
Canvassing Prospects of Glyco-Nanovaccines for Developing Cross-Presentation Mediated Anti-Tumor Immunotherapy



Abstract
1. Getting Familiar with the Concept of ‘Glyconanovaccine’
How Did We Get Here?
2. At a Glance
2.1. Antigen Presentation
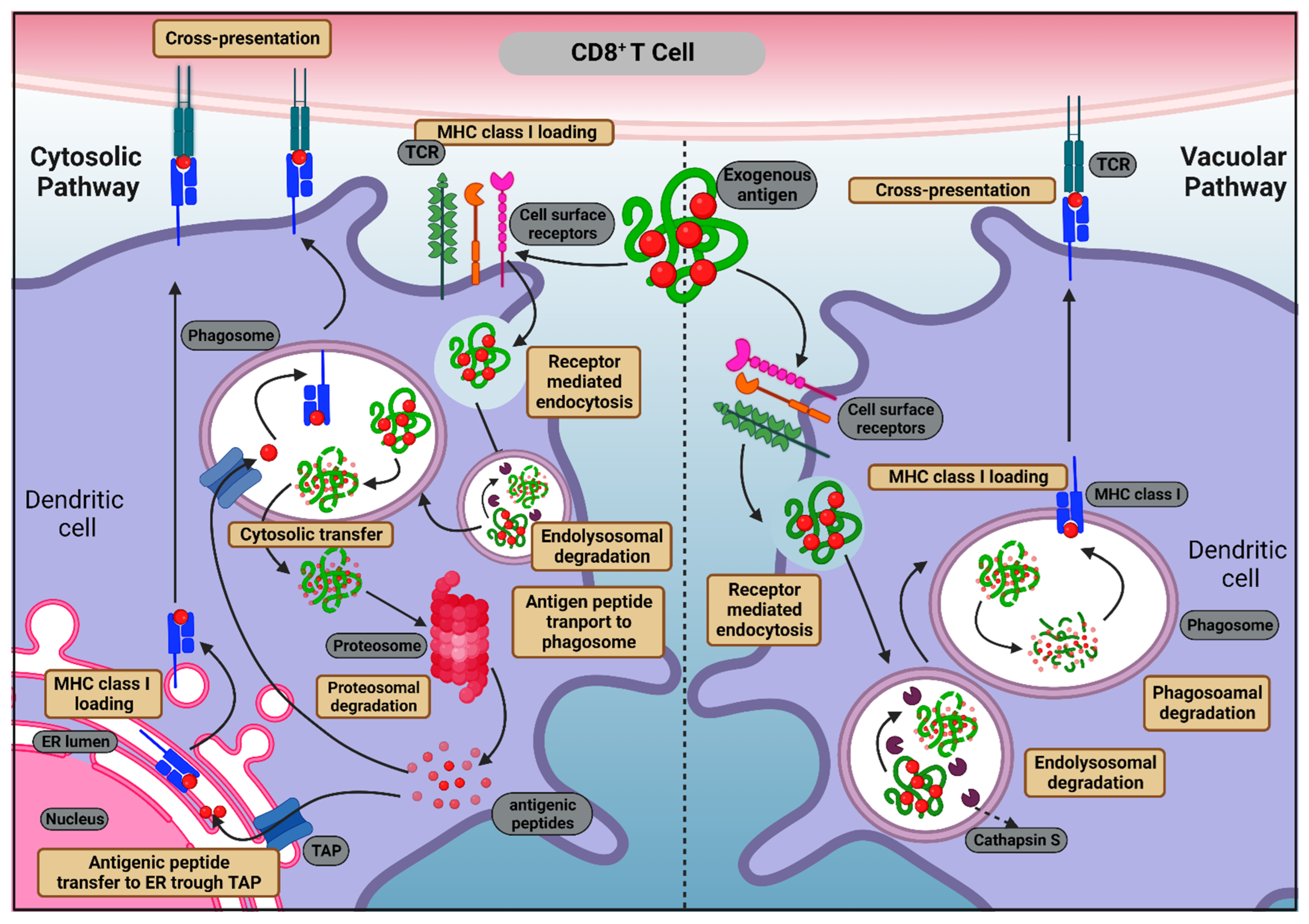
2.1.1. MHC Class I Antigen Presentation
2.1.2. MHC Class II Antigen Presentation
3. Background to The Subject Matter
3.1. Antigen Cross-Presentation
3.2. Conversing the Significance of Cross-Presentation
3.3. Comparing Different APCs Based on the Ability to Cross-Present
4. Biology of Human DCs and Role of DC Subsets in Cross-Presentation
Molecular Basis of Cross-Presentation Efficiency in Steady State DC Subsets
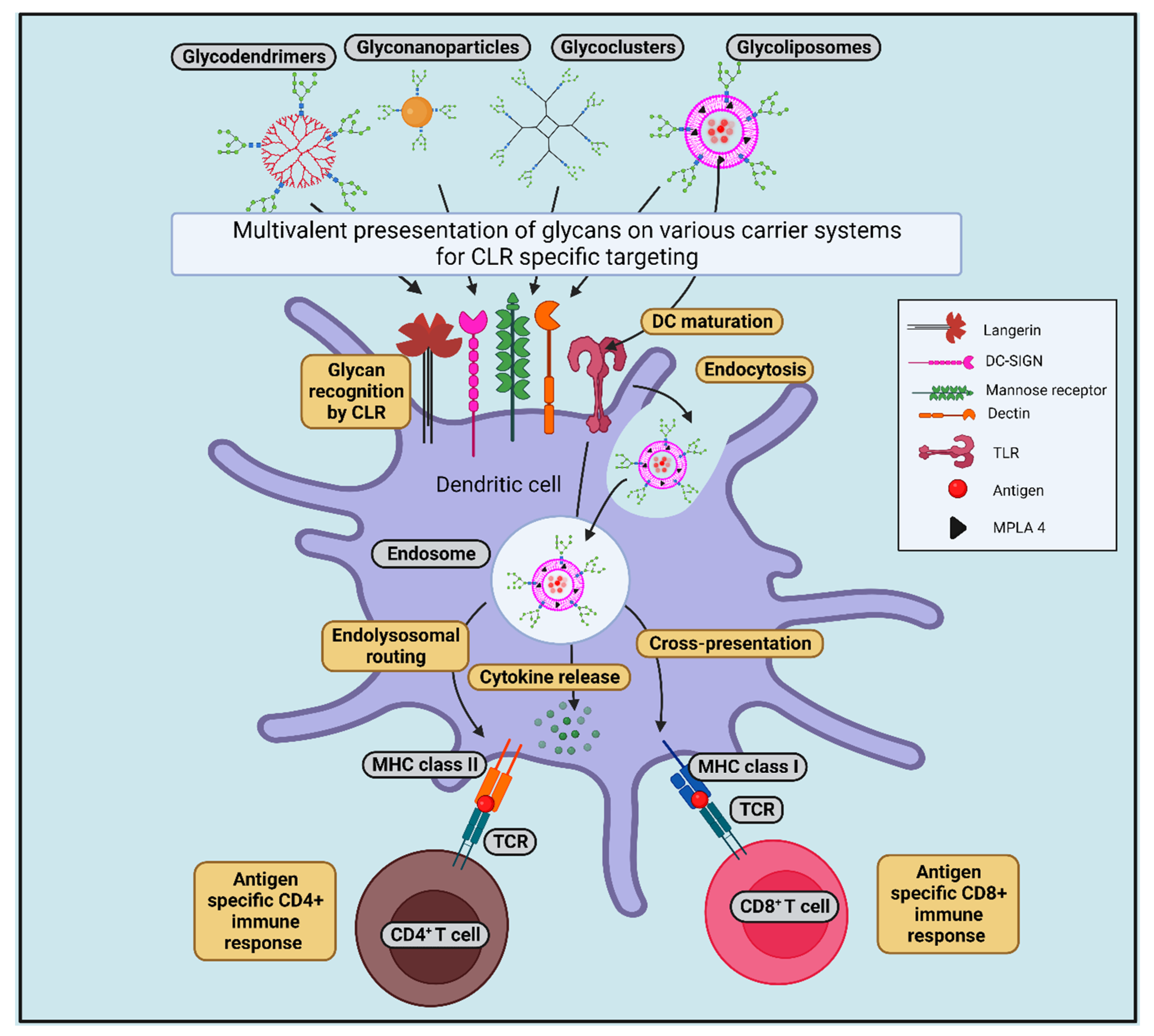
5. C-Type Lectin Receptors in Cross-Presentation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibodies Mediated CLR Targeting | GNV-Mediated CLR Targeting | Reference(s) | |
|---|---|---|---|
| Delivery to endosome type | Early or late endosome | Early endosome/endo-lysosomal compartments | [111,121,122] |
| Mode of Ag presentation | Cross-presentation | Cross-presentation | [111,123] |
| Mode of internalization into the cell | Endocytic receptor-mediated internalization | Internalized into the cell via CLR-mediated endocytosis | [2,103] |
| Type of interactions and affinity | Antibody-mediated interactions are monovalent and of high affinity | Multivalent glycan display for CLR-targeting and low affinity | [124] |
| Immunogenicity | Anti-CLR antibodies evoke immune response which leads to their elimination | Negligible immune response generated against glycans | [125] |
5.1. C-Type Lectin Receptors and Their Glycan Preferences
5.2. Toll-Like Receptors (TLRs), Adjuvant Activity and Their Role in Enhancing Cross-Presentation
| CLRs and Their Synonyms | Expression/DC Subtype | Ligands | Cross-Presentation Activity | Humoral and Cellular Response | References |
|---|---|---|---|---|---|
| DEC-205/ CD205 | Expressed by thymic epithelial cells, subsets of DCs (peripheral DCs, splenic/lymph node DCs, dermal/interstitial DCs, and LCs); homologous to MR family | Apoptotic and necrotic cell-derived antigens, CpG oligonucleotides | Effective cross-presentation of tumor- or pathogen-derived antigens | Induce efficient cellular (CD4+ and CD8+ T cell) and humoral responses; however, DCs activation by adjuvants required | [119,146,147,148] |
| Dectin-1/CD369 | Expressed by human monocytes, macrophages, DCs; mouse cDC2 | β-glucans (with β-1,3 and/or β-1,6-linked glucans) | Uptake and cross-present cellular antigens | Strong CD4+ T cell response but weak CD8+ T cell response | [149,150,151,152,153,154] |
| DC-SIGN/ CD209 | Expressed by moDCs and dermal CD14+ DCs | High-mannose- and Fucose-containing glycans, Lewis antigens | Antigen targeting to DC-SIGN leads to cross-presentation | Strong CD4+ and CD8+ T cell response | [125,155,156,157,158] |
| Langerin/CD207 | Highly expressed by LCs, dermal DCs in both mice and humans | Mannose, fucose, N-Acetylglucosamine (GlcNAc), β-glucans | Langerin mediated cross-presentation in LCs | Induce humoral response and CD8+ T cell activation | [159,160,161,162,163] |
| MR/ CD206 | Macrophages, human moDCs, mouse BMDCs | Glycoconjugates terminated with mannose, fucose, or GlcNAc. Affinity towards sulfated glycans is also present | MR-mediated targeting of antigens, directs antigens to early endosomes and leads to cross-presentation | Targeting MR elicit strong cellular and humoral immune response | [164,165,166,167] |
| MGL/ CD301 | DCs, macrophages, dDCs, murine pDC | Terminal GalNAc, Tn antigen (α–GalNAc), glycan antigen LDN, sialyl-Tn | MGL1 mediates TLR signaling independent of cross-presentation | MGL2 targeting induces Th2 skewed humoral response, Th1 skewing of CD4+ T cells and enhanced CD8+ T cell priming by glycan-modified antigen targeting | [133,168,169,170] |
| DCIR/ CD367 | DCs, monocytes, neutrophils, B cells and activated T cells | Mannotriose, Lea, Leb, and Sulfo- Lea | DCIR-mediated antigen targeting leads to cross-presentation | DCIR targeting induces CD8+ T cell response | [134,171] |
6. DC-Based Immunotherapies versus DC-Targeted Cross-Presentation
7. Orienting Antigen towards Cross-Presentation
8. Using Nanovehicles for Tumor Antigen Cross-Presentation
8.1. Glycan-Conjugated Nanovaccines Targeting DCs
8.2. Targeting CLRs by Glycans (in the Form of Glycosylated Antigens/Glycan-Modified Nanocarrier) to Induce Cross-Presentation
9. Parameters to Be Considered for Developing GNVs
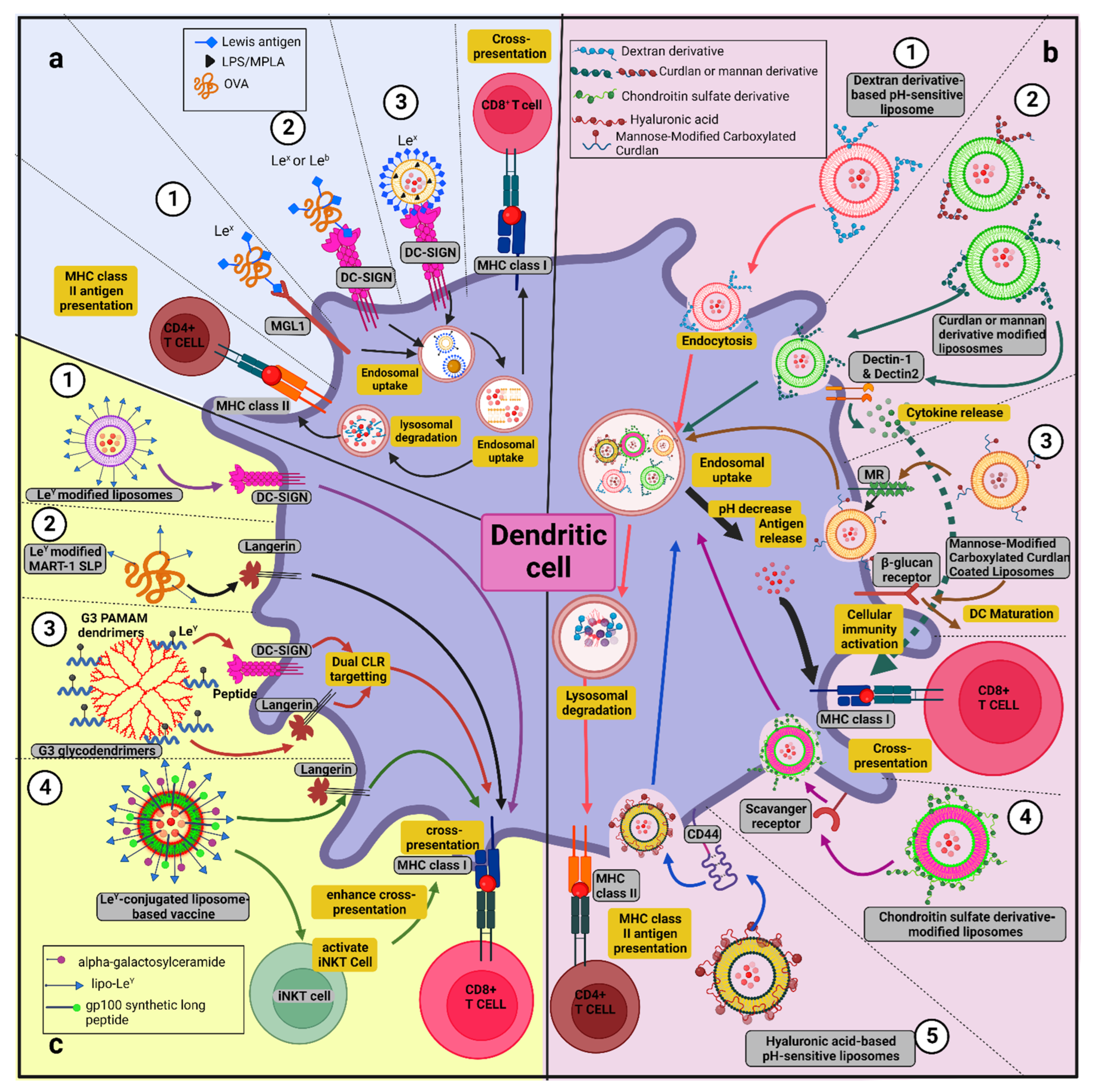
9.1. Types and Fabrication Strategies of GNVs and Their Associated Benefits in Anti-Tumor Immunotherapy
9.1.1. Glycoliposomes
9.1.2. pH-Sensitive Glycan-Modified Liposomes
9.1.3. Glycodendrimer
9.1.4. Glyconanoparticles
9.1.5. Glycan Conjugated PLGA Nanoparticle
9.1.6. Glyco-Clusters
| GNVs | Fabrication Strategy Used to Prepare GNVs | Benefits of GNV Mediated Targeting of DCs | Reference(s) |
|---|---|---|---|
| Glyco-liposome | Thio-activated glycans (Lex and Leb) were coupled to liposomes encapsulating OVA/MART-1 peptides via thiol–ene reaction with maleimide groups of MBP-PE. | Enhanced binding and internalization by human DC-SIGN-expressing BMDCs; 100-fold efficient antigen presentation was observed in the presence of LPS. | [158] |
| Inclusion of TLR ligands (MPLA, Pam3CysSK4, R484, and Poly I:C) in glycan-modified liposomes encapsulating gp100 antigenic peptide | Inclusion of TLR4 ligand MPLA induced DC maturation, pro-inflammatory cytokine production, and significantly enhanced cross-presentation. | [195] | |
| Inclusion of αGC as NKT cell activator with palmitoyl-gp100/MART-1 antigen and lipo-LeY in a single liposome. | Enhanced uptake of glycoliposome by moDC, dermal DC, and LC, and induction of strong CD8+ and iNKT cell activation. | [228] | |
| pH-sensitive glycan-modified liposomes | Glycan-modified pH-sensitive polymers were designed. Polysaccharide, such as dextrans, curdlan, and mannan, modified with 3-methylglutaric anhydride (MGlu) to form MGlu–Dex, MGlu–Curd, and MGlu–Man used in the preparation of pH-sensitive liposomes. Mannose modification of MGlu–Curd was carried out and used in the preparation of pH-sensitive liposomes. | Glycan-modified pH-sensitive liposomes showed maturation of DCs, targeting of CLR on APCs, cytosolic delivery of antigens, and antigen-specific cellular immune response. Mannose-functionalized curdlan derivatives incorporated in antigen-loaded liposomes showed superior pH-sensitivity than original curdlan derivatives. | [122,206,207] |
| Glyco-dendrimer | Branched PAMAM dendrimers used as a scaffold for gp100 long peptides and ligand LeY (for DC-SIGN and Langerin targeting) for preparing multivalent glyco-dendrimer. | Dual targeting (DC-SIGN and Langerin) by glyco-dendrimers resulted in enhanced internalization and gp100-specific CD8+ T cell activation. | [233] |
| Glyco-nanoparticles | The PLA-PEI inner core (PVax) was synthesized through nanoprecipitation. The OVA and CpG were added to PVax and mixed into mannan in a 1:5 ratio to obtain mannan-modified polymeric NPs (MPVax). | Mannan in MPVax enhances draining ability in lymph nodes and capturing by CD8+ DC, and promotes DC activation. The PLA-PEI enhances antigen endosome escape to promote cross-presentation. | [234] |
| Glycan-conjugated PLGA nanoparticle | The PLGA-NPs with the TLR7 agonist were made using an oil-in-water emulsion method and then mixed with B16-OVA membrane. Mannose-modified B16-OVA-NPs were prepared with a lipid anchor in the presence of DSPE-PEG-Man. | Mannose modification of B16-OVA-NP with TLR7 agonist R837 results in enhanced uptake and BMDC maturation. Mannose modification also enhances the MR-mediated cellular uptake of these particles by macrophages. | [237] |
| Cationic lipid membranes composed of 1,2-dioleoyl-3-trimethyl ammonium-propane DOTAP-PLGA NPs encapsulating OVA antigen with HA (HA-DOTAP-PLGA NPs) coating using double emulsion (w/o/w)/solvent evaporation method. | The HA coating of DOTAP-PLGA NPs improves the cellular uptake of these particles, which is due to HA and CD44 receptor-mediated endocytosis. Enhanced activation of DCs and upregulation of MHC, costimulatory molecules, and cytokines was also found. These particles also enhance antigen-specific CD4+ and CD8+ T cell responses. | [238,241] | |
| Glyco-cluster | Glyco-cluster–Melan-A conjugates were prepared by coupling glycosynthons. Oligosaccharyl-pyroglutamyl-β-alanine derivatives containing dimannoside (Manα-Man6) or Lewis antigens (Lea or Lex) were coupled to Melan-A(16-40) peptide. | Dimannoside and Lewis–Melan-A antigen conjugate showed enhanced binding to MR and DC-SIGN. The DC targeted with these conjugated showed efficient presentation of Melan-A antigens and CD8+ T cell response | [239] |
10. Where Are We Now and What Are the Lacunae in the Knowledge for Developing the GNVs?
11. What Do We still Need to Do and Where Are We Going Next?
12. Challenges Associated with Developing GNVs as Cancer Immunotherapy
Glycan-Lectin Interaction in the Induction of Immunosuppressive TME
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Shanmugam, M.K.; Arfuso, F.; Arumugam, S.; Chinnathambi, A.; Jinsong, B.; Warrier, S.; Wang, L.Z.; Kumar, A.P.; Ahn, K.S.; Sethi, G.; et al. Role of novel histone modifications in cancer. Oncotarget 2018, 9, 11414–11426. [Google Scholar] [CrossRef] [PubMed]
- Warrier, V.U.; Makandar, A.I.; Garg, M.; Sethi, G.; Kant, R.; Pal, J.K.; Yuba, E.; Gupta, R.K. Engineering anti-cancer nanovaccine based on antigen cross-presentation. Biosci. Rep. 2019, 39, BSR20193220. [Google Scholar] [CrossRef] [PubMed]
- Shanmugam, M.K.; Ahn, K.S.; Hsu, A.; Woo, C.C.; Yuan, Y.; Tan, K.H.B.; Chinnathambi, A.; Alahmadi, T.A.; Alharbi, S.A.; Koh, A.P.F.; et al. Thymoquinone Inhibits Bone Metastasis of Breast Cancer Cells Through Abrogation of the CXCR4 Signaling Axis. Front. Pharmacol. 2018, 9, 1294. [Google Scholar] [CrossRef] [PubMed]
- Warrier, S.; Patil, M.; Bhansali, S.; Varier, L.; Sethi, G. Designing precision medicine panels for drug refractory cancers targeting cancer stemness traits. Biochim. Biophys. Acta Rev. Cancer 2021, 1875, 188475. [Google Scholar] [CrossRef]
