
Therapeutic In Situ Cancer Vaccine Using Pulsed Stereotactic Body Radiotherapy—A Translational Model


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. The Proposed Translational Hypothesis
2.1. Definition and Components of the Proposed Hypothesis
2.2. Evidence for the Proposed Hypothesis
2.2.1. Limited Clinical Data
2.2.2. Pulsed RT Approach—Preclinical Studies
3. Basis for the Proposed Hypothesis
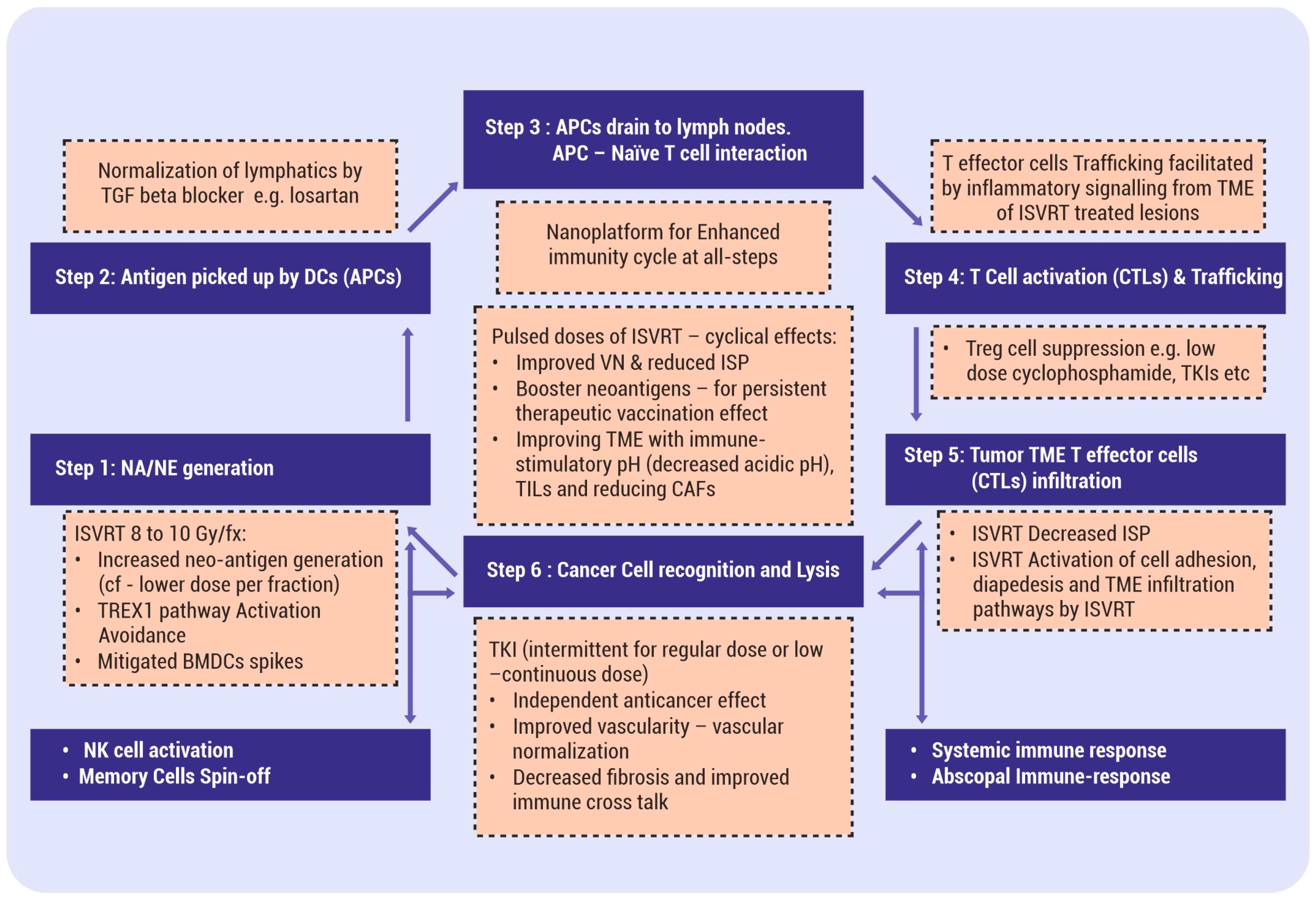
3.1. SBRT-Induced Immunity Cycle
3.1.1. SBRT In Situ Mechanism of Actions
3.1.2. Single-Dose SBRT Immunological Effects
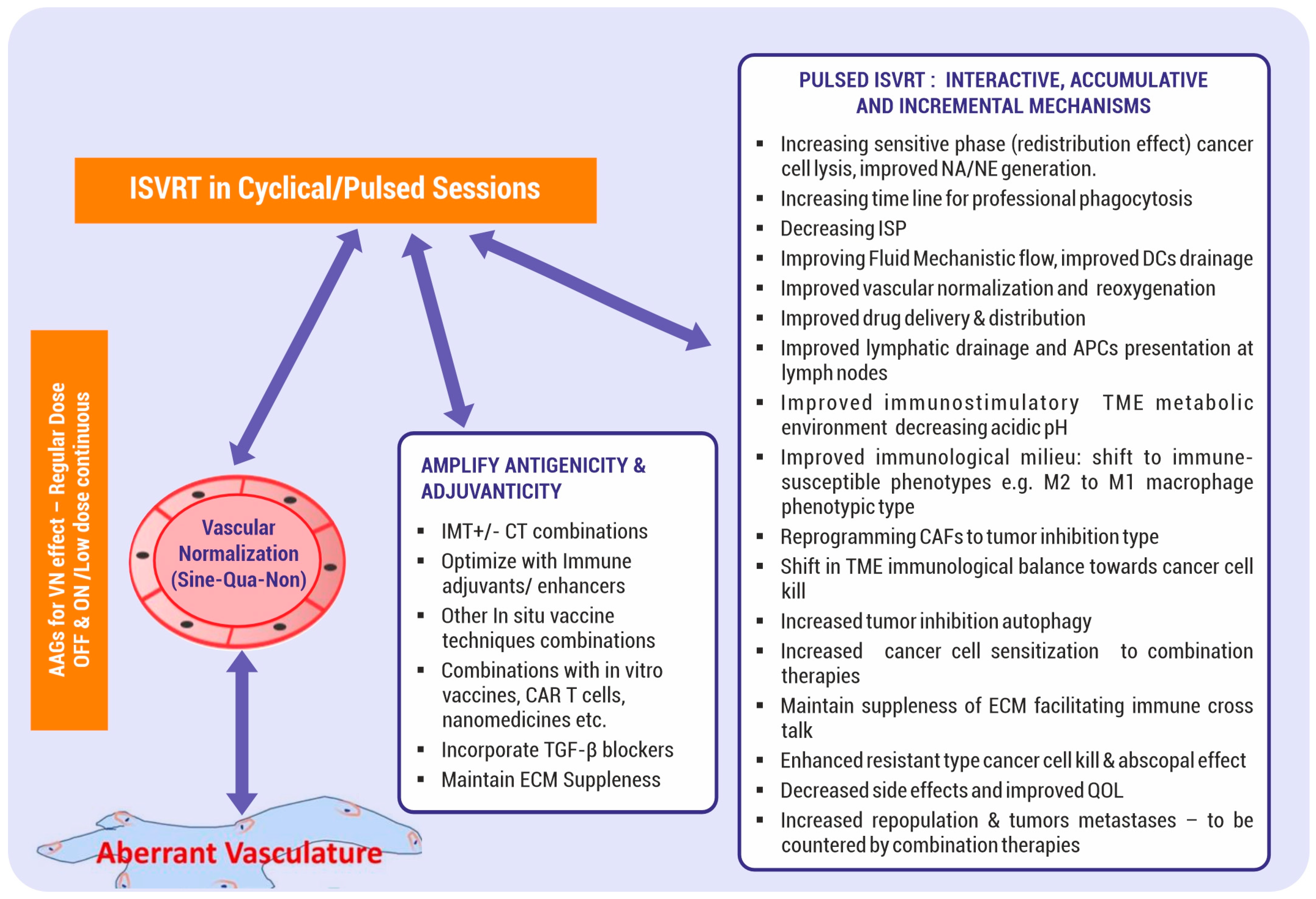
3.1.3. SBRT Immunological Effect of a Pulsed Dose
3.1.4. Single-Dose SBRT Palliative Effects
3.2. Improved Phagocytosis, Interstitial Pressure, and Lymphatic Vessel Drainage after SBRT
3.2.1. Vascular Normalization
3.2.2. The Mechanical TME
3.2.3. Essentiality of Functional Lymphatics (Not Just the UnCollapsing of the Lymphatics)
3.3. Arming of APCs
3.4. APCs—T Cell Interaction—Activation of T Cells
3.5. Cytotoxic Lymphocytes (CTLs)’ Formation and Trafficking—The Role of SBRT
3.6. SBRT-Assisted In Situ TIL Infiltration
4. Factors Mitigating the SBRT In Situ Response
4.1. Hypoxia/Anoxia
Overcoming Hypoxia
4.2. Immunosuppressive Recoil—Tregs, BMDCs
4.2.1. Importance of Concomitant Immune Tolerance from Untreated Lesions through RT
4.2.2. Overcoming Tregs and BMDCs
4.3. Activation of the Transcription and Export (TREX)1 Pathway
Preventing TREX1 Pathway Recoil
4.4. Other Immunosuppressive Recoil Pathways
Overcoming Other Immunosuppressive Pathways
4.5. Summary of Overcoming the Immunosuppressive Pathways
5. Subclinical Disease, Dormancy, ECM Suppleness, Memory Cell Cycle, and the Abscopal Cycle
5.1. Memory Cell Cycle
5.2. The Role of NK Cells in Minimal Disease and Dormancy
6. Amplification of the Antigenicity and Adjuvanticity of SBRT
6.1. Immunotherapy (+/− CT/AAGs) Complementing the In Situ Vaccination Effect of SBRT
6.1.1. Immunological Evolution of a Cancer Mass
6.1.2. The Difference That SBRT Can Make
6.1.3. Newer Avenues of RT and CAR-T Adoptive Cell Therapy
6.1.4. SBRT–IMT Timing
6.2. Further Points about Enhancing the Antigenicity and Adjuvanticity of SBRT
6.2.1. Alternative RT Therapies
6.2.2. Potential Combinations of Other In Situ Techniques with SBRT
6.2.3. Other Local Therapies
6.2.4. Making Use of Autophagy
7. Note on Minimizing Toxicities
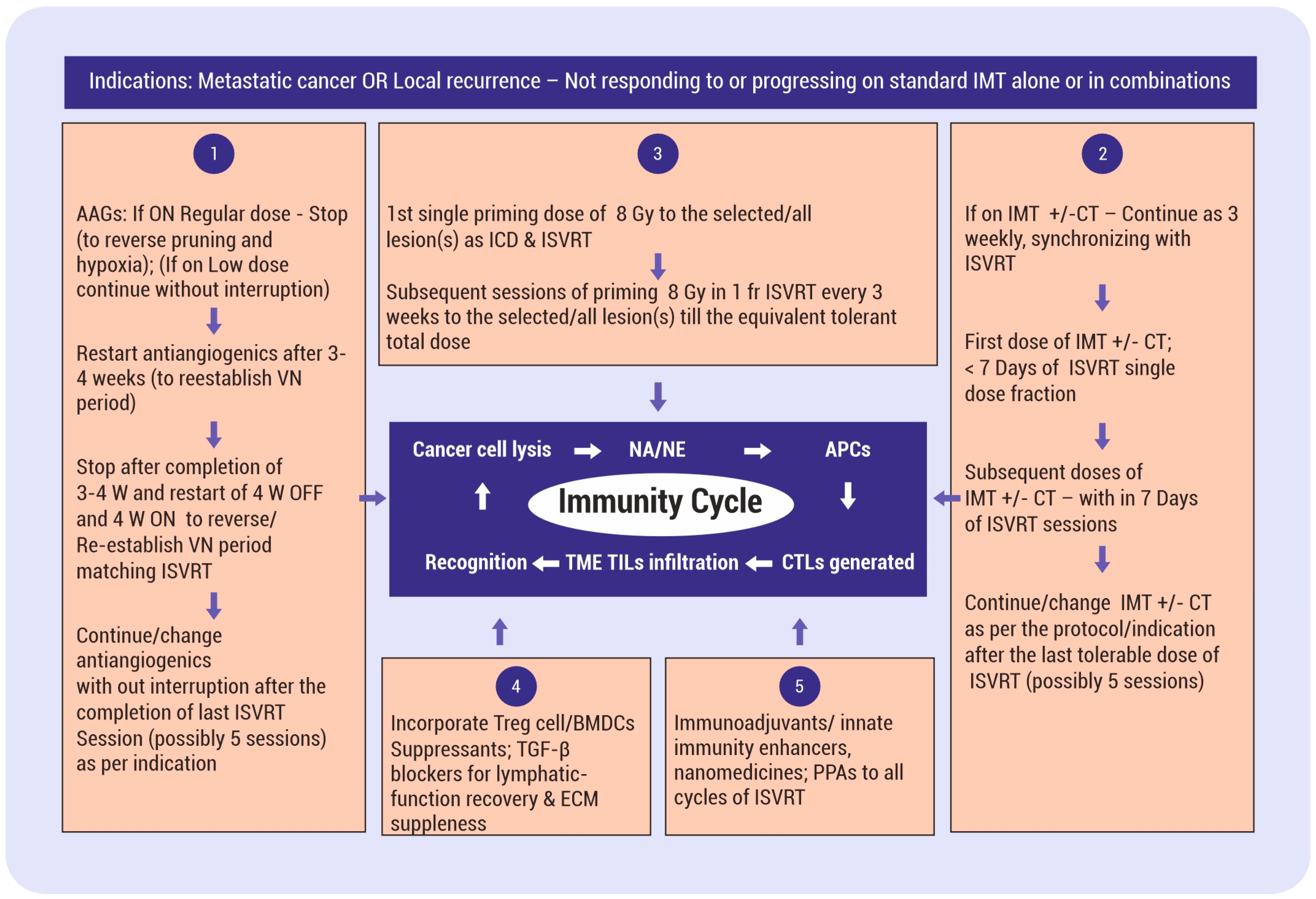
8. Hypothesis Validation Algorithm for Pulsed/Cyclical/Intermittent ISVRT with Supporting Stratagems
8.1. The Indications
8.2. Incorporate Antiangiogenics for Vascular Normalization
8.3. ISVRT Pulsed Schedule for In Situ Vaccine Effect
8.4. Integrating IMT
8.5. ISVRT Immunological Manipulations
8.6. ISVRT Immune Adjuvants Combinations
8.7. ISVRT versus Trunk and Branch Mutations
8.8. Sequencing Surgery
8.9. Criticality of ECM Suppleness
8.10. ISVRT Countering Accelerated Repopulation
8.11. Novel Unconventional and Other Radiotherapy Techniques
9. Summary and Limitations of the Article
Funding
Conflicts of Interest
References
- Bodey, B.; Bodey, B., Jr.; Siegel, S.E.; Kaiser, H.E. Failure of cancer vaccines: The significant limitations of this approach to IMT. Anticancer. Res. 2000, 20, 2665–2676. [Google Scholar] [PubMed]
- Kaczmarek, M.; Poznańska, J.; Fechner, F.; Michalska, N.; Paszkowska, S.; Napierała, A.; Mackiewicz, A. Cancer Vaccine Therapeutics: Limitations and Effectiveness—A Literature Review. Cells 2023, 12, 2159. [Google Scholar] [CrossRef] [PubMed]
