Oxidative Stress and Inflammatory Modulation of Ca2+ Handling in Metabolic HFpEF-Related Left Atrial Cardiomyopathy

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Methods
2.1. Heart Failure (HF) Model
2.2. Echocardiography
2.3. Chemicals, Solutions
2.4. Cardiomyocyte Isolation
2.5. Mitochondrial Structure
2.6. Reactive Oxygen Species (ROS)
2.7. Ca2+ Imaging
2.8. Western Blots
2.9. Statistical Analysis
3. Results
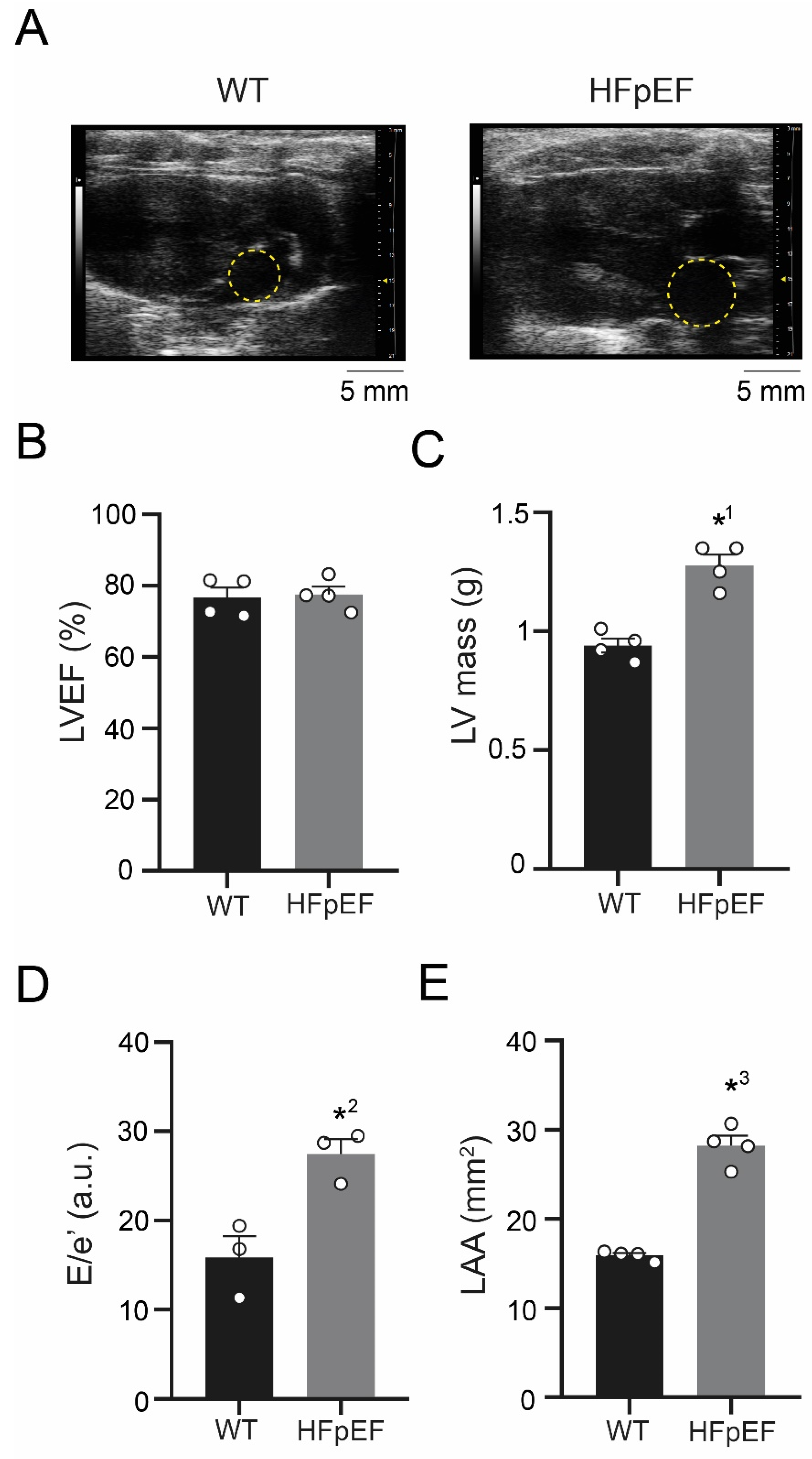
3.1. ZSF-1 Obese Rats Show Distinct Features of Metabolic HFpEF-Related Left Atrial Cardiomyopathy
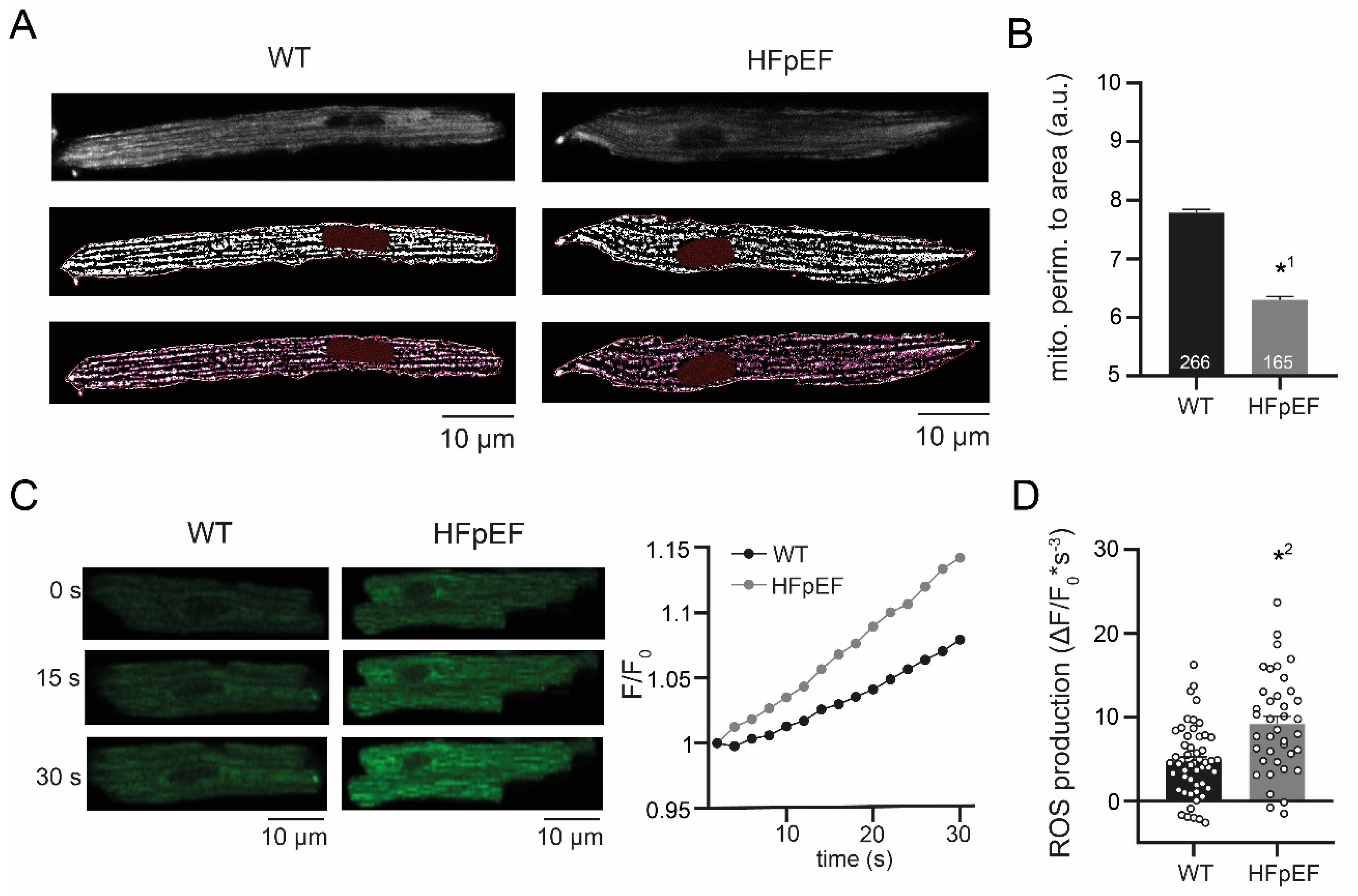
3.2. HFpEF Shows Increased Mitochondrial Fission and Production of ROS
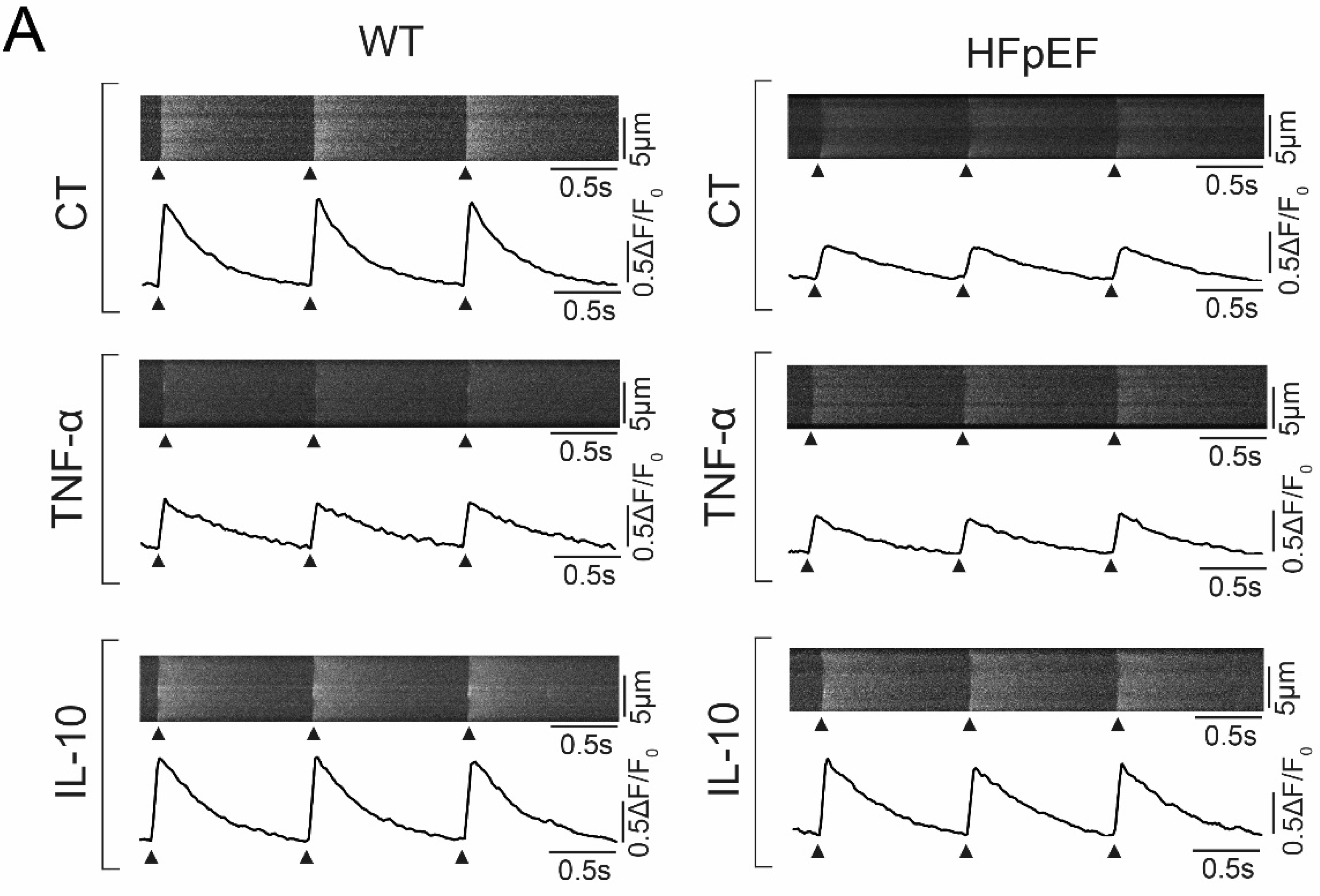
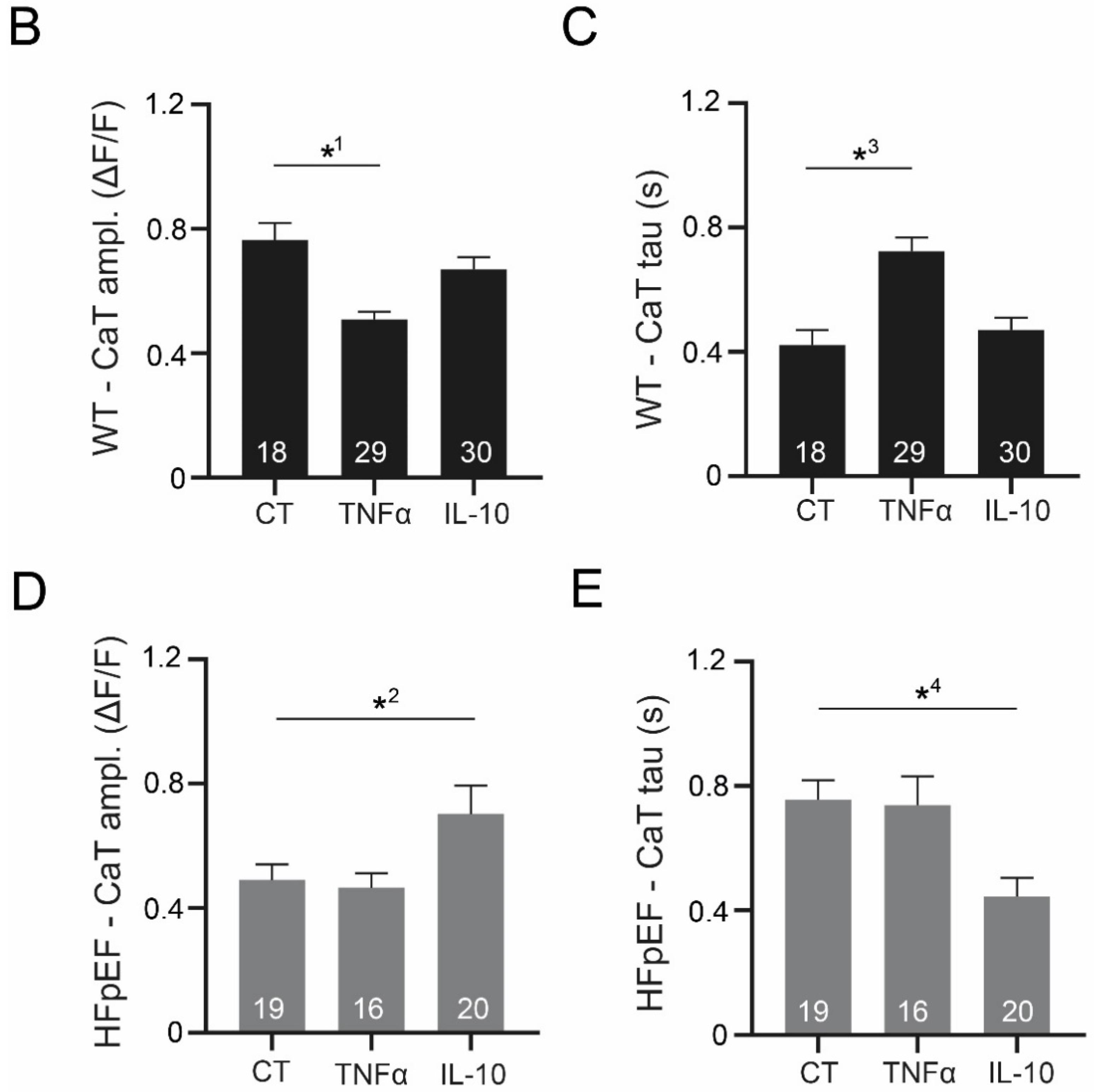
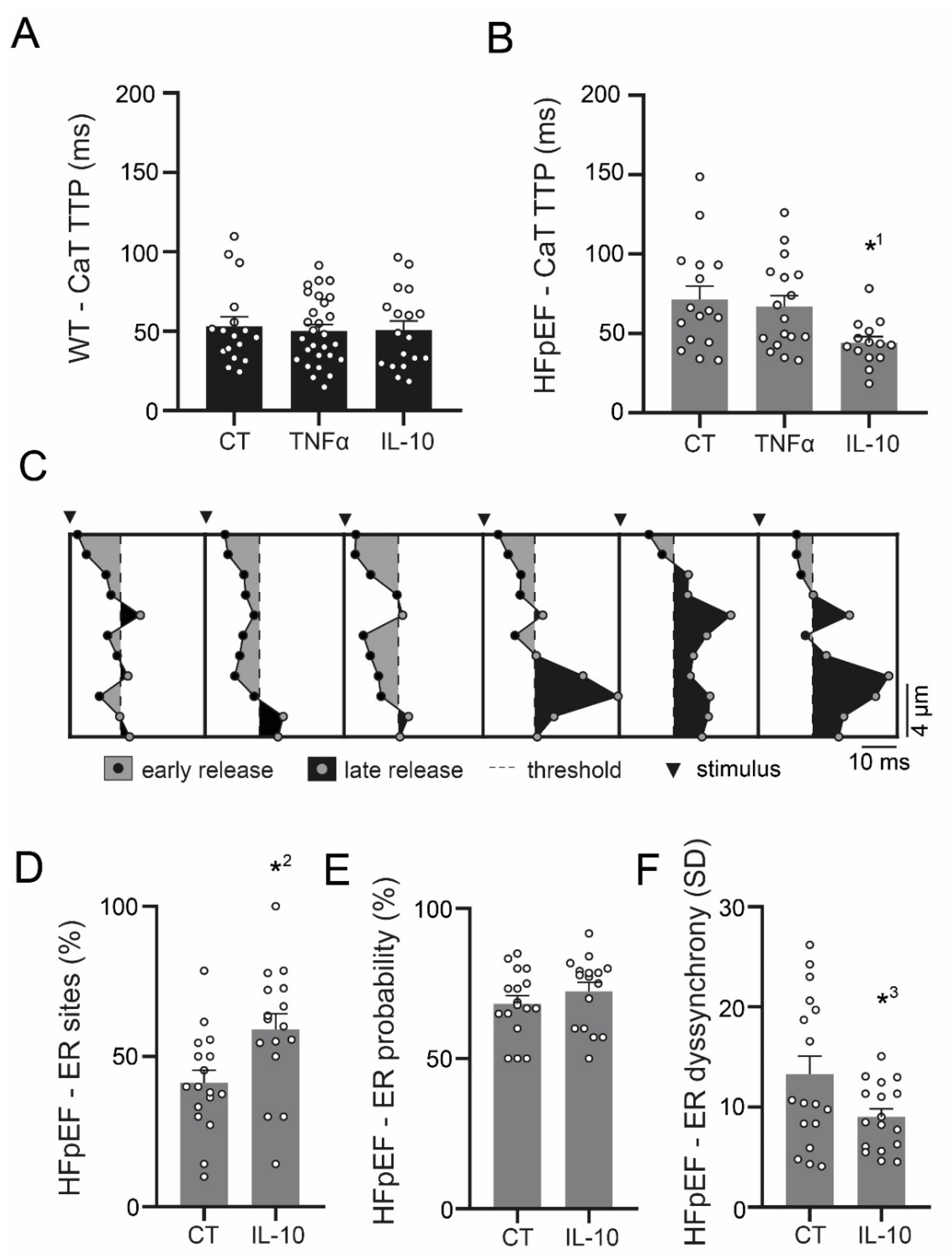
3.3. Ca2+ Signaling during Excitation-Contraction Coupling in HFpEF Is Not Affected by TNF-α, but Benefits from Exposure to IL-10
3.4. IL-10 Induces Recruitment and Synchronization of Early Ca2+ Release Sites in HFpEF
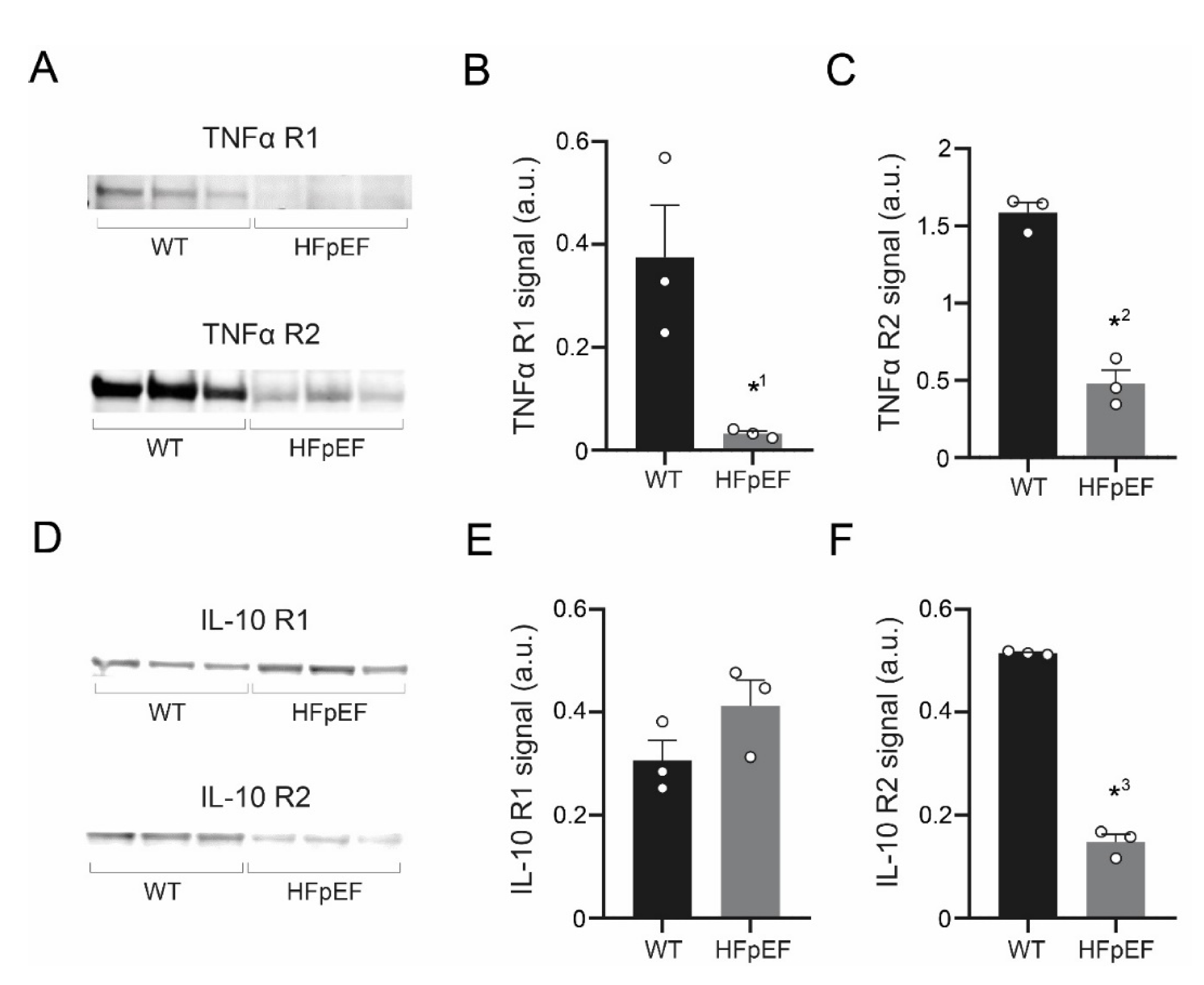
3.5. TNF-α Receptor Expression Is Reduced, IL-10 Receptor Expression Partially Maintained in HFpEF
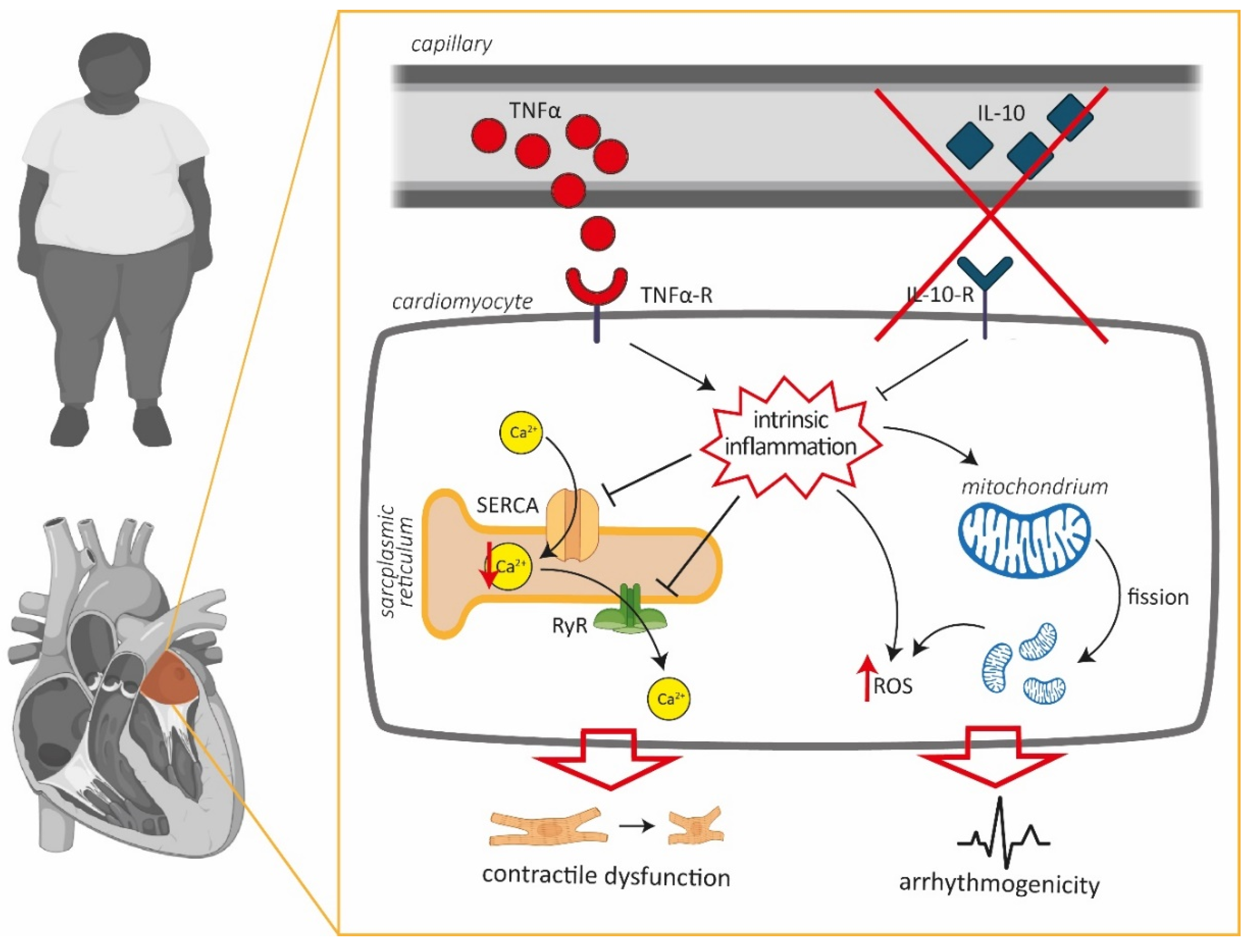
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Oktay, A.A.; Rich, J.D.; Shah, S.J. The emerging epidemic of heart failure with preserved ejection fraction. Curr. Heart Fail. Rep. 2013, 10, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Ponikowski, P.; Voors, A.A.; Anker, S.D.; Bueno, H.; Cleland, J.G.F.; Coats, A.J.S.; Falk, V.; Gonzalez-Juanatey, J.R.; Harjola, V.P.; Jankowska, E.A.; et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC)Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur. Heart J. 2016, 37, 2129–2200. [Google Scholar] [PubMed]
