Oxidative Stress in Cardiovascular Diseases


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
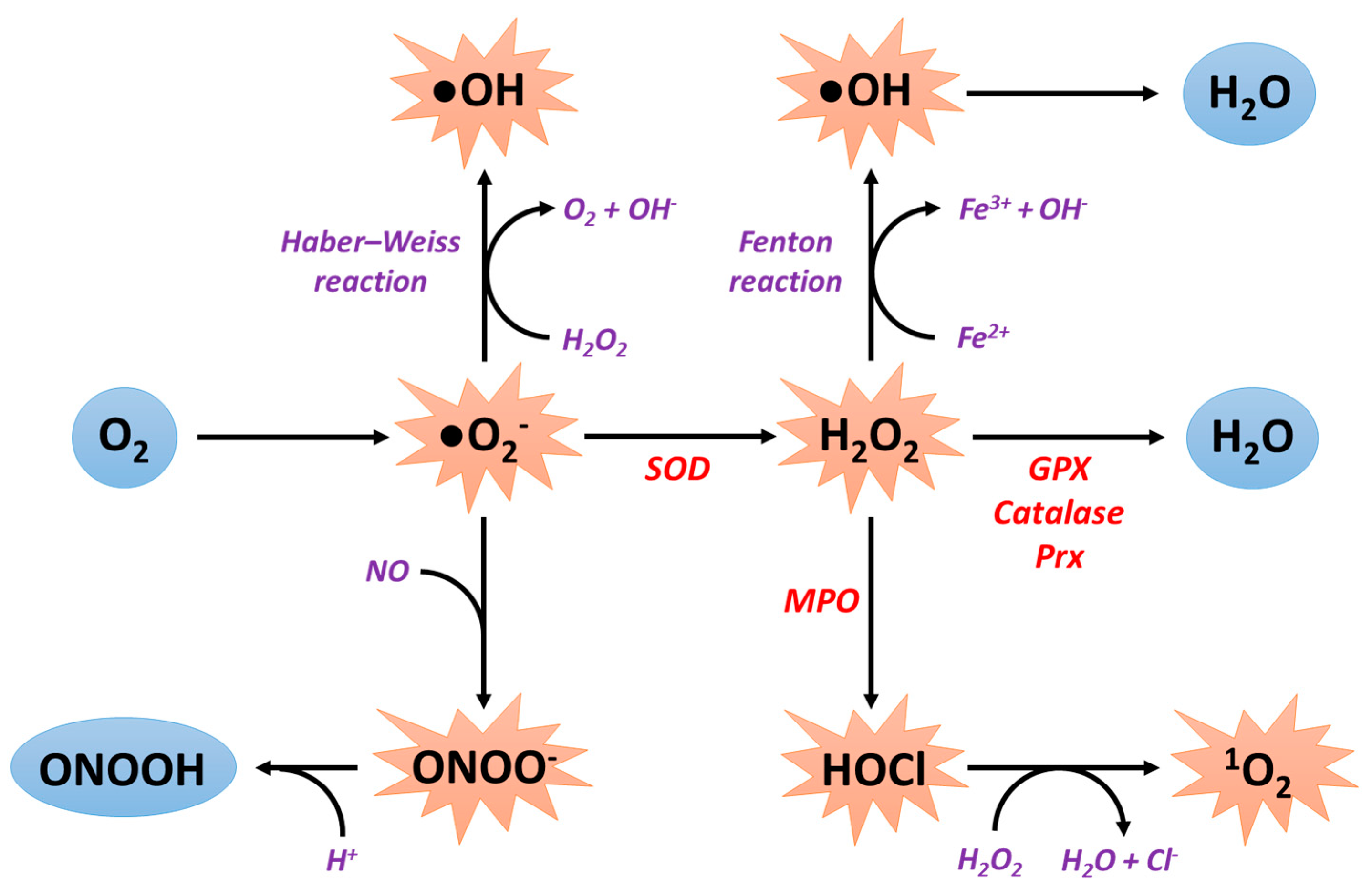
2. General Information about Reactive Oxygen Species Production
2.1. Sources of ROS Production
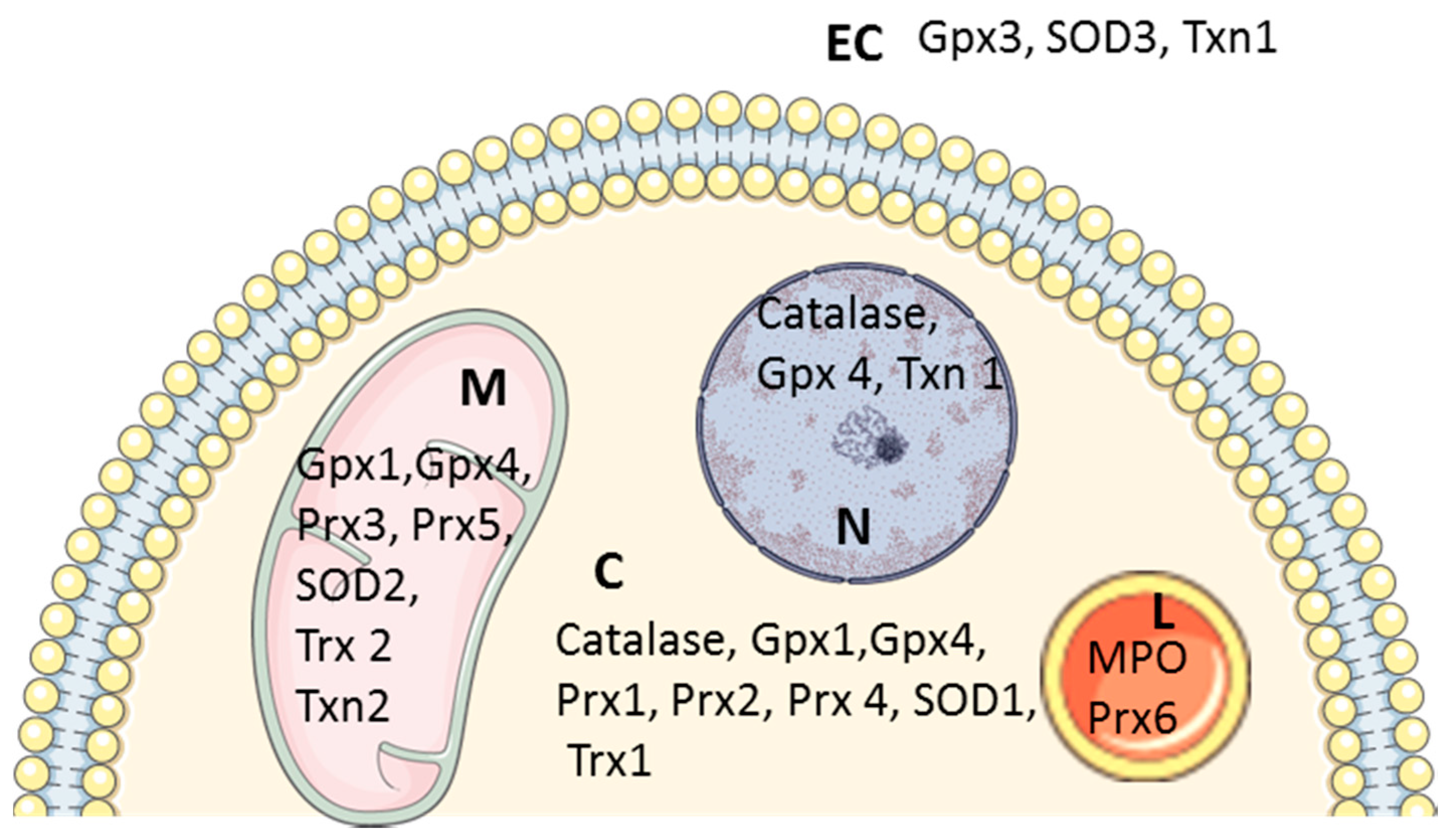
2.2. Antioxidant Systems
2.2.1. Superoxide Dismutases
2.2.2. Catalase
2.2.3. Peroxiredoxins
2.2.4. Glutathione Peroxidases
2.2.5. Non-Enzymatic Antioxidant Defense
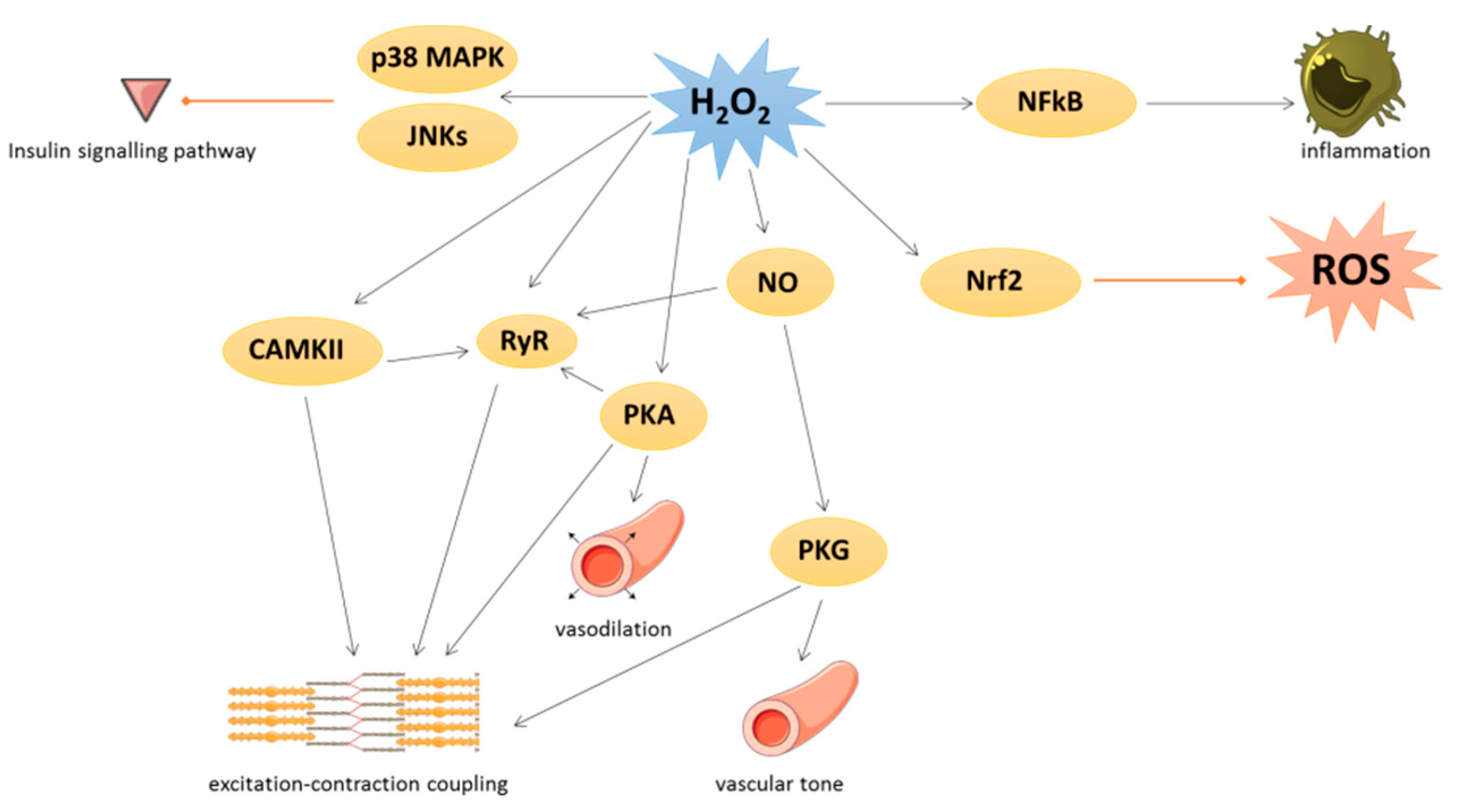
3. Physiological Roles of Oxidative Stress in Cardiovascular Tissues
4. Pathological Implications of Oxidative Stress in Cardiovascular Tissues
4.1. General Aspects
4.2. NOX-Dependent Effects
4.3. Endothelial Dysfunction
4.4. Mitochondrial Oxidative Stress
5. New Therapeutic Strategies
5.1. Vitamins
5.2. Polyphenols
5.3. Mitochondrial-Targeted Antioxidant
5.4. Clinical Trials
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Benjamin, E.J.; Blaha, M.J.; Chiuve, S.E.; Cushman, M.; Das, S.R.; Deo, R.; de Ferranti, S.D.; Floyd, J.; Fornage, M.; Gillespie, C.; et al. Heart Disease and Stroke Statistics-2017 Update: A Report From the American Heart Association. Circulation 2017, 135, e146–e603. [Google Scholar] [CrossRef]
- Al Hariri, M.; Zibara, K.; Farhat, W.; Hashem, Y.; Soudani, N.; Al Ibrahim, F.; Hamade, E.; Zeidan, A.; Husari, A.; Kobeissy, F. Cigarette sImoking-induced cardiac hypertrophy, vascular inflammation and injury are attenuated by antioxidant supplementation in an animal model. Front. Pharmacol. 2016, 7, e397. [Google Scholar] [CrossRef]
- Dikalov, S.; Itani, H.; Richmond, B.; Vergeade, A.; Rahman, S.M.J.; Boutaud, O.; Blackwell, T.; Massion, P.P.; Harrison, D.G.; Dikalova, A. Tobacco smoking induces cardiovascular mitochondrial oxidative stress, promotes endothelial dysfunction, and enhances hypertension. Am. J. Physiol.-Heart Circ. Physiol. 2019, 316, H639–H646. [Google Scholar] [CrossRef]
- Gupta, S.C.; Hevia, D.; Patchva, S.; Park, B.; Koh, W.; Aggarwal, B.B. Upsides and downsides of reactive oxygen species for Cancer: The roles of reactive oxygen species in tumorigenesis, prevention, and therapy. Antioxid. Redox Signal 2012, 16, 1295–1322. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [PubMed]
- Tsutsui, H.; Kinugawa, S.; Matsushima, S. Oxidative stress and heart failure. Am. J. Physiol.-Heart Circ. Physiol. 2011, 301, 2181–2190. [Google Scholar] [CrossRef] [PubMed]
- Liguori, I.; Russo, G.; Curcio, F.; Bulli, G.; Aran, L.; Della-Morte, D.; Gargiulo, G.; Testa, G.; Cacciatore, F.; Bonaduce, D.; et al. Oxidative stress, aging, and diseases. Clin. Interv. Aging 2018, 13, 757–772. [Google Scholar] [CrossRef] [PubMed]
- Sharifi-Rad, M.; Kumar, N.V.A.; Zucca, P.; Varoni, E.M.; Dini, L.; Panzarini, E.; Rajkovic, J.; Fokou, P.V.T.; Azzini, E.; Peluso, I.; et al. Lifestyle, Oxidative Stress, and Antioxidants: Back and Forth in the Pathophysiology of Chronic Diseases. Front. Physiol. 2020, 11, 694. [Google Scholar] [CrossRef]
- Dubois-Deruy, E.; Cuvelliez, M.; Fiedler, J.; Charrier, H.; Mulder, P.; Hebbar, E.; Pfanne, A.; Beseme, O.; Chwastyniak, M.; Amouyel, P.; et al. MicroRNAs regulating superoxide dismutase 2 are new circulating biomarkers of heart failure. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef]