- Mansoori BMohammadi, A.; Davudian, S.; Shirjang, S.; Baradaran, B. The Different Mechanisms of Cancer Drug Resistance: A Brief Review. Adv. Pharm. Bull. 2017, 7, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Cremers, C.G.; Nguyen, L.K. Network rewiring, adaptive resistance and combating strategies in breast cancer. Cancer Drug Resist. 2019, 2, 1106–1126. [Google Scholar] [CrossRef] [PubMed]
- Formenti, S.C.; Demaria, S. Radiotherapy to convert the tumor into an in-situ vaccine. J. Radiat. Res. 2012, 53, 545–556. [Google Scholar] [CrossRef][Green Version]
- Hammerich, L.; Binder, A.; Brody, J.D. In-situ vaccination: Cancer IMT both personalized and off-the-shelf. Mol. Oncol. 2015, 9, 1966–1981. [Google Scholar] [CrossRef] [PubMed]
- Ko, E.C.; Benjamin, K.T.; Formenti, S.C. Generating antitumor immunity by targeted radiation therapy: Role of dose and fractionation. Adv. Radiat. Oncol. 2018, 3, 486–493. [Google Scholar] [CrossRef]
- Schumacher, T.N.; Schreiber, R.D. Neo-antigens in cancer IMT. Science 2015, 348, 69–74. [Google Scholar] [CrossRef]
- Swamy, K. Vascular-immuno-phenotypic (VIP) model for locally advanced and oligo-metastatic cancer: A hypothesis. Med. Hypotheses 2021, 152, 110618. [Google Scholar] [CrossRef]
- Swamy, K. Stereotactic Body Radiotherapy Immunological Planning—A Review With a Proposed Theoretical Model. Front. Oncol. 2022, 12, 729250. [Google Scholar] [CrossRef]
- Swamy, K. Vascular normalization and IMT: Spawning a virtuous cycle. Front. Oncol. 2022, 12, 1002957. [Google Scholar] [CrossRef] [PubMed]
- Goedegebuure, R.S.A.; de Klerk, L.K.; Bass, A.J.; Derks, S.; Thijssen, V.L.J.L. Combining radiotherapy with anti-angiogenic therapy and IMT; a therapeutic triad for cancer? Front. Immunol. 2019, 9, 3107. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Zubiaurre, I.; Chalmers, A.J.; Hellevik, T. Radiation-induced transformation of immunoregulatory networks in the tumor stroma. Front. Immunol. 2018, 9, 167. [Google Scholar] [CrossRef] [PubMed]
- Koustas, E.; Trifylli, E.-M.; Sarantis, P.; Papadopoulos, N.; Papanikolopoulos, K.; Aloizos, G.; Damaskos, C.; Garmpis, N.; Garmpi, A.; Matthaios, D.; et al. Exploiting Autophagy-Dependent Neoantigen Presentation in Tumor Microenvironment. Genes 2023, 14, 474. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Mellman, I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Wegiel, B.; Vuerich, M.; Daneshmandi, S.; Seth, P. Metabolic switch in the tumor microenvironment determines immune responses to anticancer therapy. Front. Oncol. 2018, 8, 284. [Google Scholar] [CrossRef] [PubMed]
- Soyfer, V.; Geva, R.; Michelson, M.; Inbar, M.; Shacham-Shmueli, E.; Corn, B.W. The impact of overall radiotherapy treatment time and delay in initiation of radiotherapy on local control and distant metastases in gastric cancer. Radiat. Oncol. 2014, 9, 81. [Google Scholar] [CrossRef]
- Sanz, J.; Zhao, M.; Rodríguez, N.; Granado, R.; Foro, P.; Reig, A.; Membrive, I.; Algara, M. Once-Weekly Hypofractionated Radiotherapy for Breast Cancer in Elderly Patients: Efficacy and Tolerance in 486 Patients. BioMed. Res. Int. 2018, 2018, 8321871. [Google Scholar] [CrossRef]
- Sezen, D.; Barsoumian, H.B.; He, K.; Hu, Y.; Wang, Q.; Abana, C.O.; Puebla-Osorio, N.; Hsu, E.Y.; Wasley, M.; Masrorpour, F.; et al. ISVRT radıotherapy to mıtıgate hıgh tumor burden and generate ımmune memory. Front. Immunol. 2022, 13, 984318. [Google Scholar] [CrossRef]