- Pfeffer, M.A.; Shah, A.M.; Borlaug, B.A. Heart failure with preserved ejection fraction in perspective. Circ. Res. 2019, 124, 1598–1617. [Google Scholar] [CrossRef] [PubMed]
- Pieske, B.; Tschope, C.; de Boer, R.A.; Fraser, A.G.; Anker, S.D.; Donal, E.; Edelmann, F.; Fu, M.; Guazzi, M.; Lam, C.S.P.; et al. How to diagnose heart failure with preserved ejection fraction: The HFA-PEFF diagnostic algorithm: A consensus recommendation from the Heart Failure Association (HFA) of the European Society of Cardiology (ESC). Eur. Heart J. 2019, 40, 3297–3317. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Fang, F.; Yu, C.M. Noncardiac comorbidities in heart failure with preserved ejection fraction—Commonly ignored fact. Circ. J. 2015, 79, 954–959. [Google Scholar] [CrossRef]
- Franssen, C.; Chen, S.; Unger, A.; Korkmaz, H.I.; De Keulenaer, G.W.; Tschope, C.; Leite-Moreira, A.F.; Musters, R.; Niessen, H.W.; Linke, W.A.; et al. Myocardial microvascular inflammatory endothelial activation in heart failure with preserved ejection fraction. JACC Heart Fail. 2016, 4, 312–324. [Google Scholar] [CrossRef]
- Hamdani, N.; Bishu, K.G.; von Frieling-Salewsky, M.; Redfield, M.M.; Linke, W.A. Deranged myofilament phosphorylation and function in experimental heart failure with preserved ejection fraction. Cardiovasc. Res. 2013, 97, 464–471. [Google Scholar] [CrossRef]