- Dehshahri, A.; Ashrafizadeh, M.; Ghasemipour Afshar, E.; Pardakhty, A.; Mandegary, A.; Mohammadinejad, R.; Sethi, G. Topoisomerase inhibitors: Pharmacology and emerging nanoscale delivery systems. Pharmacol. Res. 2020, 151, 104551. [Google Scholar] [CrossRef]
- Kirtonia, A.; Gala, K.; Fernandes, S.G.; Pandya, G.; Pandey, A.K.; Sethi, G.; Khattar, E.; Garg, M. Repurposing of drugs: An attractive pharmacological strategy for cancer therapeutics. Semin. Cancer Biol. 2021, 68, 258–278. [Google Scholar] [CrossRef]
- Sever, R.; Brugge, J.S. Signal transduction in cancer. Cold Spring Harb. Perspect. Med. 2015, 5, a006098. [Google Scholar] [CrossRef]
- Gupta, B.; Sadaria, D.; Warrier, V.U.; Kirtonia, A.; Kant, R.; Awasthi, A.; Baligar, P.; Pal, J.K.; Yuba, E.; Sethi, G.; et al. Plant lectins and their usage in preparing targeted nanovaccines for cancer immunotherapy. Semin. Cancer Biol. 2022, 80, 87–106. [Google Scholar] [CrossRef]
- Feng, S.-S.; Chien, S. Chemotherapeutic engineering: Application and further development of chemical engineering principles for chemotherapy of cancer and other diseases. Chem. Eng. Sci. 2003, 58, 4087–4114. [Google Scholar] [CrossRef]
- Wu, A.; Wei, J.; Kong, L.Y.; Wang, Y.; Priebe, W.; Qiao, W.; Sawaya, R.; Heimberger, A.B. Glioma cancer stem cells induce immunosuppressive macrophages/microglia. Neuro Oncol. 2010, 12, 1113–1125. [Google Scholar] [CrossRef]
- Zhang, L.; Gu, F.X.; Chan, J.M.; Wang, A.Z.; Langer, R.S.; Farokhzad, O.C. Nanoparticles in medicine: Therapeutic applications and developments. Clin. Pharmacol. Ther. 2008, 83, 761–769. [Google Scholar] [CrossRef] [PubMed]
- Scatena, R.; Bottoni, P.; Pontoglio, A.; Mastrototaro, L.; Giardina, B. Glycolytic enzyme inhibitors in cancer treatment. Expert Opin. Investig. Drugs. 2008, 17, 1533–1545. [Google Scholar] [CrossRef] [PubMed]
- Rapoport, N.; Marin, A.; Luo, Y.; Prestwich, G.D.; Muniruzzaman, M.D. Intracellular uptake and trafficking of Pluronic micelles in drug-sensitive and MDR cells: Effect on the intracellular drug localization. J. Pharm. Sci. 2002, 91, 157–170. [Google Scholar] [CrossRef]
- Manu, K.A.; Shanmugam, M.K.; Li, F.; Chen, L.; Siveen, K.S.; Ahn, K.S.; Kumar, A.P.; Sethi, G. Simvastatin sensitizes human gastric cancer xenograft in nude mice to capecitabine by suppressing nuclear factor-kappa B-regulated gene products. J. Mol. Med. 2014, 92, 267–276. [Google Scholar] [CrossRef]
- Rajendran, P.; Ong, T.H.; Chen, L.; Li, F.; Shanmugam, M.K.; Vali, S.; Abbasi, T.; Kapoor, S.; Sharma, A.; Kumar, A.P.; et al. Suppression of signal transducer and activator of transcription 3 activation by butein inhibits growth of human hepatocellular carcinoma in vivo. Clin. Cancer Res. 2011, 17, 1425–1439. [Google Scholar] [CrossRef]
- Orth, M.; Lauber, K.; Niyazi, M.; Friedl, A.A.; Li, M.; Maihofer, C.; Schuttrumpf, L.; Ernst, A.; Niemoller, O.M.; Belka, C. Current concepts in clinical radiation oncology. Radiat. Environ. Biophys. 2014, 53, 1–29. [Google Scholar] [CrossRef]
- Baek, S.H.; Ko, J.H.; Lee, H.; Jung, J.; Kong, M.; Lee, J.W.; Lee, J.; Chinnathambi, A.; Zayed, M.E.; Alharbi, S.A.; et al. Resveratrol inhibits STAT3 signaling pathway through the induction of SOCS-1: Role in apoptosis induction and radiosensitization in head and neck tumor cells. Phytomedicine 2016, 23, 566–577. [Google Scholar] [CrossRef] [PubMed]
- Mollaei, H.; Safaralizadeh, R.; Rostami, Z. MicroRNA replacement therapy in cancer. J. Cell Physiol. 2019, 234, 12369–12384. [Google Scholar] [CrossRef]
- Ma, Z.; Xiang, X.; Li, S.; Xie, P.; Gong, Q.; Goh, B.C.; Wang, L. Targeting hypoxia-inducible factor-1, for cancer treatment: Recent advances in developing small-molecule inhibitors from natural compounds. Semin. Cancer Biol. 2022, 80, 379–390. [Google Scholar] [CrossRef]
- Zhou, Z.; Wang, X.; Zhang, H.; Huang, H.; Sun, L.; Ma, L.; Du, Y.; Pei, C.; Zhang, Q.; Li, H.; et al. Activating Layered Metal Oxide Nanomaterials via Structural Engineering as Biodegradable Nanoagents for Photothermal Cancer Therapy. Small 2021, 17, e2007486. [Google Scholar] [CrossRef]
- Wang, X.; Zhong, X.; Cheng, L. Titanium-based nanomaterials for cancer theranostics. Coord. Chem. Rev. 2021, 430, 213662. [Google Scholar] [CrossRef]
- Overwijk, W.W.; Wang, E.; Marincola, F.M.; Rammensee, H.G.; Restifo, N.P. Mining the mutanome: Developing highly personalized Immunotherapies based on mutational analysis of tumors. J. Immunother. Cancer 2013, 1, 11. [Google Scholar] [CrossRef] [PubMed]
- Gotwals, P.; Cameron, S.; Cipolletta, D.; Cremasco, V.; Crystal, A.; Hewes, B.; Mueller, B.; Quaratino, S.; Sabatos-Peyton, C.; Petruzzelli, L.; et al. Prospects for combining targeted and conventional cancer therapy with immunotherapy. Nat. Rev. Cancer 2017, 17, 286–301. [Google Scholar] [CrossRef] [PubMed]
- Khatoon, E.; Parama, D.; Kumar, A.; Alqahtani, M.S.; Abbas, M.; Girisa, S.; Sethi, G.; Kunnumakkara, A.B. Targeting PD-1/PD-L1 axis as new horizon for ovarian cancer therapy. Life Sci. 2022, 306, 120827. [Google Scholar] [CrossRef] [PubMed]
- Papaioannou, N.E.; Beniata, O.V.; Vitsos, P.; Tsitsilonis, O.; Samara, P. Harnessing the immune system to improve cancer therapy. Ann. Transl. Med. 2016, 4, 261. [Google Scholar] [CrossRef]
- Aguilar, L.K.; Guzik, B.W.; Aguilar-Cordova, E. Cytotoxic immunotherapy strategies for cancer: Mechanisms and clinical development. J. Cell Biochem. 2011, 112, 1969–1977. [Google Scholar] [CrossRef]
- Chen, P.; Liu, X.; Sun, Y.; Zhou, P.; Wang, Y.; Zhang, Y. Dendritic cell targeted vaccines: Recent progresses and challenges. Hum. Vaccin. Immunother. 2016, 12, 612–622. [Google Scholar] [CrossRef]
- Moballegh Nasery, M.; Abadi, B.; Poormoghadam, D.; Zarrabi, A.; Keyhanvar, P.; Khanbabaei, H.; Ashrafizadeh, M.; Mohammadinejad, R.; Tavakol, S.; Sethi, G. Curcumin Delivery Mediated by Bio-Based Nanoparticles: A Review. Molecules 2020, 25, 686. [Google Scholar] [CrossRef]
- Roy, N.K.; Parama, D.; Banik, K.; Bordoloi, D.; Devi, A.K.; Thakur, K.K.; Padmavathi, G.; Shakibaei, M.; Fan, L.; Sethi, G.; et al. An Update on Pharmacological Potential of Boswellic Acids against Chronic Diseases. Int. J. Mol. Sci. 2019, 20, 4101. [Google Scholar] [CrossRef]
- Ashrafizadeh, M.; Ahmadi, Z.; Kotla, N.G.; Afshar, E.G.; Samarghandian, S.; Mandegary, A.; Pardakhty, A.; Mohammadinejad, R.; Sethi, G. Nanoparticles Targeting STATs in Cancer Therapy. Cells 2019, 8, 1158. [Google Scholar] [CrossRef]
- Mirzaei, S.; Gholami, M.H.; Ang, H.L.; Hashemi, F.; Zarrabi, A.; Zabolian, A.; Hushmandi, K.; Delfi, M.; Khan, H.; Ashrafizadeh, M.; et al. Pre-Clinical and Clinical Applications of Small Interfering RNAs (siRNA) and Co-Delivery Systems for Pancreatic Cancer Therapy. Cells 2021, 10, 3348. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Noh, Y.W.; Lim, Y.T. Polymer nanoparticles for cross-presentation of exogenous antigens and enhanced cytotoxic T-lymphocyte immune response. Int. J. Nanomed. 2016, 11, 3753–3764. [Google Scholar] [CrossRef]
- Babensee, J.E. Interaction of dendritic cells with biomaterials. Semin. Immunol. 2008, 20, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Kou, P.M.; Babensee, J.E. Macrophage and dendritic cell phenotypic diversity in the context of biomaterials. J. Biomed. Mater. Res. A 2011, 96, 239–260. [Google Scholar] [CrossRef]
- Cruz, L.J.; Tacken, P.J.; Eich, C.; Rueda, F.; Torensma, R.; Figdor, C.G. Controlled release of antigen and Toll-like receptor ligands from PLGA nanoparticles enhances immunogenicity. Nanomedicine 2017, 12, 491–510. [Google Scholar] [CrossRef]
- Weis, W.I.; Drickamer, K. Structural basis of lectin-carbohydrate recognition. Annu. Rev. Biochem. 1996, 65, 441–473. [Google Scholar] [CrossRef] [PubMed]
- Unger, W.W.; Mayer, C.T.; Engels, S.; Hesse, C.; Perdicchio, M.; Puttur, F.; Streng-Ouwehand, I.; Litjens, M.; Kalay, H.; Berod, L.; et al. Antigen targeting to dendritic cells combined with transient regulatory T cell inhibition results in long-term tumor regression. Oncoimmunology 2015, 4, e970462. [Google Scholar] [CrossRef] [PubMed]
- Van Hateren, A.; Elliott, T. The role of MHC I protein dynamics in tapasin and TAPBPR-assisted immunopeptidome editing. Curr. Opin. Immunol. 2021, 70, 138–143. [Google Scholar] [CrossRef]
- Rock, K.L.; Reits, E.; Neefjes, J. Present Yourself! By MHC Class I and MHC Class II Molecules. Trends Immunol. 2016, 37, 724–737. [Google Scholar] [CrossRef]
- Yewdell, J.W. Not such a dismal science: The economics of protein synthesis, folding, degradation and antigen processing. Trends Cell Biol. 2001, 11, 294–297. [Google Scholar] [CrossRef]
- Goldberg, A.L.; Cascio, P.; Saric, T.; Rock, K.L. The importance of the proteasome and subsequent proteolytic steps in the generation of antigenic peptides. Mol. Immunol. 2002, 39, 147–164. [Google Scholar] [CrossRef] [PubMed]
- Roche, P.A.; Furuta, K. The ins and outs of MHC class II-mediated antigen processing and presentation. Nat. Rev. Immunol. 2015, 15, 203–216. [Google Scholar] [CrossRef] [PubMed]
- Perrin, P.; Jongsma, M.L.; Neefjes, J.; Berlin, I. The labyrinth unfolds: Architectural rearrangements of the endolysosomal system in antigen-presenting cells. Curr. Opin. Immunol. 2019, 58, 1–8. [Google Scholar] [CrossRef]
- Wieczorek, M.; Abualrous, E.T.; Sticht, J.; Alvaro-Benito, M.; Stolzenberg, S.; Noe, F.; Freund, C. Major Histocompatibility Complex (MHC) Class I and MHC Class II Proteins: Conformational Plasticity in Antigen Presentation. Front. Immunol. 2017, 8, 292. [Google Scholar] [CrossRef] [PubMed]
- Bevan, M.J. Pillars Article: The Major Histocompatibility Complex Determines Susceptibility to Cytotoxic T Cells Directed against Minor Histocompatibility Antigens. J. Immunol. 2005, 175, 7069–7084. [Google Scholar] [CrossRef]
- Rodriguez, A.; Regnault, A.; Kleijmeer, M.; Ricciardi-Castagnoli, P.; Amigorena, S. Selective transport of internalized antigens to the cytosol for MHC class I presentation in dendritic cells. Nat. Cell Biol. 1999, 1, 362–368. [Google Scholar] [CrossRef]
