

Macrophages as a Potential Immunotherapeutic Target in Solid Cancers
 , ,
, ,
Abstract
1. Introduction
2. Origin and Phenotypic Plasticity of TAMs
3. Prognostic Significance of TAMs in Solid Tumors
4. Pro-Tumorigenic Roles of TAMs
4.1. TAM-Mediated Inflammation and Genomic Instability
4.2. Tumor Growth Promotion
4.3. Formation of Cancer Stem Cells (CSC)
4.4. Epithelial to Mesenchymal Transition (EMT) and Metastasis
4.5. Angiogenesis
4.6. Treatment Resistance
4.7. Fibrosis and Tissue Remodeling
4.8. Immunosuppression
4.8.1. Releasing Anti-Inflammatory Cytokines
4.8.2. Metabolic Reprogramming
4.8.3. Expressing Immune Checkpoints
5. Therapeutic Targeting of Macrophages
5.1. TAM Depletion
5.2. Inhibition of Macrophage Recruitment
5.3. Inhibition of M2 Polarization and Reprogramming
5.4. Targeting Programmed Cell Removal (PrCR) and Anti-Phagocytic Checkpoints
5.5. Macrophages-Mediated Cytokine Delivery
5.6. Macrophages-Based Adoptive Cell Transfusion
6. Challenges in Therapeutic Targeting of TAMs
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ADCC | antibody-dependent cellular cytotoxicity |
| ADCP | antibody-dependent cellular phagocytosis |
| AFAP1 | actin filament-associated protein 1 |
| Akt | alpha serine/threonine-protein kinase |
| APCs | antigen-presenting cells |
| ARG1 | arginase 1 |
| AS1 | antisense RNA 1 |
| ASK1 | apoptosis signal-regulating kinase 1 |
| ATF2 | activating transcription factor 2 |
| ATP | adenosine triphosphate |
| B2M | beta-2-microglobulin |
| B7S1 | B7 superfamily member 1 |
| bFGF | basic fibroblast growth factor |
| Btk | Bruton’s tyrosine kinase |
| CAF | cancer-associated fibroblasts |
| CAR | chimeric antigen receptor |
| CAR-Ms | chimeric antigen receptor macrophages |
| CCL | C-C chemokine ligand |
| CCL3 | chemokine (C-C motif) ligand 3 |
| CCR | C-C chemokine receptor |
| CD | cluster of differentiation |
| COX-2 | cyclooxygenase-2 |
| CSC | cancer stem cell |
| CSF | colony-stimulating factor |
| CSF-1R | colony-stimulating factor-1 receptor |
| CXCL | CXC chemokine ligand |
| DC | dendritic cell |
| EC | endothelial cell |
| ECM | extracellular matrix |
| EGF | epidermal growth factor |
| EGFR | epidermal growth factor receptor |
| EMT | epithelial–mesenchymal transition |
| EP4 | prostaglandin E2 receptor 4 |
| EphA4 | EPH Receptor A4 |
| ERK | extracellular signal-regulated kinase |
| EVs | extracellular vesicles |
| FAK | focal adhesion kinase |
| FGF2 | fibroblast growth factor 2 |
| Foxp3 | forkhead box P 3 |
| FRβ | folate receptor β |
| HCC | hepatocellular carcinoma |
| HDAC | histone deacetylase |
| HER2 | human epidermal growth factor receptor 2 |
| HIF-1α | hypoxia-inducible factor 1-alpha |
| HLA | human leukocyte antigen |
| HMGB1 | high mobility group box 1 |
| IFN | interferon |
| Ifna1 | interferon Alpha 1 |
| IGF | insulin-like growth factor |
| IL | interleukin |
| iNOS | inducible nitric oxide synthase |
| JAK1 | Janus kinase 1 |
| KLF3 | Krüppel-like factor 3 |
| LILRB1 | leukocyte immunoglobulin-like receptor subfamily B member 1 |
| Lin28b | Lin-28 Homolog B |
| LncRNA | long noncoding RNA |
| LPS | lipopolysaccharide |
| mABs | monoclonal antibodies |
| MAPK M-CSF | mitogen-activated protein kinase Macrophage colony-stimulating factor |
| MDSCs | myeloid-derived suppressor cells |
| MEK1 | mitogen-activated protein kinase kinase 1 |
| MERTKR | mer tyrosine-protein kinase receptor |
| MFG-E8 | milk fat globule-EGF factor 8 protein |
| Mgat5 | alpha-1,6-Mannosylglycoprotein 6-Beta-N-Acetylglucosaminyltransferase |
| MHC | major histocompatibility complex |
| MIF | migration inhibitory factor |
| miRNA | microRNA |
| MMP | matrix metalloproteinase |
| Mreg | regulatory macrophage |
| mTOR | mammalian target of rapamycin |
| MUC1 | mucin 1 |
| MVD | micro vessel density |
| NK | natural killer |
| NO | nitric oxide |
| NSCLC | non-small cell lung cancer |
| OCT3/4 | organic cation/carnitine transporter 3/4 |
| PAMP | pathogen-associated molecular pattern |
| PCX | paclitaxel |
| PD-1 | programmed cell death protein 1 |
| PDL1 | PD ligand 1 |
| PDAC | pancreatic ductal adenocarcinoma |
| PDGF | platelet-derived growth factor |
| PGE2 | prostaglandin E2 |
| PlGF | placental growth factor |
| PI3K | phosphoinositide 3-kinase |
| PrCR | programmed cell removal |
| RBCs | red blood cells |
| ROI | reactive oxygen intermediates |
| RT | radiotherapy |
| scRNA-seq | single-cell RNA sequencing |
| SIRPα | signal regulatory protein alpha |
| SISP1 | secreted phosphoprotein 1 |
| SMAD | suppressor of mothers against decapentaplegic |
| SOX2 | SRY-box transcription factor 2 |
| STAT3 | signal transducer and activator of transcription 3 |
| TAMs | tumor-associated macrophages |
| TCR | T-cell receptor |
| TGCT | tenosynovial giant cell tumors |
| TGF | tumor growth factor |
| TGF-β | transforming growth factor-β |
| TGF-βR | transforming growth factor-β receptor |
| Th | T helper cell |
| Tie2 | angiopoietin-1 receptor |
| TLR | toll-like receptor |
| TME | tumor microenvironment |
| TNBC | triple-negative breast cancer |
| TNF | tumor necrosis factor |
| TNFR | tumor necrosis factor receptor |
| TRAIL | TNF-related apoptosis-inducing ligand |
| Treg | regulatory T-cell |
| TREM | triggering receptor expressed on myeloid cells |
| VEGF | vascular endothelial growth factor |
| VISTA | V-domain Ig-containing suppressor of T-cell activation |
| WNT7b | Wnt Family Member 7B |
References
- Kaufmann, S.H.E. Immunology’s foundation: The 100-year anniversary of the Nobel Prize to Paul Ehrlich and Elie Metchnikoff. Nat. Immunol. 2008, 9, 705–712. [Google Scholar] [CrossRef] [PubMed]
- Rosales, C. The first 100 years of phagocytosis. In Phagocytosis: Overview, History and Role in Human Health and Disease; Nova Science Publishers, Inc.: New York, NY, USA, 2018. [Google Scholar]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef] [PubMed]
- Tamura, R.; Tanaka, T.; Yamamoto, Y.; Akasaki, Y.; Sasaki, H. Dual role of macrophage in tumor immunity. Immunotherapy 2018, 10, 899–909. [Google Scholar] [CrossRef] [PubMed]
- Cendrowicz, E.; Sas, Z.; Bremer, E.; Rygiel, T. The Role of Macrophages in Cancer Development and Therapy. Cancers 2021, 13, 1946. [Google Scholar] [CrossRef]
- Hirayama, D.; Iida, T.; Nakase, H. The Phagocytic Function of Macrophage-Enforcing Innate Immunity and Tissue Homeostasis. Int. J. Mol. Sci. 2018, 19, 92. [Google Scholar] [CrossRef]
- Mantovani, A. Macrophages, Neutrophils, and Cancer: A Double Edged Sword. New J. Sci. 2014, 2014, 1–14. [Google Scholar] [CrossRef]
- Cassetta, L.; Pollard, J.W. Targeting macrophages: Therapeutic approaches in cancer. Nat. Rev. Drug Discov. 2018, 17, 887–904. [Google Scholar] [CrossRef]
- Chen, Y.; Song, Y.; Du, W.; Gong, L.; Chang, H.; Zou, Z. Tumor-associated macrophages: An accomplice in solid tumor progression. J. Biomed. Sci. 2019, 26, 78. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Geng, X.; Hou, J.; Wu, G. New insights into M1/M2 macrophages: Key modulators in cancer progression. Cancer Cell Int. 2021, 21, 389. [Google Scholar] [CrossRef]
- Salmaninejad, A.; Valilou, S.F.; Soltani, A.; Ahmadi, S.; Abarghan, Y.J.; Rosengren, R.J.; Sahebkar, A. Tumor-associated macrophages: Role in cancer development and therapeutic implications. Cell. Oncol. 2019, 42, 591–608. [Google Scholar] [CrossRef]
- Cortese, N.; Carriero, R.; Laghi, L.; Mantovani, A.; Marchesi, F. Prognostic significance of tumor-associated macrophages: Past, present and future. Semin. Immunol. 2020, 48, 101408. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zhang, L.; Yang, L.; Li, H.; Li, R.; Yu, J.; Yang, L.; Wei, F.; Yan, C.; Sun, Q.; et al. Anti-CD47 Antibody As a Targeted Therapeutic Agent for Human Lung Cancer and Cancer Stem Cells. Front. Immunol. 2017, 8, 404. [Google Scholar] [CrossRef] [PubMed]
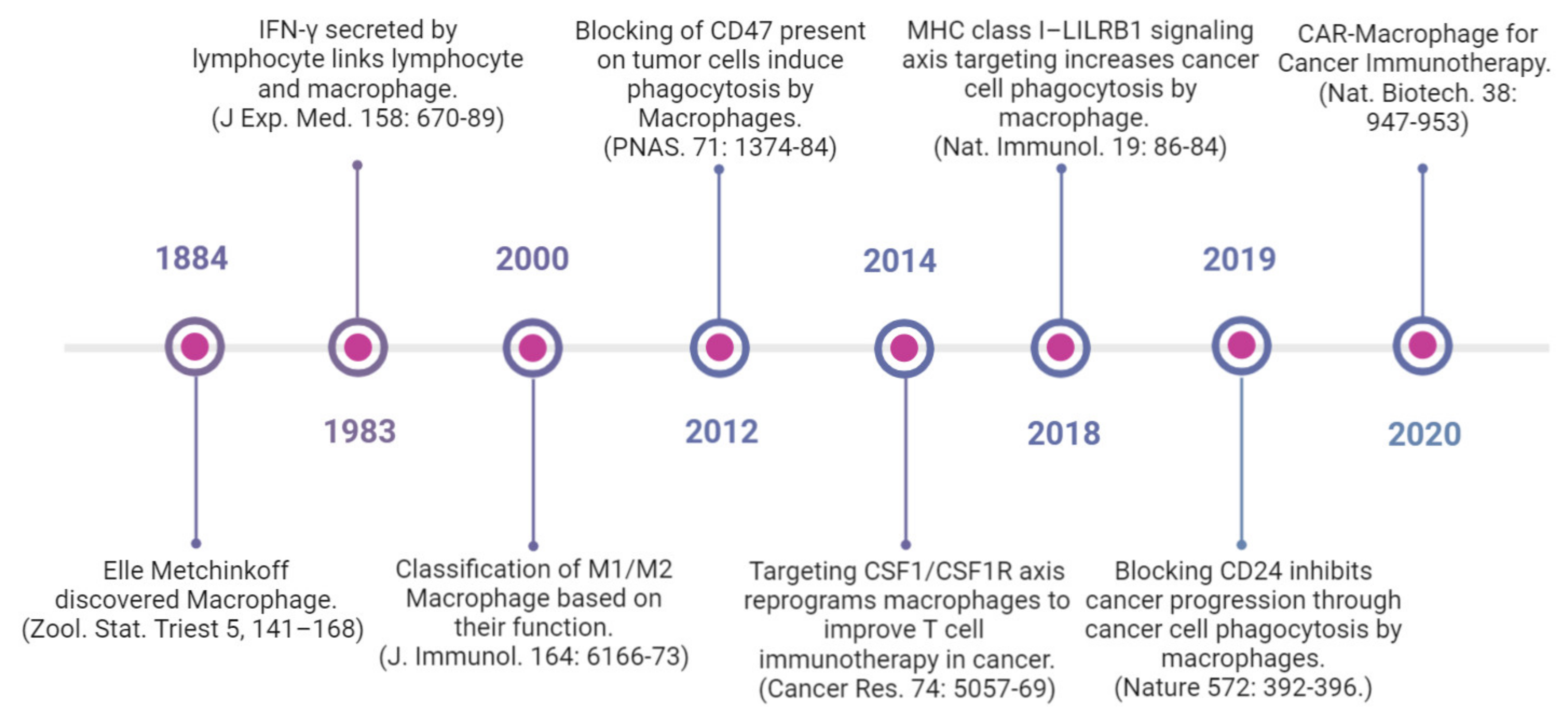
