

Introduction of an Ultraviolet C-Irradiated 4T1 Murine Breast Cancer Whole-Cell Vaccine Model

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culturing
2.2. Flow Cytometric MHCI Investigation
2.3. Whole-Cell Vaccine Generation
2.4. Resazurin Viability Assay
2.5. Flow Cytometric Viability Assay
2.6. Establishment of the 4T1 Tumor Model and Ethical License
2.7. Immunohistochemistry (IHC)
2.8. Adoptive Transfer Experiments
2.9. Detection of Anti-4T1 IgG Antibodies
2.10. Statistics
3. Results
3.1. UVC Irradiation of 4T1 Cells Used to Prepare a Whole-Cell Vaccine
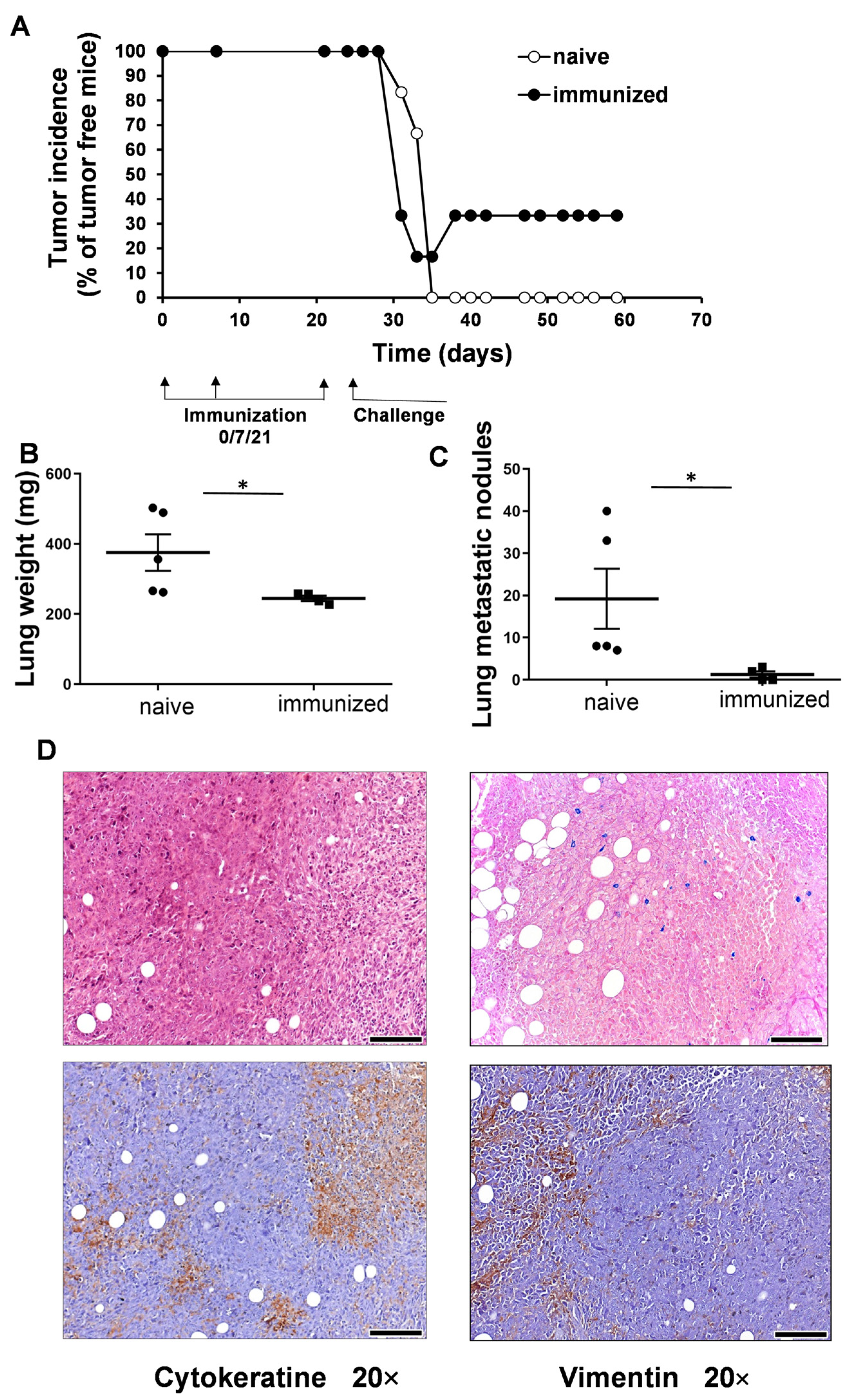
3.2. Vaccination with UVC-Irradiated 4T1 Whole-Cell Vaccine Resulted in Partial Remission and Lower Lung Metastatic Burden
3.3. Adoptive Transfer of Plasma or Splenocytes from Mice with Remission Endowed the Recipient Mice with Protective Immunity against Living 4T1 Cells
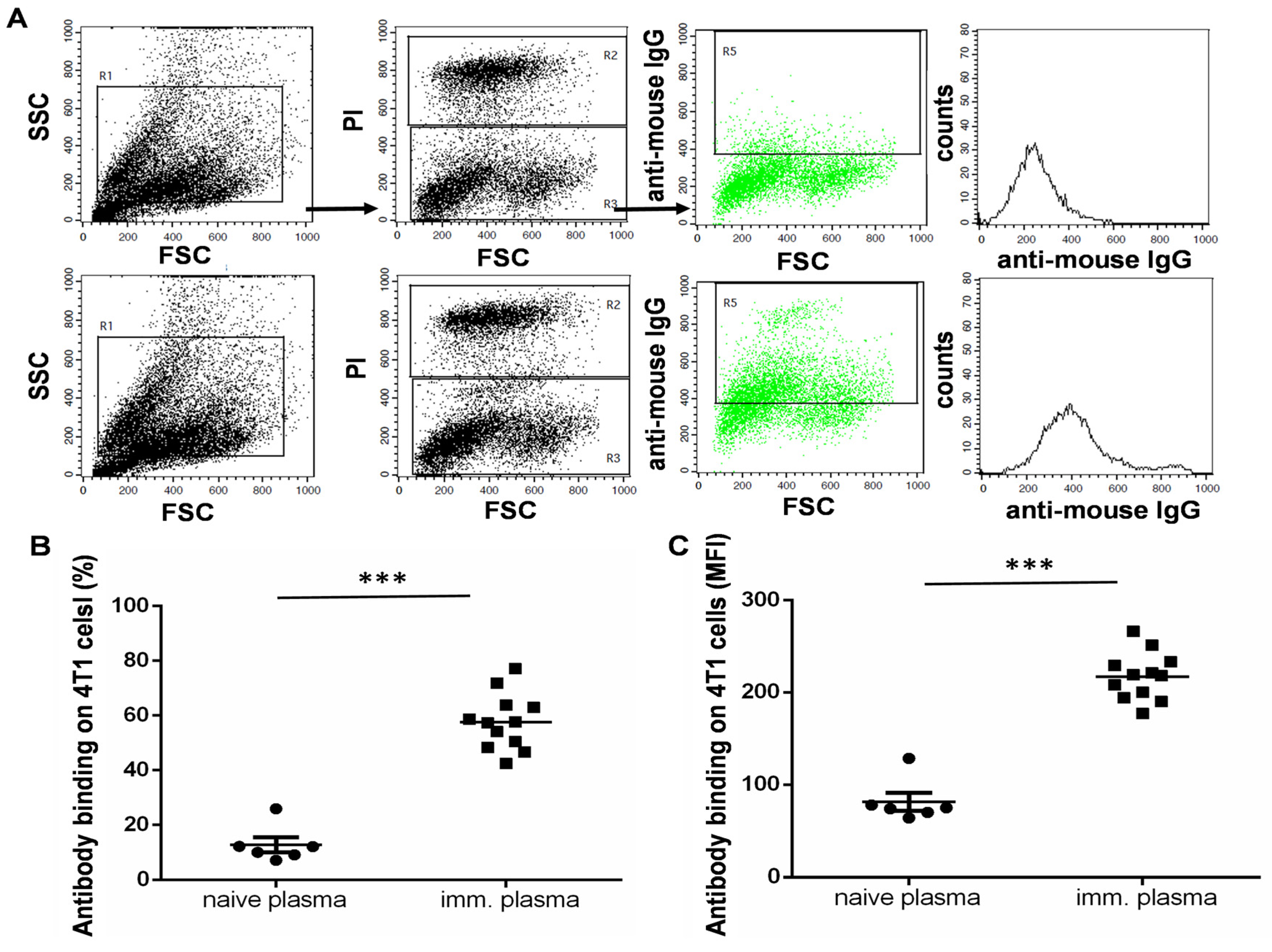
3.4. The UVC-Irradiated 4T1 Whole-Cell Vaccine Resulted in the Production of 4T1-Binding IgG Antibodies
3.5. Adoptive Transfer of Plasma or Splenocytes from the UVC-Irradiated 4T1-Whole-Cell-Vaccinated C57BL/6 Mice into Cyclophosphamide Pre-Treated Non-Syngeneic BALB/c Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Restifo, N.P.; Dudley, M.E.; Rosenberg, S.A. Adoptive immunotherapy for cancer: Harnessing the T cell response. Nat. Rev. Immunol. 2012, 12, 269–281. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Li, Y.; Zhu, B. T-cell exhaustion in the tumor microenvironment. Cell Death Dis. 2015, 6, e1792. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Liu, S.; Zhang, B.; Qiao, L.; Zhang, Y.; Zhang, Y. T Cell Dysfunction and Exhaustion in Cancer. Front. Cell Dev. Biol. 2020, 8, 17. [Google Scholar] [CrossRef]
- Kovacs-Solyom, F.; Blasko, A.; Fajka-Boja, R.; Katona, R.L.; Vegh, L.; Novak, J.; Szebeni, G.J.; Krenacs, L.; Uher, F.; Tubak, V.; et al. Mechanism of tumor cell-induced T-cell apoptosis mediated by galectin-1. Immunol. Lett. 2010, 127, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Ion, G.; Fajka-Boja, R.; Kovacs, F.; Szebeni, G.; Gombos, I.; Czibula, A.; Matko, J.; Monostori, E. Acid sphingomyelinase mediated release of ceramide is essential to trigger the mitochondrial pathway of apoptosis by galectin-1. Cell Signal. 2006, 18, 1887–1896. [Google Scholar] [CrossRef]
- Fajka-Boja, R.; Blasko, A.; Kovacs-Solyom, F.; Szebeni, G.J.; Toth, G.K.; Monostori, E. Co-localization of galectin-1 with GM1 ganglioside in the course of its clathrin- and raft-dependent endocytosis. Cell. Mol. Life Sci. 2008, 65, 2586–2593. [Google Scholar] [CrossRef]