- Chaitanya, T.S.; Patil, S.N.; Ghosh, S.; Pal, J.K.; Yuba, E.; Gupta, R.K. Chapter 18-Cross-presentation-based nanovaccine for cancer immunotherapy. In Nanotherapeutics in Cancer Vaccination and Challenges; Rahman, M., Beg, S., Almalki, W.H., Alhakamy, N.A., Choudhry, H., Eds.; Academic Press: Cambridge, MA, USA, 2022; pp. 349–396. [Google Scholar] [CrossRef]
- Heath, W.R.; Carbone, F.R. Cross-presentation in viral immunity and self-tolerance. Nat. Rev. Immunol. 2001, 1, 126–134. [Google Scholar] [CrossRef]
- Cruz, F.M.; Colbert, J.D.; Merino, E.; Kriegsman, B.A.; Rock, K.L. The Biology and Underlying Mechanisms of Cross-Presentation of Exogenous Antigens on MHC-I Molecules. Annu. Rev. Immunol. 2017, 35, 149–176. [Google Scholar] [CrossRef]
- Kurts, C. Cross-presentation: Inducing CD8 T cell immunity and tolerance. J. Mol. Med. 2000, 78, 326–332. [Google Scholar] [CrossRef]
- Heipertz, E.L.; Davies, M.L.; Lin, E.; Norbury, C.C. Prolonged antigen presentation following an acute virus infection requires direct and then cross-presentation. J. Immunol. 2014, 193, 4169–4177. [Google Scholar] [CrossRef]
- Leon, B.; Ballesteros-Tato, A.; Randall, T.D.; Lund, F.E. Prolonged antigen presentation by immune complex-binding dendritic cells programs the proliferative capacity of memory CD8 T cells. J. Exp. Med. 2014, 211, 1637–1655. [Google Scholar] [CrossRef] [PubMed]
- Bachem, A.; Hartung, E.; Guttler, S.; Mora, A.; Zhou, X.; Hegemann, A.; Plantinga, M.; Mazzini, E.; Stoitzner, P.; Gurka, S.; et al. Expression of XCR1 Characterizes the Batf3-Dependent Lineage of Dendritic Cells Capable of Antigen Cross-Presentation. Front. Immunol. 2012, 3, 214. [Google Scholar] [CrossRef]
- Fehres, C.M.; Unger, W.W.; Garcia-Vallejo, J.J.; van Kooyk, Y. Understanding the biology of antigen cross-presentation for the design of vaccines against cancer. Front. Immunol. 2014, 5, 149. [Google Scholar] [CrossRef]
- Delamarre, L.; Pack, M.; Chang, H.; Mellman, I.; Trombetta, E.S. Differential lysosomal proteolysis in antigen-presenting cells determines antigen fate. Science 2005, 307, 1630–1634. [Google Scholar] [CrossRef] [PubMed]
- Pozzi, L.A.; Maciaszek, J.W.; Rock, K.L. Both dendritic cells and macrophages can stimulate naive CD8 T cells in vivo to proliferate, develop effector function, and differentiate into memory cells. J. Immunol. 2005, 175, 2071–2081. [Google Scholar] [CrossRef] [PubMed]
- Backer, R.; Schwandt, T.; Greuter, M.; Oosting, M.; Jungerkes, F.; Tuting, T.; Boon, L.; O’Toole, T.; Kraal, G.; Limmer, A.; et al. Effective collaboration between marginal metallophilic macrophages and CD8+ dendritic cells in the generation of cytotoxic T cells. Proc. Natl. Acad. Sci. USA 2010, 107, 216–221. [Google Scholar] [CrossRef]
- Hirosue, S.; Vokali, E.; Raghavan, V.R.; Rincon-Restrepo, M.; Lund, A.W.; Corthesy-Henrioud, P.; Capotosti, F.; Halin Winter, C.; Hugues, S.; Swartz, M.A. Steady-state antigen scavenging, cross-presentation, and CD8+ T cell priming: A new role for lymphatic endothelial cells. J. Immunol. 2014, 192, 5002–5011. [Google Scholar] [CrossRef]
- Muntjewerff, E.M.; Meesters, L.D.; van den Bogaart, G. Antigen Cross-Presentation by Macrophages. Front. Immunol. 2020, 11, 1276. [Google Scholar] [CrossRef]
- Hon, H.; Oran, A.; Brocker, T.; Jacob, J. B lymphocytes participate in cross-presentation of antigen following gene gun vaccination. J. Immunol. 2005, 174, 5233–5242. [Google Scholar] [CrossRef]
- Cho, J.H.; Youn, J.W.; Sung, Y.C. Cross-priming as a predominant mechanism for inducing CD8+ T cell responses in gene gun DNA immunization. J. Immunol. 2001, 167, 5549–5557. [Google Scholar] [CrossRef]
- Heit, A.; Huster, K.M.; Schmitz, F.; Schiemann, M.; Busch, D.H.; Wagner, H. CpG-DNA aided cross-priming by cross-presenting B cells. J. Immunol. 2004, 172, 1501–1507. [Google Scholar] [CrossRef] [PubMed]
- Den Haan, J.M.; Bevan, M.J. Constitutive versus activation-dependent cross-presentation of immune complexes by CD8+ and CD8− dendritic cells in vivo. J. Exp. Med. 2002, 196, 817–827. [Google Scholar] [CrossRef] [PubMed]
- Schuurhuis, D.H.; Ioan-Facsinay, A.; Nagelkerken, B.; van Schip, J.J.; Sedlik, C.; Melief, C.J.; Verbeek, J.S.; Ossendorp, F. Antigen-antibody immune complexes empower dendritic cells to efficiently prime specific CD8+ CTL responses in vivo. J. Immunol. 2002, 168, 2240–2246. [Google Scholar] [CrossRef]
- De Jong, J.M.; Schuurhuis, D.H.; Ioan-Facsinay, A.; van der Voort, E.I.; Huizinga, T.W.; Ossendorp, F.; Toes, R.E.; Verbeek, J.S. Murine Fc receptors for IgG are redundant in facilitating presentation of immune complex derived antigen to CD8+ T cells in vivo. Mol. Immunol. 2006, 43, 2045–2050. [Google Scholar] [CrossRef]
- Zhu, Y.; Gu, H.; Yang, L.; Li, N.; Chen, Q.; Kang, D.; Lin, S.; Jing, Y.; Jiang, P.; Chen, Q.; et al. Involvement of MST1/mTORC1/STAT1 activity in the regulation of B-cell receptor signalling by chemokine receptor 2. Clin. Transl. Med. 2022, 12, e887. [Google Scholar] [CrossRef] [PubMed]
- Clayton, K.; Vallejo, A.F.; Davies, J.; Sirvent, S.; Polak, M.E. Langerhans Cells-Programmed by the Epidermis. Front. Immunol. 2017, 8, 1676. [Google Scholar] [CrossRef] [PubMed]
- Gardner, A.; de Mingo Pulido, A.; Ruffell, B. Dendritic Cells and Their Role in Immunotherapy. Front. Immunol. 2020, 11, 924. [Google Scholar] [CrossRef]
- Ashrafizadeh, M.; Zarrabi, A.; Mostafavi, E.; Aref, A.R.; Sethi, G.; Wang, L.; Tergaonkar, V. Non-coding RNA-based regulation of inflammation. In Seminars in Immunology; Academic Press: Cambridge, MA, USA, 2022; p. 101606. [Google Scholar] [CrossRef]
- Akira, S.; Takeda, K.; Kaisho, T. Toll-like receptors: Critical proteins linking innate and acquired immunity. Nat. Immunol. 2001, 2, 675–680. [Google Scholar] [CrossRef]
- Caparros, E.; Munoz, P.; Sierra-Filardi, E.; Serrano-Gomez, D.; Puig-Kroger, A.; Rodriguez-Fernandez, J.L.; Mellado, M.; Sancho, J.; Zubiaur, M.; Corbi, A.L. DC-SIGN ligation on dendritic cells results in ERK and PI3K activation and modulates cytokine production. Blood 2006, 107, 3950–3958. [Google Scholar] [CrossRef]
- Figdor, C.G.; van Kooyk, Y.; Adema, G.J. C-type lectin receptors on dendritic cells and Langerhans cells. Nat. Rev. Immunol. 2002, 2, 77–84. [Google Scholar] [CrossRef]
- Geijtenbeek, T.B.; van Vliet, S.J.; Engering, A.; t Hart, B.A.; van Kooyk, Y. Self- and nonself-recognition by C-type lectins on dendritic cells. Annu. Rev. Immunol. 2004, 22, 33–54. [Google Scholar] [CrossRef] [PubMed]
- Pyz, E.; Marshall, A.S.; Gordon, S.; Brown, G.D. C-type lectin-like receptors on myeloid cells. Ann. Med. 2006, 38, 242–251. [Google Scholar] [CrossRef] [PubMed]
- Banchereau, J.; Steinman, R.M. Dendritic cells and the control of immunity. Nature 1998, 392, 245–252. [Google Scholar] [CrossRef]
- Rissoan, M.C.; Soumelis, V.; Kadowaki, N.; Grouard, G.; Briere, F.; De Waal Malefyt, R.; Liu, Y.J. Reciprocal control of T helper cell and dendritic cell differentiation. Science 1999, 283, 1183–1186. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xiang, Y.; Xin, V.W.; Wang, X.W.; Peng, X.C.; Liu, X.Q.; Wang, D.; Li, N.; Cheng, J.T.; Lyv, Y.N.; et al. Dendritic cell biology and its role in tumor immunotherapy. J. Hematol. Oncol. 2020, 13, 107. [Google Scholar] [CrossRef]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef]
- Kim, R.; Emi, M.; Tanabe, K. Functional roles of immature dendritic cells in impaired immunity of solid tumour and their targeted strategies for provoking tumour immunity. Clin. Exp. Immunol. 2006, 146, 189–196. [Google Scholar] [CrossRef]
- Visintin, A.; Mazzoni, A.; Spitzer, J.H.; Wyllie, D.H.; Dower, S.K.; Segal, D.M. Regulation of Toll-like receptors in human monocytes and dendritic cells. J. Immunol. 2001, 166, 249–255. [Google Scholar] [CrossRef]
- Moore, C.; Bae, J.; Liu, L.; Li, H.; Fu, Y.X.; Qiao, J. Exogenous signaling repairs defective T cell signaling inside the tumor microenvironment for better immunity. JCI Insight 2022, 7, e159479. [Google Scholar] [CrossRef]
- Dhodapkar, M.V.; Dhodapkar, K.M.; Palucka, A.K. Interactions of tumor cells with dendritic cells: Balancing immunity and tolerance. Cell Death Differ. 2008, 15, 39–50. [Google Scholar] [CrossRef]
- Baar, J. Clinical applications of dendritic cell cancer vaccines. Oncologist 1999, 4, 140–144. [Google Scholar] [CrossRef] [PubMed]
- Macatonia, S.E.; Hosken, N.A.; Litton, M.; Vieira, P.; Hsieh, C.S.; Culpepper, J.A.; Wysocka, M.; Trinchieri, G.; Murphy, K.M.; O’Garra, A. Dendritic cells produce IL-12 and direct the development of Th1 cells from naive CD4+ T cells. J. Immunol. 1995, 154, 5071–5079. [Google Scholar] [PubMed]
- Zhou, L.J.; Tedder, T.F. A distinct pattern of cytokine gene expression by human CD83+ blood dendritic cells. Blood 1995, 86, 3295–3301. [Google Scholar] [CrossRef] [PubMed]
- Collin, M.; Bigley, V. Human dendritic cell subsets: An update. Immunology 2018, 154, 3–20. [Google Scholar] [CrossRef]
- Veglia, F.; Gabrilovich, D.I. Dendritic cells in cancer: The role revisited. Curr. Opin. Immunol. 2017, 45, 43–51. [Google Scholar] [CrossRef]
- Shin, K.S.; Jeon, I.; Kim, B.S.; Kim, I.K.; Park, Y.J.; Koh, C.H.; Song, B.; Lee, J.M.; Lim, J.; Bae, E.A.; et al. Monocyte-Derived Dendritic Cells Dictate the Memory Differentiation of CD8+ T Cells During Acute Infection. Front. Immunol. 2019, 10, 1887. [Google Scholar] [CrossRef]
- Steinman, R.M.; Hawiger, D.; Liu, K.; Bonifaz, L.; Bonnyay, D.; Mahnke, K.; Iyoda, T.; Ravetch, J.; Dhodapkar, M.; Inaba, K.; et al. Dendritic cell function in vivo during the steady state: A role in peripheral tolerance. Ann. N. Y. Acad. Sci. 2003, 987, 15–25. [Google Scholar] [CrossRef]
- Joffre, O.P.; Segura, E.; Savina, A.; Amigorena, S. Cross-presentation by dendritic cells. Nat. Rev. Immunol. 2012, 12, 557–569. [Google Scholar] [CrossRef]
- Bonifaz, L.; Bonnyay, D.; Mahnke, K.; Rivera, M.; Nussenzweig, M.C.; Steinman, R.M. Efficient targeting of protein antigen to the dendritic cell receptor DEC-205 in the steady state leads to antigen presentation on major histocompatibility complex class I products and peripheral CD8+ T cell tolerance. J. Exp. Med. 2002, 196, 1627–1638. [Google Scholar] [CrossRef]
- Segura, E.; Albiston, A.L.; Wicks, I.P.; Chai, S.Y.; Villadangos, J.A. Different cross-presentation pathways in steady-state and inflammatory dendritic cells. Proc. Natl. Acad. Sci. USA 2009, 106, 20377–20381. [Google Scholar] [CrossRef]