- Barkal, A.A.; Brewer, R.E.; Markovic, M.; Kowarsky, M.; Barkal, S.A.; Zaro, B.W.; Krishnan, V.; Hatakeyama, J.; Dorigo, O.; Barkal, L.J.; et al. CD24 signalling through macrophage Siglec-10 is a target for cancer immunotherapy. Nature 2019, 572, 392–396. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; He, L.; Zhu, J.; Zhang, P.; Liang, S. Targeting tumor-associated macrophages for cancer treatment. Cell Biosci. 2022, 12, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Yu, Y.; Wang, X.; Zhang, T. Tumor-Associated Macrophages in Tumor Immunity. Front. Immunol. 2020, 11, 583084. [Google Scholar] [CrossRef]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef] [PubMed]
- Larionova, I.; Cherdyntseva, N.; Liu, T.; Patysheva, M.; Rakina, M.; Kzhyshkowska, J. Interaction of tumor-associated macrophages and cancer chemotherapy. Oncoimmunology 2019, 8, e1596004. [Google Scholar] [CrossRef] [PubMed]
- Chong, B.F.; Tseng, L.-C.; Hosler, G.A.; Teske, N.M.; Zhang, S.; Karp, D.R.; Olsen, N.J.; Mohan, C. A subset of CD163+ macrophages displays mixed polarizations in discoid lupus skin. Thromb. Haemost. 2015, 17, 1–10. [Google Scholar] [CrossRef]
- Biswas, S.K.; Gangi, L.; Paul, S.; Schioppa, T.; Saccani, A.; Sironi, M.; Bottazzi, B.; Doni, A.; Vincenzo, B.; Pasqualini, F.; et al. A distinct and unique transcriptional program expressed by A distinct and unique transcriptional program expressed by tumor-associated macrophages (defective NF- B and enhanced IRF-3/STAT1 activation). Blood 2006, 107, 2112–2122. [Google Scholar] [CrossRef]
- Ryder, M.; A Ghossein, R.; Ricarte-Filho, J.C.M.; A Knauf, J.; A Fagin, J. Increased density of tumor-associated macrophages is associated with decreased survival in advanced thyroid cancer. Endocrine-Related Cancer 2008, 15, 1069–1074. [Google Scholar] [CrossRef]
- Mantovani, A.; Ponzetta, A.; Inforzato, A.; Jaillon, S. Innate immunity, inflammation and tumour progression: Double-edged swords. J. Intern. Med. 2019, 285, 524–532. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Qu, J.; Sun, Y.; Wang, J.; Liu, X.; Wang, F.; Zhang, H.; Wang, W.; Ma, X.; Gao, X.; et al. Prognostic significance of tumor-associated macrophages in breast cancer: A meta-analysis of the literature. Oncotarget 2017, 8, 30576–30586. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.W.; Liu, L.; Gong, C.-Y.; Shi, H.-S.; Zeng, Y.-H.; Wang, X.-Z.; Zhao, Y.-W.; Wei, Y.-Q. Prognostic Significance of Tumor-Associated Macrophages in Solid Tumor: A Meta-Analysis of the Literature. PLoS ONE 2012, 7, e50946. [Google Scholar] [CrossRef]
- Bingle, L.; Brown, N.; Lewis, C.E. The role of tumour-associated macrophages in tumour progression: Implications for new anticancer therapies. J. Pathol. 2002, 196, 254–265. [Google Scholar] [CrossRef] [PubMed]
- Kumari, N.; Choi, S.H. Tumor-associated macrophages in cancer: Recent advancements in cancer nanoimmunotherapies. J. Exp. Clin. Cancer Res. 2022, 41, 1–39. [Google Scholar] [CrossRef]
- Li, Z.; Ding, Y.; Liu, J.; Wang, J.; Mo, F.; Wang, Y.; Chen-Mayfield, T.-J.; Sondel, P.M.; Hong, S.; Hu, Q. Depletion of tumor associated macrophages enhances local and systemic platelet-mediated anti-PD-1 delivery for post-surgery tumor recurrence treatment. Nat. Commun. 2022, 13, 1–15. [Google Scholar] [CrossRef]
- Forssell, J.; Öberg, A.; Henriksson, M.L.; Stenling, R.; Jung, A.; Palmqvist, R. High Macrophage Infiltration along the Tumor Front Correlates with Improved Survival in Colon Cancer. Clin. Cancer Res. 2007, 13, 1472–1479. [Google Scholar] [CrossRef]
- Ohno, S.; Inagawa, H.; Dhar, D.K.; Fujii, T.; Ueda, S.; Tachibana, M.; Suzuki, N.; Inoue, M.; Soma, G.-I.; Nagasue, N. The degree of macrophage infiltration into the cancer cell nest is a significant predictor of survival in gastric cancer patients. Anticancer. Res. 2003, 23, 5015–5022. [Google Scholar]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef]
- Liu, J.Y.; Yang, X.J.; Geng, X.F.; Huang, C.Q.; Yu, Y.; Li, Y. Prognostic significance of tumor-associated macrophages density in gastric cancer: A systemic review and meta-analysis. Minerva Medica 2016, 107, 314–321. [Google Scholar]
- Balkwill, F.; Mantovani, A. Inflammation and cancer: Back to Virchow? Lancet 2001, 357, 539–545. [Google Scholar] [CrossRef] [PubMed]
- Majumder, S.; Shivaji, U.N.; Kasturi, R.; Sigamani, A.; Ghosh, S.; Iacucci, M. Inflammatory bowel disease-related colorectal cancer: Past, present and future perspectives. World J. Gastrointest. Oncol. 2022, 14, 547–567. [Google Scholar] [CrossRef] [PubMed]
- Cucarull, B.; Tutusaus, A.; Rider, P.; Hernáez-Alsina, T.; Cuño, C.; de Frutos, P.G.; Colell, A.; Marí, M.; Morales, A. Hepatocellular Carcinoma: Molecular Pathogenesis and Therapeutic Advances. Cancers 2022, 14, 621. [Google Scholar] [CrossRef] [PubMed]
- Polk, D.B.; Peek, R.M. Helicobacter pylori: Gastric cancer and beyond. Nat. Rev. Cancer 2010, 10, 403–414. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Shu, X.; Wang, J. Helicobacter pylori-Mediated Oxidative Stress and Gastric Diseases: A Review. Front. Microbiol. 2022, 13. [Google Scholar] [CrossRef]
- Nemajerova, A.; Mena, P.; Fingerle-Rowson, G.; Moll, U.M.; Petrenko, O. Impaired DNA damage checkpoint response in MIF-deficient mice. EMBO J. 2007, 26, 987–997. [Google Scholar] [CrossRef]
- Fingerle-Rowson, G.; Petrenko, O. MIF coordinates the cell cycle with DNA damage checkpoints. Lessons from knockout mouse models. Cell Div. 2007, 2, 22. [Google Scholar] [CrossRef]
- Mantovani, A.; Allavena, P.; Marchesi, F.; Garlanda, C. Macrophages as tools and targets in cancer therapy. Nat. Rev. Drug Discov. 2022, 21, 799–820. [Google Scholar] [CrossRef]
- Zhang, D.; Qiu, X.; Li, J.; Zheng, S.; Li, L.; Zhao, H. TGF-β secreted by tumor-associated macrophages promotes proliferation and invasion of colorectal cancer via miR-34a-VEGF axis. Cell Cycle 2018, 17, 2766–2778. [Google Scholar] [CrossRef]
- Kong, L.; Zhou, Y.; Bu, H.; Lv, T.; Shi, Y.; Yang, J. Deletion of interleukin-6 in monocytes/macrophages suppresses the initiation of hepatocellular carcinoma in mice. J. Exp. Clin. Cancer Res. 2016, 35, 1–11. [Google Scholar] [CrossRef]
- Li, D.; Ji, H.; Niu, X.; Yin, L.; Wang, Y.; Gu, Y.; Wang, J.; Zhou, X.; Zhang, H.; Zhang, Q. Tumor-associated macrophages secrete CC-chemokine ligand 2 and induce tamoxifen resistance by activating PI3K/Akt/mTOR in breast cancer. Cancer Sci. 2020, 111, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Xu, Z.; Guo, J.; Yang, K.; Zheng, J.; Sun, X. Tumor-associated macrophages (TAMs) depend on MMP1 for their cancer-promoting role. Cell Death Discov. 2021, 7, 1–10. [Google Scholar] [CrossRef]
- Aramini, B.; Masciale, V.; Grisendi, G.; Banchelli, F.; D’Amico, R.; Maiorana, A.; Morandi, U.; Dominici, M.; Haider, K.H. Cancer stem cells and macrophages: Molecular connections and future perspectives against cancer. Oncotarget 2021, 12, 230–250. [Google Scholar] [CrossRef] [PubMed]
- Hide, T.; Komohara, Y.; Miyasato, Y.; Nakamura, H.; Makino, K.; Takeya, M.; Kuratsu, J.-I.; Mukasa, A.; Yano, S. Oligodendrocyte Progenitor Cells and Macrophages/Microglia Produce Glioma Stem Cell Niches at the Tumor Border. Ebiomedicine 2018, 30, 94–104. [Google Scholar] [CrossRef]
- Yang, L.; Dong, Y.; Li, Y.; Wang, D.; Liu, S.; Wang, D.; Gao, Q.; Ji, S.; Chen, X.; Lei, Q.; et al. IL-10 derived from M2 macrophage promotes cancer stemness via JAK1/STAT1/NF-κB/Notch1 pathway in non-small cell lung cancer. Int. J. Cancer 2019, 145, 1099–1110. [Google Scholar] [CrossRef]
- Wan, S.; Zhao, E.; Kryczek, I.; Vatan, L.; Sadovskaya, A.; Ludema, G.; Simeone, D.M.; Zou, W.; Welling, T.H. Tumor-Associated Macrophages Produce Interleukin 6 and Signal via STAT3 to Promote Expansion of Human Hepatocellular Carcinoma Stem Cells. Gastroenterology 2014, 147, 1393–1404. [Google Scholar] [CrossRef]