- Li, C.; Jiang, P.; Wei, S.; Xu, X.; Wang, J. Regulatory T cells in tumor microenvironment: New mechanisms, potential therapeutic strategies and future prospects. Mol. Cancer 2020, 19, 116. [Google Scholar] [CrossRef]
- Scott, E.N.; Gocher, A.M.; Workman, C.J.; Vignali, D.A.A. Regulatory T Cells: Barriers of Immune Infiltration Into the Tumor Microenvironment. Front. Immunol. 2021, 12, 702726. [Google Scholar] [CrossRef]
- Devillier, R.; Chretien, A.S.; Pagliardini, T.; Salem, N.; Blaise, D.; Olive, D. Mechanisms of NK cell dysfunction in the tumor microenvironment and current clinical approaches to harness NK cell potential for immunotherapy. J. Leukoc. Biol. 2021, 109, 1071–1088. [Google Scholar] [CrossRef]
- Zhang, W.; Zhao, Z.; Li, F. Natural killer cell dysfunction in cancer and new strategies to utilize NK cell potential for cancer immunotherapy. Mol. Immunol. 2022, 144, 58–70. [Google Scholar] [CrossRef]
- Strauss, L.; Sangaletti, S.; Consonni, F.M.; Szebeni, G.; Morlacchi, S.; Totaro, M.G.; Porta, C.; Anselmo, A.; Tartari, S.; Doni, A.; et al. RORC1 Regulates Tumor-Promoting "Emergency" Granulo-Monocytopoiesis. Cancer Cell 2015, 28, 253–269. [Google Scholar] [CrossRef]
- Hegde, S.; Leader, A.M.; Merad, M. MDSC: Markers, development, states, and unaddressed complexity. Immunity 2021, 54, 875–884. [Google Scholar] [CrossRef] [PubMed]
- Szebeni, G.J.; Vizler, C.; Nagy, L.I.; Kitajka, K.; Puskas, L.G. Pro-Tumoral Inflammatory Myeloid Cells as Emerging Therapeutic Targets. Int. J. Mol. Sci. 2016, 17, 1958. [Google Scholar] [CrossRef] [PubMed]
- Szebeni, G.J.; Vizler, C.; Kitajka, K.; Puskas, L.G. Inflammation and Cancer: Extra- and Intracellular Determinants of Tumor-Associated Macrophages as Tumor Promoters. Mediat. Inflamm. 2017, 2017, 9294018. [Google Scholar] [CrossRef] [PubMed]
- Taylor, B.C.; Balko, J.M. Mechanisms of MHC-I Downregulation and Role in Immunotherapy Response. Front. Immunol. 2022, 13, 844866. [Google Scholar] [CrossRef]
- Escors, D. Tumour immunogenicity, antigen presentation and immunological barriers in cancer immunotherapy. New J. Sci. 2014, 2014, 734515. [Google Scholar] [CrossRef]
- Zagorulya, M.; Duong, E.; Spranger, S. Impact of anatomic site on antigen-presenting cells in cancer. J. Immunother. Cancer 2020, 8, e001204. [Google Scholar] [CrossRef]
- Jeong, S.; Park, S.H. Co-Stimulatory Receptors in Cancers and Their Implications for Cancer Immunotherapy. Immune Netw. 2020, 20, e3. [Google Scholar] [CrossRef]
- Tan, R.; Nie, M.; Long, W. The role of B cells in cancer development. Front. Oncol. 2022, 12, 958756. [Google Scholar] [CrossRef]