- Miller, J.C.; Brown, B.D.; Shay, T.; Gautier, E.L.; Jojic, V.; Cohain, A.; Pandey, G.; Leboeuf, M.; Elpek, K.G.; Helft, J.; et al. Deciphering the transcriptional network of the dendritic cell lineage. Nat. Immunol. 2012, 13, 888–899. [Google Scholar] [CrossRef] [PubMed]
- Shortman, K.; Heath, W.R. The CD8+ dendritic cell subset. Immunol. Rev. 2010, 234, 18–31. [Google Scholar] [CrossRef] [PubMed]
- Kamphorst, A.O.; Guermonprez, P.; Dudziak, D.; Nussenzweig, M.C. Route of antigen uptake differentially impacts presentation by dendritic cells and activated monocytes. J. Immunol. 2010, 185, 3426–3435. [Google Scholar] [CrossRef] [PubMed]
- Boltjes, A.; van Wijk, F. Human dendritic cell functional specialization in steady-state and inflammation. Front. Immunol. 2014, 5, 131. [Google Scholar] [CrossRef]
- Cella, M.; Jarrossay, D.; Facchetti, F.; Alebardi, O.; Nakajima, H.; Lanzavecchia, A.; Colonna, M. Plasmacytoid monocytes migrate to inflamed lymph nodes and produce large amounts of type I interferon. Nat. Med. 1999, 5, 919–923. [Google Scholar] [CrossRef]
- Ito, T.; Hanabuchi, S.; Wang, Y.H.; Park, W.R.; Arima, K.; Bover, L.; Qin, F.X.; Gilliet, M.; Liu, Y.J. Two functional subsets of FOXP3+ regulatory T cells in human thymus and periphery. Immunity 2008, 28, 870–880. [Google Scholar] [CrossRef]
- Martin-Gayo, E.; Sierra-Filardi, E.; Corbi, A.L.; Toribio, M.L. Plasmacytoid dendritic cells resident in human thymus drive natural Treg cell development. Blood 2010, 115, 5366–5375. [Google Scholar] [CrossRef]
- Oberkampf, M.; Guillerey, C.; Mouries, J.; Rosenbaum, P.; Fayolle, C.; Bobard, A.; Savina, A.; Ogier-Denis, E.; Enninga, J.; Amigorena, S.; et al. Mitochondrial reactive oxygen species regulate the induction of CD8+ T cells by plasmacytoid dendritic cells. Nat. Commun. 2018, 9, 2241. [Google Scholar] [CrossRef]
- Li, K.; Underhill, D.M. C-Type Lectin Receptors in Phagocytosis. Curr. Top Microbiol. Immunol. 2020, 429, 1–18. [Google Scholar] [CrossRef]
- Drickamer, K.; Taylor, M.E. Recent insights into structures and functions of C-type lectins in the immune system. Curr. Opin. Struct. Biol. 2015, 34, 26–34. [Google Scholar] [CrossRef]
- Lehmann, C.H.; Heger, L.; Heidkamp, G.F.; Baranska, A.; Luhr, J.J.; Hoffmann, A.; Dudziak, D. Direct Delivery of Antigens to Dendritic Cells via Antibodies Specific for Endocytic Receptors as a Promising Strategy for Future Therapies. Vaccines 2016, 4, 8. [Google Scholar] [CrossRef] [PubMed]
- Hawiger, D.; Inaba, K.; Dorsett, Y.; Guo, M.; Mahnke, K.; Rivera, M.; Ravetch, J.V.; Steinman, R.M.; Nussenzweig, M.C. Dendritic cells induce peripheral T cell unresponsiveness under steady state conditions in vivo. J. Exp. Med. 2001, 194, 769–779. [Google Scholar] [CrossRef] [PubMed]
- Mahnke, K.; Guo, M.; Lee, S.; Sepulveda, H.; Swain, S.L.; Nussenzweig, M.; Steinman, R.M. The dendritic cell receptor for endocytosis, DEC-205, can recycle and enhance antigen presentation via major histocompatibility complex class II-positive lysosomal compartments. J. Cell Biol. 2000, 151, 673–684. [Google Scholar] [CrossRef] [PubMed]
- Flacher, V.; Tripp, C.H.; Stoitzner, P.; Haid, B.; Ebner, S.; Del Frari, B.; Koch, F.; Park, C.G.; Steinman, R.M.; Idoyaga, J.; et al. Epidermal Langerhans cells rapidly capture and present antigens from C-type lectin-targeting antibodies deposited in the dermis. J. Investig. Dermatol. 2010, 130, 755–762. [Google Scholar] [CrossRef]
- Ni, L.; Gayet, I.; Zurawski, S.; Duluc, D.; Flamar, A.L.; Li, X.H.; O’Bar, A.; Clayton, S.; Palucka, A.K.; Zurawski, G.; et al. Concomitant activation and antigen uptake via human dectin-1 results in potent antigen-specific CD8+ T cell responses. J. Immunol. 2010, 185, 3504–3513. [Google Scholar] [CrossRef]
- Pereira, C.F.; Torensma, R.; Hebeda, K.; Kretz-Rommel, A.; Faas, S.J.; Figdor, C.G.; Adema, G.J. In vivo targeting of DC-SIGN-positive antigen-presenting cells in a nonhuman primate model. J. Immunother. 2007, 30, 705–714. [Google Scholar] [CrossRef]
- Tsuji, T.; Matsuzaki, J.; Kelly, M.P.; Ramakrishna, V.; Vitale, L.; He, L.Z.; Keler, T.; Odunsi, K.; Old, L.J.; Ritter, G.; et al. Antibody-targeted NY-ESO-1 to mannose receptor or DEC-205 in vitro elicits dual human CD8+ and CD4+ T cell responses with broad antigen specificity. J. Immunol. 2011, 186, 1218–1227. [Google Scholar] [CrossRef]
- Unger, W.W.; van Kooyk, Y. ‘Dressed for success’ C-type lectin receptors for the delivery of glyco-vaccines to dendritic cells. Curr. Opin. Immunol. 2011, 23, 131–137. [Google Scholar] [CrossRef]
- Tacken, P.J.; Ginter, W.; Berod, L.; Cruz, L.J.; Joosten, B.; Sparwasser, T.; Figdor, C.G.; Cambi, A. Targeting DC-SIGN via its neck region leads to prolonged antigen residence in early endosomes, delayed lysosomal degradation, and cross-presentation. Blood 2011, 118, 4111–4119. [Google Scholar] [CrossRef]
- Iborra, S.; Sancho, D. Signalling versatility following self and non-self sensing by myeloid C-type lectin receptors. Immunobiology 2015, 220, 175–184. [Google Scholar] [CrossRef]
- Johnson, J.L.; Jones, M.B.; Ryan, S.O.; Cobb, B.A. The regulatory power of glycans and their binding partners in immunity. Trends Immunol. 2013, 34, 290–298. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Nakata, K.; Kato, Y.; Shima, M.; Ishii, N.; Koji, T.; Taketa, K.; Endo, Y.; Nagataki, S. Early recognition of hepatocellular carcinoma based on altered profiles of alpha-fetoprotein. N. Engl. J. Med. 1993, 328, 1802–1806. [Google Scholar] [CrossRef] [PubMed]
- McGreal, E.P.; Miller, J.L.; Gordon, S. Ligand recognition by antigen-presenting cell C-type lectin receptors. Curr. Opin. Immunol. 2005, 17, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Streng-Ouwehand, I.; Litjens, M.; Weelij, D.R.; Garcia-Vallejo, J.J.; van Vliet, S.J.; Saeland, E.; van Kooyk, Y. Characterization of murine MGL1 and MGL2 C-type lectins: Distinct glycan specificities and tumor binding properties. Mol. Immunol. 2009, 46, 1240–1249. [Google Scholar] [CrossRef]
- Van Vliet, S.J.; Saeland, E.; van Kooyk, Y. Sweet preferences of MGL: Carbohydrate specificity and function. Trends Immunol. 2008, 29, 83–90. [Google Scholar] [CrossRef]
- Hossain, M.K.; Wall, K.A. Use of Dendritic Cell Receptors as Targets for Enhancing Anti-Cancer Immune Responses. Cancers 2019, 11, 418. [Google Scholar] [CrossRef]
- Stoitzner, P.; Romani, N.; Rademacher, C.; Probst, H.C.; Mahnke, K. Antigen targeting to dendritic cells: Still a place in future immunotherapy? Eur. J. Immunol. 2022. [Google Scholar] [CrossRef]
- Carter, R.W.; Thompson, C.; Reid, D.M.; Wong, S.Y.; Tough, D.F. Preferential induction of CD4+ T cell responses through in vivo targeting of antigen to dendritic cell-associated C-type lectin-1. J. Immunol. 2006, 177, 2276–2284. [Google Scholar] [CrossRef]
- Ahmadi, M.; Bryson, C.J.; Cloake, E.A.; Welch, K.; Filipe, V.; Romeijn, S.; Hawe, A.; Jiskoot, W.; Baker, M.P.; Fogg, M.H. Small amounts of sub-visible aggregates enhance the immunogenic potential of monoclonal antibody therapeutics. Pharm. Res. 2015, 32, 1383–1394. [Google Scholar] [CrossRef]
- Yuba, E.; Fukaya, Y.; Yanagihara, S.; Kasho, N.; Harada, A. Development of Mannose-Modified Carboxylated Curdlan-Coated Liposomes for Antigen Presenting Cell Targeted Antigen Delivery. Pharmaceutics 2020, 12, 754. [Google Scholar] [CrossRef]
- Chiba, M.; Fujita, S.; Suzuki, T. Pharmacokinetic correlation between in vitro hepatic microsomal enzyme kinetics and in vivo metabolism of imipramine and desipramine in rats. J. Pharm. Sci. 1990, 79, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Lepenies, B.; Lee, J.; Sonkaria, S. Targeting C-type lectin receptors with multivalent carbohydrate ligands. Adv. Drug Deliv. Rev. 2013, 65, 1271–1281. [Google Scholar] [CrossRef] [PubMed]
- Van Kooyk, Y.; Unger, W.W.; Fehres, C.M.; Kalay, H.; Garcia-Vallejo, J.J. Glycan-based DC-SIGN targeting vaccines to enhance antigen cross-presentation. Mol. Immunol. 2013, 55, 143–145. [Google Scholar] [CrossRef] [PubMed]
- Fasting, C.; Schalley, C.A.; Weber, M.; Seitz, O.; Hecht, S.; Koksch, B.; Dernedde, J.; Graf, C.; Knapp, E.W.; Haag, R. Multivalency as a chemical organization and action principle. Angew. Chem. Int. Ed. Engl. 2012, 51, 10472–10498. [Google Scholar] [CrossRef] [PubMed]
- Engering, A.; Geijtenbeek, T.B.; van Vliet, S.J.; Wijers, M.; van Liempt, E.; Demaurex, N.; Lanzavecchia, A.; Fransen, J.; Figdor, C.G.; Piguet, V.; et al. The dendritic cell-specific adhesion receptor DC-SIGN internalizes antigen for presentation to T cells. J. Immunol. 2002, 168, 2118–2126. [Google Scholar] [CrossRef] [PubMed]
- Engering, A.J.; Cella, M.; Fluitsma, D.M.; Hoefsmit, E.C.; Lanzavecchia, A.; Pieters, J. Mannose receptor mediated antigen uptake and presentation in human dendritic cells. Adv. Exp. Med. Biol. 1997, 417, 183–187. [Google Scholar] [CrossRef]
- Martinez-Pomares, L. The mannose receptor. J. Leukoc. Biol. 2012, 92, 1177–1186. [Google Scholar] [CrossRef]
- Guo, Y.; Feinberg, H.; Conroy, E.; Mitchell, D.A.; Alvarez, R.; Blixt, O.; Taylor, M.E.; Weis, W.I.; Drickamer, K. Structural basis for distinct ligand-binding and targeting properties of the receptors DC-SIGN and DC-SIGNR. Nat. Struct. Mol. Biol. 2004, 11, 591–598. [Google Scholar] [CrossRef]
- van Liempt, E.; Bank, C.M.; Mehta, P.; Garcia-Vallejo, J.J.; Kawar, Z.S.; Geyer, R.; Alvarez, R.A.; Cummings, R.D.; Kooyk, Y.; van Die, I. Specificity of DC-SIGN for mannose-and fucose-containing glycans. FEBS Lett. 2006, 580, 6123–6131. [Google Scholar] [CrossRef]
- Stambach, N.S.; Taylor, M.E. Characterization of carbohydrate recognition by langerin, a C-type lectin of Langerhans cells. Glycobiology 2003, 13, 401–410. [Google Scholar] [CrossRef]
- Van Vliet, S.J.; van Liempt, E.; Saeland, E.; Aarnoudse, C.A.; Appelmelk, B.; Irimura, T.; Geijtenbeek, T.B.; Blixt, O.; Alvarez, R.; van Die, I.; et al. Carbohydrate profiling reveals a distinctive role for the C-type lectin MGL in the recognition of helminth parasites and tumor antigens by dendritic cells. Int. Immunol. 2005, 17, 661–669. [Google Scholar] [CrossRef]