- Strassburger, M.; Bloch, W.; Sulyok, S.; Schüller, J.; Keist, A.F.; Schmidt, A.; Wenk, J.; Peters, T.; Wlaschek, M.; Krieg, T.; et al. Heterozygous deficiency of manganese superoxide dismutase results in severe lipid peroxidation and spontaneous apoptosis in murine myocardium in vivo. Free Radic. Biol. Med. 2005, 38, 1458–1470. [Google Scholar] [CrossRef]
- Li, X.; Lin, Y.; Wang, S.; Zhou, S.; Ju, J.; Wang, X.; Chen, Y.; Xia, M. Extracellular Superoxide Dismutase Is Associated With Left Ventricular Geometry and Heart Failure in Patients with Cardiovascular Disease. J. Am. Heart Assoc. 2020, 9, e016862. [Google Scholar] [CrossRef] [PubMed]
- Tehrani, H.S.; Moosavi-Movahedi, A.A. Catalase and its mysteries. Prog. Biophys. Mol. Biol. 2018, 140, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Detienne, G.; De Haes, W.; Mergan, L.; Edwards, S.L.; Temmerman, L.; Van Bael, S. Beyond ROS clearance: Peroxiredoxins in stress signaling and aging. Ageing Res. Rev. 2018, 44, 33–48. [Google Scholar] [CrossRef] [PubMed]
- Kuzuya, K.; Ichihara, S.; Suzuki, Y.; Inoue, C.; Ichihara, G.; Kurimoto, S.; Oikawa, S. Proteomics analysis identified peroxiredoxin 2 involved in early-phase left ventricular impairment in hamsters with cardiomyopathy. PLoS ONE 2018, 13, e0192624. [Google Scholar] [CrossRef] [PubMed]
- Ibarrola, J.; Arrieta, V.; Sádaba, R.; Martinez-Martinez, E.; Garcia-Peña, A.; Alvarez, V.; Fernández-Celis, A.; Gainza, A.; Santamaría, E.; Fernández-Irigoyen, J.; et al. Galectin-3 down-regulates antioxidant peroxiredoxin-4 in human cardiac fibroblasts: A new pathway to induce cardiac damage. Clin. Sci. 2018, 132, 1471–1485. [Google Scholar] [CrossRef]
- Cieniewski-Bernard, C.; Mulder, P.; Henry, J.-P.; Drobecq, H.; Dubois, E.; Pottiez, G.; Thuillez, C.; Amouyel, P.; Richard, V.; Pinet, F. Proteomic Analysis of Left Ventricular Remodeling in an Experimental Model of Heart Failure. J. Proteome Res. 2008, 7, 5004–5016. [Google Scholar] [CrossRef]
- Kalyanaraman, B. Teaching the basics of redox biology to medical and graduate students: Oxidants, antioxidants and disease mechanisms. Redox Biol. 2013, 1, 244–257. [Google Scholar] [CrossRef]
- Park, T.J.; Park, J.H.; Lee, G.S.; Lee, J.Y.; Shin, J.H.; Kim, M.W.; Kim, Y.S.; Kim, J.Y.; Oh, K.J.; Han, B.S.; et al. Quantitative proteomic analyses reveal that GPX4 downregulation during myocardial infarction contributes to ferroptosis in cardiomyocytes. Cell Death Dis. 2019, 10, 835. [Google Scholar] [CrossRef]
- Tsutsui, H.; Kinugawa, S.; Matsushima, S. Mitochondrial oxidative stress and dysfunction in myocardial remodelling. Cardiovasc. Res. 2008, 81, 449–456. [Google Scholar] [CrossRef]
- Sack, M.N.; Fyhrquist, F.Y.; Saijonmaa, O.J.; Fuster, V.; Kovacic, J.C. Basic Biology of Oxidative Stress and the Cardiovascular System: Part 1 of a 3-Part Series. J. Am. Coll. Cardiol. 2017, 70, 196–211. [Google Scholar] [CrossRef]
- Burgoyne, J.R.; Mongue-Din, H.; Eaton, P.; Shah, A.M. Redox signaling in cardiac physiology and pathology. Circ. Res. 2012, 111, 1091–1106. [Google Scholar] [CrossRef] [PubMed]
- Moris, D.; Spartalis, M.; Tzatzaki, E.; Spartalis, E.; Karachaliou, G.S.; Triantafyllis, A.S.; Karaolanis, G.I.; Tsilimigras, D.I.; Theocharis, S. The role of reactive oxygen species in myocardial redox signaling and regulation. Ann. Transl. Med. 2017, 5, 324. [Google Scholar] [CrossRef] [PubMed]
- Lismont, C.; Revenco, I.; Fransen, M. Peroxisomal hydrogen peroxide metabolism and signaling in health and disease. Int. J. Mol. Sci. 2019, 20, 3673. [Google Scholar] [CrossRef]
