The Role of Hydrogen Peroxide and Peroxiredoxins throughout the Cell Cycle



Abstract
1. Introduction
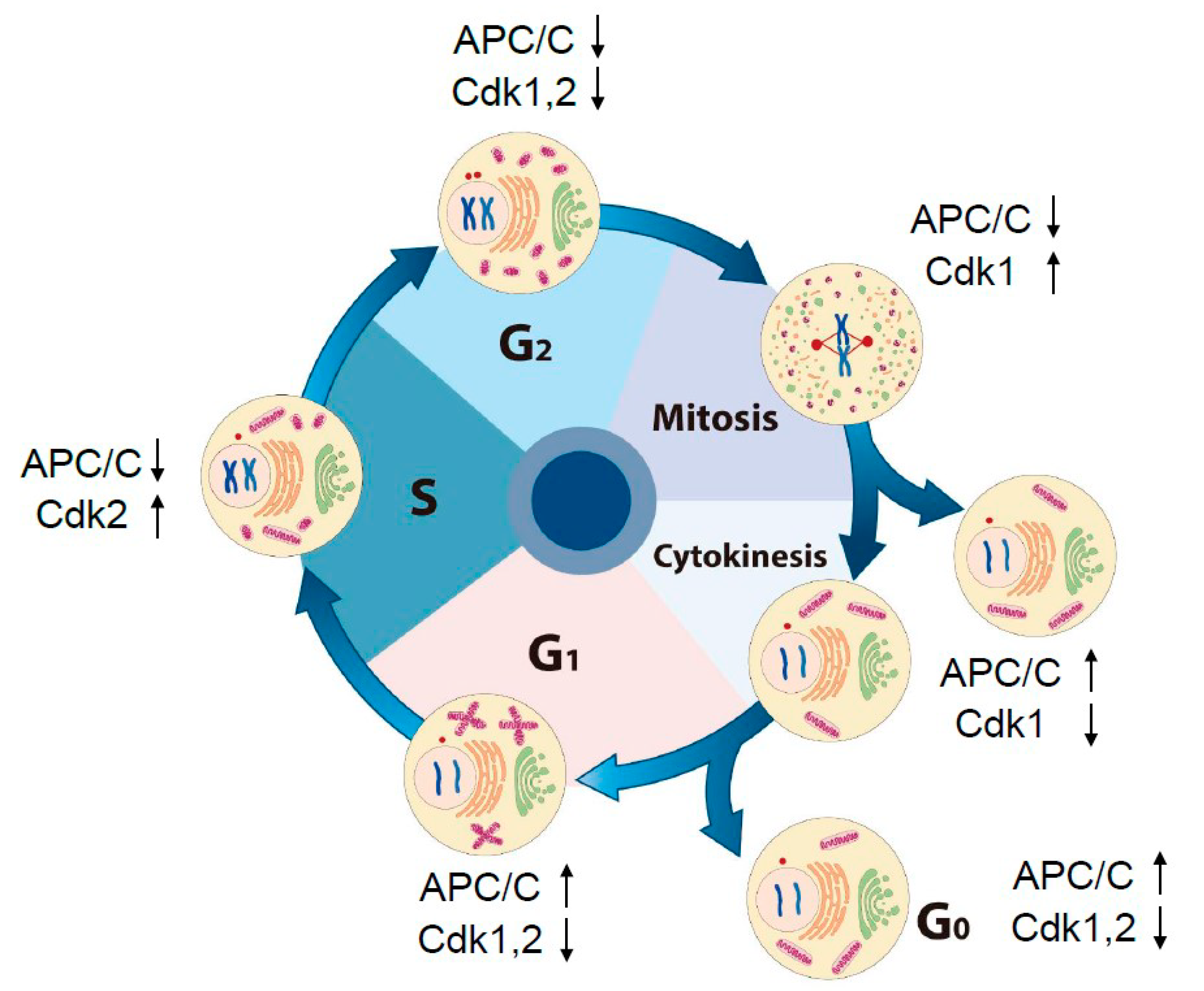
2. Dynamic Change in Intracellular Morphology and Oscillation of Activities of Cdks and APC/C through the Cell Cycle
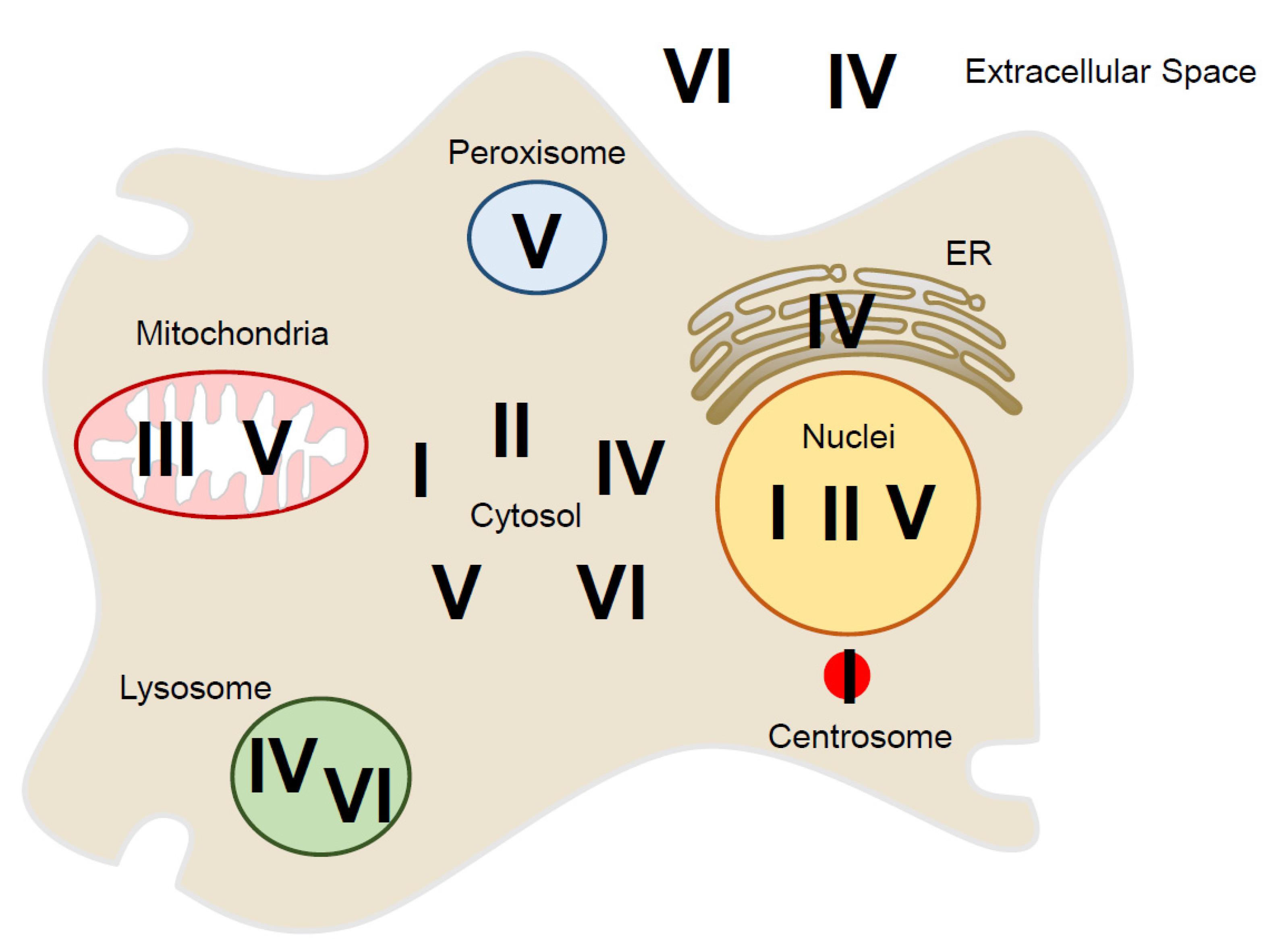
3. Localization of Peroxiredoxin Proteins Inside and Outside of the Cell
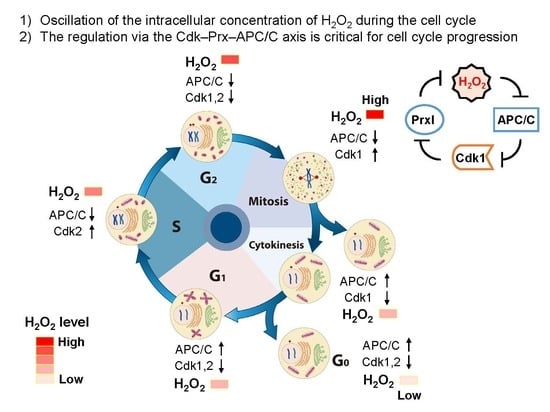
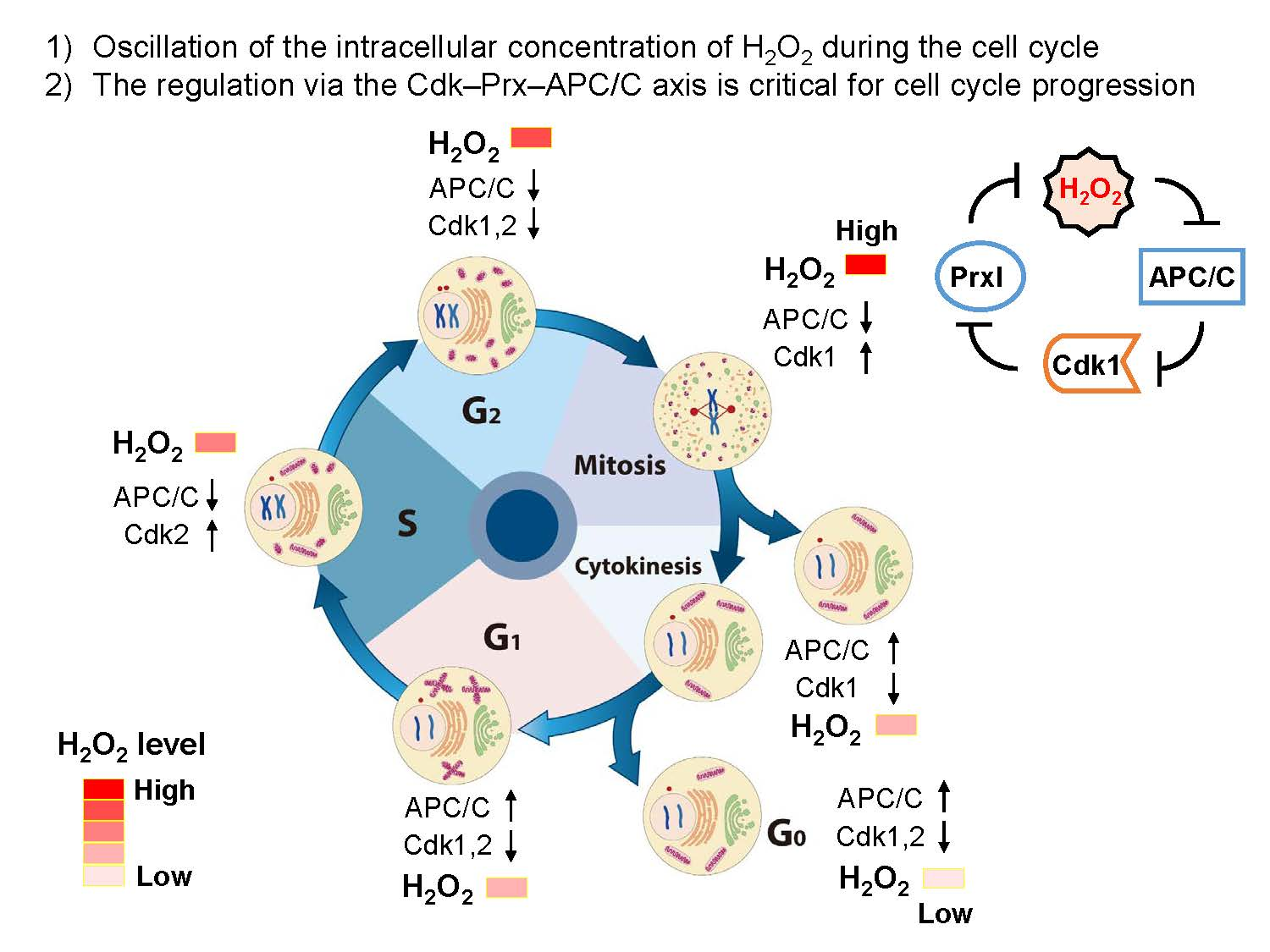
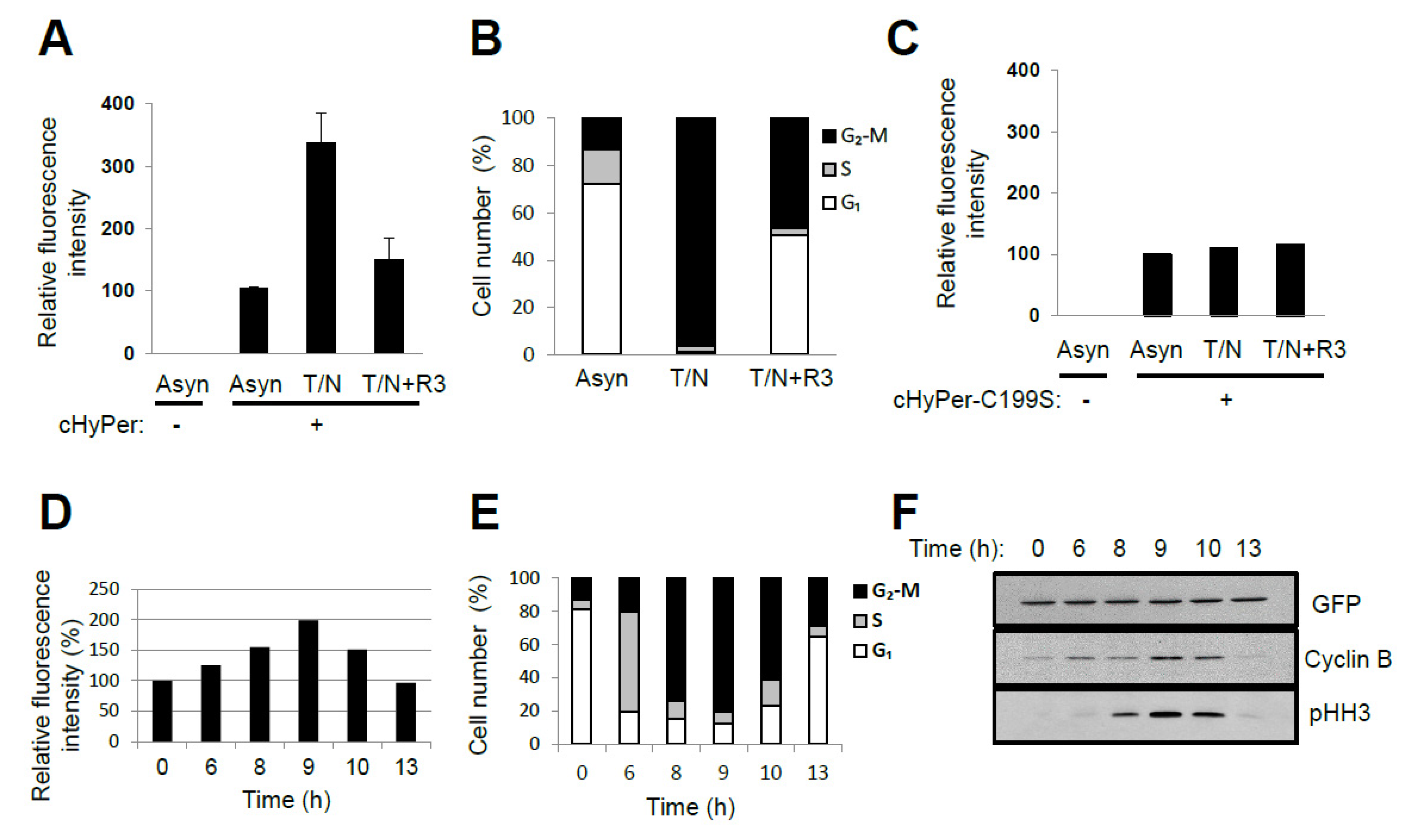
4. Oscillation of the Intracellular Concentration of H2O2 during the Cell Cycle
5. Source for H2O2 Generation throughout the Cell Cycle
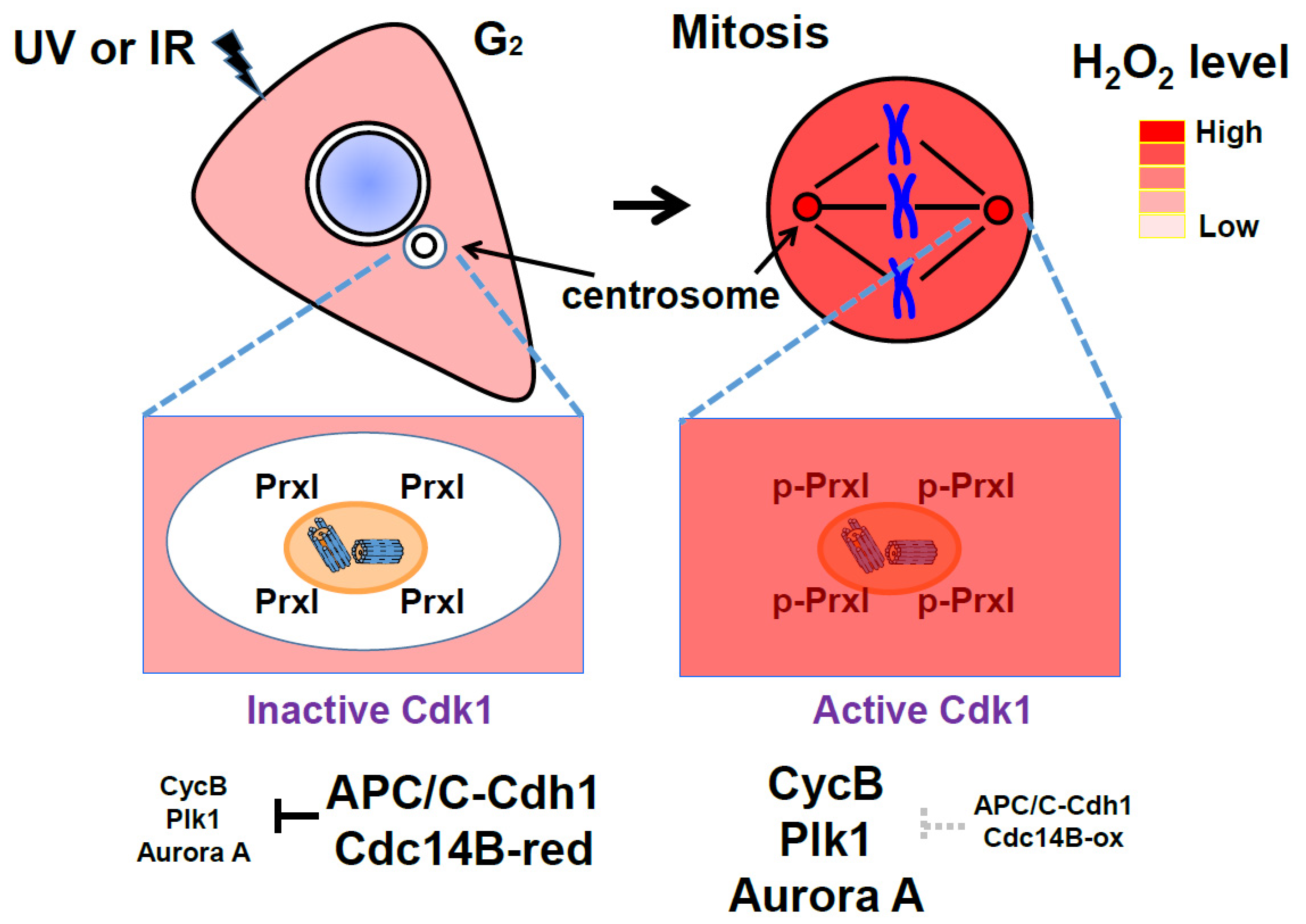
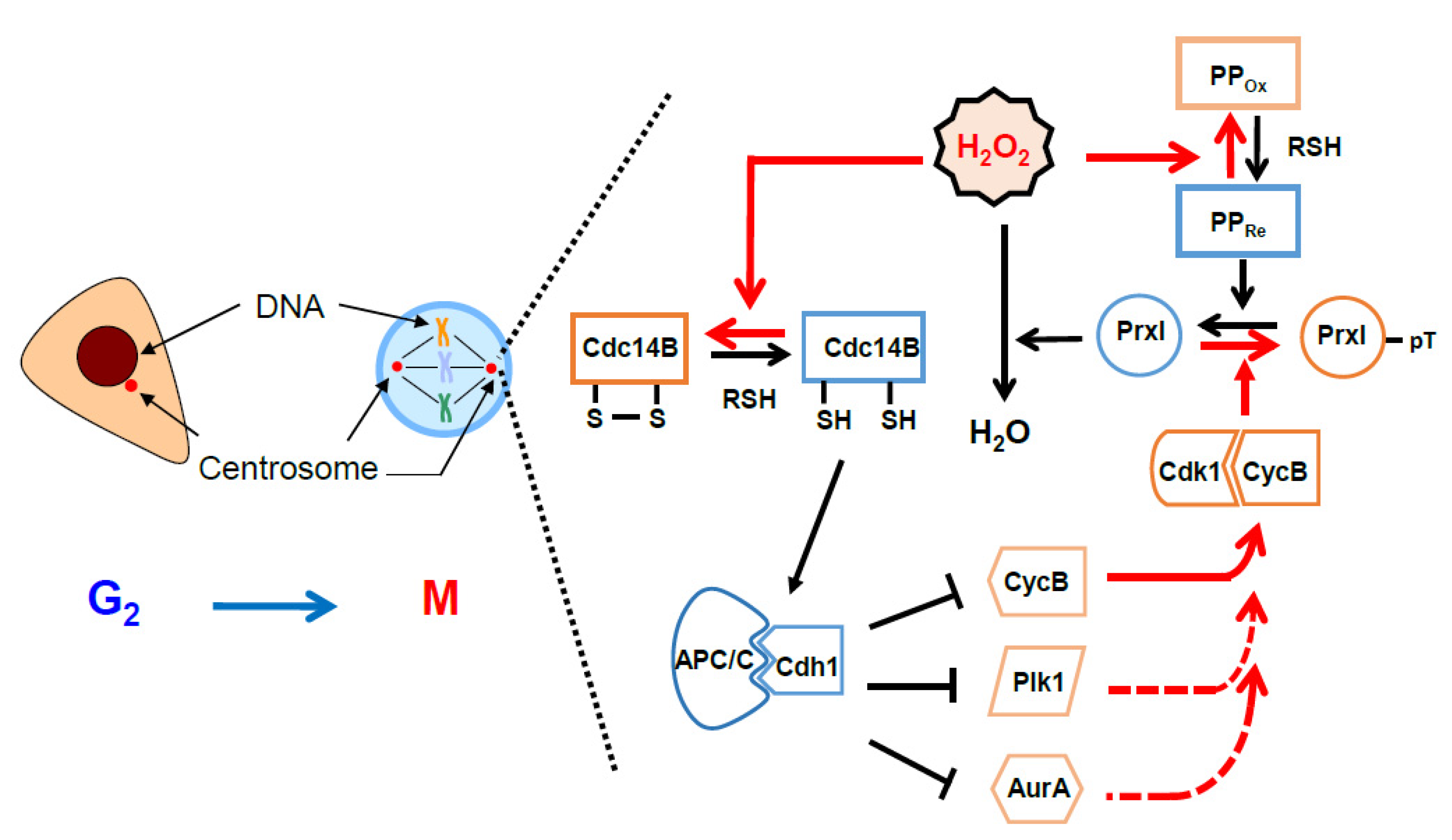
6. Protective and Signaling Role of Peroxiredoxin around the Centrosomes