- Bloem, K.; Vuist, I.M.; van den Berk, M.; Klaver, E.J.; van Die, I.; Knippels, L.M.; Garssen, J.; Garcia-Vallejo, J.J.; van Vliet, S.J.; van Kooyk, Y. DCIR interacts with ligands from both endogenous and pathogenic origin. Immunol. Lett. 2014, 158, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Jegouzo, S.A.; Feinberg, H.; Dungarwalla, T.; Drickamer, K.; Weis, W.I.; Taylor, M.E. A Novel Mechanism for Binding of Galactose-terminated Glycans by the C-type Carbohydrate Recognition Domain in Blood Dendritic Cell Antigen 2. J. Biol. Chem. 2015, 290, 16759–16771. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.D.; Gordon, S. Immune recognition. A new receptor for beta-glucans. Nature 2001, 413, 36–37. [Google Scholar] [CrossRef] [PubMed]
- Ujita, M.; Nagayama, H.; Kanie, S.; Koike, S.; Ikeyama, Y.; Ozaki, T.; Okumura, H. Carbohydrate binding specificity of recombinant human macrophage beta-glucan receptor dectin-1. Biosci. Biotechnol. Biochem. 2009, 73, 237–240. [Google Scholar] [CrossRef][Green Version]
- Medzhitov, R. Toll-like receptors and innate immunity. Nat. Rev. Immunol. 2001, 1, 135–145. [Google Scholar] [CrossRef]
- Ignacio, B.J.; Albin, T.J.; Esser-Kahn, A.P.; Verdoes, M. Toll-like Receptor Agonist Conjugation: A Chemical Perspective. Bioconjug. Chem. 2018, 29, 587–603. [Google Scholar] [CrossRef]
- Khan, S.; Bijker, M.S.; Weterings, J.J.; Tanke, H.J.; Adema, G.J.; van Hall, T.; Drijfhout, J.W.; Melief, C.J.; Overkleeft, H.S.; van der Marel, G.A.; et al. Distinct uptake mechanisms but similar intracellular processing of two different toll-like receptor ligand-peptide conjugates in dendritic cells. J. Biol. Chem. 2007, 282, 21145–21159. [Google Scholar] [CrossRef]
- Fritz, J.H.; Girardin, S.E.; Fitting, C.; Werts, C.; Mengin-Lecreulx, D.; Caroff, M.; Cavaillon, J.M.; Philpott, D.J.; Adib-Conquy, M. Synergistic stimulation of human monocytes and dendritic cells by Toll-like receptor 4 and NOD1- and NOD2-activating agonists. Eur. J. Immunol. 2005, 35, 2459–2470. [Google Scholar] [CrossRef]
- Gringhuis, S.I.; den Dunnen, J.; Litjens, M.; van Het Hof, B.; van Kooyk, Y.; Geijtenbeek, T.B. C-type lectin DC-SIGN modulates Toll-like receptor signaling via Raf-1 kinase-dependent acetylation of transcription factor NF-kappaB. Immunity 2007, 26, 605–616. [Google Scholar] [CrossRef]
- Kim, H.; Griffith, T.S.; Panyam, J. Poly(d,l-lactide-co-glycolide) Nanoparticles as Delivery Platforms for TLR7/8 Agonist-Based Cancer Vaccine. J. Pharmacol. Exp. Ther. 2019, 370, 715–724. [Google Scholar] [CrossRef] [PubMed]
- Panda, A.K. Induction of anti-tumor immunity and T-cell responses using nanodelivery systems engrafting TLR-5 ligand. Expert Rev. Vaccines 2011, 10, 155–157. [Google Scholar] [CrossRef] [PubMed]
- Steinhagen, F.; Kinjo, T.; Bode, C.; Klinman, D.M. TLR-based immune adjuvants. Vaccine 2011, 29, 3341–3355. [Google Scholar] [CrossRef]
- Sartorius, R.; Bettua, C.; D’Apice, L.; Caivano, A.; Trovato, M.; Russo, D.; Zanoni, I.; Granucci, F.; Mascolo, D.; Barba, P.; et al. Vaccination with filamentous bacteriophages targeting DEC-205 induces DC maturation and potent anti-tumor T-cell responses in the absence of adjuvants. Eur. J. Immunol. 2011, 41, 2573–2584. [Google Scholar] [CrossRef] [PubMed]
- Shrimpton, R.E.; Butler, M.; Morel, A.S.; Eren, E.; Hue, S.S.; Ritter, M.A. CD205 (DEC-205): A recognition receptor for apoptotic and necrotic self. Mol. Immunol. 2009, 46, 1229–1239. [Google Scholar] [CrossRef]
- Stylianou, E.; Pepponi, I.; van Dolleweerd, C.J.; Paul, M.J.; Ma, J.K.; Reljic, R. Exploring the vaccine potential of Dec-205 targeting in Mycobacterium tuberculosis infection in mice. Vaccine 2011, 29, 2279–2286. [Google Scholar] [CrossRef]
- Brown, G.D. Dectin-1: A signalling non-TLR pattern-recognition receptor. Nat. Rev. Immunol. 2006, 6, 33–43. [Google Scholar] [CrossRef]
- Drummond, R.A.; Dambuza, I.M.; Vautier, S.; Taylor, J.A.; Reid, D.M.; Bain, C.C.; Underhill, D.M.; Masopust, D.; Kaplan, D.H.; Brown, G.D. CD4+ T-cell survival in the GI tract requires dectin-1 during fungal infection. Mucosal Immunol. 2016, 9, 492–502. [Google Scholar] [CrossRef]
- Goedhart, M.; Slot, E.; Pascutti, M.F.; Geerman, S.; Rademakers, T.; Nota, B.; Huveneers, S.; Buul, J.D.V.; MacNamara, K.C.; Voermans, C.; et al. Bone Marrow Harbors a Unique Population of Dendritic Cells with the Potential to Boost Neutrophil Formation upon Exposure to Fungal Antigen. Cells 2021, 11, 55. [Google Scholar] [CrossRef]
- Guo, Y.; Kasahara, S.; Jhingran, A.; Tosini, N.L.; Zhai, B.; Aufiero, M.A.; Mills, K.A.M.; Gjonbalaj, M.; Espinosa, V.; Rivera, A.; et al. During Aspergillus Infection, Monocyte-Derived DCs, Neutrophils, and Plasmacytoid DCs Enhance Innate Immune Defense through CXCR3-Dependent Crosstalk. Cell Host Microbe 2020, 28, 104–116.e104. [Google Scholar] [CrossRef]
- Mukhopadhaya, A.; Hanafusa, T.; Jarchum, I.; Chen, Y.G.; Iwai, Y.; Serreze, D.V.; Steinman, R.M.; Tarbell, K.V.; DiLorenzo, T.P. Selective delivery of beta cell antigen to dendritic cells in vivo leads to deletion and tolerance of autoreactive CD8+ T cells in NOD mice. Proc. Natl. Acad. Sci. USA 2008, 105, 6374–6379. [Google Scholar] [CrossRef] [PubMed]
- Ren, A.; Li, Z.; Zhang, X.; Deng, R.; Ma, Y. Inhibition of Dectin-1 on Dendritic Cells Prevents Maturation and Prolongs Murine Islet Allograft Survival. J. Inflamm. Res. 2021, 14, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Tiburcio, R.; Melo, L.D.; Nunes, S.; Barbosa, A.L.A.; de Oliveira, E.C.; Suarez, M.; Borges, V.M.; Tavares, N.; Brodskyn, C.I. DC-SIGN Mediates the Interaction Between Neutrophils and Leishmania amazonensis-Infected Dendritic Cells to Promote DC Maturation and Parasite Elimination. Front. Immunol. 2021, 12, 750648. [Google Scholar] [CrossRef] [PubMed]
- Holla, A.; Skerra, A. Comparative analysis reveals selective recognition of glycans by the dendritic cell receptors DC-SIGN and Langerin. Protein Eng. Des. Sel. 2011, 24, 659–669. [Google Scholar] [CrossRef] [PubMed]
- Nonaka, M.; Ma, B.Y.; Imaeda, H.; Kawabe, K.; Kawasaki, N.; Hodohara, K.; Kawasaki, N.; Andoh, A.; Fujiyama, Y.; Kawasaki, T. Dendritic cell-specific intercellular adhesion molecule 3-grabbing non-integrin (DC-SIGN) recognizes a novel ligand, Mac-2-binding protein, characteristically expressed on human colorectal carcinomas. J. Biol. Chem. 2011, 286, 22403–22413. [Google Scholar] [CrossRef]
- Unger, W.W.; van Beelen, A.J.; Bruijns, S.C.; Joshi, M.; Fehres, C.M.; van Bloois, L.; Verstege, M.I.; Ambrosini, M.; Kalay, H.; Nazmi, K.; et al. Glycan-modified liposomes boost CD4+ and CD8+ T-cell responses by targeting DC-SIGN on dendritic cells. J. Control. Release 2012, 160, 88–95. [Google Scholar] [CrossRef]
- Bouteau, A.; Kervevan, J.; Su, Q.; Zurawski, S.M.; Contreras, V.; Dereuddre-Bosquet, N.; Le Grand, R.; Zurawski, G.; Cardinaud, S.; Levy, Y.; et al. DC Subsets Regulate Humoral Immune Responses by Supporting the Differentiation of Distinct Tfh Cells. Front. Immunol. 2019, 10, 1134. [Google Scholar] [CrossRef]
- de Jong, M.A.; Vriend, L.E.; Theelen, B.; Taylor, M.E.; Fluitsma, D.; Boekhout, T.; Geijtenbeek, T.B. C-type lectin Langerin is a beta-glucan receptor on human Langerhans cells that recognizes opportunistic and pathogenic fungi. Mol. Immunol. 2010, 47, 1216–1225. [Google Scholar] [CrossRef]
- Fehres, C.M.; Duinkerken, S.; Bruijns, S.C.; Kalay, H.; van Vliet, S.J.; Ambrosini, M.; de Gruijl, T.D.; Unger, W.W.; Garcia-Vallejo, J.J.; van Kooyk, Y. Langerin-mediated internalization of a modified peptide routes antigens to early endosomes and enhances cross-presentation by human Langerhans cells. Cell Mol. Immunol. 2017, 14, 360–370. [Google Scholar] [CrossRef]
- Rajesh, A.; Stuart, G.; Real, N.; Ahn, J.; Tschirley, A.; Wise, L.; Hibma, M. Depletion of langerin(+) cells enhances cutaneous wound healing. Immunology 2020, 160, 366–381. [Google Scholar] [CrossRef]
- Valladeau, J.; Duvert-Frances, V.; Pin, J.J.; Dezutter-Dambuyant, C.; Vincent, C.; Massacrier, C.; Vincent, J.; Yoneda, K.; Banchereau, J.; Caux, C.; et al. The monoclonal antibody DCGM4 recognizes Langerin, a protein specific of Langerhans cells, and is rapidly internalized from the cell surface. Eur. J. Immunol. 1999, 29, 2695–2704. [Google Scholar] [CrossRef]
- Feinberg, H.; Jegouzo, S.A.F.; Lasanajak, Y.; Smith, D.F.; Drickamer, K.; Weis, W.I.; Taylor, M.E. Structural analysis of carbohydrate binding by the macrophage mannose receptor CD206. J. Biol. Chem. 2021, 296, 100368. [Google Scholar] [CrossRef] [PubMed]
- McKenzie, E.J.; Taylor, P.R.; Stillion, R.J.; Lucas, A.D.; Harris, J.; Gordon, S.; Martinez-Pomares, L. Mannose receptor expression and function define a new population of murine dendritic cells. J. Immunol. 2007, 178, 4975–4983. [Google Scholar] [CrossRef] [PubMed]
- Vassilaros, S.; Tsibanis, A.; Tsikkinis, A.; Pietersz, G.A.; McKenzie, I.F.; Apostolopoulos, V. Up to 15-year clinical follow-up of a pilot Phase III immunotherapy study in stage II breast cancer patients using oxidized mannan-MUC1. Immunotherapy 2013, 5, 1177–1182. [Google Scholar] [CrossRef]
- Zehner, M.; Burgdorf, S. Regulation of antigen transport into the cytosol for cross-presentation by ubiquitination of the mannose receptor. Mol. Immunol. 2013, 55, 146–148. [Google Scholar] [CrossRef] [PubMed]
- Denda-Nagai, K.; Aida, S.; Saba, K.; Suzuki, K.; Moriyama, S.; Oo-Puthinan, S.; Tsuiji, M.; Morikawa, A.; Kumamoto, Y.; Sugiura, D.; et al. Distribution and function of macrophage galactose-type C-type lectin 2 (MGL2/CD301b): Efficient uptake and presentation of glycosylated antigens by dendritic cells. J. Biol. Chem. 2010, 285, 19193–19204. [Google Scholar] [CrossRef]
- Murakami, R.; Denda-Nagai, K.; Hashimoto, S.; Nagai, S.; Hattori, M.; Irimura, T. A unique dermal dendritic cell subset that skews the immune response toward Th2. PLoS ONE 2013, 8, e73270. [Google Scholar] [CrossRef]
- Streng-Ouwehand, I.; Ho, N.I.; Litjens, M.; Kalay, H.; Boks, M.A.; Cornelissen, L.A.; Kaur Singh, S.; Saeland, E.; Garcia-Vallejo, J.J.; Ossendorp, F.A.; et al. Glycan modification of antigen alters its intracellular routing in dendritic cells, promoting priming of T cells. Elife 2016, 5, e11765. [Google Scholar] [CrossRef]