- Kasai, S.; Shimizu, S.; Tatara, Y.; Mimura, J.; Itoh, K. Regulation of Nrf2 by mitochondrial reactive oxygen species in physiology and pathology. Biomolecules 2020, 10, 320. [Google Scholar] [CrossRef]
- Loyer, X.; Heymes, C.; Samuel, J.L. Constitutive nitric oxide synthases in the heart from hypertrophy to failure. Clin. Exp. Pharmacol. Physiol. 2008, 35, 483–488. [Google Scholar] [CrossRef]
- Hammond, J.; Balligand, J.L. Nitric oxide synthase and cyclic GMP signaling in cardiac myocytes: From contractility to remodeling. J. Mol. Cell. Cardiol. 2012, 52, 330–340. [Google Scholar] [CrossRef] [PubMed]
- Lancel, S.; Qin, F.; Lennon, S.L.; Zhang, J.; Tong, X.; Mazzini, M.J.; Kang, Y.J.; Siwik, D.A.; Cohen, R.A.; Colucci, W.S. Oxidative posttranslational modifications mediate decreased SERCA activity and myocyte dysfunction in Galphaq-overexpressing mice. Circ. Res. 2010, 107, 228–232. [Google Scholar] [CrossRef]
- Steinberg, S.F. Oxidative stress and sarcomeric proteins. Circ. Res. 2013, 112, 393–405. [Google Scholar] [CrossRef]
- Hermida, N.; Michel, L.; Esfahani, H.; Dubois-Deruy, E.; Hammond, J.; Bouzin, C.; Markl, A.; Colin, H.; Van Steenbergen, A.; De Meester, C.; et al. Cardiac myocyte β3-adrenergic receptors prevent myocardial fibrosis by modulating oxidant stress-dependent paracrine signaling. Eur. Heart J. 2018, 39, 888–897. [Google Scholar] [CrossRef]
- Serpillon, S.; Floyd, B.C.; Gupte, R.S.; George, S.; Kozicky, M.; Neito, V.; Recchia, F.; Stanley, W.; Wolin, M.S.; Gupte, S.A. Superoxide production by NAD(P)H oxidase and mitochondria is increased in genetically obese and hyperglycemic rat heart and aorta before the development of cardiac dysfunction. The role of glucose-6-phosphate dehydrogenase-derived NADPH. Am. J. Physiol.-Heart Circ. Physiol. 2009, 297, H153. [Google Scholar] [CrossRef]
- Anderson, E.J.; Kypson, A.P.; Rodriguez, E.; Anderson, C.A.; Lehr, E.J.; Neufer, P.D. Substrate-Specific Derangements in Mitochondrial Metabolism and Redox Balance in the Atrium of the Type 2 Diabetic Human Heart. J. Am. Coll. Cardiol. 2009, 54, 1891–1898. [Google Scholar] [CrossRef] [PubMed]
- Fortuño, A.; José, G.S.; Moreno, M.U.; Beloqui, O.; Díez, J.; Zalba, G. Phagocytic NADPH oxidase overactivity underlies oxidative stress in metabolic syndrome. Diabetes 2006, 55, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Sverdlov, A.L.; Elezaby, A.; Qin, F.; Behring, J.B.; Luptak, I.; Calamaras, T.D.; Siwik, D.A.; Miller, E.J.; Liesa, M.; Shirihai, O.S.; et al. Mitochondrial reactive oxygen species mediate cardiac structural, functional, and mitochondrial consequences of diet-induced metabolic heart disease. J. Am. Heart Assoc. 2016, 5, e002555. [Google Scholar] [CrossRef]
- Jeong, E.M.; Chung, J.; Liu, H.; Go, Y.; Gladstein, S.; Farzaneh-Far, A.; Lewandowski, E.D.; Dudley, S.C. Role of Mitochondrial Oxidative Stress in Glucose Tolerance, Insulin Resistance, and Cardiac Diastolic Dysfunction. J. Am. Heart Assoc. 2016, 5, e003046. [Google Scholar] [CrossRef] [PubMed]
- Niemann, B.; Chen, Y.; Teschner, M.; Li, L.; Silber, R.E.; Rohrbach, S. Obesity induces signs of premature cardiac aging in younger patients: The role of mitochondria. J. Am. Coll. Cardiol. 2011, 57, 577–585. [Google Scholar] [CrossRef]
- Jiménez-González, S.; Marín-Royo, G.; Jurado-López, R.; Bartolomé, M.V.; Romero-Miranda, A.; Luaces, M.; Islas, F.; Nieto, M.L.; Martínez-Martínez, E.; Cachofeiro, V. The Crosstalk between Cardiac Lipotoxicity and Mitochondrial Oxidative Stress in the Cardiac Alterations in Diet-Induced Obesity in Rats. Cells 2020, 9, 451. [Google Scholar] [CrossRef]