- van Heerebeek, L.; Hamdani, N.; Falcao-Pires, I.; Leite-Moreira, A.F.; Begieneman, M.P.; Bronzwaer, J.G.; van der Velden, J.; Stienen, G.J.; Laarman, G.J.; Somsen, A.; et al. Low myocardial protein kinase G activity in heart failure with preserved ejection fraction. Circulation 2012, 126, 830–839. [Google Scholar] [CrossRef]
- van der Pol, A.; van Gilst, W.H.; Voors, A.A.; van der Meer, P. Treating oxidative stress in heart failure: Past, present and future. Eur. J. Heart Fail. 2019, 21, 425–435. [Google Scholar] [CrossRef]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive oxygen species in inflammation and tissue injury. Antioxid. Redox Signal. 2014, 20, 1126–1167. [Google Scholar] [CrossRef]
- Deswal, A.; Petersen, N.J.; Feldman, A.M.; Young, J.B.; White, B.G.; Mann, D.L. Cytokines and cytokine receptors in advanced heart failure: An analysis of the cytokine database from the Vesnarinone trial (VEST). Circulation 2001, 103, 2055–2059. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Gamad, N.; Shafiq, N.; Malhotra, S. Effect size in CANTOS trial. BMJ Evid.-Based Med. 2018, 23, 44. [Google Scholar] [CrossRef] [PubMed]
- Melenovsky, V.; Hwang, S.J.; Redfield, M.M.; Zakeri, R.; Lin, G.; Borlaug, B.A. Left atrial remodeling and function in advanced heart failure with preserved or reduced ejection fraction. Circ. Heart Fail. 2015, 8, 295–303. [Google Scholar] [CrossRef]
- Hohendanner, F.; Messroghli, D.; Bode, D.; Blaschke, F.; Parwani, A.; Boldt, L.H.; Heinzel, F.R. Atrial remodelling in heart failure: Recent developments and relevance for heart failure with preserved ejection fraction. ESC Heart Fail. 2018, 5, 211–221. [Google Scholar] [CrossRef]