- Moore, C.; Hsu, C.C.; Chen, W.M.; Chen, B.P.C.; Han, C.; Story, M.; Aguilera, T.; Pop, L.M.; Hannan, R.; Fu, Y.-X.; et al. Personalized ultrafractionated stereotactic adaptive radiotherapy (PULSAR) in preclinical models enhances single-agent immune checkpoint blockade. Int. J. Radiat. Oncol. Biol. Phys. 2021, 110, 1306–1316. [Google Scholar] [CrossRef]
- He, K.; Barsoumian, H.B.; Sezen, D.; Puebla-Osorio, N.; Hsu, E.Y.; Verma, V.; Abana, C.O.; Chen, D.; Patel, R.R.; Gu, M.; et al. ISVRT radiation therapy to improve systemic control of metastatic cancer. Front. Oncol. 2021, 11, 737425. [Google Scholar] [CrossRef] [PubMed]
- Vanpouille-Box, C.; Pilones, K.A.; Wennerberg, E.; Formenti, S.C.; Demaria, S. In-situ vaccination by radiotherapy to improve responses to anti-CTLA-4 treatment. Vaccine 2015, 33, 7415–7422. [Google Scholar] [CrossRef] [PubMed]
- Richard, G.; Princiotta, M.F.; Bridona, D.; Martin, W.D.; Steinberg, G.D.; De Groot, A.S. Neoantigen-based personalized cancer vaccines: The emergence of precision cancer immunotherapy. Expert Rev. Vaccines 2022, 21, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Rock, K.L.; Reits, E.; Neefjes, J. Present Yourself! By MHC Class I and MHC Class II Molecules. Trends Immunol. 2016, 37, 724–737. [Google Scholar] [CrossRef] [PubMed]
- Ko, E.C.; Formenti, S.C. Radiation therapy to enhance tumor IMT: A novel application for an established modality. Int. J. Radiat. Oncol. Biol. Phys. 2019, 104, 3–4. [Google Scholar] [CrossRef]
- Ahmed, M.M.; Hodge, J.W.; Guha, C.; Bernhard, E.J.; Vikram, B.; Coleman, C.N. Harnessing the potential of radiation-induced immune modulation for cancer therapy. Cancer Immunol. Res. 2013, 1, 280–284. [Google Scholar] [CrossRef]
- Sharabi, A.B.; Nirschl, C.J.; Kochel, C.M.; Nirschl, T.R.; Francica, B.J.; Velarde, E.; Deweese, T.L.; Drake, C.G. Stereotactic radiation therapy augments antigen-specific PD-1-mediated antitumor immune responses via cross-presentation of tumor antigen. Cancer Immunol. Res. 2015, 3, 345–355. [Google Scholar] [CrossRef]
- He, Q.; Jiang, X.; Zhou, X.; Weng, J. Targeting cancers through TCR-peptide/MHC interactions. J. Hematol. Oncol. 2019, 12, 139. [Google Scholar] [CrossRef]
- Appleton, E.; Hassan, J.; Hak, C.C.W.; Sivamanoharan, N.; Wilkins, A.; Samson, A.; Ono, M.; Harrington, K.J.; Melcher, A.; Wennerberg, E. Kickstarting Immunity in Cold Tumours: Localised Tumour Therapy Combinations With Immune Checkpoint Blockade. Front. Immunol. 2021, 12, 754436. [Google Scholar] [CrossRef]
- Benoit, A.; Vogin, G.; Duhem, C.; Berchem, G.; Janji, B. Lighting Up the Fire in the Microenvironment of Cold Tumors: A Major Challenge to Improve Cancer IMT. Cells 2023, 12, 1787. [Google Scholar] [CrossRef]
- Lussier, D.M.; Alspach, E.; Ward, J.P.; Miceli, A.P.; Runci, D.; White, J.M.; Mpoy, C.; Arthur, C.D.; Kohlmiller, H.N.; Jacks, T.; et al. Radiation-induced neoantigens broaden the immunotherapeutic window of cancers with low mutational loads. Proc. Natl. Acad. Sci. USA 2022, 119, e2102611118. [Google Scholar] [CrossRef] [PubMed]
- Jagodinsky, J.C.; Morris, Z.S. Priming and propagating antitumor immunity: Focal hypofractionated radiation for in-situ vaccination and systemic targeted radionuclide theranostics for immunomodulation of tumor microenvironments. Semin. Radiat. Oncol. 2020, 30, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Moding, E.J.; Castle, K.D.; Perez, B.A.; Oh, P.; Min, H.D.; Norris, H.; Ma, Y.; Cardona, D.M.; Lee, C.-L.; Kirsch, D.G. Tumor cells, but not endothelial cells, mediate the eradication of primary sarcomas by stereotactic body radiation therapy. Sci. Transl. Med. 2015, 7, 278ra34. [Google Scholar] [CrossRef] [PubMed]