- Li, J.M.; Gall, N.P.; Grieve, D.J.; Chen, M.; Shah, A.M. Activation of NADPH oxidase during progression of cardiac hypertrophy to failure. Hypertension 2002, 40, 477–484. [Google Scholar] [CrossRef]
- Dai, D.F.; Johnson, S.C.; Villarin, J.J.; Chin, M.T.; Nieves-Cintrón, M.; Chen, T.; Marcinek, D.J.; Dorn, G.W.; Kang, Y.J.; Prolla, T.A.; et al. Mitochondrial oxidative stress mediates angiotensin II-induced cardiac hypertrophy and gαq overexpression-induced heart failure. Circ. Res. 2011, 108, 837–846. [Google Scholar] [CrossRef]
- Dai, D.F.; Hsieh, E.J.; Liu, Y.; Chen, T.; Beyer, R.P.; Chin, M.T.; MacCoss, M.J.; Rabinovitch, P.S. Mitochondrial proteome remodelling in pressure overload-induced heart failure: The role of mitochondrial oxidative stress. Cardiovasc. Res. 2012, 93, 79–88. [Google Scholar] [CrossRef]
- Ide, T.; Tsutsui, H.; Hayashidani, S.; Kang, D.; Suematsu, N.; Nakamura, K.I.; Utsumi, H.; Hamasaki, N.; Takeshita, A. Mitochondrial DNA damage and dysfunction associated with oxidative stress in failing hearts after myocardial infarction. Circ. Res. 2001, 88, 529–535. [Google Scholar] [CrossRef]
- Merabet, N.; Bellien, J.; Glevarec, E.; Nicol, L.; Lucas, D.; Remy-Jouet, I.; Bounoure, F.; Dreano, Y.; Thuillez, C.; Mulder, P. Soluble epoxide hydrolase inhibition improves myocardial perfusion and function in experimental heart failure. J. Mol. Cell. Cardiol. 2012, 52, 660–666. [Google Scholar] [CrossRef]
- Santillo, M.; Colantuoni, A.; Mondola, P.; Guida, B.; Damiano, S. NOX signaling in molecular cardiovascular mechanisms involved in the blood pressure homeostasis. Front. Physiol. 2015, 6, 194. [Google Scholar] [CrossRef] [PubMed]
- Weng, X.; Yu, L.; Liang, P.; Li, L.; Dai, X.; Zhou, B.; Wu, X.; Xu, H.; Fang, M.; Chen, Q.; et al. A crosstalk between chromatin remodeling and histone H3K4 methyltransferase complexes in endothelial cells regulates angiotensin II-induced cardiac hypertrophy. J. Mol. Cell. Cardiol. 2015, 82, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Sorescu, D.; Weiss, D.; Lassègue, B.; Clempus, R.E.; Szöcs, K.; Sorescu, G.P.; Valppu, L.; Quinn, M.T.; Lambeth, J.D.; Vega, J.D.; et al. Superoxide production and expression of Nox family proteins in human atherosclerosis. Circulation 2002, 105, 1429–1435. [Google Scholar] [CrossRef] [PubMed]
- Erickson, J.R.; Mei-ling, A.J.; Guan, X.; Kutschke, W.; Yang, J.; Oddis, C.V.; Bartlett, R.K.; Lowe, J.S.; O’Donnell, S.E.; Aykin-Burns, N.; et al. A Dynamic Pathway for Calcium-Independent Activation of CaMKII by Methionine Oxidation. Cell 2008, 133, 462–474. [Google Scholar] [CrossRef] [PubMed]
- Scioli, M.G.; Storti, G.; D’Amico, F.; Rodríguez Guzmán, R.; Centofanti, F.; Doldo, E.; Miranda, E.M.C.; Orlandi, A. Oxidative Stress and New Pathogenetic Mechanisms in Endothelial Dysfunction: Potential Diagnostic Biomarkers and Therapeutic Targets. J. Clin. Med. 2020, 9, 1995. [Google Scholar] [CrossRef] [PubMed]
- Umar, S.; Van Der Laarse, A. Nitric oxide and nitric oxide synthase isoforms in the normal, hypertrophic, and failing heart. Mol. Cell. Biochem. 2010, 333, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Harrison, D.G. Endothelial dysfunction in cardiovascular diseases: The role of oxidant stress. Circ. Res. 2000, 87, 840–844. [Google Scholar] [CrossRef]
