.; Brinig, M.M.; Relman, D.A.; Mooi, F.R. Bordetella pertussis, the causative agent of whooping cough, evolved from a distinct, human-associated lineage of B. bronchiseptica. PLoS Pathog. 2005, 1, e45. [Google Scholar] [CrossRef] [PubMed]
- Mooi, F.R. Bordetella pertussis and vaccination: The persistence of a genetically monomorphic pathogen. Infect. Genet. Evol. 2010, 10, 36–49. [Google Scholar] [CrossRef] [PubMed]
- Hegerle, N.; Paris, A.S.; Brun, D.; Dore, G.; Njamkepo, E.; Guillot, S.; Guiso, N. Evolution of French Bordetella pertussis and Bordetella parapertussis isolates: Increase of Bordetellae not expressing pertactin. Clin. Microbiol. Infect. 2012, 18, E340–E346. [Google Scholar] [CrossRef] [PubMed]
- Mooi, F.R.; van der Maas, N.A.; de Melker, H.E. Pertussis resurgence: Waning immunity and pathogen adaptation—Two sides of the same coin. Epidemiol. Infect. 2014, 142, 685–694. [Google Scholar] [CrossRef] [PubMed]
- Van Loo, I.H.; Heuvelman, K.J.; King, A.J.; Mooi, F.R. Multilocus sequence typing of Bordetella pertussis based on surface protein genes. J. Clin. Microbiol. 2002, 40, 1994–2001. [Google Scholar] [CrossRef] [PubMed]
- Chenal-Francisque, V.; Caro, V.; Boursaux-Eude, C.; Guiso, N. Genomic analysis of the adenylate cyclase-hemolysin C-terminal region of Bordetella pertussis, Bordetella parapertussis and Bordetella bronchiseptica. Res. Microbiol. 2009, 160, 330–336. [Google Scholar] [CrossRef] [PubMed]
- Bart, M.J.; van Gent, M.; van der Heide, H.G.; Boekhorst, J.; Hermans, P.; Parkhill, J.; Mooi, F.R. Comparative genomics of prevaccination and modern Bordetella pertussis strains. BMC Genom. 2010. [Google Scholar] [CrossRef] [PubMed]
- Van Gent, M.; Bart, M.J.; van der Heide, H.G.; Heuvelman, K.J.; Mooi, F.R. Small mutations in Bordetella pertussis are associated with selective sweeps. PLoS ONE 2012, 7, e46407. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Zhang, Y.; Buboltz, A.M.; Zhang, X.; Schuster, S.C.; Ahuja, U.; Liu, M.; Miller, J.F.; Sebaihia, M.; Bentley, S.D.; et al. Comparative genomics of the classical Bordetella subspecies: The evolution and exchange of virulence-associated diversity amongst closely related pathogens. BMC Genom. 2012. [Google Scholar] [CrossRef] [PubMed]
- Bart, M.J.; Harris, S.R.; Advani, A.; Arakawa, Y.; Bottero, D.; Bouchez, V.; Cassiday, P.K.; Chiang, C.S.; Dalby, T.; Fry, N.K.; et al. Global population structure and evolution of Bordetella pertussis and their relationship with vaccination. MBIO 2014, 5, e01074. [Google Scholar] [CrossRef] [PubMed]
- Hegerle, N.; Guiso, N. Bordetella pertussis and pertactin-deficient clinical isolates: Lessons for pertussis vaccines. Expert Rev. Vaccines 2014, 13, 1135–1146. [Google Scholar] [CrossRef] [PubMed]
- Bouchez, V.; Brun, D.; Cantinelli, T.; Dore, G.; Njamkepo, E.; Guiso, N. First report and detailed characterization of B. pertussis isolates not expressing Pertussis Toxin or Pertactin. Vaccine 2009, 27, 6034–6041. [Google Scholar] [CrossRef] [PubMed]
- Bodilis, H.; Guiso, N. Virulence of pertactin-negative Bordetella pertussis isolates from infants, France. Emerg. Infect. Dis. 2013, 19, 471–474. [Google Scholar] [CrossRef] [PubMed]