- Uppelschoten, J.M.; Wanders, S.L.; de Jong, J.M. Single-dose radiotherapy (6 Gy): Palliation in painful bone metastases. Radiother. Oncol. 1995, 36, 198–202. [Google Scholar] [CrossRef] [PubMed]
- Goel, S.; Duda, D.G.; Xu, L.; Munn, L.L.; Boucher, Y.; Fukumura, D.; Jain, R.K.; Riera-Domingo, C.; Audigé, A.; Granja, S.; et al. Normalization of the Vasculature for Treatment of Cancer and Other Diseases. Physiol. Rev. 2011, 91, 1071–1121. [Google Scholar] [CrossRef]
- Zhou, H. Functions and clinical significance of mechanical tumor microenvironment: Cancer cell sensing, mechanobiology, and metastasis. Cancer Commun. 2022, 42, 374–400. [Google Scholar] [CrossRef]
- Macià, I.; Garau, M. Radiobiology of stereotactic body radiation therapy (SBRT). Rep. Pract. Oncol. Radiother. 2017, 22, 86–95. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, X.; Chen, D.; Yu, J. Radiotherapy combined with IMT: The dawn of cancer treatment. Signal Transduct. Target. Ther. 2022, 7, 258. [Google Scholar] [CrossRef]
- Li, S.Y.; Guo, Y.L.; Tian, J.W.; Zhang, H.J.; Li, R.F.; Gong, P.; Yu, Z.L. Antitumor Strategies by Harnessing the Phagocytosis of Macrophages. Cancers 2023, 15, 2717. [Google Scholar] [CrossRef]
- Yu, W.B.; Ye, Z.H.; Chen, X.; Shi, J.J.; Lu, J.J. The development of small-molecule inhibitors targeting CD47. Drug Discov. Today 2021, 26, 561–568. [Google Scholar] [CrossRef]
- Lian, S.; Xie, R.; Ye, Y.; Lu, Y.; Cheng, Y.; Xie, X.; Li, S.; Jia, L. Dual blockage of both PD-L1 and CD47 enhances IMT against circulating tumor cells. Sci. Rep. 2019, 9, 4532. [Google Scholar] [CrossRef] [PubMed]
- Hagendoorn, J.; Tong, R.; Fukumura, D.; Lin, Q.; Lobo, J.; Padera, T.P.; Xu, L.; Kucherlapati, R.; Jain, R.K. Onset of abnormal blood and lymphatic vessel function and interstitial hypertension in early stages of carcinogenesis. Cancer Res. 2006, 66, 3360–3364. [Google Scholar] [CrossRef] [PubMed]
- Padera, T.P.; Stoll, B.R.; Tooredman, J.B.; Capen, D.; di Tomaso, E.; Jain, R.K. Pathology: Cancer cells compress intratumour vessels. Nature 2004, 427, 695. [Google Scholar] [CrossRef] [PubMed]
- Liao, S.; Liu, J.; Lin, P.; Shi, T.; Jain, R.K.; Xu, L. TGF- blockade controls ascites by preventing abnormalization of lymphatic vessels in orthotopic human ovarian carcinoma models. Clin. Cancer Res. 2011, 17, 1415–1424. [Google Scholar] [CrossRef] [PubMed]
- Avraham, T.; Daluvoy, S.; Zampell, J.; Yan, A.; Haviv, Y.S.; Rockson, S.G.; Mehrara, B.J. Blockade of transforming growth factor-beta1 accelerates lymphatic regeneration during wound repair. Am. J. Pathol. 2010, 177, 3202–3214. [Google Scholar] [CrossRef] [PubMed]
- Chi, A.; Nguyen, N.P. Mechanistic rationales for combining IMT with radiotherapy. Front. Immunol. 2023, 14, 1125905. [Google Scholar] [CrossRef]
- Kratky, W.; Reis e Sousa, C.; Oxenius, A.; Spörri, R. Direct activation of antigen-presenting cells is required for CD8+ T-cell priming and tumor vaccination. Proc. Natl. Acad. Sci. USA 2011, 108, 17414–17419. [Google Scholar] [CrossRef]
- Rotman, J.; Koster, B.D.; Jordanova, E.S.; Heeren, A.M.; de Gruijl, T.D. Unlocking the therapeutic potential of primary tumor-draining lymph nodes. Cancer Immunol. Immunother. 2019, 68, 1681–1688. [Google Scholar] [CrossRef]