- Nasrollahzadeh, E.; Razi, S.; Keshavarz-Fathi, M.; Mazzone, M.; Rezaei, N. Pro-tumorigenic functions of macrophages at the primary, invasive and metastatic tumor site. Cancer Immunol. Immunother. 2020, 69, 1673–1697. [Google Scholar] [CrossRef]
- Singh, J.K.; Farnie, G.; Bundred, N.J.; Simões, B.M.; Shergill, A.; Landberg, G.; Howell, S.J.; Clarke, R.B. Targeting CXCR1/2 Significantly Reduces Breast Cancer Stem Cell Activity and Increases the Efficacy of Inhibiting HER2 via HER2-Dependent and -Independent Mechanisms. Clin. Cancer Res. 2013, 19, 643–656. [Google Scholar] [CrossRef]
- Huang, W.-C.; Chan, M.-L.; Chen, M.-J.; Tsai, T.-H.; Chen, Y.-J. Modulation of macrophage polarization and lung cancer cell stemness by MUC1 and development of a related small-molecule inhibitor pterostilbene. Oncotarget 2016, 7, 39363–39375. [Google Scholar] [CrossRef]
- Zhang, F.; Li, P.; Liu, S.; Yang, M.; Zeng, S.; Deng, J.; Chen, D.; Yi, Y.; Liu, H. β-Catenin-CCL2 feedback loop mediates crosstalk between cancer cells and macrophages that regulates breast cancer stem cells. Oncogene 2021, 40, 5854–5865. [Google Scholar] [CrossRef]
- Wang, D.; Fu, L.; Sun, H.; Guo, L.; Dubois, R.N. Prostaglandin E2 Promotes Colorectal Cancer Stem Cell Expansion and Metastasis in Mice. Gastroenterology 2015, 149, 1884–1895.e4. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Clauser, K.R.; Tam, W.L.; Frose, J.; Ye, X.; Eaton, E.N.; Reinhardt, F.; Donnenberg, V.S.; Bhargava, R.; Carr, S.A.; et al. A breast cancer stem cell niche supported by juxtacrine signalling from monocytes and macrophages. Nat. Cell Biol. 2014, 16, 1105–1117. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Li, H.; Zhu, Z.; Mei, P.; Hu, W.; Xiong, X.; Tao, J. microRNA-21-5p from M2 macrophage-derived extracellular vesicles promotes the differentiation and activity of pancreatic cancer stem cells by mediating KLF. Cell Biol. Toxicol. 2022, 38, 577–590. [Google Scholar] [CrossRef] [PubMed]
- Mitchem, J.B.; Brennan, D.J.; Knolhoff, B.L.; Belt, B.A.; Zhu, Y.; Sanford, D.E.; Belaygorod, L.; Carpenter, D.; Collins, L.; Piwnica-Worms, D.; et al. Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res. 2013, 73, 1128–1141. [Google Scholar] [CrossRef] [PubMed]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef]
- Egberts, J.-H.; Cloosters, V.; Noack, A.; Schniewind, B.; Thon, L.; Klose, S.; Kettler, B.; von Forstner, C.; Kneitz, C.; Tepel, J.; et al. Anti–Tumor Necrosis Factor Therapy Inhibits Pancreatic Tumor Growth and Metastasis. Cancer Res 2008, 68, 1443–1450. [Google Scholar] [CrossRef]
- Liu, P.; He, K.; Song, H.; Ma, Z.; Yin, W.; Xu, L.X. Deferoxamine-induced increase in the intracellular iron levels in highly aggressive breast cancer cells leads to increased cell migration by enhancing TNF-α-dependent NF-κB signaling and TGF-β signaling. J. Inorg. Biochem. 2016, 160, 40–48. [Google Scholar] [CrossRef]
- Li, C.-W.; Xia, W.; Huo, L.; Lim, S.-O.; Wu, Y.; Hsu, J.L.; Chao, C.-H.; Yamaguchi, H.; Yang, N.-K.; Ding, Q.; et al. Epithelial–Mesenchymal Transition Induced by TNF-α Requires NF-κB–Mediated Transcriptional Upregulation of Twist. Cancer Res 2012, 72, 1290–1300. [Google Scholar] [CrossRef]
- Lin, X.; Chen, L.; Yao, Y.; Zhao, R.; Cui, X.; Chen, J.; Hou, K.; Zhang, M.; Su, F.; Chen, J.; et al. CCL18-mediated down-regulation of miR98 and miR27b promotes breast cancer metastasis. Oncotarget 2015, 6, 20485–20499. [Google Scholar] [CrossRef]
- Wen, J.; Zhao, Z.; Huang, L.; Wang, L.; Miao, Y.; Wu, J. IL-8 promotes cell migration through regulating EMT by activating the Wnt/β-catenin pathway in ovarian cancer. J. Cell. Mol. Med. 2020, 24, 1588–1598. [Google Scholar] [CrossRef]
- Deng, F.; Weng, Y.; Li, X.; Wang, T.; Fan, M.; Shi, Q. Overexpression of IL-8 promotes cell migration via PI3K-Akt signaling pathway and EMT in triple-negative breast cancer. Pathol.—Res. Pr. 2021, 223, 152824. [Google Scholar] [CrossRef]
- Che, D.; Zhang, S.; Jing, Z.; Shang, L.; Jin, S.; Liu, F.; Shen, J.; Li, Y.; Hu, J.; Meng, Q.; et al. Macrophages induce EMT to promote invasion of lung cancer cells through the IL-6-mediated COX-2/PGE 2 /β-catenin signalling pathway. Mol. Immunol. 2017, 90, 197–210. [Google Scholar] [CrossRef]
- Liu, C.Y.; Xu, J.Y.; Shi, X.Y.; Huang, W.; Ruan, T.Y.; Xie, P.; Ding, J.L. M2-polarized tumor-associated macrophages promoted epithelial–mesenchymal transition in pancreatic cancer cells, partially through TLR4/IL-10 signaling pathway. Lab. Investig. 2013, 93, 844–854. [Google Scholar] [CrossRef] [PubMed]
- Yao, R.-R.; Li, J.-H.; Zhang, R.; Chen, R.-X.; Wang, Y.-H. M2-polarized tumor-associated macrophages facilitated migration and epithelial-mesenchymal transition of HCC cells via the TLR4/STAT3 signaling pathway. World J. Surg. Oncol. 2018, 16, 9. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, Q.; Lou, Y.; Fu, Q.; Chen, Q.; Wei, T.; Yang, J.; Tang, J.; Wang, J.; Chen, Y.; et al. Hypoxia-inducible factor-1α/interleukin-1β signaling enhances hepatoma epithelial–mesenchymal transition through macrophages in a hypoxic-inflammatory microenvironment. Hepatology 2018, 67, 1872–1889. [Google Scholar] [CrossRef]
- Gao, L.; Zhang, W.; Zhong, W.-Q.; Liu, Z.-J.; Li, H.-M.; Yu, Z.-L.; Zhao, Y.-F. Tumor associated macrophages induce epithelial to mesenchymal transition via the EGFR/ERK1/2 pathway in head and neck squamous cell carcinoma. Oncol. Rep. 2018, 40, 2558–2572. [Google Scholar] [CrossRef]
- Samaniego, R.; Gutiérrez-González, A.; Gutiérrez-Seijo, A.; Sánchez-Gregorio, S.; García-Giménez, J.; Mercader, E.; Marquez-Rodas, I.; Avilés, J.A.; Relloso, M.; Sánchez-Mateos, P. CCL20 Expression by Tumor-Associated Macrophages Predicts Progression of Human Primary Cutaneous Melanoma. Cancer Immunol. Res. 2018, 6, 267–275. [Google Scholar] [CrossRef]
- Mi, X.; Xu, R.; Hong, S.; Xu, T.; Zhang, W.; Liu, M. M2 Macrophage-Derived Exosomal lncRNA AFAP1-AS1 and MicroRNA-26a Affect Cell Migration and Metastasis in Esophageal Cancer. Mol. Ther.—Nucleic Acids 2020, 22, 779–790. [Google Scholar] [CrossRef]
- Carmeliet, P. VEGF as a Key Mediator of Angiogenesis in Cancer. Oncology 2005, 69, 4–10. [Google Scholar] [CrossRef]
- Lin, E.Y.; Pollard, J.W. Tumor-Associated Macrophages Press the Angiogenic Switch in Breast Cancer. Cancer Res 2007, 67, 5064–5066. [Google Scholar] [CrossRef]
- Valkovic, T.; Dobrila, F.; Melato, M.; Sasso, F.; Rizzardi, C.; Jonjić, N. Correlation between vascular endothelial growth factor, angiogenesis, and tumor-associated macrophages in invasive ductal breast carcinoma. Virchows Arch. 2002, 440, 583–588. [Google Scholar] [CrossRef]
- Tsutsui, S.; Yasuda, K.; Suzuki, K.; Tahara, K.; Higashi, H.; Era, S. Macrophage infiltration and its prognostic implications in breast cancer: The relationship with VEGF expression and microvessel density. Oncol. Rep. 2005, 14, 425–431. [Google Scholar] [CrossRef] [PubMed]
- Okikawa, S.; Morine, Y.; Saito, Y.; Yamada, S.; Tokuda, K.; Teraoku, H.; Miyazaki, K.; Yamashita, S.; Ikemoto, T.; Imura, S.; et al. Inhibition of the VEGF signaling pathway attenuates tumor-associated macrophage activity in liver cancer. Oncol. Rep. 2022, 47, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Reusser, N.M.; Dalton, H.J.; Pradeep, S.; Gonzalez-Villasana, V.; Jennings, N.B.; Vasquez, H.G.; Wen, Y.; Rupaimoole, R.; Nagaraja, A.S.; Gharpure, K.; et al. Clodronate inhibits tumor angiogenesis in mouse models of ovarian cancer. Cancer Biol. Ther. 2014, 15, 1061–1067. [Google Scholar] [CrossRef] [PubMed]
- Badawi, M.A.; Abouelfadl, D.M.; El-Sharkawy, S.L.; El-Aal, W.E.A.; Abbas, N.F. Tumor-Associated Macrophage (TAM) and Angiogenesis in Human Colon Carcinoma. Open Access Maced. J. Med Sci. 2015, 3, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Riabov, V.; Gudima, A.; Wang, N.; Mickley, A.; Orekhov, A.; Kzhyshkowska, J. Role of tumor associated macrophages in tumor angiogenesis and lymphangiogenesis. Front. Physiol. 2014, 5, 75. [Google Scholar] [CrossRef]