- Wu, H.; Chen, C.; Gu, L.; Li, J.; Yue, Y.; Lyu, M.; Cui, Y.; Zhang, X.; Liu, Y.; Zhu, H.; et al. B cell deficiency promotes the initiation and progression of lung cancer. Front. Oncol. 2022, 12, 1006477. [Google Scholar] [CrossRef]
- Balog, J.A.; Hackler, L., Jr.; Kovacs, A.K.; Neuperger, P.; Alfoldi, R.; Nagy, L.I.; Puskas, L.G.; Szebeni, G.J. Single Cell Mass Cytometry Revealed the Immunomodulatory Effect of Cisplatin Via Downregulation of Splenic CD44+, IL-17A+ MDSCs and Promotion of Circulating IFN-gamma+ Myeloid Cells in the 4T1 Metastatic Breast Cancer Model. Int. J. Mol. Sci. 2019, 21, 170. [Google Scholar] [CrossRef]
- Kerr, W.G.; Chisholm, J.D. The Next Generation of Immunotherapy for Cancer: Small Molecules Could Make Big Waves. J. Immunol. 2019, 202, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Yang, Z.; Cheng, K.; Bi, H.; Chen, J. Small molecule-based immunomodulators for cancer therapy. Acta Pharm. Sin. B 2022, 12, 4287–4308. [Google Scholar] [CrossRef] [PubMed]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.; Reckamp, K.L.; Baas, P.; Crino, L.; Eberhardt, W.E.; Poddubskaya, E.; Antonia, S.; Pluzanski, A.; Vokes, E.E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Squamous-Cell Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Baas, P.; Kim, D.W.; Felip, E.; Perez-Gracia, J.L.; Han, J.Y.; Molina, J.; Kim, J.H.; Arvis, C.D.; Ahn, M.J.; et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): A randomised controlled trial. Lancet 2016, 387, 1540–1550. [Google Scholar] [CrossRef]
- Hopkins, A.C.; Yarchoan, M.; Durham, J.N.; Yusko, E.C.; Rytlewski, J.A.; Robins, H.S.; Laheru, D.A.; Le, D.T.; Lutz, E.R.; Jaffee, E.M. T cell receptor repertoire features associated with survival in immunotherapy-treated pancreatic ductal adenocarcinoma. JCI Insight 2018, 3, e122092. [Google Scholar] [CrossRef]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef] [PubMed]
- Shiravand, Y.; Khodadadi, F.; Kashani, S.M.A.; Hosseini-Fard, S.R.; Hosseini, S.; Sadeghirad, H.; Ladwa, R.; O’Byrne, K.; Kulasinghe, A. Immune Checkpoint Inhibitors in Cancer Therapy. Curr. Oncol. 2022, 29, 3044–3060. [Google Scholar] [CrossRef]
- Tan, S.; Day, D.; Nicholls, S.J.; Segelov, E. Immune Checkpoint Inhibitor Therapy in Oncology: Current Uses and Future Directions: JACC: CardioOncology State-of-the-Art Review. JACC CardioOncol. 2022, 4, 579–597. [Google Scholar] [CrossRef]