- Klechevsky, E.; Flamar, A.L.; Cao, Y.; Blanck, J.P.; Liu, M.; O’Bar, A.; Agouna-Deciat, O.; Klucar, P.; Thompson-Snipes, L.; Zurawski, S.; et al. Cross-priming CD8+ T cells by targeting antigens to human dendritic cells through DCIR. Blood 2010, 116, 1685–1697. [Google Scholar] [CrossRef]
- Palmer, D.H.; Midgley, R.S.; Mirza, N.; Torr, E.E.; Ahmed, F.; Steele, J.C.; Steven, N.M.; Kerr, D.J.; Young, L.S.; Adams, D.H. A phase II study of adoptive immunotherapy using dendritic cells pulsed with tumor lysate in patients with hepatocellular carcinoma. Hepatology 2009, 49, 124–132. [Google Scholar] [CrossRef]
- Wang, Q.T.; Nie, Y.; Sun, S.N.; Lin, T.; Han, R.J.; Jiang, J.; Li, Z.; Li, J.Q.; Xiao, Y.P.; Fan, Y.Y.; et al. Tumor-associated antigen-based personalized dendritic cell vaccine in solid tumor patients. Cancer Immunol. Immunother. 2020, 69, 1375–1387. [Google Scholar] [CrossRef] [PubMed]
- Paulis, L.E.; Mandal, S.; Kreutz, M.; Figdor, C.G. Dendritic cell-based nanovaccines for cancer immunotherapy. Curr. Opin. Immunol. 2013, 25, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Alloatti, A.; Kotsias, F.; Magalhaes, J.G.; Amigorena, S. Dendritic cell maturation and cross-presentation: Timing matters! Immunol. Rev. 2016, 272, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Davey, G.M.; Sutherland, R.M.; Kurts, C.; Lew, A.M.; Hirst, C.; Carbone, F.R.; Heath, W.R. Cell-associated ovalbumin is cross-presented much more efficiently than soluble ovalbumin in vivo. J. Immunol. 2001, 166, 6099–6103. [Google Scholar] [CrossRef] [PubMed]
- Falo, L.D., Jr.; Kovacsovics-Bankowski, M.; Thompson, K.; Rock, K.L. Targeting antigen into the phagocytic pathway in vivo induces protective tumour immunity. Nat. Med. 1995, 1, 649–653. [Google Scholar] [CrossRef] [PubMed]
- Kovacsovics-Bankowski, M.; Clark, K.; Benacerraf, B.; Rock, K.L. Efficient major histocompatibility complex class I presentation of exogenous antigen upon phagocytosis by macrophages. Proc. Natl. Acad. Sci. USA 1993, 90, 4942–4946. [Google Scholar] [CrossRef]
- Rock, K.L.; Gamble, S.; Rothstein, L. Presentation of exogenous antigen with class I major histocompatibility complex molecules. Science 1990, 249, 918–921. [Google Scholar] [CrossRef]
- Heath, W.R.; Belz, G.T.; Behrens, G.M.; Smith, C.M.; Forehan, S.P.; Parish, I.A.; Davey, G.M.; Wilson, N.S.; Carbone, F.R.; Villadangos, J.A. Cross-presentation, dendritic cell subsets, and the generation of immunity to cellular antigens. Immunol. Rev. 2004, 199, 9–26. [Google Scholar] [CrossRef]
- Burgdorf, S.; Kautz, A.; Bohnert, V.; Knolle, P.A.; Kurts, C. Distinct pathways of antigen uptake and intracellular routing in CD4 and CD8 T cell activation. Science 2007, 316, 612–616. [Google Scholar] [CrossRef]
- Burgdorf, S.; Scholz, C.; Kautz, A.; Tampe, R.; Kurts, C. Spatial and mechanistic separation of cross-presentation and endogenous antigen presentation. Nat. Immunol. 2008, 9, 558–566. [Google Scholar] [CrossRef]
- Cullen, S.P.; Brunet, M.; Martin, S.J. Granzymes in cancer and immunity. Cell Death Differ. 2010, 17, 616–623. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Lostao, L.; Anel, A.; Pardo, J. How Do Cytotoxic Lymphocytes Kill Cancer Cells? Clin. Cancer Res. 2015, 21, 5047–5056. [Google Scholar] [CrossRef] [PubMed]
- Shroff, K.; Vidyasagar, A. Polymer Nanoparticles: Newer Strategies towards Targeted Cancer Therapy. J. Phys. Chem. Biophys. 2013, 3, 2161–2398. [Google Scholar]
- Shi, J.; Votruba, A.R.; Farokhzad, O.C.; Langer, R. Nanotechnology in drug delivery and tissue engineering: From discovery to applications. Nano Lett. 2010, 10, 3223–3230. [Google Scholar] [CrossRef]
- Duncan, R. Polymer conjugates as anticancer nanomedicines. Nat. Rev. Cancer 2006, 6, 688–701. [Google Scholar] [CrossRef]
- Peer, D.; Karp, J.M.; Hong, S.; Farokhzad, O.C.; Margalit, R.; Langer, R. Nanocarriers as an emerging platform for cancer therapy. Nat. Nanotechnol. 2007, 2, 751–760. [Google Scholar] [CrossRef]
- Sun, X.; Liu, Z.; Welsher, K.; Robinson, J.T.; Goodwin, A.; Zaric, S.; Dai, H. Nano-Graphene Oxide for Cellular Imaging and Drug Delivery. Nano Res. 2008, 1, 203–212. [Google Scholar] [CrossRef]
- Moon, J.J.; Suh, H.; Li, A.V.; Ockenhouse, C.F.; Yadava, A.; Irvine, D.J. Enhancing humoral responses to a malaria antigen with nanoparticle vaccines that expand Tfh cells and promote germinal center induction. Proc. Natl. Acad. Sci. USA 2012, 109, 1080–1085. [Google Scholar] [CrossRef]
- Singh, S.S.; Yap, W.N.; Arfuso, F.; Kar, S.; Wang, C.; Cai, W.; Dharmarajan, A.M.; Sethi, G.; Kumar, A.P. Targeting the PI3K/Akt signaling pathway in gastric carcinoma: A reality for personalized medicine? World J. Gastroenterol. 2015, 21, 12261–12273. [Google Scholar] [CrossRef]
- Arina, A.; Tirapu, I.; Alfaro, C.; Rodriguez-Calvillo, M.; Mazzolini, G.; Inoges, S.; Lopez, A.; Feijoo, E.; Bendandi, M.; Melero, I. Clinical implications of antigen transfer mechanisms from malignant to dendritic cells: Exploiting cross-priming. Exp. Hematol. 2002, 30, 1355–1364. [Google Scholar] [CrossRef]
- Mayer, C.T.; Berod, L.; Sparwasser, T. Layers of dendritic cell-mediated T cell tolerance, their regulation and the prevention of autoimmunity. Front. Immunol. 2012, 3, 183. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Stephani, J.; Schaefer, M.; Kalay, H.; Garcia-Vallejo, J.J.; den Haan, J.; Saeland, E.; Sparwasser, T.; van Kooyk, Y. Targeting glycan modified OVA to murine DC-SIGN transgenic dendritic cells enhances MHC class I and II presentation. Mol. Immunol. 2009, 47, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Boks, M.A.; Ambrosini, M.; Bruijns, S.C.; Kalay, H.; van Bloois, L.; Storm, G.; Garcia-Vallejo, J.J.; van Kooyk, Y. MPLA incorporation into DC-targeting glycoliposomes favours anti-tumour T cell responses. J. Control. Release 2015, 216, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Boks, M.A.; Bruijns, S.C.M.; Ambrosini, M.; Kalay, H.; van Bloois, L.; Storm, G.; Gruijl, T.; van Kooyk, Y. In situ Delivery of Tumor Antigen- and Adjuvant-Loaded Liposomes Boosts Antigen-Specific T-Cell Responses by Human Dermal Dendritic Cells. J. Investig. Dermatol. 2015, 135, 2697–2704. [Google Scholar] [CrossRef] [PubMed]
- Fehres, C.M.; Kalay, H.; Bruijns, S.C.; Musaafir, S.A.; Ambrosini, M.; van Bloois, L.; van Vliet, S.J.; Storm, G.; Garcia-Vallejo, J.J.; van Kooyk, Y. Cross-presentation through langerin and DC-SIGN targeting requires different formulations of glycan-modified antigens. J. Control. Release 2015, 203, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Vallejo, J.J.; Ambrosini, M.; Overbeek, A.; van Riel, W.E.; Bloem, K.; Unger, W.W.; Chiodo, F.; Bolscher, J.G.; Nazmi, K.; Kalay, H.; et al. Multivalent glycopeptide dendrimers for the targeted delivery of antigens to dendritic cells. Mol. Immunol. 2013, 53, 387–397. [Google Scholar] [CrossRef]
- Anderluh, M.; Berti, F.; Bzducha-Wrobel, A.; Chiodo, F.; Colombo, C.; Compostella, F.; Durlik, K.; Ferhati, X.; Holmdahl, R.; Jovanovic, D.; et al. Emerging glyco-based strategies to steer immune responses. FEBS J. 2021, 288, 4746–4772. [Google Scholar] [CrossRef]
- Glaffig, M.; Stergiou, N.; Hartmann, S.; Schmitt, E.; Kunz, H. A Synthetic MUC1 Anticancer Vaccine Containing Mannose Ligands for Targeting Macrophages and Dendritic Cells. ChemMedChem 2018, 13, 25–29. [Google Scholar] [CrossRef]
- Rauen, J.; Kreer, C.; Paillard, A.; van Duikeren, S.; Benckhuijsen, W.E.; Camps, M.G.; Valentijn, A.R.; Ossendorp, F.; Drijfhout, J.W.; Arens, R.; et al. Enhanced cross-presentation and improved CD8+ T cell responses after mannosylation of synthetic long peptides in mice. PLoS ONE 2014, 9, e103755. [Google Scholar] [CrossRef]
- Apostolopoulos, V.; Pietersz, G.A.; Gordon, S.; Martinez-Pomares, L.; McKenzie, I.F. Aldehyde-mannan antigen complexes target the MHC class I antigen-presentation pathway. Eur. J. Immunol. 2000, 30, 1714–1723. [Google Scholar] [CrossRef]
- Apostolopoulos, V.; Barnes, N.; Pietersz, G.A.; McKenzie, I.F. Ex vivo targeting of the macrophage mannose receptor generates anti-tumor CTL responses. Vaccine 2000, 18, 3174–3184. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, M.; Yuba, E.; Hayashi, H.; Harada, A.; Kono, K. Development of pH-Responsive Hyaluronic Acid-Based Antigen Carriers for Induction of Antigen-Specific Cellular Immune Responses. ACS Biomater. Sci. Eng. 2019, 5, 5790–5797. [Google Scholar] [CrossRef] [PubMed]
- Okubo, M.; Miyazaki, M.; Yuba, E.; Harada, A. Chondroitin Sulfate-Based pH-Sensitive Polymer-Modified Liposomes for Intracellular Antigen Delivery and Induction of Cancer Immunity. Bioconjug. Chem. 2019, 30, 1518–1529. [Google Scholar] [CrossRef] [PubMed]
- Yuba, E.; Uesugi, S.; Miyazaki, M.; Kado, Y.; Harada, A.; Kono, K. Development of pH-sensitive Dextran Derivatives with Strong Adjuvant Function and Their Application to Antigen Delivery. Membranes 2017, 7, 41. [Google Scholar] [CrossRef] [PubMed]
- Yuba, E.; Yamaguchi, A.; Yoshizaki, Y.; Harada, A.; Kono, K. Bioactive polysaccharide-based pH-sensitive polymers for cytoplasmic delivery of antigen and activation of antigen-specific immunity. Biomaterials 2017, 120, 32–45. [Google Scholar] [CrossRef]
- Menon, S.; Rosenberg, K.; Graham, S.A.; Ward, E.M.; Taylor, M.E.; Drickamer, K.; Leckband, D.E. Binding-site geometry and flexibility in DC-SIGN demonstrated with surface force measurements. Proc. Natl. Acad. Sci. USA 2009, 106, 11524–11529. [Google Scholar] [CrossRef]
- Li, R.E.; van Vliet, S.J.; van Kooyk, Y. Using the glycan toolbox for pathogenic interventions and glycan immunotherapy. Curr. Opin. Biotechnol. 2018, 51, 24–31. [Google Scholar] [CrossRef]
- Martinez, J.D.; Infantino, A.S.; Valverde, P.; Diercks, T.; Delgado, S.; Reichardt, N.C.; Arda, A.; Canada, F.J.; Oscarson, S.; Jimenez-Barbero, J. The Interaction of Fluorinated Glycomimetics with DC-SIGN: Multiple Binding Modes Disentangled by the Combination of NMR Methods and MD Simulations. Pharmaceuticals 2020, 13, 179. [Google Scholar] [CrossRef]
- Parmiani, G.; De Filippo, A.; Novellino, L.; Castelli, C. Unique human tumor antigens: Immunobiology and use in clinical trials. J. Immunol. 2007, 178, 1975–1979. [Google Scholar] [CrossRef]