- Hegerle, N.; Guiso, N. Antibody-mediated inhibition of Bordetella pertussis adenylate cyclase-haemolysin-induced macrophage cytotoxicity is influenced by variations in the bacterial population. Microbiol. (Reading Engl.) 2014, 160, 962–969. [Google Scholar] [CrossRef] [PubMed]
- SMartin, W.; Pawloski, L.; Williams, M.; Weening, K.; DeBolt, C.; Qin, X.; Reynolds, L.; Kenyon, C.; Giambrone, G.; Kudish, K.; et al. Pertactin-negative Bordetella pertussis strains: Evidence for a possible selective advantage. Clin. Infect. Dis. 2015, 60, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Stainer, D.W.; Scholte, M.J. A simple chemically defined medium for the production of phase I Bordetella pertussis. J. Gen. Microbiol. 1970, 63, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Van de Waterbeemd, B.; Streefland, M.; Pennings, J.; van der Pol, L.; Beuvery, C.; Tramper, J.; Martens, D. Gene-expression-based quality scores indicate optimal harvest point in Bordetella pertussis cultivation for vaccine production. Biotechnol. Bioeng. 2009, 103, 900–908. [Google Scholar] [CrossRef] [PubMed]
- Joly, N.; Biology IT Center, Institute Pasteur, France. Unpublished tools. 2015.
- Rio, D.C.; Ares, M., Jr.; Hannon, G.J.; Nilsen, T.W. Purification of RNA using TRIzol (TRI reagent). Cold Spring Harb. Protoc. 2010. [Google Scholar] [CrossRef] [PubMed]
- Pfaffl, M.W. The ongoing evolution of qPCR. Methods (San Diego Calif) 2010, 50, 215–216. [Google Scholar] [CrossRef] [PubMed]
- R Development Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2012. [Google Scholar]
- Bouchez, V.; Institut Pasteur, Paris, France. Unpublished data. 2015.
- Caro, V.; Bouchez, V.; Guiso, N. Is the Sequenced Bordetella pertussis strain Tohama I representative of the species? J. Clin. Microbiol. 2008, 46, 2125–2128. [Google Scholar] [CrossRef] [PubMed]
- De Gouw, D.; Diavatopoulos, D.A.; Bootsma, H.J.; Hermans, P.W.; Mooi, F.R. Pertussis: A matter of immune modulation. FEMS Microbiol. Rev. 2011, 35, 441–474. [Google Scholar] [CrossRef] [PubMed]
- Sealey, K.L.; Harris, S.R.; Fry, N.K.; Hurst, L.D.; Gorringe, A.R.; Parkhill, J.; Preston, A. Genomic analysis of isolates from the United Kingdom 2012 pertussis outbreak reveals That vaccine antigen genes are unusually fast evolving. J. Infect. Dis. 2015, 212, 294–301. [Google Scholar] [CrossRef] [PubMed]
- van Gent, M.; Bart, M.J.; van der Heide, H.G.; Heuvelman, K.J.; Kallonen, T.; He, Q.; Mertsola, J.; Advani, A.; Hallander, H.O.; Janssens, K.; et al. SNP-based typing: A useful tool to study Bordetella pertussis populations. PLoS ONE 2011, 6, e20340. [Google Scholar] [CrossRef] [PubMed]
- Tsang, R.S.; Shuel, M.; Jamieson, F.B.; Drews, S.; Hoang, L.; Horsman, G.; Lefebvre, B.; Desai, S.; St-Laurent, M. Pertactin-negative Bordetella pertussis strains in Canada: Characterization of a dozen isolates based on a survey of 224 samples collected in different parts of the country over the last 20 years. Int. J. Infect. Dis. 2014, 28, 65–69. [Google Scholar] [CrossRef] [PubMed]