- Buchwald, Z.S.; Nasti, T.H.; Lee, J.; Eberhardt, C.S.; Wieland, A.; Im, S.J.; Lawson, D.; Curran, W.; Ahmed, R.; Khan, M.K. Tumor-draining lymph node is important for a robust abscopal effect stimulated by radiotherapy. J. Immunother. Cancer. 2020, 8, e000867. [Google Scholar] [CrossRef]
- Liu, Z.; Yu, Z.; Chen, D.; Verma, V.; Yuan, C.; Wang, M.; Wang, F.; Fan, Q.; Wang, X.; Li, Y.; et al. Pivotal roles of tumor-draining lymph nodes in the abscopal effects from combined IMT and radiotherapy. Cancer Commun. 2022, 42, 971–986. [Google Scholar] [CrossRef]
- Baxevanis, C.N.; Gritzapis, A.D.; Voutsas, I.F.; Batsaki, P.; Goulielmaki, M.; Adamaki, M.; Zoumpourlis, V.; Fortis, S.P. T-Cell Repertoire in Tumor Radiation: The Emerging Frontier as a Radiotherapy Biomarker. Cancers 2022, 14, 2674. [Google Scholar] [CrossRef]
- Demaria, S.; Formenti, S.C. Role of T lymphocytes in tumor response to radiotherapy. Front. Oncol. 2012, 2, 95. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Dong, Y.; Kong, L.; Shi, F.; Zhu, H.; Yu, J. Abscopal effect of radiotherapy combined with immune checkpoint inhibitors. J. Hematol. Oncol. 2018, 11, 104. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Mowday, A.M.; Smaill, J.B.; Hermans, I.F.; Patterson, A.V. Tumour Hypoxia-Mediated Immunosuppression: Mechanisms and Therapeutic Approaches to Improve Cancer IMT. Cells 2021, 10, 1006. [Google Scholar] [CrossRef] [PubMed]
- Eckert, F.; Zwirner, K.; Boeke, S.; Thorwarth, D.; Zips, D.; Huber, S.M. Rationale for combining radiotherapy and immune checkpoint inhibition for patients with hypoxic tumors. Front. Immunol. 2019, 10, 407. [Google Scholar] [CrossRef] [PubMed]
- Milano, M.T.; Constine, L.S.; Okunieff, P. Normal tissue toxicity after small field hypofractionated stereotactic body radiation. Radiat. Oncol. 2008, 3, 36. [Google Scholar] [CrossRef]
- Withers, H.R. The Four R’s of Radiotherapy. In Advances in Radiation Biology; Elsevier: Amsterdam, The Netherlands, 1975; Volume 5, pp. 241–271. [Google Scholar] [CrossRef]
- Fowler, J.F. Rapid repopulation in radiotherapy: A debate on mechanism. The phantom of tumor treatment - continually rapid proliferation unmasked. Radiother. Oncol. 1991, 22, 156–158. [Google Scholar] [CrossRef]
- Willers, H.; Azzoli, C.G.; Santivasi, W.L.; Xia, F. Basic mechanisms of therapeutic resistance to radiation and CT in lung cancer. Cancer J. 2013, 19, 200–207. [Google Scholar] [CrossRef]
- Shibamoto, Y.; Miyakawa, A.; Otsuka, S.; Iwata, H. Radiobiology of hypofractionated stereotactic radiotherapy: What are the optimal fractionation schedules? J. Radiat. Res. 2016, 57, i76–i82. [Google Scholar] [CrossRef]
- Morris, Z.S.; Guy, E.I.; Francis, D.M.; Gressett, M.M.; Werner, L.R.; Carmichael, L.L.; Yang, R.K.; Armstrong, E.A.; Huang, S.; Navid, F.; et al. In-situ tumor vaccination by combining local radiation and tumor-specific antibody or immunocytokine treatments. Cancer Res. 2016, 76, 3929–3941. [Google Scholar] [CrossRef]
- Ohue, Y.; Nishikawa, H. Regulatory T (Treg) cells in cancer: Can Treg cells be a new therapeutic target? Cancer Sci. 2019, 110, 2080–2089. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Sakaguchi, S. Targeting Treg cells in cancer IMT. Eur. J. Immunol. 2019, 49, 1140–1146. [Google Scholar] [CrossRef] [PubMed]
- Darragh, L.B.; Oweida, A.J.; Karam, S.D. Overcoming resistance to combination radiation IMT: A focus on contributing pathways within the tumor microenvironment. Front. Immunol. 2019, 9, 3154. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Shi, Z.; Zhang, F.; Zeng, W.; Zhu, D.; Mei, L. Symphony of nanomaterials and IMT based on the cancer-immunity cycle. Acta Pharm. Sin. B 2022, 12, 107–134. [Google Scholar] [CrossRef] [PubMed]