- Ma, L.; Wang, K.; Shang, J.; Cao, C.; Zhen, P.; Liu, X.; Wang, W.; Zhang, H.; Du, Y.; Liu, H. Anti-peroxynitrite treatment ameliorated vasorelaxation of resistance arteries in aging rats: Involvement with NO-sGC-cGKs pathway. PLoS ONE 2014, 9, e104788. [Google Scholar] [CrossRef]
- Mollnau, H.; Oelze, M.; August, M.; Wendt, M.; Daiber, A.; Schulz, E.; Baldus, S.; Kleschyov, A.L.; Materne, A.; Wenzel, P.; et al. Mechanisms of increased vascular superoxide production in an experimental model of idiopathic dilated cardiomyopathy. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 2554–2559. [Google Scholar] [CrossRef]
- Scialò, F.; Fernández-Ayala, D.J.; Sanz, A. Role of mitochondrial reverse electron transport in ROS signaling: Potential roles in health and disease. Front. Physiol. 2017, 8, 428. [Google Scholar] [CrossRef] [PubMed]
- Mazat, J.P.; Devin, A.; Ransac, S. Modelling mitochondrial ROS production by the respiratory chain. Cell. Mol. Life Sci. 2020, 77, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Bhatti, J.S.; Bhatti, G.K.; Reddy, P.H. Mitochondrial dysfunction and oxidative stress in metabolic disorders—A step towards mitochondria based therapeutic strategies. Biochim. Biophys. Acta-Mol. Basis Dis. 2017, 1863, 1066–1077. [Google Scholar] [CrossRef] [PubMed]
- Niemann, B.; Rohrbach, S.; Miller, M.R.; Newby, D.E.; Fuster, V.; Kovacic, J.C. Oxidative Stress and Cardiovascular Risk: Obesity, Diabetes, Smoking, and Pollution: Part 3 of a 3-Part Series. J. Am. Coll. Cardiol. 2017, 70, 230–251. [Google Scholar] [CrossRef] [PubMed]
- Bugger, H.; Pfeil, K. Mitochondrial ROS in myocardial ischemia reperfusion and remodeling. Biochim. Biophys. Acta-Mol. Basis Dis. 2020, 1866, 165768. [Google Scholar] [CrossRef]
- Rababa’h, A.M.; Guillory, A.N.; Mustafa, R.; Hijjawi, T. Oxidative Stress and Cardiac Remodeling: An Updated Edge. Curr. Cardiol. Rev. 2018, 14, 53–59. [Google Scholar] [CrossRef]
- Gori, T.; Münzel, T. Oxidative stress and endothelial dysfunction: Therapeutic implications. Ann. Med. 2011, 43, 259–272. [Google Scholar] [CrossRef]
- Stamler, J.S.; Osborne, J.A.; Jaraki, O.; Rabbani, L.E.; Mullins, M.; Singel, D.; Loscalzo, J. Adverse vascular effects of homocysteine are modulated by endothelium-derived relaxing factor and related oxides of nitrogen. J. Clin. Investig. 1993, 91, 308–318. [Google Scholar] [CrossRef]
- McCully, K.S. Homocystinuria, Arteriosclerosis, Methylmalonic Aciduria, and Methyltransferase Deficiency: A Key Case Revisited. Nutr. Rev. 1992, 50, 7–12. [Google Scholar] [CrossRef]
- Cianciolo, G.; De Pascalis, A.; Di Lullo, L.; Ronco, C.; Zannini, C.; La Manna, G. Folic acid and homocysteine in chronic kidney disease and cardiovascular disease progression: Which comes first? CardioRenal Med. 2017, 7, 255–266. [Google Scholar] [CrossRef]
- Stroes, E.S.G.; Van Faassen, E.E.; Yo, M.; Martasek, P.; Boer, P.; Govers, R.; Rabelink, T.J. Folic acid reverts dysfunction of endothelial nitric oxide synthase. Circ. Res. 2000, 86, 1129–1134. [Google Scholar] [CrossRef] [PubMed]
- Hagar, H.H. Folic acid and vitamin B12 supplementation attenuates isoprenaline-induced myocardial infarction in experimental hyperhomocysteinemic rats. Pharmacol. Res. 2002, 46, 213–219. [Google Scholar] [CrossRef]