- Van Broekhoven, C.L.; Parish, C.R.; Demangel, C.; Britton, W.J.; Altin, J.G. Targeting dendritic cells with antigen-containing liposomes: A highly effective procedure for induction of antitumor immunity and for tumor immunotherapy. Cancer Res. 2004, 64, 4357–4365. [Google Scholar] [CrossRef]
- Wang, X.; Zhong, X.; Li, J.; Liu, Z.; Cheng, L. Inorganic nanomaterials with rapid clearance for biomedical applications. Chem. Soc. Rev. 2021, 50, 8669–8742. [Google Scholar] [CrossRef] [PubMed]
- Molino, N.M.; Anderson, A.K.; Nelson, E.L.; Wang, S.W. Biomimetic protein nanoparticles facilitate enhanced dendritic cell activation and cross-presentation. ACS Nano 2013, 7, 9743–9752. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyay, S.; Chen, J.Y.; Chen, H.W.; Hu, C.J. Nanoparticle Vaccines Adopting Virus-like Features for Enhanced Immune Potentiation. Nanotheranostics 2017, 1, 244–260. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Vallejo, J.J.; Unger, W.W.; Kalay, H.; van Kooyk, Y. Glycan-based DC-SIGN targeting to enhance antigen cross-presentation in anticancer vaccines. Oncoimmunology 2013, 2, e23040. [Google Scholar] [CrossRef]
- Van Kooyk, Y.; Rabinovich, G.A. Protein-glycan interactions in the control of innate and adaptive immune responses. Nat. Immunol. 2008, 9, 593–601. [Google Scholar] [CrossRef]
- Wilson, J.T.; Keller, S.; Manganiello, M.J.; Cheng, C.; Lee, C.C.; Opara, C.; Convertine, A.; Stayton, P.S. pH-Responsive nanoparticle vaccines for dual-delivery of antigens and immunostimulatory oligonucleotides. ACS Nano 2013, 7, 3912–3925. [Google Scholar] [CrossRef]
- Liu, Q.; Jia, J.; Yang, T.; Fan, Q.; Wang, L.; Ma, G. Pathogen-Mimicking Polymeric Nanoparticles based on Dopamine Polymerization as Vaccines Adjuvants Induce Robust Humoral and Cellular Immune Responses. Small 2016, 12, 1744–1757. [Google Scholar] [CrossRef]
- Reuven, E.M.; Leviatan Ben-Arye, S.; Yu, H.; Duchi, R.; Perota, A.; Conchon, S.; Bachar Abramovitch, S.; Soulillou, J.P.; Galli, C.; Chen, X.; et al. Biomimetic Glyconanoparticle Vaccine for Cancer Immunotherapy. ACS Nano 2019, 13, 2936–2947. [Google Scholar] [CrossRef]
- Joshi, M.D.; Unger, W.J.; Storm, G.; van Kooyk, Y.; Mastrobattista, E. Targeting tumor antigens to dendritic cells using particulate carriers. J. Control. Release 2012, 161, 25–37. [Google Scholar] [CrossRef]
- Yuba, E. Development of functional liposomes by modification of stimuli-responsive materials and their biomedical applications. J. Mater. Chem. B 2020, 8, 1093–1107. [Google Scholar] [CrossRef]
- Maji, M.; Mazumder, S.; Bhattacharya, S.; Choudhury, S.T.; Sabur, A.; Shadab, M.; Bhattacharya, P.; Ali, N. A Lipid Based Antigen Delivery System Efficiently Facilitates MHC Class-I Antigen Presentation in Dendritic Cells to Stimulate CD8+ T Cells. Sci. Rep. 2016, 6, 27206. [Google Scholar] [CrossRef] [PubMed]
- Zho, F.; Neutra, M.R. Antigen delivery to mucosa-associated lymphoid tissues using liposomes as a carrier. Biosci. Rep. 2002, 22, 355–369. [Google Scholar] [CrossRef][Green Version]
- Yuba, E. Liposome-based immunity-inducing systems for cancer immunotherapy. Mol. Immunol. 2018, 98, 8–12. [Google Scholar] [CrossRef] [PubMed]
- Ahsan, F.; Rivas, I.P.; Khan, M.A.; Torres Suarez, A.I. Targeting to macrophages: Role of physicochemical properties of particulate carriers—Liposomes and microspheres—On the phagocytosis by macrophages. J. Control. Release 2002, 79, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Joshi, M.D.; Unger, W.W.; van Beelen, A.J.; Bruijns, S.C.; Litjens, M.; van Bloois, L.; Kalay, H.; van Kooyk, Y.; Storm, G. DC-SIGN mediated antigen-targeting using glycan-modified liposomes: Formulation considerations. Int. J. Pharm. 2011, 416, 426–432. [Google Scholar] [CrossRef] [PubMed]
- Stolk, D.A.; de Haas, A.; Vree, J.; Duinkerken, S.; Lubbers, J.; van de Ven, R.; Ambrosini, M.; Kalay, H.; Bruijns, S.; van der Vliet, H.J.; et al. Lipo-Based Vaccines as an Approach to Target Dendritic Cells for Induction of T-and iNKT Cell Responses. Front. Immunol. 2020, 11, 990. [Google Scholar] [CrossRef]
- Affandi, A.J.; Grabowska, J.; Olesek, K.; Lopez Venegas, M.; Barbaria, A.; Rodriguez, E.; Mulder, P.P.G.; Pijffers, H.J.; Ambrosini, M.; Kalay, H.; et al. Selective tumor antigen vaccine delivery to human CD169+ antigen-presenting cells using ganglioside-liposomes. Proc. Natl. Acad. Sci. USA 2020, 117, 27528–27539. [Google Scholar] [CrossRef]
- Nijen Twilhaar, M.K.; Czentner, L.; Bouma, R.G.; Olesek, K.; Grabowska, J.; Wang, A.Z.; Affandi, A.J.; Belt, S.C.; Kalay, H.; van Nostrum, C.F.; et al. Incorporation of Toll-Like Receptor Ligands and Inflammasome Stimuli in GM3 Liposomes to Induce Dendritic Cell Maturation and T Cell Responses. Front. Immunol. 2022, 13, 842241. [Google Scholar] [CrossRef]
- Yuba, E.; Kado, Y.; Kasho, N.; Harada, A. Cationic lipid potentiated the adjuvanticity of polysaccharide derivative-modified liposome vaccines. J. Control. Release 2022. [Google Scholar] [CrossRef]
- Biricova, V.; Laznickova, A. Dendrimers: Analytical characterization and applications. Bioorg. Chem. 2009, 37, 185–192. [Google Scholar] [CrossRef]
- Duinkerken, S.; Horrevorts, S.K.; Kalay, H.; Ambrosini, M.; Rutte, L.; de Gruijl, T.D.; Garcia-Vallejo, J.J.; van Kooyk, Y. Glyco-Dendrimers as Intradermal Anti-Tumor Vaccine Targeting Multiple Skin DC Subsets. Theranostics 2019, 9, 5797–5809. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Ma, S.; Zhao, J.; Chen, H.; Si, X.; Huang, Z.; Yu, Z.; Song, W.; Tang, Z.; Chen, X. Mannan-decorated pathogen-like polymeric nanoparticles as nanovaccine carriers for eliciting superior anticancer immunity. Biomaterials 2022, 284, 121489. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Ackerman, A.L.; Cody, V.; Giodini, A.; Hinson, E.R.; Cresswell, P.; Edelson, R.L.; Saltzman, W.M.; Hanlon, D.J. Enhanced and prolonged cross-presentation following endosomal escape of exogenous antigens encapsulated in biodegradable nanoparticles. Immunology 2006, 117, 78–88. [Google Scholar] [CrossRef]
- Wang, B.; Chen, G.; Mao, Z.; Zhang, Y.; Yu, D.; Gao, C. Preparation and cellular uptake of PLGA particles loaded with lamivudine. Chin. Sci. Bull. 2012, 57, 3985–3993. [Google Scholar] [CrossRef][Green Version]
- Yang, R.; Xu, J.; Xu, L.; Sun, X.; Chen, Q.; Zhao, Y.; Peng, R.; Liu, Z. Cancer Cell Membrane-Coated Adjuvant Nanoparticles with Mannose Modification for Effective Anticancer Vaccination. ACS Nano 2018, 12, 5121–5129. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Cao, F.; Liu, X.; Wang, H.; Zhang, C.; Sun, H.; Wang, C.; Leng, X.; Song, C.; Kong, D.; et al. Hyaluronic Acid-Modified Cationic Lipid-PLGA Hybrid Nanoparticles as a Nanovaccine Induce Robust Humoral and Cellular Immune Responses. ACS Appl. Mater. Interfaces 2016, 8, 11969–11979. [Google Scholar] [CrossRef] [PubMed]
- Srinivas, O.; Larrieu, P.; Duverger, E.; Boccaccio, C.; Bousser, M.T.; Monsigny, M.; Fonteneau, J.F.; Jotereau, F.; Roche, A.C. Synthesis of glycocluster-tumor antigenic peptide conjugates for dendritic cell targeting. Bioconjug. Chem. 2007, 18, 1547–1554. [Google Scholar] [CrossRef]
- Quétard, C.; Bourgerie, S.; Normand-Sdiqui, N.; Mayer, R.; Strecker, G.; Midoux, P.; Roche, A.-C.; Monsigny, M. Novel Glycosynthons for Glycoconjugate Preparation: Oligosaccharylpyroglutamylanilide Derivatives. Bioconjug. Chem. 1998, 9, 268–276. [Google Scholar] [CrossRef]
- Liu, C.; Chu, X.; Yan, M.; Qi, J.; Liu, H.; Gao, F.; Gao, R.; Ma, G.; Ma, Y. Encapsulation of Poly I:C and the natural phosphodiester CpG ODN enhanced the efficacy of a hyaluronic acid-modified cationic lipid-PLGA hybrid nanoparticle vaccine in TC-1-grafted tumors. Int. J. Pharm. 2018, 553, 327–337. [Google Scholar] [CrossRef]
- Fuss, M.; Luna, M.; Alcantara, D.; Fuente, J.M.; Penades, S.; Briones, F. Supramolecular self-assembled arrangements of maltose glyconanoparticles. Langmuir 2008, 24, 5124–5128. [Google Scholar] [CrossRef]
- Matheoud, D.; Perie, L.; Hoeffel, G.; Vimeux, L.; Parent, I.; Maranon, C.; Bourdoncle, P.; Renia, L.; Prevost-Blondel, A.; Lucas, B.; et al. Cross-presentation by dendritic cells from live cells induces protective immune responses in vivo. Blood 2010, 115, 4412–4420. [Google Scholar] [CrossRef] [PubMed]
- Harshyne, L.A.; Watkins, S.C.; Gambotto, A.; Barratt-Boyes, S.M. Dendritic cells acquire antigens from live cells for cross-presentation to CTL. J. Immunol. 2001, 166, 3717–3723. [Google Scholar] [CrossRef] [PubMed]
- Garcia, I.; Marradi, M.; Penades, S. Glyconanoparticles: Multifunctional nanomaterials for biomedical applications. Nanomedicine 2010, 5, 777–792. [Google Scholar] [CrossRef] [PubMed]
- Avci, F.Y.; Li, X.; Tsuji, M.; Kasper, D.L. A mechanism for glycoconjugate vaccine activation of the adaptive immune system and its implications for vaccine design. Nat. Med. 2011, 17, 1602–1609. [Google Scholar] [CrossRef]
- Mestas, J.; Hughes, C.C. Of mice and not men: Differences between mouse and human immunology. J. Immunol. 2004, 172, 2731–2738. [Google Scholar] [CrossRef] [PubMed]
- Segura, E.; Amigorena, S. Cross-Presentation in Mouse and Human Dendritic Cells. Adv. Immunol. 2015, 127, 1–31. [Google Scholar] [CrossRef]
- Met, O.; Buus, S.; Claesson, M.H. Peptide-loaded dendritic cells prime and activate MHC-class I-restricted T cells more efficiently than protein-loaded cross-presenting DC. Cell Immunol. 2003, 222, 126–133. [Google Scholar] [CrossRef]
- Ramasubramanian, M.K.; Barham, O.M.; Swaminathan, V. Mechanics of a mosquito bite with applications to microneedle design. Bioinspir. Biomim. 2008, 3, 046001. [Google Scholar] [CrossRef]
- Prausnitz, M.R. Microneedles for transdermal drug delivery. Adv. Drug Deliv. Rev. 2004, 56, 581–587. [Google Scholar] [CrossRef]
- Henry, S.; McAllister, D.V.; Allen, M.G.; Prausnitz, M.R. Microfabricated microneedles: A novel approach to transdermal drug delivery. J. Pharm. Sci. 1998, 87, 922–925. [Google Scholar] [CrossRef]
- Nestle, F.O.; Zheng, X.G.; Thompson, C.B.; Turka, L.A.; Nickoloff, B.J. Characterization of dermal dendritic cells obtained from normal human skin reveals phenotypic and functionally distinctive subsets. J. Immunol. 1993, 151, 6535–6545. [Google Scholar] [PubMed]