- Alnefaie, A.; Albogami, S.; Asiri, Y.; Ahmad, T.; Alotaibi, S.S.; Al-Sanea, M.M.; Althobaiti, H. Chimeric Antigen Receptor T-Cells: An Overview of Concepts, Applications, Limitations, and Proposed Solutions. Front. Bioeng. Biotechnol. 2022, 10, 797440. [Google Scholar] [CrossRef] [PubMed]
- Yan, T.; Zhu, L.; Chen, J. Current advances and challenges in CAR T-Cell therapy for solid tumors: Tumor-associated antigens and the tumor microenvironment. Exp. Hematol. Oncol. 2023, 12, 14. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Meng, Y.; Feng, X.; Han, Z. CAR-NK cells for cancer immunotherapy: From bench to bedside. Biomark. Res. 2022, 10, 12. [Google Scholar] [CrossRef]
- Circelli, L.; Tornesello, M.; Buonaguro, F.M.; Buonaguro, L. Use of adjuvants for immunotherapy. Hum. Vaccin Immunother. 2017, 13, 1774–1777. [Google Scholar] [CrossRef]
- Diao, L.; Liu, M. Rethinking Antigen Source: Cancer Vaccines Based on Whole Tumor Cell/tissue Lysate or Whole Tumor Cell. Adv. Sci. 2023, e2300121. [Google Scholar] [CrossRef]
- Chen, Y.; Guo, S.S.; Zhu, W. Autologous whole-cell cancer vaccination: Cryogenic silicified vaccines. Matter UK 2022, 5, 2434–2436. [Google Scholar] [CrossRef]
- Najafabadi, S.A.S.; Bolhassani, A.; Aghasadeghi, M.R. Tumor cell-based vaccine: An effective strategy for eradication of cancer cells. Immunother. UK 2022, 14, 639–654. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Fu, M.; Wang, M.; Wan, D.; Wei, Y.; Wei, X. Cancer vaccines as promising immuno-therapeutics: Platforms and current progress. J. Hematol. Oncol. 2022, 15, 28. [Google Scholar] [CrossRef]
- Lin, M.J.; Svensson-Arvelund, J.; Lubitz, G.S.; Marabelle, A.; Melero, I.; Brown, B.D.; Brody, J.D. Cancer vaccines: The next immunotherapy frontier. Nat. Cancer 2022, 3, 911–926. [Google Scholar] [CrossRef]
- Grimmett, E.; Al-Share, B.; Alkassab, M.B.; Zhou, R.W.; Desai, A.; Rahim, M.M.A.; Woldie, I. Cancer vaccines: Past, present and future; a review article. Discov. Oncol. 2022, 13, 31. [Google Scholar] [CrossRef]
- Hu, H.G.; Li, Y.M. Emerging Adjuvants for Cancer Immunotherapy. Front. Chem. 2020, 8, 601. [Google Scholar] [CrossRef]
- Cuzzubbo, S.; Mangsbo, S.; Nagarajan, D.; Habra, K.; Pockley, A.G.; McArdle, S.E.B. Cancer Vaccines: Adjuvant Potency, Importance of Age, Lifestyle, and Treatments. Front. Immunol. 2020, 11, 615240. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yang, B.; Ni, Q.; Chen, X. Materials engineering strategies for cancer vaccine adjuvant development. Chem. Soc. Rev. 2023, 52, 2886–2910. [Google Scholar] [CrossRef] [PubMed]