- Silva, D.; Quintas, C.; Gonçalves, J.; Fresco, P. Contribution of adrenergic mechanisms for the stress-induced breast cancer carcinogenesis. J. Cell. Physiol. 2022, 237, 2107–2127. [Google Scholar] [CrossRef]
- Sierra, J.R.; Corso, S.; Caione, L.; Cepero, V.; Conrotto, P.; Cignetti, A.; Piacibello, W.; Kumanogoh, A.; Kikutani, H.; Comoglio, P.; et al. Tumor angiogenesis and progression are enhanced by Sema4D produced by tumor-associated macrophages. J. Exp. Med. 2008, 205, 1673–1685. [Google Scholar] [CrossRef]
- Gomes, F.G.; Nedel, F.; Alves, A.M.; Nör, J.E.; Tarquinio, S.B.C. Tumor angiogenesis and lymphangiogenesis: Tumor/endothelial crosstalk and cellular/microenvironmental signaling mechanisms. Life Sci. 2013, 92, 101–107. [Google Scholar] [CrossRef]
- Leek, R.D.; Landers, R.; Fox, S.B.; Ng, F.; Harris, A.L.; Lewis, C.E. Association of tumour necrosis factor alpha and its receptors with thymidine phosphorylase expression in invasive breast carcinoma. Br. J. Cancer 1998, 77, 2246–2251. [Google Scholar] [CrossRef]
- Albini, A.; Bruno, A.; Noonan, D.M.; Mortara, L. Contribution to Tumor Angiogenesis From Innate Immune Cells Within the Tumor Microenvironment: Implications for Immunotherapy. Front. Immunol. 2018, 9, 527. [Google Scholar] [CrossRef]
- Chanmee, T.; Ontong, P.; Konno, K.; Itano, N. Tumor-Associated Macrophages as Major Players in the Tumor Microenvironment. Cancers 2014, 6, 1670–1690. [Google Scholar] [CrossRef] [PubMed]
- Ruan, H.; Leibowitz, B.J.; Zhang, L.; Yu, J. Immunogenic cell death in colon cancer prevention and therapy. Mol. Carcinog. 2020, 59, 783–793. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Galluzzi, L.; Kepp, O.; Zitvogel, L. Immunogenic Cell Death in Cancer Therapy. Annu. Rev. Immunol. 2013, 31, 51–72. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Allavena, P. The interaction of anticancer therapies with tumor-associated macrophages. J. Exp. Med. 2015, 212, 435–445. [Google Scholar] [CrossRef]
- Kes, M.M.; Bossche, J.V.D.; Griffioen, A.W.; Huijbers, E.J. Oncometabolites lactate and succinate drive pro-angiogenic macrophage response in tumors. Biochim. Biophys. Acta (BBA) Bioenerg. 2020, 1874, 188427. [Google Scholar] [CrossRef]
- Puthenveetil, A.; Dubey, S. Metabolic reprograming of tumor-associated macrophages. Ann. Transl. Med. 2020, 8, 1030. [Google Scholar] [CrossRef]
- Shu, Y.; Cheng, P. Targeting tumor-associated macrophages for cancer immunotherapy. Biochim. et Biophys. Acta 2020, 1874, 188434. [Google Scholar] [CrossRef]
- Gomez, S.; Tabernacki, T.; Kobyra, J.; Roberts, P.; Chiappinelli, K.B. Combining epigenetic and immune therapy to overcome cancer resistance. Semin. Cancer Biol. 2020, 65, 99–113. [Google Scholar] [CrossRef]
- Arlauckas, S.P.; Garris, C.S.; Kohler, R.H.; Kitaoka, M.; Cuccarese, M.F.; Yang, K.S.; Miller, M.A.; Carlson, J.C.; Freeman, G.J.; Anthony, R.M.; et al. In vivo imaging reveals a tumor-associated macrophage–mediated resistance pathway in anti–PD-1 therapy. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef]
- Binnewies, M.; Pollack, J.L.; Rudolph, J.; Dash, S.; Abushawish, M.; Lee, T.; Jahchan, N.S.; Canaday, P.; Lu, E.; Norng, M.; et al. Targeting TREM2 on Tumor Associated Macrophages Enhances Efficacious Immunotherapy. SSRN Electron. J. 2021, 37, 109844. [Google Scholar] [CrossRef]
- Duong, L.; Radley-Crabb, H.G.; Gardner, J.K.; Tomay, F.; Dye, D.E.; Grounds, M.; Pixley, F.; Nelson, D.J.; Jackaman, C. Macrophage Depletion in Elderly Mice Improves Response to Tumor Immunotherapy, Increases Anti-tumor T Cell Activity and Reduces Treatment-Induced Cachexia. Front. Genet. 2018, 9, 526. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef] [PubMed]
- Chomarat, P.; Banchereau, J.; Davoust, J.; Palucka, A.K. IL-6 switches the differentiation of monocytes from dendritic cells to macrophages. Nat. Immunol. 2000, 1, 510–514. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.-H.; Hong, H.-C.; Hong, T.-M.; Chiang, W.-F.; Jin, Y.-T.; Chen, Y.-L. Targeting Galectin-1 in Carcinoma-Associated Fibroblasts Inhibits Oral Squamous Cell Carcinoma Metastasis by Downregulating MCP-1/CCL2 Expression. Clin. Cancer Res. 2011, 17, 1306–1316. [Google Scholar] [CrossRef] [PubMed]
- Mathew, E.; Brannon, A.L.; Del Vecchio, A.; Garcia, P.E.; Penny, M.K.; Kane, K.T.; Vinta, A.; Buckanovich, R.J.; di Magliano, M.P. Mesenchymal Stem Cells Promote Pancreatic Tumor Growth by Inducing Alternative Polarization of Macrophages. Neoplasia 2016, 18, 142–151. [Google Scholar] [CrossRef]
- Mace, T.A.; Ameen, Z.; Collins, A.; Wojcik, S.; Mair, M.; Young, G.S.; Fuchs, J.R.; Eubank, T.D.; Frankel, W.L.; Bekaii-Saab, T.; et al. Pancreatic Cancer-Associated Stellate Cells Promote Differentiation of Myeloid-Derived Suppressor Cells in a STAT3-Dependent Manner. Cancer Res. 2013, 73, 3007–3018. [Google Scholar] [CrossRef]
- Wang, Q.; He, Z.; Huang, M.; Liu, T.; Wang, Y.; Xu, H.; Duan, H.; Ma, P.; Zhang, L.; Zamvil, S.S.; et al. Vascular niche IL-6 induces alternative macrophage activation in glioblastoma through HIF-2α. Nat. Commun. 2018, 9, 559. [Google Scholar] [CrossRef]
- Stahl, M.; Schupp, J.; Jäger, B.; Schmid, M.; Zissel, G.; Müller-Quernheim, J.; Prasse, A. Lung Collagens Perpetuate Pulmonary Fibrosis via CD204 and M2 Macrophage Activation. PLoS ONE 2013, 8, e81382. [Google Scholar] [CrossRef]
- Tang, M.; Diao, J.; Gu, H.; Khatri, I.; Zhao, J.; Cattral, M.S. Toll-like Receptor 2 Activation Promotes Tumor Dendritic Cell Dysfunction by Regulating IL-6 and IL-10 Receptor Signaling. Cell Rep. 2015, 13, 2851–2864. [Google Scholar] [CrossRef]
- Kim, S.; Takahashi, H.; Lin, W.-W.; Descargues, P.; Grivennikov, S.; Kim, Y.; Luo, J.-L.; Karin, M. Carcinoma-produced factors activate myeloid cells through TLR2 to stimulate metastasis. Nature 2009, 457, 102–106. [Google Scholar] [CrossRef]
- Kobayashi, N.; Miyoshi, S.; Mikami, T.; Koyama, H.; Kitazawa, M.; Takeoka, M.; Sano, K.; Amano, J.; Isogai, Z.; Niida, S.; et al. Hyaluronan Deficiency in Tumor Stroma Impairs Macrophage Trafficking and Tumor Neovascularization. Cancer Res 2010, 70, 7073–7083. [Google Scholar] [CrossRef] [PubMed]
- Lee-Sayer, S.S.; Dong, Y.; Arif, A.A.; Olsson, M.; Brown, K.L.; Johnson, P. The where, when, how and why of hyaluronan binding by immune cells. Front. Immunol. 2015, 6, 150. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.B.; Yeh, E.S.; Soloff, A.C. Tumor-associated macrophages: Unwitting accomplices in breast cancer malignancy. NPJ Breast Cancer 2016, 2, 15025. [Google Scholar] [CrossRef] [PubMed]
- Hughes, R.; Qian, B.-Z.; Rowan, C.; Muthana, M.; Keklikoglou, I.; Olson, O.C.; Tazzyman, S.; Danson, S.; Addison, C.; Clemons, M.; et al. Perivascular M2 Macrophages Stimulate Tumor Relapse after Chemotherapy. Cancer Res. 2015, 75, 3479–3491. [Google Scholar] [CrossRef] [PubMed]
- Yeo, E.-J.; Cassetta, L.; Qian, B.-Z.; Lewkowich, I.; Li, J.-F.; Stefater, J.A., 3rd; Smith, A.N.; Wiechmann, L.S.; Wang, Y.; Pollard, J.W.; et al. Myeloid WNT7b Mediates the Angiogenic Switch and Metastasis in Breast Cancer. Cancer Res. 2014, 74, 2962–2973. [Google Scholar] [CrossRef] [PubMed]
- Werno, C.; Menrad, H.; Weigert, A.; Dehne, N.; Goerdt, S.; Schledzewski, K.; Kzhyshkowska, J.; Brüne, B. Knockout of HIF-1 in tumor-associated macrophages enhances M2 polarization and attenuates their pro-angiogenic responses. Carcinog. 2010, 31, 1863–1872. [Google Scholar] [CrossRef]
- Ceci, C.; Atzori, M.G.; Lacal, P.M.; Graziani, G. Targeting Tumor-Associated Macrophages to Increase the Efficacy of Immune Checkpoint Inhibitors: A Glimpse into Novel Therapeutic Approaches for Metastatic Melanoma. Cancers 2020, 12, 3401. [Google Scholar] [CrossRef]
- Oft, M. IL-10: Master Switch from Tumor-Promoting Inflammation to Antitumor Immunity. Cancer Immunol. Res. 2014, 2, 194–199. [Google Scholar] [CrossRef]