- Teran-Navarro, H.; Zeoli, A.; Salines-Cuevas, D.; Marradi, M.; Montoya, N.; Gonzalez-Lopez, E.; Ocejo-Vinyals, J.G.; Dominguez-Esteban, M.; Gutierrez-Banos, J.L.; Campos-Juanatey, F.; et al. Gold Glyconanoparticles Combined with 91-99 Peptide of the Bacterial Toxin, Listeriolysin O, Are Efficient Immunotherapies in Experimental Bladder Tumors. Cancers 2022, 14, 2413. [Google Scholar] [CrossRef] [PubMed]
- Chi, G.; Lv, Y.; Chao, S.; Hou, C.; Pei, Y.; Pei, Z. Glyconanoparticles with Activatable Near-Infrared Probes for Tumor-Cell Imaging and Targeted Drug Delivery. Int. J. Nanomed. 2022, 17, 1567–1575. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Zhao, Q.; Dong, H.; Xiao, M.; Huang, X.; Wu, X. Developing Acid-Responsive Glyco-Nanoplatform Based Vaccines for Enhanced Cytotoxic T-lymphocyte Responses Against Cancer and SARS-CoV-2. Adv. Funct. Mater. 2021, 31, 2105059. [Google Scholar] [CrossRef]
- Belizaire, R.; Unanue, E.R. Targeting proteins to distinct subcellular compartments reveals unique requirements for MHC class I and II presentation. Proc. Natl. Acad. Sci. USA 2009, 106, 17463–17468. [Google Scholar] [CrossRef]
- Li, H.; Li, Y.; Jiao, J.; Hu, H.M. Alpha-alumina nanoparticles induce efficient autophagy-dependent cross-presentation and potent antitumour response. Nat. Nanotechnol. 2011, 6, 645–650. [Google Scholar] [CrossRef]
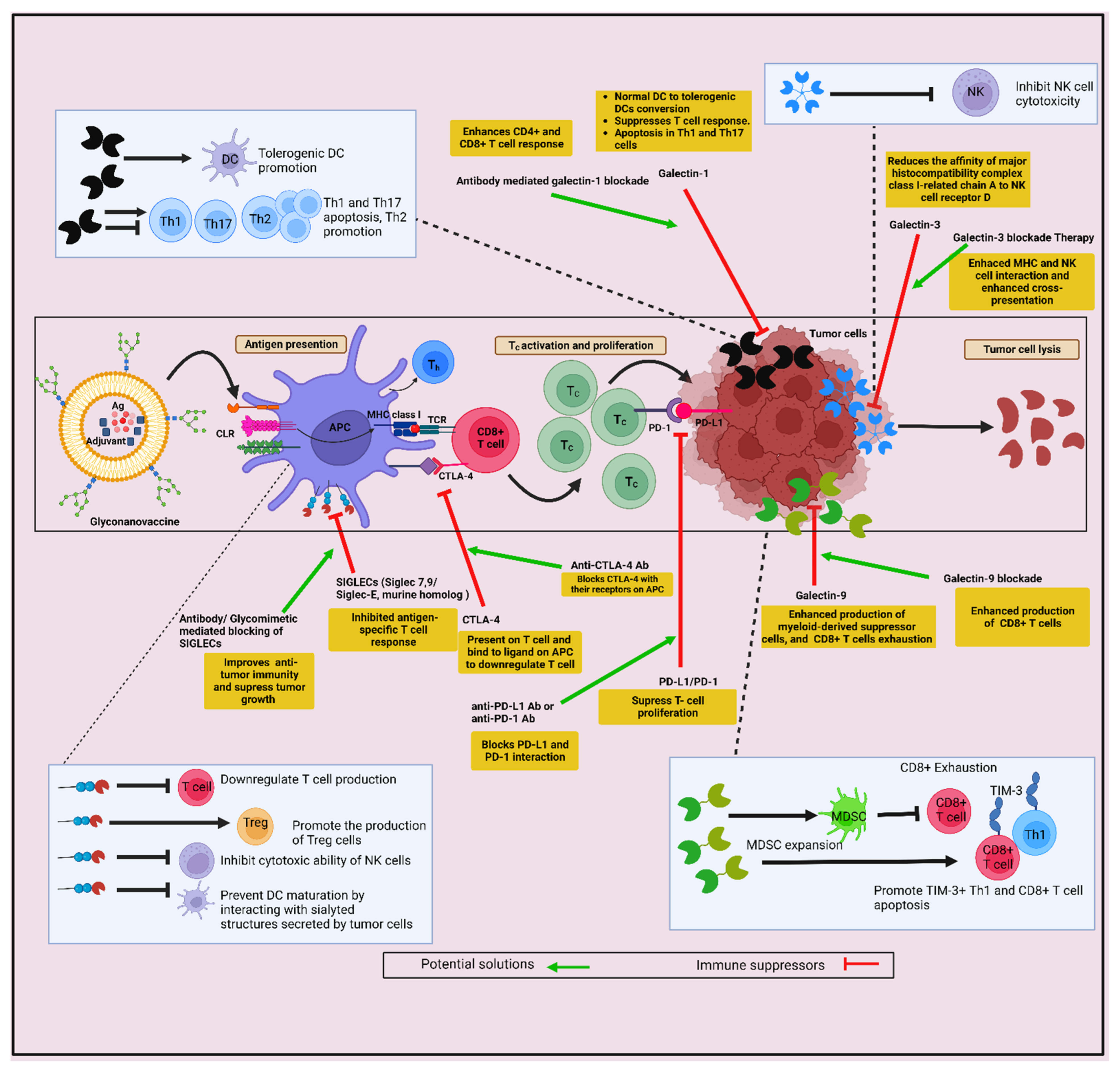
- RodrIguez, E.; Schetters, S.T.T.; van Kooyk, Y. The tumour glyco-code as a novel immune checkpoint for immunotherapy. Nat. Rev. Immunol. 2018, 18, 204–211. [Google Scholar] [CrossRef]
- Park, R.; Winnicki, M.; Liu, E.; Chu, W.M. Immune checkpoints and cancer in the immunogenomics era. Brief. Funct. Genom. 2019, 18, 133–139. [Google Scholar] [CrossRef]
- Darvin, P.; Toor, S.M.; Sasidharan Nair, V.; Elkord, E. Immune checkpoint inhibitors: Recent progress and potential biomarkers. Exp. Mol. Med. 2018, 50, 1–11. [Google Scholar] [CrossRef]
- Qureshi, O.S.; Zheng, Y.; Nakamura, K.; Attridge, K.; Manzotti, C.; Schmidt, E.M.; Baker, J.; Jeffery, L.E.; Kaur, S.; Briggs, Z.; et al. Trans-endocytosis of CD80 and CD86: A molecular basis for the cell-extrinsic function of CTLA-4. Science 2011, 332, 600–603. [Google Scholar] [CrossRef]
- Keir, M.E.; Butte, M.J.; Freeman, G.J.; Sharpe, A.H. PD-1 and its ligands in tolerance and immunity. Annu. Rev. Immunol. 2008, 26, 677–704. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; You, L.; Nepovimova, E.; Heger, Z.; Wu, W.; Kuca, K.; Adam, V. Hypoxia-inducible factors: Master regulators of hypoxic tumor immune escape. J. Hematol. Oncol. 2022, 15, 77. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.; Hwu, W.J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef] [PubMed]
- Iwama, S.; De Remigis, A.; Callahan, M.K.; Slovin, S.F.; Wolchok, J.D.; Caturegli, P. Pituitary expression of CTLA-4 mediates hypophysitis secondary to administration of CTLA-4 blocking antibody. Sci. Transl. Med. 2014, 6, 230ra245. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Zhu, Y.; Dong, Z.; Li, W.; Yang, N.; Wang, X.; Feng, L.; Liu, Z. Tumor-killing nanoreactors fueled by tumor debris can enhance radiofrequency ablation therapy and boost antitumor immune responses. Nat. Commun. 2021, 12, 4299. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Liu, K.; Gong, Y.; Zhu, W.; Zhu, J.; Pan, F.; Chao, Y.; Xiao, Z.; Liu, Y.; Wang, X.; et al. Vitamin C supramolecular hydrogel for enhanced cancer immunotherapy. Biomaterials 2022, 287, 121673. [Google Scholar] [CrossRef]
- Pinho, S.S.; Reis, C.A. Glycosylation in cancer: Mechanisms and clinical implications. Nat. Rev. Cancer 2015, 15, 540–555. [Google Scholar] [CrossRef]
- Perdicchio, M.; Cornelissen, L.A.; Streng-Ouwehand, I.; Engels, S.; Verstege, M.I.; Boon, L.; Geerts, D.; van Kooyk, Y.; Unger, W.W. Tumor sialylation impedes T cell mediated anti-tumor responses while promoting tumor associated-regulatory T cells. Oncotarget 2016, 7, 8771–8782. [Google Scholar] [CrossRef]
- Jandus, C.; Boligan, K.F.; Chijioke, O.; Liu, H.; Dahlhaus, M.; Demoulins, T.; Schneider, C.; Wehrli, M.; Hunger, R.E.; Baerlocher, G.M.; et al. Interactions between Siglec-7/9 receptors and ligands influence NK cell-dependent tumor immunosurveillance. J. Clin. Investig. 2014, 124, 1810–1820. [Google Scholar] [CrossRef]
- Wang, J.; Sun, J.; Liu, L.N.; Flies, D.B.; Nie, X.; Toki, M.; Zhang, J.; Song, C.; Zarr, M.; Zhou, X.; et al. Siglec-15 as an immune suppressor and potential target for normalization cancer immunotherapy. Nat. Med. 2019, 25, 656–666. [Google Scholar] [CrossRef] [PubMed]
- Nandi, S.; Ghosh, S.; Ranjan, A.; Sood, R.S.; Pal, J.K.; Hajela, K.; Gupta, R.K. Lectins in Health and Diseases: Galectins and Cancer. In Lectins: Innate Immune Defense and Therapeutics; Elumalai, P., Lakshmi, S., Eds.; Springer: Singapore, 2021; pp. 215–271. [Google Scholar] [CrossRef]
- Liu, F.T.; Rabinovich, G.A. Galectins as modulators of tumour progression. Nat. Rev. Cancer 2005, 5, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Mendez-Huergo, S.P.; Blidner, A.G.; Rabinovich, G.A. Galectins: Emerging regulatory checkpoints linking tumor immunity and angiogenesis. Curr. Opin. Immunol. 2017, 45, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Toscano, M.A.; Bianco, G.A.; Ilarregui, J.M.; Croci, D.O.; Correale, J.; Hernandez, J.D.; Zwirner, N.W.; Poirier, F.; Riley, E.M.; Baum, L.G.; et al. Differential glycosylation of TH1, TH2 and TH-17 effector cells selectively regulates susceptibility to cell death. Nat. Immunol. 2007, 8, 825–834. [Google Scholar] [CrossRef]
- Ilarregui, J.M.; Croci, D.O.; Bianco, G.A.; Toscano, M.A.; Salatino, M.; Vermeulen, M.E.; Geffner, J.R.; Rabinovich, G.A. Tolerogenic signals delivered by dendritic cells to T cells through a galectin-1-driven immunoregulatory circuit involving interleukin 27 and interleukin 10. Nat. Immunol. 2009, 10, 981–991. [Google Scholar] [CrossRef]
- Rubinstein, N.; Alvarez, M.; Zwirner, N.W.; Toscano, M.A.; Ilarregui, J.M.; Bravo, A.; Mordoh, J.; Fainboim, L.; Podhajcer, O.L.; Rabinovich, G.A. Targeted inhibition of galectin-1 gene expression in tumor cells results in heightened T cell-mediated rejection; A potential mechanism of tumor-immune privilege. Cancer Cell 2004, 5, 241–251. [Google Scholar] [CrossRef]
- Baker, G.J.; Chockley, P.; Yadav, V.N.; Doherty, R.; Ritt, M.; Sivaramakrishnan, S.; Castro, M.G.; Lowenstein, P.R. Natural killer cells eradicate galectin-1-deficient glioma in the absence of adaptive immunity. Cancer Res. 2014, 74, 5079–5090. [Google Scholar] [CrossRef]
- Tsuboi, S.; Sutoh, M.; Hatakeyama, S.; Hiraoka, N.; Habuchi, T.; Horikawa, Y.; Hashimoto, Y.; Yoneyama, T.; Mori, K.; Koie, T.; et al. A novel strategy for evasion of NK cell immunity by tumours expressing core2 O-glycans. EMBO J. 2011, 30, 3173–3185. [Google Scholar] [CrossRef]
- Dardalhon, V.; Anderson, A.C.; Karman, J.; Apetoh, L.; Chandwaskar, R.; Lee, D.H.; Cornejo, M.; Nishi, N.; Yamauchi, A.; Quintana, F.J.; et al. Tim-3/galectin-9 pathway: Regulation of Th1 immunity through promotion of CD11b+Ly-6G+ myeloid cells. J. Immunol. 2010, 185, 1383–1392. [Google Scholar] [CrossRef]
- Sakuishi, K.; Jayaraman, P.; Behar, S.M.; Anderson, A.C.; Kuchroo, V.K. Emerging Tim-3 functions in antimicrobial and tumor immunity. Trends Immunol. 2011, 32, 345–349. [Google Scholar] [CrossRef]
- Apostolopoulos, V.; Yuriev, E.; Ramsland, P.A.; Halton, J.; Osinski, C.; Li, W.; Plebanski, M.; Paulsen, H.; McKenzie, I.F. A glycopeptide in complex with MHC class I uses the GalNAc residue as an anchor. Proc. Natl. Acad. Sci USA 2003, 100, 15029–15034. [Google Scholar] [CrossRef] [PubMed]
- Vlad, A.M.; Muller, S.; Cudic, M.; Paulsen, H.; Otvos, L., Jr.; Hanisch, F.G.; Finn, O.J. Complex carbohydrates are not removed during processing of glycoproteins by dendritic cells: Processing of tumor antigen MUC1 glycopeptides for presentation to major histocompatibility complex class II-restricted T cells. J. Exp. Med. 2002, 196, 1435–1446. [Google Scholar] [CrossRef] [PubMed]
- Malaker, S.A.; Penny, S.A.; Steadman, L.G.; Myers, P.T.; Loke, J.C.; Raghavan, M.; Bai, D.L.; Shabanowitz, J.; Hunt, D.F.; Cobbold, M. Identification of Glycopeptides as Posttranslationally Modified Neoantigens in Leukemia. Cancer Immunol. Res. 2017, 5, 376–384. [Google Scholar] [CrossRef] [PubMed]




| Pathogens | Imitative Approach | Reference(s) | |
|---|---|---|---|
| Size | Viruses (20–200 nm) | DC-targeted nanoparticle of the same size as viruses (20 nm to 200 nm) | [214,215] |
| Interactions | Interactions between the carbohydrates present on the pathogen surface and the APC receptor | Nanoparticle coated with glycan interacts with specific CLRs | [216,217] |
| Escaping endo/lysosomal trafficking pathways into the cytosol | Using pH-dependent mechanisms | pH-responsive endosomal escape | [218] |
| Immune cell recruitment | Achieved through cytokine secretion | Increased cytokine secretion and immune cell recruitment on administration | [219] |
| Surface properties | Pathogen-associated molecular patterns (PAMPs) | Glycan-coated nanoparticles | [78,220] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Makandar, A.I.; Jain, M.; Yuba, E.; Sethi, G.; Gupta, R.K. Canvassing Prospects of Glyco-Nanovaccines for Developing Cross-Presentation Mediated Anti-Tumor Immunotherapy. Vaccines 2022, 10, 2049. https://doi.org/10.3390/vaccines10122049
Makandar AI, Jain M, Yuba E, Sethi G, Gupta RK. Canvassing Prospects of Glyco-Nanovaccines for Developing Cross-Presentation Mediated Anti-Tumor Immunotherapy. Vaccines. 2022; 10(12):2049. https://doi.org/10.3390/vaccines10122049
Chicago/Turabian StyleMakandar, Amina I., Mannat Jain, Eiji Yuba, Gautam Sethi, and Rajesh Kumar Gupta. 2022. "Canvassing Prospects of Glyco-Nanovaccines for Developing Cross-Presentation Mediated Anti-Tumor Immunotherapy" Vaccines 10, no. 12: 2049. https://doi.org/10.3390/vaccines10122049
APA StyleMakandar, A. I., Jain, M., Yuba, E., Sethi, G., & Gupta, R. K. (2022). Canvassing Prospects of Glyco-Nanovaccines for Developing Cross-Presentation Mediated Anti-Tumor Immunotherapy. Vaccines, 10(12), 2049. https://doi.org/10.3390/vaccines10122049