- Baniel, C.C.; Heinze, C.M.; Hoefges, A.; Sumiec, E.G.; Hank, J.A.; Carlson, P.M.; Jin, W.J.; Patel, R.B.; Sriramaneni, R.N.; Gillies, S.D.; et al. In-situ vaccine plus checkpoint blockade induces Memory humoral response. Front. Immunol. 2020, 11, 1610. [Google Scholar] [CrossRef] [PubMed]
- Bald, T.; Krummel, M.F.; Smyth, M.J.; Barry, K.C. The NK cell-cancer cycle: Advances and new challenges in NK cell-based immunotherapies. Nat. Immunol. 2020, 21, 835–847. [Google Scholar] [CrossRef] [PubMed]
- Boopathi, E.; Den, R.B.; Thangavel, C. Innate Immune System in the Context of Radiation Therapy for Cancer. Cancers 2022, 15, 3972. [Google Scholar] [CrossRef] [PubMed]
- Lakatos, E.; Williams, M.J.; Schenck, R.O.; Cross, W.C.H.; Househam, J.; Zapata, L.; Werner, B.; Gatenbee, C.; Robertson-Tessi, M.; Barnes, C.P.; et al. Evolutionary dynamics of neoantigens in growing tumors. Nat. Genet. 2019, 51, 734–742. [Google Scholar] [CrossRef]
- Iliadi, C.; Verset, L.; Bouchart, C.; Martinive, P.; Gestel, D.V.; Mohammad Krayem, M. The current understanding of the immune landscape relative to radiotherapy across tumor types. Front. Immunol. 2023, 14, 1148692. [Google Scholar] [CrossRef]
- Boustani, J.; Grapin, M.; Laurent, P.A.; Apetoh, L.; Mirjolet, C. The 6th R of Radiobiology: Reactivation of Anti-Tumor Immune Response. Cancers 2019, 11, 860. [Google Scholar] [CrossRef]
- Wirth, T.C.; Kühnel, F. Neoantigen Targeting—Dawn of a New Era in CancerIMT? Front. Immunol. 2017, 8, 1848. [Google Scholar] [CrossRef] [PubMed]
- Nelson, B.E.; Adashek, J.J.; Sheth, A.A.; Subbiah, V. Predicting the Abscopal Effect: Associated Tumor Histologic Subtypes and Biomarkers. Mol. Cancer Ther. 2023, 22, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Takeshima, T.; Chamoto, K.; Wakita, D.; Ohkuri, T.; Togashi, Y.; Shirato, H.; Kitamura, H.; Nishimura, T. Local radiation therapy inhibits tumor growth through the generation of tumor-specific CTL: Its potentiation by combination with Th1 cell therapy. Cancer Res. 2010, 70, 2697–2706. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.; Li, Y.; Muluh, T.; Wang, Y. Combination of CAR-T cell therapy and radiotherapy: Opportunities and challenges in solid tumors (Review). Oncol. Lett. 2023, 25, 13867. [Google Scholar] [CrossRef] [PubMed]
- Jiaqiang Wang, J.; Ge, H.; Tian, Z. IMT Plus Radiotherapy for the Treatment of Sarcomas: Is There a Potential for Synergism? OncoTargets Ther. 2023, 16, 385–397. [Google Scholar] [CrossRef]
- Breen, W.G.; Leventakos, K.; Dong, H.; Merrell, K.W. Radiation and IMT: Emerging Mechanisms of Synergy. J. Thorac. Dis. 2020, 12 (Suppl. S1), S41–S51. [Google Scholar] [CrossRef]
- Lurje, I.; Werner, W.; Mohr, R.; Roderburg, C.; Tacke, F.; Hammerich, L. In Situ Vaccination as a Strategy to Modulate the Immune Microenvironment of Hepatocellular Carcinoma. Front. Immunol. 2021, 12, 650486. [Google Scholar] [CrossRef]
- Sheen, M.R.; Fiering, S. In-situ vaccination: Harvesting low-hanging fruit on the cancer IMT tree. WIREs Nanomed. Nanobiotechnol. 2018, 10, e1524. [Google Scholar] [CrossRef]
- Van Den Bijgaart, R.J.E.; Schuurmans, F.; Fütterer, J.J.; Verheij, M.; Cornelissen, L.A.M.; Adema, G.J. Immune modulation plus tumor ablation: Adjuvants and antibodies to prime and boost antitumor immunity in-situ. Front Immunol. 2021, 12, 617365. [Google Scholar] [CrossRef]