- Gratchev, A. TGF-β signalling in tumour associated macrophages. Immunobiology 2017, 222, 75–81. [Google Scholar] [CrossRef]
- MaruYama, T.; Chen, W.J.; Shibata, H. TGF-β and Cancer Immunotherapy. Biol. Pharm. Bull. 2022, 45, 155–161. [Google Scholar] [CrossRef]
- Wu, W.-K.; Llewellyn, O.P.; Bates, D.O.; Nicholson, L.B.; Dick, A.D. IL-10 regulation of macrophage VEGF production is dependent on macrophage polarisation and hypoxia. Immunobiology 2010, 215, 796–803. [Google Scholar] [CrossRef] [PubMed]
- Curiel, T.J.; Coukos, G.; Zou, L.; Alvarez, X.; Cheng, P.; Mottram, P.; Evdemon-Hogan, M.; Conejo-Garcia, J.R.; Zhang, L.; Burow, M.; et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat. Med. 2004, 10, 942–949. [Google Scholar] [CrossRef] [PubMed]
- Petty, A.J.; Li, A.; Wang, X.; Dai, R.; Heyman, B.; Hsu, D.; Huang, X.; Yang, Y. Hedgehog signaling promotes tumor-associated macrophage polarization to suppress intratumoral CD8+ T cell recruitment. J. Clin. Investig. 2019, 129, 5151–5162. [Google Scholar] [CrossRef] [PubMed]
- Zelenay, S.; van der Veen, A.G.; Böttcher, J.P.; Snelgrove, K.J.; Rogers, N.; Acton, S.E.; Chakravarty, P.; Girotti, M.R.; Marais, R.; Quezada, S.A.; et al. Cyclooxygenase-Dependent Tumor Growth through Evasion of Immunity. Cell 2015, 162, 1257–1270. [Google Scholar] [CrossRef]
- Wang, D.; Dubois, R.N. An inflammatory mediator, prostaglandin e2, in colorectal cancer. Cancer J. (United States) 2013, 19, 502. [Google Scholar] [CrossRef]
- Holt, D.; Ma, X.; Kundu, N.; Fulton, A. Prostaglandin E2 (PGE2) suppresses natural killer cell function primarily through the PGE2 receptor EP. Cancer Immunol. Immunother. 2011, 60, 1577–1586. [Google Scholar] [CrossRef]
- Muthuswamy, R.; Mueller-Berghaus, J.; Haberkorn, U.; Reinhart, T.A.; Schadendorf, D.; Kalinski, P. PGE2 transiently enhances DC expression of CCR7 but inhibits the ability of DCs to produce CCL19 and attract naive T cells. Blood 2010, 116, 1454–1459. [Google Scholar] [CrossRef]
- Nakanishi, M.; Rosenberg, D.W. Multifaceted roles of PGE2 in inflammation and cancer. Semin. Immunopathol. 2013, 35, 123–137. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, L.; Kang, D.; Yang, D.; Tang, Y. Activation of PGE2/EP2 and PGE2/EP4 signaling pathways positively regulate the level of PD-1 in infiltrating CD8+ T cells in patients with lung cancer. Oncol. Lett. 2018, 15, 552–558. [Google Scholar] [CrossRef]
- Betz, M.; Fox, B.S. Prostaglandin E2 inhibits production of Th1 lymphokines but not of Th2 lymphokines. J. Immunol. 1991, 146, 108–113. [Google Scholar]
- Condamine, T.; Gabrilovich, D.I. Molecular mechanisms regulating myeloid-derived suppressor cell differentiation and function. Trends Immunol. 2011, 32, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Colegio, O.R.; Chu, N.-Q.; Szabo, A.L.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Cyrus, N.; Brokowski, C.E.; Eisenbarth, S.C.; Phillips, G.M.; et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 2014, 513, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Cook, R.S.; Jacobsen, K.M.; Wofford, A.M.; DeRyckere, D.; Stanford, J.; Prieto, A.L.; Redente, E.; Sandahl, M.; Hunter, D.M.; Strunk, K.E.; et al. MerTK inhibition in tumor leukocytes decreases tumor growth and metastasis. J. Clin. Investig. 2013, 123, 3231–3242. [Google Scholar] [CrossRef] [PubMed]
- Graham, D.K.; DeRyckere, D.; Davies, K.D.; Earp, H.S. The TAM family: Phosphatidylserine-sensing receptor tyrosine kinases gone awry in cancer. Nat. Rev. Cancer 2014, 14, 769–785. [Google Scholar] [CrossRef]
- DeNardo, D.G.; Brennan, D.J.; Rexhepaj, E.; Ruffell, B.; Shiao, S.L.; Madden, S.F.; Gallagher, W.M.; Wadhwani, N.; Keil, S.D.; Junaid, S.A.; et al. Leukocyte Complexity Predicts Breast Cancer Survival and Functionally Regulates Response to Chemotherapy. Cancer Discov. 2011, 1, 54–67. [Google Scholar] [CrossRef]
- Ruffell, B.; Chang-Strachan, D.; Chan, V.; Rosenbusch, A.; Ho, C.M.; Pryer, N.; Daniel, D.; Hwang, E.S.; Rugo, H.S.; Coussens, L.M. Macrophage IL-10 Blocks CD8+ T Cell-Dependent Responses to Chemotherapy by Suppressing IL-12 Expression in Intratumoral Dendritic Cells. Cancer Cell 2014, 26, 623–637. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef]
- Geiger, R.; Rieckmann, J.C.; Wolf, T.; Basso, C.; Feng, Y.; Fuhrer, T.; Kogadeeva, M.; Picotti, P.; Meissner, F.; Mann, M.; et al. L-Arginine Modulates T Cell Metabolism and Enhances Survival and Anti-tumor Activity. Cell 2016, 167, 829–842.e13. [Google Scholar] [CrossRef]
- Mazzone, M.; Menga, A.; Castegna, A. Metabolism and TAM functions-it takes two to tango. FEBS J. 2018, 285, 700–716. [Google Scholar] [CrossRef]
- Rath, M.; Müller, I.; Kropf, P.; Closs, E.I.; Munder, M. Metabolism via Arginase or Nitric Oxide Synthase: Two Competing Arginine Pathways in Macrophages. Front. Immunol. 2014, 5, 532. [Google Scholar] [CrossRef]
- Rodriguez, P.C.; Quiceno, D.G.; Zabaleta, J.; Ortiz, B.; Zea, A.H.; Piazuelo, M.B.; Delgado, A.; Correa, P.; Brayer, J.; Sotomayor, E.M.; et al. Arginase I Production in the Tumor Microenvironment by Mature Myeloid Cells Inhibits T-Cell Receptor Expression and Antigen-Specific T-Cell Responses. Cancer Res 2004, 64, 5839–5849. [Google Scholar] [CrossRef] [PubMed]
- Szefel, J.; Danielak, A.; Kruszewski, W.J. Metabolic pathways of L-arginine and therapeutic consequences in tumors. Adv. Med Sci. 2019, 64, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Liu, D.; Li, L. PD-1/PD-L1 pathway: Current researches in cancer. Am. J. Cancer Res. 2020, 10, 727–742. [Google Scholar]
- Yu, H.; Boyle, T.A.; Zhou, C.; Rimm, D.; Hirsch, F.R. PD-L1 expression in lung cancer. J. Thorac. Oncol. 2016, 11. [Google Scholar] [CrossRef]
- Kuang, D.-M.; Zhao, Q.; Peng, C.; Xu, J.; Zhang, J.-P.; Wu, C.; Zheng, L. Activated monocytes in peritumoral stroma of hepatocellular carcinoma foster immune privilege and disease progression through PD-L. J. Exp. Med. 2009, 206, 1327–1337. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Jin, T.; Zhu, Y.; Dai, C. Immune checkpoint therapy in liver cancer. J. Exp. Clin. Cancer Res. 2018, 37, 110. [Google Scholar] [CrossRef]
- Prasad, D.V.; Richards, S.; Mai, X.M.; Dong, C. B7S1, a Novel B7 Family Member that Negatively Regulates T Cell Activation. Immunity 2003, 18, 863–873. [Google Scholar] [CrossRef] [PubMed]
- Lines, J.L.; Pantazi, E.; Mak, J.; Sempere, L.F.; Wang, L.; O’Connell, S.; Ceeraz, S.; Suriawinata, A.A.; Yan, S.; Ernstoff, M.S.; et al. VISTA Is an Immune Checkpoint Molecule for Human T Cells. Cancer Res. 2014, 74, 1924–1932. [Google Scholar] [CrossRef] [PubMed]
- Green, K.A.; Wang, L.; Noelle, R.J.; Green, W.R. Selective Involvement of the Checkpoint Regulator VISTA in Suppression of B-Cell, but Not T-Cell, Responsiveness by Monocytic Myeloid-Derived Suppressor Cells from Mice Infected with an Immunodeficiency-Causing Retrovirus. J. Virol. 2015, 89, 9693–9698. [Google Scholar] [CrossRef]
- Bharaj, P.; Chahar, H.S.; Alozie, O.K.; Rodarte, L.; Bansal, A.; Goepfert, P.A.; Dwivedi, A.; Manjunath, N.; Shankar, P. Characterization of Programmed Death-1 Homologue-1 (PD-1H) Expression and Function in Normal and HIV Infected Individuals. PLoS ONE 2014, 9, e109103. [Google Scholar] [CrossRef]
- Nowak, E.C.; Lines, J.L.; Varn, F.S.; Deng, J.; Sarde, A.; Mabaera, R.; Kuta, A.; Le Mercier, I.; Cheng, C.; Noelle, R.J. Immunoregulatory functions of VISTA. Immunol. Rev. 2017, 276, 66–79. [Google Scholar] [CrossRef] [PubMed]
- Le Mercier, I.; Chen, W.; Lines, J.L.; Day, M.; Li, J.; Sergent, P.; Noelle, R.J.; Wang, L. VISTA Regulates the Development of Protective Antitumor Immunity. Cancer Res 2014, 74, 1933–1944. [Google Scholar] [CrossRef] [PubMed]
- Metschnikoff, E. Untersuchung ueber die intracellulare verdauung bei wirbellosen thieren. Arb. Zool. Inst. Univ. Wien. Zool. Stat. Triest. 1884, 5, 141–168. [Google Scholar]