7. Possible Role of Peroxiredoxin around the Golgi, ER, and Mitochondria
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Giorgio, M.; Trinei, M.; Migliaccio, E.; Pelicci, P.G. Hydrogen peroxide: A metabolic by-product or a common mediator of ageing signals? Nat. Rev. Mol. Cell Biol. 2007, 8, 722–728. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.G. Cell signaling. H2O2, a necessary evil for cell signaling. Science 2006, 312, 1882–1883. [Google Scholar] [CrossRef] [PubMed]
- Suh, Y.A.; Arnold, R.S.; Lassegue, B.; Shi, J.; Xu, X.; Sorescu, D.; Chung, A.B.; Griendling, K.K.; Lambeth, J.D. Cell transformation by the superoxide-generating oxidase Mox1. Nature 1999, 401, 79–82. [Google Scholar] [CrossRef] [PubMed]
- van der Vliet, A.; Hristova, M.; Cross, C.E.; Eiserich, J.P.; Goldkorn, T. Peroxynitrite induces covalent dimerization of epidermal growth factor receptors in A431 epidermoid carcinoma cells. J. Biol. Chem. 1998, 273, 31860–31866. [Google Scholar] [CrossRef] [PubMed]
- Clempus, R.E.; Griendling, K.K. Reactive oxygen species signaling in vascular smooth muscle cells. Cardiovasc. Res. 2006, 71, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Lambeth, J.D. NOX enzymes and the biology of reactive oxygen. Nat. Rev. Immunol. 2004, 4, 181–189. [Google Scholar] [CrossRef]
- Yuan, Z.; Schellekens, H.; Warner, L.; Janssen-Heininger, Y.; Burch, P.; Heintz, N.H. Reactive nitrogen species block cell cycle re-entry through sustained production of hydrogen peroxide. Am. J. Respir. Cell Mol. Biol. 2003, 28, 705–712. [Google Scholar] [CrossRef]