- Czapla, J.; Drzyzga, A.; Matuszczak, S.; Cichoń, T.; Rusin, M.; Jarosz-Biej, M.; Pilny, E.; Smolarczyk, R. Antitumor effect of anti-vascular therapy with STING agonist depends on the tumor microenvironment context. Front. Oncol. 2023, 13, 1249524. [Google Scholar] [CrossRef]
- Zhao, Y.; Yu, X.; Li, J. Manipulation of immune-vascular crosstalk: New strategies towards cancer treatment. Acta Pharm. Sin. B 2020, 10, 2018–2036. [Google Scholar] [CrossRef] [PubMed]
- Wirsdörfer, F.; De Leve, S.; Jendrossek, V. Combining radiotherapy and IMT in lung cancer: Can we expect limitations due to altered normal tissue toxicity? Int. J. Mol. Sci. 2019, 20, 24. [Google Scholar] [CrossRef] [PubMed]
- Tubin, S.; Popper, H.H.; Brcic, L. Novel stereotactic body radiation therapy (SBRT)-based partial tumor irradiation targeting hypoxic segment of bulky tumors (SBRT-PATHY): Improvement of the radiotherapy outcome by exploiting the bystander and abscopal effects. Radiat. Oncol. 2019, 14, 21. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, M.R.; Davis, R.; Norberg, S.M.; O’brien, S.; Sennino, B.; Nakahara, T.; Yao, V.J.; Inai, T.; Brooks, P.; Freimark, B.; et al. Rapid vascular regrowth in tumors after reversal of VEGF inhibition. J. Clin. Investig. 2006, 116, 2610–2621. [Google Scholar] [CrossRef] [PubMed]
- Fukumura, F.D.; Kloepper, J.; Amoozgar, Z.; Duda, D.G.; Jain, R.K. Enhancing cancer IMT using antiangiogenics: Opportunities and challenges. Nat. Rev. Clin. Oncol. 2018, 15, 325–340. [Google Scholar] [CrossRef] [PubMed]
- Fan, P.; Qiang, H.; Liu, Z.; Zhao, Q.; Wang, Y.; Liu, T.; Wang, X.; Chu, T.; Huang, Y.; Xu, W.; et al. Effective low-dose Anlotinib induces long-term tumor vascular normalization and improves anti-PD-1 therapy. Front. Immunol. 2022, 13, 937924. [Google Scholar] [CrossRef]
- Gao, Z.; Zhao, Q.; Xu, Y.; Wang, L. Improving the efficacy of combined radiotherapy and IMT: Focusing on the effects of radiosensitivity. Radiat. Oncol. 2023, 17, 89. [Google Scholar] [CrossRef]
- Wuensch, K.E.; Tan, A.C. Trunk or branch? Identifying and targeting intratumoral heterogeneity in hepatocellular carcinoma using genomics and patient-derived primary cancer cells. Transl. Cancer Res. 2017, 6 (Suppl. S5), S903–S915. [Google Scholar] [CrossRef]
- Gao, Q.; Wang, Z.C.; Duan, M.; Lin, Y.H.; Zhou, X.Y.; Worthley, D.L. Cell culture system for analysis of genetic heterogeneity within hepatocellular carcinoma and response to pharmacological agents. Gastroenterology 2017, 152, 232–242. [Google Scholar] [CrossRef]
- Kievit, H.; Muntinghe-Wagenaar, M.B.; Hijmering-Kappelle, L.B.M.; Hiddinga, B.I.; Ubbels, J.F.; Wijsman, R.; Slingers, G.; de Vries, R.; Groen, H.J.M.; Kerstjens, H.A.M.; et al. Safety and tolerability of stereotactic radiotherapy combined with durvalumab with or without tremelimumab in advanced non-small cell lung cancer, the phase I SICI trial. Lung Cancer 2023, 178, 96–102. [Google Scholar] [CrossRef]
- Martin, J.D.; Cabral, H.; Stylianopoulos, T.; Jain, R.K. Improving cancer IMT using nanomedicines: Progress, opportunities, and challenges. Nat. Rev. Clin. Oncol. 2020, 17, 251–266. [Google Scholar] [CrossRef]
- Joyce, J.A.; Pollard, J.W. Microenvironmental regulation of metastasis. Nat. Rev. Cancer. 2009, 9, 239–252. [Google Scholar] [CrossRef] [PubMed]
- Diop-Frimpong, B.; Chauhan, V.P.; Krane, S.; Boucher, Y.; Jain, R.K. Losartan inhibits collagen I synthesis and improves the distribution and efficacy of nanotherapeutics in tumors. Proc. Natl. Acad. Sci. USA 2011, 108, 2909–2914. [Google Scholar] [CrossRef] [PubMed]