- Nathan, C.F.; Murray, H.W.; Wiebe, M.E.; Rubin, B.Y. Identification of interferon-gamma as the lymphokine that activates human macrophage oxidative metabolism and antimicrobial activity. J. Exp. Med. 1983, 158, 670–689. [Google Scholar] [CrossRef] [PubMed]
- Mills, C.D.; Kincaid, K.; Alt, J.M.; Heilman, M.J.; Hill, A.M. M-1/M-2 macrophages and the Th1/Th2 paradigm. J. Immunol. 2000, 164, 166–173. [Google Scholar] [CrossRef]
- Willingham, S.B.; Volkmer, J.P.; Gentles, A.J.; Sahoo, D.; Dalerba, P.; Mitra, S.S.; Wang, J.; Contreras-Trujillo, H.; Martin, R.; Cohen, J.D.; et al. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc. Natl. Acad. Sci. USA 2012, 109, 6662–66627. [Google Scholar] [CrossRef]
- Zhu, Y.; Knolhoff, B.L.; Meyer, M.A.; Nywening, T.M.; West, B.L.; Luo, J.; Wang-Gillam, A.; Goedegebuure, S.P.; Linehan, D.C.; DeNardo, D.G. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. 2014, 74, 5057–5069. [Google Scholar] [CrossRef]
- Barkal, A.A.; Weiskopf, K.; Kao, K.S.; Gordon, S.R.; Rosental, B.; Yiu, Y.Y.; George, B.M.; Markovic, M.; Ring, N.G. Engagement of MHC class I by the inhibitory receptor LILRB1 suppresses macrophages and is a target of cancer immunotherapy. Nat. Immunol. 2019, 76–84. [Google Scholar] [CrossRef]
- Klichinsky, M.; Ruella, M.; Shestova, O.; Lu, X.M.; Best, A.; Zeeman, M.; Schmierer, M.; Gabrusiewicz, K.; Anderson, N.R.; Petty, N.E.; et al. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat. Biotechnol. 2020, 38, 947–953. [Google Scholar] [CrossRef]
- Opperman, K.S.; Vandyke, K.; Clark, K.C.; Coulter, E.A.; Hewett, D.R.; Mrozik, K.M.; Schwarz, N.; Evdokiou, A.; I Croucher, P.; Psaltis, P.J.; et al. Clodronate-Liposome Mediated Macrophage Depletion Abrogates Multiple Myeloma Tumor Establishment In Vivo. Neoplasia 2019, 21, 777–787. [Google Scholar] [CrossRef]
- Borgoni, S.; Iannello, A.; Cutrupi, S.; Allavena, P.; D’Incalci, M.; Novelli, F.; Cappello, P. Depletion of tumor-associated macrophages switches the epigenetic profile of pancreatic cancer infiltrating T cells and restores their anti-tumor phenotype. OncoImmunology 2018, 7, e1393596. [Google Scholar] [CrossRef] [PubMed]
- Dawson, J.C.; Serrels, A.; Stupack, D.G.; Schlaepfer, D.D.; Frame, M.C. Targeting FAK in anticancer combination therapies. Nat. Rev. Cancer 2021, 21, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Chuang, H.-H.; Zhen, Y.-Y.; Tsai, Y.-C.; Chuang, C.-H.; Hsiao, M.; Huang, M.-S.; Yang, C.-J. FAK in Cancer: From Mechanisms to Therapeutic Strategies. Int. J. Mol. Sci. 2022, 23, 1726. [Google Scholar] [CrossRef] [PubMed]
- Grandjean, C.L.; Garcia, Z.; Lemaître, F.; Bréart, B.; Bousso, P. Imaging the mechanisms of anti-CD20 therapy in vivo uncovers spatiotemporal bottlenecks in antibody-dependent phagocytosis. Sci. Adv. 2021, 7. [Google Scholar] [CrossRef] [PubMed]
- Pierpont, T.M.; Limper, C.B.; Richards, K.L. Past, present, and future of Rituximab-The world’s first oncology monoclonal antibody therapy. Front. Oncol. 2018, 8, 163. [Google Scholar] [CrossRef]
- Upton, R.; Banuelos, A.; Feng, D.; Biswas, T.; Kao, K.; McKenna, K.; Willingham, S.; Ho, P.Y.; Rosental, B.; Tal, M.C.; et al. Combining CD47 blockade with trastuzumab eliminates HER2-positive breast cancer cells and overcomes trastuzumab tolerance. Proc. Natl. Acad. Sci. USA 2021, 118, e2026849118. [Google Scholar] [CrossRef]
- Shi, Y.; Fan, X.; Deng, H.; Brezski, R.J.; Rycyzyn, M.; Jordan, R.E.; Strohl, W.; Zou, Q.; Zhang, N.; An, Z. Trastuzumab Triggers Phagocytic Killing of High HER2 Cancer Cells In Vitro and In Vivo by Interaction with Fcγ Receptors on Macrophages. J. Immunol. 2015, 194, 4379–4386. [Google Scholar] [CrossRef]
- de Louche CD, Roghanian, A. Human inhibitory leukocyte Ig-like receptors: From immunotolerance to immunotherapy. JCI Insight 2022, 7.
- Cabrales, P. RRx-001 Acts as a Dual Small Molecule Checkpoint Inhibitor by Downregulating CD47 on Cancer Cells and SIRP-α on Monocytes/Macrophages. Transl. Oncol. 2019, 12, 626–632. [Google Scholar] [CrossRef]
- Logtenberg, M.E.W.; Jansen, J.H.M.; Raaben, M.; Toebes, M.; Franke, K.; Brandsma, A.M.; Matlung, H.L.; Fauster, A.; Gomez-Eerland, R.; Bakker, N.A.M.; et al. Glutaminyl cyclase is an enzymatic modifier of the CD47- SIRPα axis and a target for cancer immunotherapy. Nat. Med. 2019, 25, 612–619. [Google Scholar] [CrossRef]
- Nam, G.-H.; Lee, E.J.; Kim, Y.K.; Hong, Y.; Choi, Y.; Ryu, M.-J.; Woo, J.; Cho, Y.; Ahn, D.J.; Yang, Y.; et al. Combined Rho-kinase inhibition and immunogenic cell death triggers and propagates immunity against cancer. Nat. Commun. 2018, 9, 1–15. [Google Scholar] [CrossRef]
- Kroemer, G.; Galassi, C.; Zitvogel, L.; Galluzzi, L. Immunogenic cell stress and death. Nat. Immunol. 2022, 23, 487–500. [Google Scholar] [CrossRef] [PubMed]
- Bonomi, M.; Ahmed, T.; Addo, S.; Kooshki, M.; Palmieri, D.; Levine, B.J.; Ruiz, J.; Grant, S.; Petty, W.J.; Triozzi, P.L. Circulating immune biomarkers as predictors of the response to pembrolizumab and weekly low dose carboplatin and paclitaxel in NSCLC and poor PS: An interim analysis. Oncol. Lett. 2019, 17, 1349–1356. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhao, Z.; Qiu, N.; Zhou, Q.; Wang, G.; Jiang, H.; Piao, Y.; Zhou, Z.; Tang, J.; Shen, Y. Co-delivery of IOX1 and doxorubicin for antibody-independent cancer chemo-immunotherapy. Nat. Commun. 2021, 12, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Chu, Z.; Zhu, L.; Yang, T.; Wang, P.; Liu, F.; Huang, Y.; Zhang, F.; Zhang, X.; Ding, W.; et al. 2-Deoxy-d-Glucose Treatment Decreases Anti-inflammatory M2 Macrophage Polarization in Mice with Tumor and Allergic Airway Inflammation. Front. Immunol. 2017, 8, 637. [Google Scholar] [CrossRef]
- De Palma, M.; Mazzieri, R.; Politi, L.S.; Pucci, F.; Zonari, E.; Sitia, G.; Mazzoleni, S.; Moi, D.; Venneri, M.A.; Indraccolo, S.; et al. Tumor-Targeted Interferon-α Delivery by Tie2-Expressing Monocytes Inhibits Tumor Growth and Metastasis. Cancer Cell 2008, 14, 299–311. [Google Scholar] [CrossRef]
- Shields, C.W.; Evans, M.A.; Wang, L.L.-W.; Baugh, N.; Iyer, S.; Wu, D.; Zhao, Z.; Pusuluri, A.; Ukidve, A.; Pan, D.C.; et al. Cellular backpacks for macrophage immunotherapy. Sci. Adv. 2020, 6, eaaz6579. [Google Scholar] [CrossRef]
- Kang, H.; Zhang, J.; Wang, B.; Liu, M.; Zhao, J.; Yang, M.; Li, Y. Puerarin inhibits M2 polarization and metastasis of tumor-associated macrophages from NSCLC xenograft model via inactivating MEK/ERK 1/2 pathway. Int. J. Oncol. 2017, 50, 545–554. [Google Scholar] [CrossRef]
- Li, X.; Su, X.; Liu, R.; Pan, Y.; Fang, J.; Cao, L.; Feng, C.; Shang, Q.; Chen, Y.; Shao, C.; et al. HDAC inhibition potentiates anti-tumor activity of macrophages and enhances anti-PD-L1-mediated tumor suppression. Oncogene 2021, 40, 1836–1850. [Google Scholar] [CrossRef]
- Han, Y.; Sun, J.; Yang, Y.; Liu, Y.; Lou, J.; Pan, H.; Yao, J.; Han, W. TMP195 Exerts Antitumor Effects on Colorectal Cancer by Promoting M1 Macrophages Polarization. Int. J. Biol. Sci. 2022, 18, 5653–5666. [Google Scholar] [CrossRef]
- He, L.; Marneros, A.G. Doxycycline Inhibits Polarization of Macrophages to the Proangiogenic M2-type and Subsequent Neovascularization. J. Biol. Chem. 2014, 289, 8019–8028. [Google Scholar] [CrossRef]
- Jin, J.; Li, Y.; Zhao, Q.; Chen, Y.; Fu, S.; Wu, J. Coordinated regulation of immune contexture: Crosstalk between STAT3 and immune cells during breast cancer progression. Cell Commun. Signal. 2021, 19, 1–19. [Google Scholar] [CrossRef]
- Jaynes, J.M.; Sable, R.; Ronzetti, M.; Bautista, W.; Knotts, Z.; Abisoye-Ogunniyan, A.; Li, D.; Calvo, R.; Dashnyam, M.; Singh, A.; et al. Mannose receptor (CD206) activation in tumor-associated macrophages enhances adaptive and innate antitumor immune responses. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Horwitz, S.M.; Koch, R.; Porcu, P.; Oki, Y.; Moskowitz, A.; Perez, M.; Myskowski, P.; Officer, A.; Jaffe, J.D.; Morrow, S.N.; et al. Activity of the PI3K-δ,γ inhibitor duvelisib in a phase 1 trial and preclinical models of T-cell lymphoma. Blood 2018, 131, 888–898. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Xu, X.; Wei, S.; Jiang, P.; Xue, L.; Wang, J. Tumor-associated macrophages: Potential therapeutic strategies and future prospects in cancer. J. Immunother. Cancer 2021, 9, e001341. [Google Scholar] [CrossRef]