- Stronen, E.; Toebes, M.; Kelderman, S.; van Buuren, M.M.; Yang, W.; van Rooij, N.; Donia, M.; Boschen, M.L.; Lund-Johansen, F.; Olweus, J.; et al. Targeting of cancer neoantigens with donor-derived T cell receptor repertoires. Science 2016, 352, 1337–1341. [Google Scholar] [CrossRef] [PubMed]
- Matera, L.; Garetto, S. Cross-presentation of tumor antigens is increased by UVC light tumor treatment. Clin. Cancer Res. 2009, 15, 7447. [Google Scholar] [CrossRef]
- Gorelik, E.; Begovic, M.; Duty, L.; Herberman, R.B. Effect of ultraviolet irradiation on MCA102 tumor cell immunogenicity and sensitivity to tumor necrosis factor. Cancer Res. 1991, 51, 1521–1528. [Google Scholar]
- Gullo, C.A.; Hwang, W.Y.; Poh, C.K.; Au, M.; Cow, G.; Teoh, G. Use of ultraviolet-light irradiated multiple myeloma cells as immunogens to generate tumor-specific cytolytic T lymphocytes. J. Immune Based Ther. Vaccines 2008, 6, 2. [Google Scholar] [CrossRef]
- Chiang, C.L.; Coukos, G.; Kandalaft, L.E. Whole Tumor Antigen Vaccines: Where Are We? Vaccines 2015, 3, 344–372. [Google Scholar] [CrossRef]
- Zhang, Y.; Palmer, G.H.; Abbott, J.R.; Howard, C.J.; Hope, J.C.; Brown, W.C. CpG ODN 2006 and IL-12 are comparable for priming Th1 lymphocyte and IgG responses in cattle immunized with a rickettsial outer membrane protein in alum. Vaccine 2003, 21, 3307–3318. [Google Scholar] [CrossRef]
- Conforti, V.A.; de Avila, D.M.; Cummings, N.S.; Wells, K.J.; Ulker, H.; Reeves, J.J. The effectiveness of a CpG motif-based adjuvant (CpG ODN 2006) for LHRH immunization. Vaccine 2007, 25, 6537–6543. [Google Scholar] [CrossRef]
- Szebeni, G.J.; Balog, J.A.; Demjen, A.; Alfoldi, R.; Vegi, V.L.; Feher, L.Z.; Man, I.; Kotogany, E.; Guban, B.; Batar, P.; et al. Imidazo[1,2-b]pyrazole-7-carboxamides Induce Apoptosis in Human Leukemia Cells at Nanomolar Concentrations. Molecules 2018, 23, 2845. [Google Scholar] [CrossRef]
- Demjen, A.; Alfoldi, R.; Angyal, A.; Gyuris, M.; Hackler, L., Jr.; Szebeni, G.J.; Wolfling, J.; Puskas, L.G.; Kanizsai, I. Synthesis, cytotoxic characterization, and SAR study of imidazo[1,2-b]pyrazole-7-carboxamides. Arch. Pharm. 2018, 351, e1800062. [Google Scholar] [CrossRef]
- Gemes, N.; Makra, Z.; Neuperger, P.; Szabo, E.; Balog, J.A.; Flink, L.B.; Kari, B.; Hackler, L., Jr.; Puskas, L.G.; Kanizsai, I.; et al. A cytotoxic survey on 2-amino-1H-imidazol based synthetic marine sponge alkaloid analogues. Drug Dev. Res. 2022, 83, 1906–1922. [Google Scholar] [CrossRef]
- Kulmany, A.E.; Frank, E.; Papp, D.; Szekeres, A.; Szebeni, G.J.; Zupko, I. Biological evaluation of antiproliferative and anti-invasive properties of an androstadiene derivative on human cervical cancer cell lines. J. Steroid Biochem. Mol. Biol. 2021, 214, 105990. [Google Scholar] [CrossRef] [PubMed]