- Farhangi, M.A.; Nameni, G.; Hajiluian, G.; Mesgari-Abbasi, M. Cardiac tissue oxidative stress and inflammation after vitamin D administrations in high fat- diet induced obese rats. BMC Cardiovasc. Disord. 2017, 17, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Leme Goto, P.; Cinato, M.; Merachli, F.; Vons, B.; Jimenez, T.; Marsal, D.; Todua, N.; Loi, H.; Santin, Y.; Cassel, S.; et al. In vitro and in vivo cardioprotective and metabolic efficacy of vitamin E TPGS/Apelin. J. Mol. Cell. Cardiol. 2020, 138, 165–174. [Google Scholar] [CrossRef]
- Wallert, M.; Ziegler, M.; Wang, X.; Maluenda, A.; Xu, X.; Yap, M.L.; Witt, R.; Giles, C.; Kluge, S.; Hortmann, M.; et al. α-Tocopherol preserves cardiac function by reducing oxidative stress and inflammation in ischemia/reperfusion injury. Redox Biol. 2019, 26, 101292. [Google Scholar] [CrossRef]
- Banez, M.J.; Geluz, M.I.; Chandra, A.; Hamdan, T.; Biswas, O.S.; Bryan, N.S.; Von Schwarz, E.R. A systemic review on the antioxidant and anti-inflammatory effects of resveratrol, curcumin, and dietary nitric oxide supplementation on human cardiovascular health. Nutr. Res. 2020, 78, 11–26. [Google Scholar] [CrossRef]
- Agouni, A.; Lagrue-Lak-Hal, A.H.; Mostefai, H.A.; Tesse, A.; Mulder, P.; Rouet, P.; Desmoulin, F.; Heymes, C.; Martínez, M.C.; Andriantsitohaina, R. Red wine polyphenols prevent metabolic and cardiovascular alterations associated with obesity in Zucker fatty rats (Fa/Fa). PLoS ONE 2009, 4, e5557. [Google Scholar] [CrossRef]
- Farkhondeh, T.; Folgado, S.L.; Pourbagher-Shahri, A.M.; Ashrafizadeh, M.; Samarghandian, S. The therapeutic effect of resveratrol: Focusing on the Nrf2 signaling pathway. Biomed. Pharmacother. 2020, 127, 110234. [Google Scholar] [CrossRef]
- Dyck, G.J.B.; Raj, P.; Zieroth, S.; Dyck, J.R.B.; Ezekowitz, J.A. The effects of resveratrol in patients with cardiovascular disease and heart failure: A narrative review. Int. J. Mol. Sci. 2019, 20, 904. [Google Scholar] [CrossRef]
- Huang, J.P.; Hsu, S.C.; Li, D.E.; Chen, K.H.; Kuo, C.Y.; Hung, L.M. Resveratrol mitigates high-fat diet-induced vascular dysfunction by activating the Akt/eNOS/NO and Sirt1/ER pathway. J. Cardiovasc. Pharmacol. 2018, 72, 231–241. [Google Scholar] [CrossRef]
- Chan, A.Y.M.; Dolinsky, V.W.; Soltys, C.-L.M.; Viollet, B.; Baksh, S.; Light, P.E.; Dyck, J.R.B. Resveratrol inhibits cardiac hypertrophy via AMP-activated protein kinase and Akt. J. Biol. Chem. 2008, 283, 24194–24201. [Google Scholar] [CrossRef] [PubMed]
- Sung, M.M.; Das, S.K.; Levasseur, J.; Byrne, N.J.; Fung, D.; Kim, T.T.; Masson, G.; Boisvenue, J.; Soltys, C.L.; Oudit, G.Y.; et al. Resveratrol treatment of mice with pressure-overloadinduced heart failure improves diastolic function and cardiac energy metabolism. Circ. Heart Fail. 2015, 8, 128–137. [Google Scholar] [CrossRef] [PubMed]
- Tanno, M.; Kuno, A.; Yano, T.; Miura, T.; Hisahara, S.; Ishikawa, S.; Shimamoto, K.; Horio, Y. Induction of manganese superoxide dismutase by nuclear translocation and activation of SIRT1 promotes cell survival in chronic heart failure. J. Biol. Chem. 2010, 285, 8375–8382. [Google Scholar] [CrossRef]
- Sabbah, H.N. Targeting mitochondrial dysfunction in the treatment of heart failure. Expert Rev. Cardiovasc. Ther. 2016, 14, 1305–1313. [Google Scholar] [CrossRef] [PubMed]
- Senoner, T.; Dichtl, W. Oxidative stress in cardiovascular diseases: Still a therapeutic target? Nutrients 2019, 11, 2090. [Google Scholar] [CrossRef]