- Beatty, G.L.; Chiorean, E.G.; Fishman, M.P.; Saboury, B.; Teitelbaum, U.R.; Sun, W.; Huhn, R.D.; Song, W.; Li, D.; Sharp, L.L.; et al. CD40 Agonists Alter Tumor Stroma and Show Efficacy Against Pancreatic Carcinoma in Mice and Humans. Science 2011, 331, 1612–1616. [Google Scholar] [CrossRef] [PubMed]
- Molgora, M.; Colonna, M. Turning enemies into allies—Reprogramming tumor-associated macrophages for cancer therapy. Med 2021, 2, 666–681. [Google Scholar] [CrossRef] [PubMed]
- Gunderson, A.J.; Kaneda, M.M.; Tsujikawa, T.; Nguyen, A.V.; Affara, N.I.; Ruffell, B.; Gorjestani, S.; Liudahl, S.M.; Truitt, M.; Olson, P.; et al. Bruton Tyrosine Kinase–Dependent Immune Cell Cross-Talk Drives Pancreas Cancer. Cancer Discov. 2016, 6, 270–285. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Gu, H. Emerging Nanoparticle Strategies for Modulating Tumor-Associated Macrophage Polarization. Biomolecules 2021, 11, 1912. [Google Scholar] [CrossRef] [PubMed]
- Yin, M.; Zhou, H.J.; Zhang, J.; Lin, C.; Li, H.; Li, X.; Li, Y.; Zhang, H.; Breckenridge, D.G.; Ji, W.; et al. ASK1-dependent endothelial cell activation is critical in ovarian cancer growth and metastasis. J. Clin. Investig. 2017, 2. [Google Scholar] [CrossRef] [PubMed]
- Zeisberger, S.M.; Odermatt, B.; Marty, C.; Zehnder-Fjällman, A.H.M.; Ballmer-Hofer, K.; A Schwendener, R. Clodronate-liposome-mediated depletion of tumour-associated macrophages: A new and highly effective antiangiogenic therapy approach. Br. J. Cancer 2006, 95, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Junankar, S.; Shay, G.; Jurczyluk, J.; Ali, N.; Down, J.; Pocock, N.; Parker, A.; Nguyen, A.; Sun, S.; Kashemirov, B.; et al. Real-Time Intravital Imaging Establishes Tumor-Associated Macrophages as the Extraskeletal Target of Bisphosphonate Action in Cancer. Cancer Discov. 2015, 5, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Sabatino, R.; Battistelli, S.; Magnani, M.; Rossi, L. Preclinical evaluation of an innovative anti-TAM approach based on zoledronate-loaded erythrocytes. Drug Deliv. Transl. Res. 2018, 8, 1355–1364. [Google Scholar] [CrossRef] [PubMed]
- Zang, X.; Zhang, X.; Hu, H.; Qiao, M.; Zhao, X.; Deng, Y.; Chen, D. Targeted Delivery of Zoledronate to Tumor-Associated Macrophages for Cancer Immunotherapy. Mol. Pharm. 2019, 16, 2249–2258. [Google Scholar] [CrossRef] [PubMed]
- Scott, E.M.; Jacobus, E.; Lyons, B.; Frost, S.; Freedman, J.D.; Dyer, A.; Khalique, H.; Taverner, W.K.; Carr, A.; Champion, B.R.; et al. Bi- and tri-valent T cell engagers deplete tumour-associated macrophages in cancer patient samples. J. Immunother. Cancer 2019, 7, 320. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Paulete, A.R.; Mateus-Tique, J.; Mollaoglu, G.; Nielsen, S.R.; Marks, A.; Lakshmi, A.; Khan, J.A.; Wilk, C.M.; Pia, L.; Baccarini, A.; et al. Targeting Macrophages with CAR T Cells Delays Solid Tumor Progression and Enhances Antitumor Immunity. Cancer Immunol. Res. 2022, 10, 1354–1369. [Google Scholar] [CrossRef]
- Canè, S.; Ugel, S.; Trovato, R.; Marigo, I.; De Sanctis, F.; Sartoris, S.; Bronte, V. The Endless Saga of Monocyte Diversity. Front. Immunol. 2019, 10, 1786. [Google Scholar] [CrossRef]
- Tap, W.D.; Gelderblom, H.; Palmerini, E.; Desai, J.; Bauer, S.; Blay, J.-Y.; Alcindor, T.; Ganjoo, K.; Martín-Broto, J.; Ryan, C.W.; et al. Pexidartinib versus placebo for advanced tenosynovial giant cell tumour (ENLIVEN): A randomised phase 3 trial. Lancet 2019, 394, 478–487. [Google Scholar] [CrossRef]
- Wesolowski, R.; Sharma, N.; Reebel, L.; Rodal, M.B.; Peck, A.; West, B.L.; Marimuthu, A.; Severson, P.; Karlin, D.A.; Dowlati, A.; et al. Phase Ib study of the combination of pexidartinib (PLX3397), a CSF-1R inhibitor, and paclitaxel in patients with advanced solid tumors. Ther. Adv. Med. Oncol. 2019, 11. [Google Scholar] [CrossRef]
- Li, X.; Yao, W.; Yuan, Y.; Chen, P.; Li, B.; Li, J.; Chu, R.; Song, H.; Xie, D.; Jiang, X.; et al. Targeting of tumour-infiltrating macrophages via CCL2/CCR2 signalling as a therapeutic strategy against hepatocellular carcinoma. Gut 2017, 66, 157–167. [Google Scholar] [CrossRef]
- Argyle, D.; Kitamura, T. Targeting Macrophage-Recruiting Chemokines as a Novel Therapeutic Strategy to Prevent the Progression of Solid Tumors. Front. Immunol. 2018, 9, 2629. [Google Scholar] [CrossRef]
- Cho, H.R.; Kumari, N.; Vu, H.T.; Kim, H.; Park, C.-K.; Choi, S.H. Increased Antiangiogenic Effect by Blocking CCL2-dependent Macrophages in a Rodent Glioblastoma Model: Correlation Study with Dynamic Susceptibility Contrast Perfusion MRI. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef]
- Flores-Toro, J.A.; Luo, D.; Gopinath, A.; Sarkisian, M.R.; Campbell, J.J.; Charo, I.F.; Singh, R.; Schall, T.J.; Datta, M.; Jain, R.K.; et al. CCR2 inhibition reduces tumor myeloid cells and unmasks a checkpoint inhibitor effect to slow progression of resistant murine gliomas. Proc. Natl. Acad. Sci. USA 2020, 117, 1129–1138. [Google Scholar] [CrossRef] [PubMed]
- Yao, W.; Ba, Q.; Li, X.; Li, H.; Zhang, S.; Yuan, Y.; Wang, F.; Duan, X.; Li, J.; Zhang, W.; et al. A Natural CCR2 Antagonist Relieves Tumor-associated Macrophage-mediated Immunosuppression to Produce a Therapeutic Effect for Liver Cancer. eBioMedicine 2017, 22, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Brana, I.; Calles, A.; Lorusso, P.M.; Yee, L.K.; Puchalski, T.A.; Seetharam, S.; Zhong, B.; De Boer, C.J.; Tabernero, J.; Calvo, E. Carlumab, an anti-C-C chemokine ligand 2 monoclonal antibody, in combination with four chemotherapy regimens for the treatment of patients with solid tumors: An open-label, multicenter phase 1b study. Target. Oncol. 2015, 10, 111–123. [Google Scholar] [CrossRef]
- Nywening, T.M.; Wang-Gillam, A.; E Sanford, D.; A Belt, B.; Panni, R.Z.; Cusworth, B.M.; Toriola, A.T.; Nieman, R.K.; A Worley, L.; Yano, M.; et al. Targeting tumour-associated macrophages with CCR2 inhibition in combination with FOLFIRINOX in patients with borderline resectable and locally advanced pancreatic cancer: A single-centre, open-label, dose-finding, non-randomised, phase 1b trial. Lancet Oncol. 2016, 17, 651–662. [Google Scholar] [CrossRef]
- Tu, M.M.; Abdel-Hafiz, H.A.; Jones, R.T.; Jean, A.; Hoff, K.J.; Duex, J.E.; Chauca-Diaz, A.; Costello, J.C.; Dancik, G.M.; Tamburini, B.A.J.; et al. Inhibition of the CCL2 receptor, CCR2, enhances tumor response to immune checkpoint therapy. Commun. Biol. 2020, 3, 1–12. [Google Scholar] [CrossRef]
- Feng, M.; Chen, J.Y.; Weissman-Tsukamoto, R.; Volkmer, J.-P.; Ho, P.Y.; McKenna, K.M.; Cheshier, S.; Zhang, M.; Guo, N.; Gip, P.; et al. Macrophages eat cancer cells using their own calreticulin as a guide: Roles of TLR and Btk. Proc. Natl. Acad. Sci. USA 2015, 112, 2145–2150. [Google Scholar] [CrossRef]
- Sheng, D.; Ma, W.; Zhang, R.; Zhou, L.; Deng, Q.; Tu, J.; Chen, W.; Zhang, F.; Gao, N.; Dong, M.; et al. Ccl3 enhances docetaxel chemosensitivity in breast cancer by triggering proinflammatory macrophage polarization. J. Immunother. Cancer 2022, 10, e003793. [Google Scholar] [CrossRef]
- Flieswasser, T.; Van Loenhout, J.; Boullosa, L.F.; Eynde, A.V.D.; De Waele, J.; Van Audenaerde, J.; Lardon, F.; Smits, E.; Pauwels, P.; Jacobs, J. Clinically Relevant Chemotherapeutics Have the Ability to Induce Immunogenic Cell Death in Non-Small Cell Lung Cancer. Cells 2020, 9, 1474. [Google Scholar] [CrossRef]
- Byrne, J.C.; Gabhann, J.N.; Stacey, K.B.; Coffey, B.M.; McCarthy, E.; Thomas, W.; Jefferies, C.A. Bruton’s Tyrosine Kinase Is Required for Apoptotic Cell Uptake via Regulating the Phosphorylation and Localization of Calreticulin. J. Immunol. 2013, 190, 5207–5215. [Google Scholar] [CrossRef]