- Szebeni, G.J.; Kriston-Pal, E.; Blazso, P.; Katona, R.L.; Novak, J.; Szabo, E.; Czibula, A.; Fajka-Boja, R.; Hegyi, B.; Uher, F.; et al. Identification of galectin-1 as a critical factor in function of mouse mesenchymal stromal cell-mediated tumor promotion. PLoS ONE 2012, 7, e41372. [Google Scholar] [CrossRef]
- Puskas, L.G.; Man, I.; Szebeni, G.; Tiszlavicz, L.; Tsai, S.; James, M.A. Novel Anti-CRR9/CLPTM1L Antibodies with Antitumorigenic Activity Inhibit Cell Surface Accumulation, PI3K Interaction, and Survival Signaling. Mol. Cancer Ther. 2016, 15, 985–997. [Google Scholar] [CrossRef]
- Neuperger, P.; Balog, J.A.; Tiszlavicz, L.; Furak, J.; Gemes, N.; Kotogany, E.; Szalontai, K.; Puskas, L.G.; Szebeni, G.J. Analysis of the Single-Cell Heterogeneity of Adenocarcinoma Cell Lines and the Investigation of Intratumor Heterogeneity Reveals the Expression of Transmembrane Protein 45A (TMEM45A) in Lung Adenocarcinoma Cancer Patients. Cancers 2021, 14, 144. [Google Scholar] [CrossRef]
- Liu, X.; Li, J.; Cadilha, B.L.; Markota, A.; Voigt, C.; Huang, Z.; Lin, P.P.; Wang, D.D.; Dai, J.; Kranz, G.; et al. Epithelial-type systemic breast carcinoma cells with a restricted mesenchymal transition are a major source of metastasis. Sci. Adv. 2019, 5, eaav4275. [Google Scholar] [CrossRef] [PubMed]
- Takai, K.; Le, A.; Weaver, V.M.; Werb, Z. Targeting the cancer-associated fibroblasts as a treatment in triple-negative breast cancer. Oncotarget 2016, 7, 82889–82901. [Google Scholar] [CrossRef]
- Hicks, A.M.; Riedlinger, G.; Willingham, M.C.; Alexander-Miller, M.A.; Von Kap-Herr, C.; Pettenati, M.J.; Sanders, A.M.; Weir, H.M.; Du, W.; Kim, J.; et al. Transferable anticancer innate immunity in spontaneous regression/complete resistance mice. Proc. Natl. Acad. Sci. USA 2006, 103, 7753–7758. [Google Scholar] [CrossRef] [PubMed]
- Carmi, Y.; Spitzer, M.H.; Linde, I.L.; Burt, B.M.; Prestwood, T.R.; Perlman, N.; Davidson, M.G.; Kenkel, J.A.; Segal, E.; Pusapati, G.V.; et al. Allogeneic IgG combined with dendritic cell stimuli induce antitumour T-cell immunity. Nature 2015, 521, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Lo Nigro, C.; Macagno, M.; Sangiolo, D.; Bertolaccini, L.; Aglietta, M.; Merlano, M.C. NK-mediated antibody-dependent cell-mediated cytotoxicity in solid tumors: Biological evidence and clinical perspectives. Ann. Transl. Med. 2019, 7, 105. [Google Scholar] [CrossRef] [PubMed]