- Zeddeman, A.; van Gent, M.; Heuvelman, C.J.; van der Heide, H.G.; Bart, M.J.; Advani, A.; Hallander, H.O.; Wirsing von Konig, C.H.; Riffelman, M.; Storsaeter, J.; et al. Investigations into the emergence of pertactin-deficient Bordetella pertussis isolates in six European countries, 1996 to 2012. Euro Surveill. Bull. Eur. Mal. Transm. 2014, 19, 17–27. [Google Scholar] [CrossRef]
- Bowden, K.E.; Williams, M.M.; Cassiday, P.K.; Milton, A.; Pawloski, L.; Harrison, M.; Martin, S.W.; Meyer, S.; Qin, X.; de Bolt, C.; et al. Molecular epidemiology of the pertussis epidemic in Washington State in 2012. J. Clin. Microbiol. 2014, 52, 3549–3557. [Google Scholar] [CrossRef] [PubMed]
- Schmidtke, A.J.; Boney, K.O.; Martin, S.W.; Skoff, T.H.; Tondella, M.L.; Tatti, K.M. Population diversity among Bordetella pertussis isolates, United States, 1935–2009. Emerg. Infect. Dis. 2012, 18, 1248–1255. [Google Scholar] [CrossRef] [PubMed]
- King, A.J.; Berbers, G.; van Oirschot, H.F.; Hoogerhout, P.; Knipping, K.; Mooi, F.R. Role of the polymorphic region 1 of the Bordetella pertussis protein pertactin in immunity. Microbiol. (Reading Engl.) 2001, 147, 2885–2895. [Google Scholar] [CrossRef] [PubMed]
- King, A.J.; van der Lee, S.; Mohangoo, A.; van Gent, M.; van der Ark, A.; van de Waterbeemd, B. Genome-wide gene expression analysis of Bordetella pertussis isolates associated with a resurgence in pertussis: Elucidation of factors involved in the increased fitness of epidemic strains. PLoS ONE 2013, 8, e66150. [Google Scholar] [CrossRef] [PubMed]
- De Gouw, D.; Hermans, P.W.; Bootsma, H.J.; Zomer, A.; Heuvelman, K.; Diavatopoulos, D.A.; Mooi, F.R. Differentially expressed genes in Bordetella pertussis strains belonging to a lineage which recently spread globally. PLoS ONE 2014, 9, e84523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mooi, F.R.; van Loo, I.H.; van Gent, M.; He, Q.; Bart, M.J.; Heuvelman, K.J.; de Greeff, S.C.; Diavatopoulos, D.; Teunis, P.; Nagelkerke, N.; et al. Bordetella pertussis strains with increased toxin production associated with pertussis resurgence. Emerg. Infect. Dis. 2009, 15, 1206–1213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stibitz, S. The BVG Regulon. In Bordetella Molecular Microbiology; Locht, C., Ed.; Horizon Bioscience: Norfolk, UK, 2007; pp. 47–67. [Google Scholar]
- Bibova, I.; Skopova, K.; Masin, J.; Cerny, O.; Hot, D.; Sebo, P.; Vecerek, B. The RNA chaperone Hfq is required for virulence of Bordetella pertussis. Infect. Immun. 2013, 81, 4081–4090. [Google Scholar] [CrossRef] [PubMed]
- Bibova, I.; Hot, D.; Keidel, K.; Amman, F.; Slupek, S.; Cerny, O.; Gross, R.; Vecerek, B. Transcriptional profiling of Bordetella pertussis reveals requirement of RNA chaperone Hfq for Type III secretion system functionality. RNA Biol. 2015, 12, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Hot, D.; Slupek, S.; Wulbrecht, B.; D’Hondt, A.; Hubans, C.; Antoine, R.; Locht, C.; Lemoine, Y. Detection of small RNAs in Bordetella pertussis and identification of a novel repeated genetic element. BMC Genom. 2011. [Google Scholar] [CrossRef] [PubMed]
- Leininger, E.; Ewanowich, C.A.; Bhargava, A.; Peppler, M.S.; Kenimer, J.G.; Brennan, M.J. Comparative roles of the Arg-Gly-Asp sequence present in the Bordetella pertussis adhesins pertactin and filamentous hemagglutinin. Infect. Immun. 1992, 60, 2380–2385. [Google Scholar] [PubMed]