- Dikalova, A.E.; Bikineyeva, A.T.; Budzyn, K.; Nazarewicz, R.R.; McCann, L.; Lewis, W.; Harrison, D.G.; Dikalov, S.I. Therapeutic targeting of mitochondrial superoxide in hypertension. Circ. Res. 2010, 107, 106–116. [Google Scholar] [CrossRef]
- Ni, R.; Cao, T.; Xiong, S.; Ma, J.; Fan, G.C.; Lacefield, J.C.; Lu, Y.; Tissier, S.L.; Peng, T. Therapeutic inhibition of mitochondrial reactive oxygen species with mito-TEMPO reduces diabetic cardiomyopathy. Free Radic. Biol. Med. 2016, 90, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Kelso, G.F.; Porteous, C.M.; Coulter, C.V.; Hughes, G.; Porteous, W.K.; Ledgerwood, E.C.; Smith, R.A.J.; Murphy, M.P. Selective targeting of a redox-active ubiquinone to mitochondria within cells: Antioxidant and antiapoptotic properties. J. Biol. Chem. 2001, 276, 4588–4596. [Google Scholar] [CrossRef]
- Kim, S.; Song, J.; Ernst, P.; Latimer, M.N.; Ha, C.M.; Goh, K.Y.; Ma, W.; Rajasekaran, N.S.; Zhang, J.; Liu, X.; et al. MitoQ regulates redox-related noncoding RNAs to preserve mitochondrial network integrity in pressure-overload heart failure. Am. J. Physiol. Heart Circ. Physiol. 2020, 318, H682–H695. [Google Scholar] [CrossRef]
- Rao, V.A.; Klein, S.R.; Bonar, S.J.; Zielonka, J.; Mizuno, N.; Dickey, J.S.; Keller, P.W.; Joseph, J.; Kalyanaraman, B.; Shacter, E. The antioxidant transcription factor Nrf2 negatively regulates autophagy and growth arrest induced by the anticancer redox agent mitoquinone. J. Biol. Chem. 2010, 285, 34447–34459. [Google Scholar] [CrossRef]
- Doughan, A.K.; Dikalov, S.I. Mitochondrial redox cycling of mitoquinone leads to superoxide production and cellular apoptosis. Antioxid. Redox Signal 2007, 9, 1825–1836. [Google Scholar] [CrossRef] [PubMed]
- Pokrzywinski, K.L.; Biel, T.G.; Kryndushkin, D.; Rao, V.A. Therapeutic targeting of the mitochondria initiates excessive superoxide production and mitochondrial depolarization causing decreased mtDNA integrity. PLoS ONE 2016, 11, e0168283. [Google Scholar] [CrossRef]
- Lee, I.M.; Cook, N.R.; Gaziano, J.M.; Gordon, D.; Ridker, P.M.; Manson, J.A.E.; Hennekens, C.H.; Buring, J.E. Vitamin E in the primary prevention of cardiovascular disease and cancer. The women’s health study: A randomized controlled trial. J. Am. Med. Assoc. 2005, 294, 56–65. [Google Scholar] [CrossRef]
- Toole, J.F.; Malinow, M.R.; Chambless, L.E.; Spence, J.D.; Pettigrew, L.C.; Howard, V.J.; Sides, E.G.; Wang, C.H.; Stampfer, M. Lowering Homocysteine in Patients with Ischemic Stroke to Prevent Recurrent Stroke, Myocardial Infarction, and Death: The Vitamin Intervention for Stroke Prevention (VISP) Randomized Controlled Trial. J. Am. Med. Assoc. 2004, 291, 565–575. [Google Scholar] [CrossRef]
- Lonn, E. Effects of long-term vitamin E supplementation on cardiovascular events and cancer: A randomized controlled trial. J. Am. Med. Assoc. 2005, 293, 1338–1347. [Google Scholar]
- Bønaa, K.H.; Njølstad, I.; Ueland, P.M.; Schirmer, H.; Tverdal, A.; Steigen, T.; Wang, H.; Nordrehaug, J.E.; Arnesen, E.; Rasmussen, K. Homocysteine lowering and cardiovascular events after acute myocardial infarction. N. Engl. J. Med. 2006, 354, 1578–1588. [Google Scholar] [CrossRef]
- Münzel, T.; Camici, G.G.; Maack, C.; Bonetti, N.R.; Fuster, V.; Kovacic, J.C. Impact of Oxidative Stress on the Heart and Vasculature: Part 2 of a 3-Part Series. J. Am. Coll. Cardiol. 2017, 70, 212–229. [Google Scholar] [CrossRef]