- Habibi, M.; Chahal, H.; Opdahl, A.; Gjesdal, O.; Helle-Valle, T.M.; Heckbert, S.R.; McClelland, R.; Wu, C.; Shea, S.; Hundley, G.; et al. Association of CMR-measured LA function with heart failure development: Results from the MESA study. JACC Cardiovasc. Imaging 2014, 7, 570–579. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Matulevicius, S.A.; Ayers, C.R.; Berry, J.D.; Patel, P.C.; Markham, D.W.; Levine, B.D.; Chin, K.M.; de Lemos, J.A.; Peshock, R.M.; et al. Left atrial structure and function and clinical outcomes in the general population. Eur. Heart J. 2013, 34, 278–285. [Google Scholar] [CrossRef]
- Mihm, M.J.; Yu, F.; Carnes, C.A.; Reiser, P.J.; McCarthy, P.M.; Van Wagoner, D.R.; Bauer, J.A. Impaired myofibrillar energetics and oxidative injury during human atrial fibrillation. Circulation 2001, 104, 174–180. [Google Scholar] [CrossRef]
- Bode, D.; Lindner, D.; Schwarzl, M.; Westermann, D.; Deissler, P.; Primessnig, U.; Hegemann, N.; Blatter, L.A.; van Linthout, S.; Tschope, C.; et al. The role of fibroblast—Cardiomyocyte interaction for atrial dysfunction in HFpEF and hypertensive heart disease. J. Mol. Cell. Cardiol. 2019, 131, 53–65. [Google Scholar] [CrossRef]
- Hohendanner, F.; Bode, D.; Primessnig, U.; Guthof, T.; Doerr, R.; Jeuthe, S.; Reimers, S.; Zhang, K.; Bach, D.; Wakula, P.; et al. Cellular mechanisms of metabolic syndrome-related atrial decompensation in a rat model of HFpEF. J. Mol. Cell. Cardiol. 2018, 115, 10–19. [Google Scholar] [CrossRef]