- Phalen, T.J.; Weirather, K.; Deming, P.B.; Anathy, V.; Howe, A.K.; van der Vliet, A.; Jonsson, T.J.; Poole, L.B.; Heintz, N.H. Oxidation state governs structural transitions in peroxiredoxin II that correlate with cell cycle arrest and recovery. J. Cell Biol. 2006, 175, 779–789. [Google Scholar] [CrossRef]
- Havens, C.G.; Ho, A.; Yoshioka, N.; Dowdy, S.F. Regulation of late G1/S phase transition and APC Cdh1 by reactive oxygen species. Mol. Cell Biol. 2006, 26, 4701–4711. [Google Scholar] [CrossRef]
- Yamaura, M.; Mitsushita, J.; Furuta, S.; Kiniwa, Y.; Ashida, A.; Goto, Y.; Shang, W.H.; Kubodera, M.; Kato, M.; Takata, M.; et al. NADPH oxidase 4 contributes to transformation phenotype of melanoma cells by regulating G2-M cell cycle progression. Cancer Res. 2009, 69, 2647–2654. [Google Scholar] [CrossRef]
- Hall, A.; Nelson, K.; Poole, L.B.; Karplus, P.A. Structure-based insights into the catalytic power and conformational dexterity of peroxiredoxins. Antioxid. Redox Signal. 2011, 15, 795–815. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.R.; Kwon, K.S.; Kim, S.R.; Rhee, S.G. Reversible inactivation of protein-tyrosine phosphatase 1B in A431 cells stimulated with epidermal growth factor. J. Biol. Chem. 1998, 273, 15366–15372. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.G.; Kil, I.S. Multiple Functions and Regulation of Mammalian Peroxiredoxins. Annu. Rev. Biochem. 2017, 86, 749–775. [Google Scholar] [CrossRef] [PubMed]
- Peskin, A.V.; Pace, P.E.; Behring, J.B.; Paton, L.N.; Soethoudt, M.; Bachschmid, M.M.; Winterbourn, C.C. Glutathionylation of the Active Site Cysteines of Peroxiredoxin 2 and Recycling by Glutaredoxin. J. Biol. Chem. 2016, 291, 3053–3062. [Google Scholar] [CrossRef] [PubMed]