- Mankowski, R.T.; You, L.; Buford, T.W.; Leeuwenburgh, C.; Manini, T.M.; Schneider, S.; Qiu, P.; Anton, S.D. Higher dose of resveratrol elevated cardiovascular disease risk biomarker levels in overweight older adults–A pilot study. Exp. Gerontol. 2020, 131, 110821. [Google Scholar] [CrossRef]
- Tomé-Carneiro, J.; Gonzálvez, M.; Larrosa, M.; Yáñez-Gascón, M.J.; García-Almagro, F.J.; Ruiz-Ros, J.A.; García-Conesa, M.T.; Tomás-Barberán, F.A.; Espín, J.C. One-year consumption of a grape nutraceutical containing resveratrol improves the inflammatory and fibrinolytic status of patients in primary prevention of cardiovascular disease. Am. J. Cardiol. 2012, 110, 356–363. [Google Scholar] [CrossRef]
- Macedo, R.C.S.; Vieira, A.; Marin, D.P.; Otton, R. Effects of chronic resveratrol supplementation in military firefighters undergo a physical fitness test-A placebo-controlled, double blind study. Chem. Biol. Interact. 2015, 227, 89–95. [Google Scholar] [CrossRef]
- Timmers, S.; Konings, E.; Bilet, L.; Houtkooper, R.H.; Van De Weijer, T.; Goossens, G.H.; Hoeks, J.; Van Der Krieken, S.; Ryu, D.; Kersten, S.; et al. Calorie restriction-like effects of 30 days of resveratrol supplementation on energy metabolism and metabolic profile in obese humans. Cell Metab. 2011, 14, 612–622. [Google Scholar] [CrossRef] [PubMed]
- Fujitaka, K.; Otani, H.; Jo, F.; Jo, H.; Nomura, E.; Iwasaki, M.; Nishikawa, M.; Iwasaka, T.; Das, D.K. Modified resveratrol Longevinex improves endothelial function in adults with metabolic syndrome receiving standard treatment. Nutr. Res. 2011, 31, 842–847. [Google Scholar] [CrossRef] [PubMed]
- Imamura, H.; Yamaguchi, T.; Nagayama, D.; Saiki, A.; Shirai, K.; Tatsuno, I. Resveratrol ameliorates arterial stiffness assessed by cardio-ankle vascular index in patients with type 2 diabetes mellitus. Int. Heart J. 2017, 58, 577–583. [Google Scholar] [CrossRef] [PubMed]
- Van Der Made, S.M.; Plat, J.; Mensink, R.P. Resveratrol does not influence metabolic risk markers related to cardiovascular health in overweight and slightly obese subjects: A randomized, placebo-controlled crossover trial. PLoS ONE 2015, 10, e0118393. [Google Scholar] [CrossRef] [PubMed]
- Olesen, J.; Gliemann, L.; Biensø, R.; Schmidt, J.; Hellsten, Y.; Pilegaard, H. Exercise training, but not resveratrol, improves metabolic and inflammatory status in skeletal muscle of aged men. J. Physiol. 2014, 592, 1873–1886. [Google Scholar] [CrossRef]
- Agarwal, B.; Campen, M.J.; Channell, M.M.; Wherry, S.J.; Varamini, B.; Davis, J.G.; Baur, J.A.; Smoliga, J.M. Resveratrol for primary prevention of atherosclerosis: Clinical trial evidence for improved gene expression in vascular endothelium. Int. J. Cardiol. 2013, 166, 246–248. [Google Scholar] [CrossRef]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dubois-Deruy, E.; Peugnet, V.; Turkieh, A.; Pinet, F. Oxidative Stress in Cardiovascular Diseases. Antioxidants 2020, 9, 864. https://doi.org/10.3390/antiox9090864
Dubois-Deruy E, Peugnet V, Turkieh A, Pinet F. Oxidative Stress in Cardiovascular Diseases. Antioxidants. 2020; 9(9):864. https://doi.org/10.3390/antiox9090864
Chicago/Turabian StyleDubois-Deruy, Emilie, Victoriane Peugnet, Annie Turkieh, and Florence Pinet. 2020. "Oxidative Stress in Cardiovascular Diseases" Antioxidants 9, no. 9: 864. https://doi.org/10.3390/antiox9090864
APA StyleDubois-Deruy, E., Peugnet, V., Turkieh, A., & Pinet, F. (2020). Oxidative Stress in Cardiovascular Diseases. Antioxidants, 9(9), 864. https://doi.org/10.3390/antiox9090864