- Hamdani, N.; Franssen, C.; Lourenco, A.; Falcao-Pires, I.; Fontoura, D.; Leite, S.; Plettig, L.; Lopez, B.; Ottenheijm, C.A.; Becher, P.M.; et al. Myocardial titin hypophosphorylation importantly contributes to heart failure with preserved ejection fraction in a rat metabolic risk model. Circ. Heart Fail. 2013, 6, 1239–1249. [Google Scholar] [CrossRef] [PubMed]
- Bode, D.; Guthof, T.; Pieske, B.M.; Heinzel, F.R.; Hohendanner, F. Isolation of atrial cardiomyocytes from a rat model of metabolic syndrome-related heart failure with preserved ejection fraction. J. Vis. Exp. 2018, 137, e57953. [Google Scholar] [CrossRef] [PubMed]
- Hohendanner, F.; Ljubojevic, S.; MacQuaide, N.; Sacherer, M.; Sedej, S.; Biesmans, L.; Wakula, P.; Platzer, D.; Sokolow, S.; Herchuelz, A.; et al. Intracellular dyssynchrony of diastolic cytosolic [Ca2+] decay in ventricular cardiomyocytes in cardiac remodeling and human heart failure. Circ. Res. 2013, 113, 527–538. [Google Scholar] [CrossRef] [PubMed]
- Heinzel, F.R.; Bito, V.; Biesmans, L.; Wu, M.; Detre, E.; von Wegner, F.; Claus, P.; Dymarkowski, S.; Maes, F.; Bogaert, J.; et al. Remodeling of T-tubules and reduced synchrony of Ca2+ release in myocytes from chronically ischemic myocardium. Circ. Res. 2008, 102, 338–346. [Google Scholar] [CrossRef]
- Heinzel, F.R.; Bito, V.; Volders, P.G.; Antoons, G.; Mubagwa, K.; Sipido, K.R. Spatial and temporal inhomogeneities during Ca2+ release from the sarcoplasmic reticulum in pig ventricular myocytes. Circ. Res. 2002, 91, 1023–1030. [Google Scholar] [CrossRef]