- Iori, F.; Cappelli, A.; D’Angelo, E.; Cozzi, S.; Ghersi, S.F.; De Felice, F.; Ciammella, P.; Bruni, A.; Iotti, C. Lattice Radiation Therapy in clinical practice: A systematic review. Clin. Transl. Radiat. Oncol. 2022, 39, 100569. [Google Scholar] [CrossRef] [PubMed]
- Wittenstein, O.; Krause, F.; Fischer, M.; Domschikowski, J.; Nitsche, M.; Henkenberens, C.; Habermehl, D.; Dunst, J. The tumor core boost study: A feasibility study of radical dose escalation to the central part of large tumors with an integrated boost in the palliative treatment setting. Strahlenther. Onkol. 2023, 199, 258–267. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.; Li, Y.; Huang, W.; Lu, W.; Yi, X. Escalation of radiotherapy dose in large locally advanced drug-resistant gastrointestinal stromal tumors by multishell simultaneous integrated boost intensity-modulated technique: A feasibility study. Radiat. Oncol. 2022, 17, 216. [Google Scholar] [CrossRef] [PubMed]
- Tubin, S.; Vozenin, M.C.; Prezado, Y.; Durante, M.; Prise, K.M.; Lara, P.C.; Greco, C.; Massaccesi, M.; Guha, C.; Wu, X.; et al. Novel unconventional radiotherapy techniques: Current status and future perspectives—Report from the 2nd international radiation oncology online seminar. Clin. Transl. Radiat. Oncol. 2023, 40, 100605. [Google Scholar] [CrossRef]
- Marcus, D.; Lieverse, R.I.Y.; Klein, C.; Abdollahi, A.; Lambin, P.; Dubois, L.J.; Yaromina, A. Charged Particle and Conventional Radiotherapy: Current Implications as Partner for Immunotherapy. Cancers 2021, 13, 1468. [Google Scholar] [CrossRef]
- Hageman, E.; Che, P.P.; Dahele, M.; Slotman, B.J.; Sminia, P. Radiobiological Aspects of FLASH Radiotherapy. Biomolecules 2022, 12, 1376. [Google Scholar] [CrossRef]
- Morris, Z.S.; Guy, E.I.; Werner, L.R.; Carlson, P.M.; Heinze, C.M.; Kler, J.S.; Busche, S.M.; Jaquish, A.A.; Sriramaneni, R.N.; Carmichael, L.L.; et al. Tumor-specific inhibition of in-situ vaccination by distant untreated tumor sites. Cancer Immunol. Res. 2017, 5, 517–523. [Google Scholar] [CrossRef]
- Magirr, D.; Jaki, T.; Whitehead, J. A generalized Dunnett test for multi-arm multi-stage clinical studies with treatment selection. Biometrika 2012, 99, 494–501. [Google Scholar] [CrossRef]
- Guan, H.; Wu, Y.; Li, L.U.; Yang, Y.; Qiu, S.; Zhao, Z.; Chu, X.; He, J.; Chen, Z.; Zhang, Y.; et al. Tumor neoantigens: Novel strategies for application of cancer immunotherapy. Oncol. Res. 2023, 31, 437–448. [Google Scholar] [CrossRef] [PubMed]
- Swamy, K. Adoptive In Situ Vaccination with Cyclical Intermittent Stereotactic Body Radiotherapy (SBRT) and IMT—A Review and Proposed Strategy. Preprints 2023, 2023080648. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Swamy, K. Therapeutic In Situ Cancer Vaccine Using Pulsed Stereotactic Body Radiotherapy—A Translational Model. Vaccines 2024, 12, 7. https://doi.org/10.3390/vaccines12010007
Swamy K. Therapeutic In Situ Cancer Vaccine Using Pulsed Stereotactic Body Radiotherapy—A Translational Model. Vaccines. 2024; 12(1):7. https://doi.org/10.3390/vaccines12010007
Chicago/Turabian StyleSwamy, Kumara. 2024. "Therapeutic In Situ Cancer Vaccine Using Pulsed Stereotactic Body Radiotherapy—A Translational Model" Vaccines 12, no. 1: 7. https://doi.org/10.3390/vaccines12010007
APA StyleSwamy, K. (2024). Therapeutic In Situ Cancer Vaccine Using Pulsed Stereotactic Body Radiotherapy—A Translational Model. Vaccines, 12(1), 7. https://doi.org/10.3390/vaccines12010007