- Inatsuka, C.S.; Xu, Q.; Vujkovic-Cvijin, I.; Wong, S.; Stibitz, S.; Miller, J.F.; Cotter, P.A. Pertactin is required for Bordetella species to resist neutrophil-mediated clearance. Infection and immunity 2010, 78, 2901–2909. [Google Scholar] [CrossRef] [PubMed]
- Stefanelli, P.; Fazio, C.; Fedele, G.; Spensieri, F.; Ausiello, C.M.; Mastrantonio, P. A natural pertactin deficient strain of Bordetella pertussis shows improved entry in human monocyte-derived dendritic cells. New Microbiol. 2009, 32, 159–166. [Google Scholar] [PubMed]
- Bassinet, L.; Gueirard, P.; Maitre, B.; Housset, B.; Gounon, P.; Guiso, N. Role of adhesins and toxins in invasion of human tracheal epithelial cells by Bordetella pertussis. Infect. Immun. 2000, 68, 1934–1941. [Google Scholar] [CrossRef] [PubMed]
- Lamberti, Y.; Gorgojo, J.; Massillo, C.; Rodriguez, M.E. Bordetella pertussis entry into respiratory epithelial cells and intracellular survival. Pathog. Dis. 2013, 69, 194–204. [Google Scholar] [CrossRef] [PubMed]
- Biological diagnosis of whooping cough by isolation of the Bordet-Gengou bacillus (H. pertussis) on modified Bordet medium. Ann. Biol. Clin. 1950, 8, 533–534.
- Merkel, T.J.; Halperin, S.A. Nonhuman primate and human challenge models of pertussis. J. Infect. Dis. 2014, 209, S20–S23. [Google Scholar] [CrossRef] [PubMed]
- Ross, P.J.; Sutton, C.E.; Higgins, S.; Allen, A.C.; Walsh, K.; Misiak, A.; Lavelle, E.C.; McLoughlin, R.M.; Mills, K.H. Relative contribution of Th1 and Th17 cells in adaptive immunity to Bordetella pertussis: Towards the rational design of an improved acellular pertussis vaccine. PLoS Pathog. 2013, 9, e1003264. [Google Scholar] [CrossRef] [PubMed]
- Boursaux-Eude, C.; Thiberge, S.; Carletti, G.; Guiso, N. Intranasal murine model of Bordetella pertussis infection: II. Sequence variation and protection induced by a tricomponent acellular vaccine. Vaccine 1999, 17, 2651–2660. [Google Scholar] [CrossRef]
- Denoel, P.; Godfroid, F.; Guiso, N.; Hallander, H.; Poolman, J. Comparison of acellular pertussis vaccines-induced immunity against infection due to Bordetella pertussis variant isolates in a mouse model. Vaccine 2005, 23, 5333–5341. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bouchez, V.; Hegerle, N.; Strati, F.; Njamkepo, E.; Guiso, N. New Data on Vaccine Antigen Deficient Bordetella pertussis Isolates. Vaccines 2015, 3, 751-770. https://doi.org/10.3390/vaccines3030751
Bouchez V, Hegerle N, Strati F, Njamkepo E, Guiso N. New Data on Vaccine Antigen Deficient Bordetella pertussis Isolates. Vaccines. 2015; 3(3):751-770. https://doi.org/10.3390/vaccines3030751
Chicago/Turabian StyleBouchez, Valérie, Nicolas Hegerle, Francesco Strati, Elisabeth Njamkepo, and Nicole Guiso. 2015. "New Data on Vaccine Antigen Deficient Bordetella pertussis Isolates" Vaccines 3, no. 3: 751-770. https://doi.org/10.3390/vaccines3030751
APA StyleBouchez, V., Hegerle, N., Strati, F., Njamkepo, E., & Guiso, N. (2015). New Data on Vaccine Antigen Deficient Bordetella pertussis Isolates. Vaccines, 3(3), 751-770. https://doi.org/10.3390/vaccines3030751