- Bertero, E.; Maack, C. Calcium signaling and reactive oxygen species in mitochondria. Circ. Res. 2018, 122, 1460–1478. [Google Scholar] [CrossRef]
- Hohendanner, F.; Bode, D. Mitochondrial Calcium in heart failure with preserved ejection fraction-friend or foe? Acta Physiol. 2020, 228, e13415. [Google Scholar] [CrossRef]
- Rossi, A.; Pizzo, P.; Filadi, R. Calcium, mitochondria and cell metabolism: A functional triangle in bioenergetics. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2019, 1866, 1068–1078. [Google Scholar] [CrossRef]
- Ai, X.; Curran, J.W.; Shannon, T.R.; Bers, D.M.; Pogwizd, S.M. Ca2+/calmodulin-dependent protein kinase modulates cardiac ryanodine receptor phosphorylation and sarcoplasmic reticulum Ca2+ leak in heart failure. Circ. Res. 2005, 97, 1314–1322. [Google Scholar] [CrossRef]
- Thomas, G.P.; Sims, S.M.; Cook, M.A.; Karmazyn, M. Hydrogen peroxide-induced stimulation of L-type calcium current in guinea pig ventricular myocytes and its inhibition by adenosine A1 receptor activation. J. Pharmacol. Exp. Ther. 1998, 286, 1208–1214. [Google Scholar]
- Zafrir, B.; Lund, L.H.; Laroche, C.; Ruschitzka, F.; Crespo-Leiro, M.G.; Coats, A.J.S.; Anker, S.D.; Filippatos, G.; Seferovic, P.M.; Maggioni, A.P.; et al. Prognostic implications of atrial fibrillation in heart failure with reduced, mid-range, and preserved ejection fraction: A report from 14 964 patients in the European Society of Cardiology Heart Failure Long-Term Registry. Eur. Heart J. 2018, 39, 4277–4284. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.T.; Wu, C.K.; Lee, J.K.; Chang, S.N.; Kuo, Y.M.; Wang, Y.C.; Lai, L.P.; Chiang, F.T.; Hwang, J.J.; Lin, J.L. TNF-alpha down-regulates sarcoplasmic reticulum Ca2+ ATPase expression and leads to left ventricular diastolic dysfunction through binding of NF-kappaB to promoter response element. Cardiovasc. Res. 2015, 105, 318–329. [Google Scholar] [CrossRef] [PubMed]
- Dhingra, S.; Sharma, A.K.; Arora, R.C.; Slezak, J.; Singal, P.K. IL-10 attenuates TNF-alpha-induced NF kappaB pathway activation and cardiomyocyte apoptosis. Cardiovasc. Res. 2009, 82, 59–66. [Google Scholar] [CrossRef]
- Dhingra, S.; Sharma, A.K.; Singla, D.K.; Singal, P.K. p38 and ERK1/2 MAPKs mediate the interplay of TNF-alpha and IL-10 in regulating oxidative stress and cardiac myocyte apoptosis. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H3524–H3531. [Google Scholar] [CrossRef] [PubMed]
- Mann, D.L.; McMurray, J.J.; Packer, M.; Swedberg, K.; Borer, J.S.; Colucci, W.S.; Djian, J.; Drexler, H.; Feldman, A.; Kober, L.; et al. Targeted anticytokine therapy in patients with chronic heart failure: Results of the Randomized Etanercept Worldwide Evaluation (RENEWAL). Circulation 2004, 109, 1594–1602. [Google Scholar] [CrossRef] [PubMed]
- Chung, E.S.; Packer, M.; Lo, K.H.; Fasanmade, A.A.; Willerson, J.T. Randomized, double-blind, placebo-controlled, pilot trial of infliximab, a chimeric monoclonal antibody to tumor necrosis factor-alpha, in patients with moderate-to-severe heart failure: Results of the anti-TNF Therapy Against Congestive Heart Failure (ATTACH) trial. Circulation 2003, 107, 3133–3140. [Google Scholar]