- Mishra, A.K.; Ali, A.; Dutta, S.; Banday, S.; Malonia, S.K. Emerging Trends in Immunotherapy for Cancer. Diseases 2022, 10, 60. [Google Scholar] [CrossRef] [PubMed]
- Kortylewski, M.; Moreira, D. Myeloid cells as a target for oligonucleotide therapeutics: Turning obstacles into opportunities. Cancer Immunol. Immunother. 2017, 66, 979–988. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Zhang, Z.; Jiang, Y.; Zhang, D.; Chen, J.; Dong, L.; Zhang, J. Targeted delivery of oligonucleotides into tumor-associated macrophages for cancer immunotherapy. J. Control. Release 2012, 158, 286–292. [Google Scholar] [CrossRef] [PubMed]
- Tong, N.; He, Z.; Ma, Y.; Wang, Z.; Huang, Z.; Cao, H.; Xu, L.; Zou, Y.; Wang, W.; Yi, C.; et al. Tumor Associated Macrophages, as the Dominant Immune Cells, Are an Indispensable Target for Immunologically Cold Tumor—Glioma Therapy? Front. Cell Dev. Biol. 2021, 9, 706286. [Google Scholar] [CrossRef] [PubMed]
- Zou, L.; Tao, Y.; Payne, G.; Do, L.; Thomas, T.; Rodriguez, J.; Dou, H. Targeted delivery of nano-PTX to the brain tumor-associated macrophages. Oncotarget 2017, 8, 6564–6578. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.J.; Lin, Y.-C.; Yao, P.-L.; Yuan, A.; Chen, H.-Y.; Shun, C.-T.; Tsai, M.-F.; Chen, C.-H.; Yang, P.-C. Tumor-Associated Macrophages: The Double-Edged Sword in Cancer Progression. J. Clin. Oncol. 2005, 23, 953–964. [Google Scholar] [CrossRef]
- Boström, M.M.; Irjala, H.; Mirtti, T.; Taimen, P.; Kauko, T.; Ålgars, A.; Jalkanen, S.; Boström, P.J. Tumor-Associated Macrophages Provide Significant Prognostic Information in Urothelial Bladder Cancer. PLoS ONE 2015, 10, e0133552. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Targeting TAMs Strategies | Name | Targets | Cancer Types | Phases | Clin. Trial |
|---|---|---|---|---|---|
| Phagocytosis | RRX-001 | CD47, SIRPα | Non-small cell lung cancer | Phase III | NCT03699956 |
| Hu5F9-G4 | CD47 | Advanced tumors | Phase I | NCT02216409 | |
| JTX8064 | LILRB2 | Advanced refractory solid tumors | Phase I | NCT04669899 | |
| Depletion of M2-like TAMs | Zoledronate | NA | Mammary carcinoma | Phase III | NCT00320710 |
| TAMs recruitment | Pexidatintinib | CSF-1R | Advanced solid tumors | Phase III | NCT02371369 |
| D2923 | CSF-1R | Myelogenous leukemia | Phase II | NCT04989283 | |
| Emactuzumab | CSF-1R | Advanced solid tumors | Phase III | NCT05417789 | |
| 3D185 | CSF-1R | Colorectal cancer | Phase II | NCT05039892 | |
| PLX3397 (Pexidarnitib) | CSF-1R | Melanoma | Phase II | NCT02071940 | |
| CSF-1R | Advanced solid tumors | Phase I | NCT02734433 | ||
| CSF-1R | PVNS or GCT-TS | Phase III | NCT02371369 | ||
| CSF-1R | Leukemia, sarcoma, or neurofibroma | Phase I/II | NCT02390752 | ||
| CSF-1R | Acute myeloid leukemia | Phase I/II | NCT01349049 | ||
| PLX7486 (Plexxikon) | CSF-1R | Phase I | NCT01804530 | ||
| DCC-3014 | CSF-1R CSF-1R | Advanced-stage or metastatic solid tumors | Phase I | NCT03069469 | |
| ARRY-382 | Phase I | NCT01316822 | |||
| LY3022855 mAb (IMC-CS4) | CSF-1R | Solid tumors | Phase I | NCT02265536 | |
| Phase I | NCT01346358 | ||||
| AMG820 mAb | CSF-1R | Solid tumors | Phase I/II | NCT01444404 | |
| MLN1202 | CSF-1R | Bone metastasis | Phase I/II | NCT01015560 | |
| AMG820 mAb/Pembrolizumab | CSF-1R | Solid tumors | Phase I/II | NCT02713529 | |
| BLZ945/PRD001 | CSF-1R | Advanced solid tumors | Phase I/II | NCT02829723 | |
| Cabiralizumab/Nivolumab | CSF-1R | Advanced solid tumors | Phase I | NCT02526017 | |
| MLN1202 | CSF-1R | Bone metastasis | Phase I/II | NCT01015560 | |
| RO5509554/RG7155 (Emactuzumab)/Paclitaxel | CSF-1R | Advanced solid tumors | Phase I | NCT01494688 | |
| PD-0360324 mAb/Cyclophosphamide | CSF-1R | Ovarian cancer | Phase II | NCT02948101 | |
| Eribulin | CSF-1R | Metastatic breast cancer | Phase I/II | NCT01596751 | |
| PF-04136309 | CCR2 | Pancreatic cancer | Phase II | NCT02732938 | |
| CCX872 | CCR2 | Pancreatic cancer | Phase I | NCT02345408 | |
| CCR2i | CCR2 | Cutaneous t-cell lymphoma | Phase II | NCT02732938 | |
| mNOX-E36 | CCL2 | Glioblastoma | Phase I | NCT00976729 | |
| Carlumab (anti-CCL2 antibodies Centocor) | CCL2 | Prostate cancer | Phase II | NCT00992186 | |
| CNTO 888 (Carlumab) | CCR2 | Prostate cancer | Phase II | NCT00992186 | |
| PF-04136309 | CCR2 | Pancreatic cancer | Phase I/II | NCT02732938 | |
| TAMs reprogramming | R848 | TLR7/8 | Colorectal cancer | Phase II | NCT00960752 |
| lefitolimod | TLR9 | Small-cell lung cancer | Phase I | NCT02668770 | |
| RP6530 | PI3Kδ/γ | Hodgkin lymphoma | Phase I/II | NCT03770000 | |
| Cell-based therapies | CAR-M | HER2 | Solid cancers | Phase I | NCT04660929 |
| TEMFERON | Tie-2 | Glioblastoma multiforme | (Phase I/IIa) | NCT03866109 |
| CSF-1R Inhibitors + Checkpoint Immunotherapy | ||||
|---|---|---|---|---|
| Drug Name | Combination Drugs | Cancer Types | Phasess | Clin. Trials |
| PLX3397 (Pexidarnitib) | Pembrolizumab | Solid tumors | Phase I/II | NCT02452424 |
| Durvalumab | Advanced tumors | Phase I | NCT02777710 | |
| LY3022855 mAb (IMC-CS4) | Pembrolizumab | Pancreatic cancer | Phase I | NCT03153410 |
| Durvalumab | ||||
| Tremelimumab | Advanced solid tumors | Phase I | NCT02718911 | |
| RO5509554/RG7155 (Emactuzumab) | Atezolizumab | Solid tumors | Phase I | NCT02323191 |
| CSF-1R Inhibitors + Chemotherapy | ||||
| PLX3397 (Pexidarnitib) | Paclitaxel | Advanced solid tumors | Phase I/II | NCT01525602 |
| Standard Chemotherapy | NCT01042379 | |||
| CSF-1R Inhibitors + Targeted Therapy | ||||
| PLX3397 (Pexidarnitib) | Sirolimus (Rapamycin) | Sarcoma | NCT02584647 | |
| CSF-1R Inhibitors + Radiotherapy | ||||
| PLX3397 (Pexidarnitib) | RT + ADT | Prostate cancer | Phase I | NCT02472275 |
| RT + Temozolomide | Glioblastoma | Phase I/II | NCT01790503 | |
| CCR2/CCR5 Inhibitors + Checkpoint Immunotherapy | ||||
| BMS-813160 (CCR2/CCR5 antagonist) | Nivolumab/Nabpaclitaxel | Advanced solid tumors | Phase I/II | NCT03184870 |
| Nivolumab/iplimumab | Phase II | NCT0299611 | ||
| Nivolumab | Hepatocellular carcinoma | Phase II | NCT04123379 | |
| CCR5 antagonist | Pembrolizumab | CRC | Phase I | NCT03274804 |
| Nivolumab plus Ipilimumab | Pancreatic cancer, CRC | Phase I | NCT04721301 | |
| CCR2 Inhibitors + Chemotherapy | ||||
| CNTO 888 (Carlumab) | Gemcitabine/paclitaxel | Advanced solid tumors | Phase II | NCT01204996 |
| Carboplatin/doxorubicin | ||||
| PF-04136309 | FOLFIRINOX | Advanced solid tumors | Phase I/II | NCT01413022 |
| Anti-CD47/SIRPα antibodies+ Other immunotherapies | ||||
| Hu5F9-G4 | Pembrolizumab | Solid tumors | Phase I/II | NCT03869190 |
| Multiple immunotherapy | Urothelial and bladder cancer | Phase I/II | NCT03869190 | |
| BI 754,091 (OSE Immunotherapeutics) | BI 754,091 (anti-PD1) | Solid tumors | Phase I | NCT03990233 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mishra, A.K.; Banday, S.; Bharadwaj, R.; Ali, A.; Rashid, R.; Kulshreshtha, A.; Malonia, S.K. Macrophages as a Potential Immunotherapeutic Target in Solid Cancers. Vaccines 2023, 11, 55. https://doi.org/10.3390/vaccines11010055
Mishra AK, Banday S, Bharadwaj R, Ali A, Rashid R, Kulshreshtha A, Malonia SK. Macrophages as a Potential Immunotherapeutic Target in Solid Cancers. Vaccines. 2023; 11(1):55. https://doi.org/10.3390/vaccines11010055
Chicago/Turabian StyleMishra, Alok K., Shahid Banday, Ravi Bharadwaj, Amjad Ali, Romana Rashid, Ankur Kulshreshtha, and Sunil K. Malonia. 2023. "Macrophages as a Potential Immunotherapeutic Target in Solid Cancers" Vaccines 11, no. 1: 55. https://doi.org/10.3390/vaccines11010055
APA StyleMishra, A. K., Banday, S., Bharadwaj, R., Ali, A., Rashid, R., Kulshreshtha, A., & Malonia, S. K. (2023). Macrophages as a Potential Immunotherapeutic Target in Solid Cancers. Vaccines, 11(1), 55. https://doi.org/10.3390/vaccines11010055