- Morotti, M.; Albukhari, A.; Alsaadi, A.; Artibani, M.; Brenton, J.D.; Curbishley, S.M.; Dong, T.; Dustin, M.L.; Hu, Z.; McGranahan, N.; et al. Promises and challenges of adoptive T-cell therapies for solid tumours. Br. J. Cancer 2021, 124, 1759–1776. [Google Scholar] [CrossRef]
- Li, Q.; Lao, X.; Pan, Q.; Ning, N.; Yet, J.; Xu, Y.; Li, S.; Chang, A.E. Adoptive transfer of tumor reactive B cells confers host T-cell immunity and tumor regression. Clin. Cancer Res. 2011, 17, 4987–4995. [Google Scholar] [CrossRef] [PubMed]
- Bracci, L.; Moschella, F.; Sestili, P.; La Sorsa, V.; Valentini, M.; Canini, I.; Baccarini, S.; Maccari, S.; Ramoni, C.; Belardelli, F.; et al. Cyclophosphamide enhances the antitumor efficacy of adoptively transferred immune cells through the induction of cytokine expression, B-cell and T-cell homeostatic proliferation, and specific tumor infiltration. Clin. Cancer Res. 2007, 13, 644–653. [Google Scholar] [CrossRef] [PubMed]
- Hughes, E.; Scurr, M.; Campbell, E.; Jones, E.; Godkin, A.; Gallimore, A. T-cell modulation by cyclophosphamide for tumour therapy. Immunology 2018, 154, 62–68. [Google Scholar] [CrossRef]
- Luznik, L.; Jones, R.J.; Fuchs, E.J. High-dose cyclophosphamide for graft-versus-host disease prevention. Curr. Opin. Hematol. 2010, 17, 493–499. [Google Scholar] [CrossRef]
- Nash, A.M.; Jarvis, M.I.; Aghlara-Fotovat, S.; Mukherjee, S.; Hernandez, A.; Hecht, A.D.; Rios, P.D.; Ghani, S.; Joshi, I.; Isa, D.; et al. Clinically translatable cytokine delivery platform for eradication of intraperitoneal tumors. Sci. Adv. 2022, 8, eabm1032. [Google Scholar] [CrossRef]
- West, E.E.; Jin, H.T.; Rasheed, A.U.; Penaloza-Macmaster, P.; Ha, S.J.; Tan, W.G.; Youngblood, B.; Freeman, G.J.; Smith, K.A.; Ahmed, R. PD-L1 blockade synergizes with IL-2 therapy in reinvigorating exhausted T cells. J. Clin. Investig. 2013, 123, 2604–2615. [Google Scholar] [CrossRef]
- Schwaab, T.; Schwarzer, A.; Wolf, B.; Crocenzi, T.S.; Seigne, J.D.; Crosby, N.A.; Cole, B.F.; Fisher, J.L.; Uhlenhake, J.C.; Mellinger, D.; et al. Clinical and immunologic effects of intranodal autologous tumor lysate-dendritic cell vaccine with Aldesleukin (Interleukin 2) and IFN-alpha2a therapy in metastatic renal cell carcinoma patients. Clin. Cancer Res. 2009, 15, 4986–4992. [Google Scholar] [CrossRef]
- Saberian, C.; Amaria, R.N.; Najjar, A.M.; Radvanyi, L.G.; Haymaker, C.L.; Forget, M.A.; Bassett, R.L.; Faria, S.C.; Glitza, I.C.; Alvarez, E.; et al. Randomized phase II trial of lymphodepletion plus adoptive cell transfer of tumor-infiltrating lymphocytes, with or without dendritic cell vaccination, in patients with metastatic melanoma. J. Immunother. Cancer 2021, 9. [Google Scholar] [CrossRef]