- Bickston, S.J.; Cominelli, F. Recombinant interleukin 10 for the treatment of active Crohn’s disease: Lessons in biologic therapy. Gastroenterology 2000, 119, 1781–1783. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bode, D.; Wen, Y.; Hegemann, N.; Primessnig, U.; Parwani, A.; Boldt, L.-H.; M. Pieske, B.; R. Heinzel, F.; Hohendanner, F. Oxidative Stress and Inflammatory Modulation of Ca2+ Handling in Metabolic HFpEF-Related Left Atrial Cardiomyopathy. Antioxidants 2020, 9, 860. https://doi.org/10.3390/antiox9090860
Bode D, Wen Y, Hegemann N, Primessnig U, Parwani A, Boldt L-H, M. Pieske B, R. Heinzel F, Hohendanner F. Oxidative Stress and Inflammatory Modulation of Ca2+ Handling in Metabolic HFpEF-Related Left Atrial Cardiomyopathy. Antioxidants. 2020; 9(9):860. https://doi.org/10.3390/antiox9090860
Chicago/Turabian StyleBode, David, Yan Wen, Niklas Hegemann, Uwe Primessnig, Abdul Parwani, Leif-Hendrik Boldt, Burkert M. Pieske, Frank R. Heinzel, and Felix Hohendanner. 2020. "Oxidative Stress and Inflammatory Modulation of Ca2+ Handling in Metabolic HFpEF-Related Left Atrial Cardiomyopathy" Antioxidants 9, no. 9: 860. https://doi.org/10.3390/antiox9090860
APA StyleBode, D., Wen, Y., Hegemann, N., Primessnig, U., Parwani, A., Boldt, L.-H., M. Pieske, B., R. Heinzel, F., & Hohendanner, F. (2020). Oxidative Stress and Inflammatory Modulation of Ca2+ Handling in Metabolic HFpEF-Related Left Atrial Cardiomyopathy. Antioxidants, 9(9), 860. https://doi.org/10.3390/antiox9090860