- Flesner, B.K.; Wood, G.W.; Gayheart-Walsten, P.; Sonderegger, F.L.; Henry, C.J.; Tate, D.J.; Bechtel, S.M.; Donnelly, L.L.; Johnson, G.C.; Kim, D.Y.; et al. Autologous cancer cell vaccination, adoptive T-cell transfer, and interleukin-2 administration results in long-term survival for companion dogs with osteosarcoma. J. Vet. Intern. Med. 2020, 34, 2056–2067. [Google Scholar] [CrossRef] [PubMed]
- Reilley, M.J.; Morrow, B.; Ager, C.R.; Liu, A.; Hong, D.S.; Curran, M.A. TLR9 activation cooperates with T cell checkpoint blockade to regress poorly immunogenic melanoma. J. Immunother. Cancer 2019, 7, 323. [Google Scholar] [CrossRef] [PubMed]
- Arrieta, A.; Galvis, A.E.; Osborne, S.; Morphew, T.; Imfeld, K.; Enriquez, C.; Hoang, J.; Swearingen, M.; Nieves, D.J.; Ashouri, N.; et al. Use of COVID-19 Convalescent Plasma for Treatment of Symptomatic SARS-CoV-2 Infection at a Children’s Hospital: A Contribution to a Still Inadequate Body of Evidence. Children 2023, 10, 350. [Google Scholar] [CrossRef] [PubMed]
- Marconato, M.; Abela, I.A.; Hauser, A.; Schwarzmuller, M.; Katzensteiner, R.; Braun, D.L.; Epp, S.; Audige, A.; Weber, J.; Rusert, P.; et al. Antibodies from convalescent plasma promote SARS-CoV-2 clearance in individuals with and without endogenous antibody response. J. Clin. Investig. 2022, 132, e158190. [Google Scholar] [CrossRef]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szebeni, G.J.; Alföldi, R.; Nagy, L.I.; Neuperger, P.; Gémes, N.; Balog, J.Á.; Tiszlavicz, L.; Puskás, L.G. Introduction of an Ultraviolet C-Irradiated 4T1 Murine Breast Cancer Whole-Cell Vaccine Model. Vaccines 2023, 11, 1254. https://doi.org/10.3390/vaccines11071254
Szebeni GJ, Alföldi R, Nagy LI, Neuperger P, Gémes N, Balog JÁ, Tiszlavicz L, Puskás LG. Introduction of an Ultraviolet C-Irradiated 4T1 Murine Breast Cancer Whole-Cell Vaccine Model. Vaccines. 2023; 11(7):1254. https://doi.org/10.3390/vaccines11071254
Chicago/Turabian StyleSzebeni, Gábor J., Róbert Alföldi, Lajos I. Nagy, Patrícia Neuperger, Nikolett Gémes, József Á. Balog, László Tiszlavicz, and László G. Puskás. 2023. "Introduction of an Ultraviolet C-Irradiated 4T1 Murine Breast Cancer Whole-Cell Vaccine Model" Vaccines 11, no. 7: 1254. https://doi.org/10.3390/vaccines11071254
APA StyleSzebeni, G. J., Alföldi, R., Nagy, L. I., Neuperger, P., Gémes, N., Balog, J. Á., Tiszlavicz, L., & Puskás, L. G. (2023). Introduction of an Ultraviolet C-Irradiated 4T1 Murine Breast Cancer Whole-Cell Vaccine Model. Vaccines, 11(7), 1254. https://doi.org/10.3390/vaccines11071254