- Goswami, P.C.; Sheren, J.; Albee, L.D.; Parsian, A.; Sim, J.E.; Ridnour, L.A.; Higashikubo, R.; Gius, D.; Hunt, C.R.; Spitz, D.R. Cell cycle-coupled variation in topoisomerase IIalpha mRNA is regulated by the 3′-untranslated region. Possible role of redox-sensitive protein binding in mRNA accumulation. J. Biol. Chem. 2000, 275, 38384–38392. [Google Scholar] [CrossRef]
- Sarsour, E.H.; Kumar, M.G.; Chaudhuri, L.; Kalen, A.L.; Goswami, P.C. Redox control of the cell cycle in health and disease. Antioxid. Redox Signal. 2009, 11, 2985–3011. [Google Scholar] [CrossRef]
- Lim, J.M.; Lee, K.S.; Woo, H.A.; Kang, D.; Rhee, S.G. Control of the pericentrosomal H2O2 level by peroxiredoxin I is critical for mitotic progression. J. Cell Bio.l 2015, 210, 23–33. [Google Scholar] [CrossRef]
- Pesin, J.A.; Orr-Weaver, T.L. Regulation of APC/C activators in mitosis and meiosis. Annu. Rev. Cell Dev. Biol. 2008, 24, 475–499. [Google Scholar] [CrossRef]
- Peters, J.M. The anaphase promoting complex/cyclosome: A machine designed to destroy. Nat. Rev. Mol. Cell Biol. 2006, 7, 644–656. [Google Scholar] [CrossRef]
- Skaar, J.R.; Pagano, M. Cdh1: A master G0/G1 regulator. Nat. Cell Biol. 2008, 10, 755–757. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.S.; Jeong, W.; Lee, D.Y.; Cho, C.S.; Rhee, S.G. The RING-H2-finger protein APC11 as a target of hydrogen peroxide. Free Radic. Biol. Med. 2004, 37, 521–530. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sherr, C.J.; Roberts, J.M. Inhibitors of mammalian G1 cyclin-dependent kinases. Genes Dev. 1995, 9, 1149–1163. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J.; Roberts, J.M. CDK inhibitors: Positive and negative regulators of G1-phase progression. Genes Dev. 1999, 13, 1501–1512. [Google Scholar] [CrossRef] [PubMed]
- Obaya, A.J.; Sedivy, J.M. Regulation of cyclin-Cdk activity in mammalian cells. Cell Mol. Life Sci. 2002, 59, 126–142. [Google Scholar] [CrossRef]
- Hsu, J.Y.; Reimann, J.D.; Sorensen, C.S.; Lukas, J.; Jackson, P.K. E2F-dependent accumulation of hEmi1 regulates S phase entry by inhibiting APC(Cdh1). Nat. Cell Biol. 2002, 4, 358–366. [Google Scholar] [CrossRef]
- Reimann, J.D.; Gardner, B.E.; Margottin-Goguet, F.; Jackson, P.K. Emi1 regulates the anaphase-promoting complex by a different mechanism than Mad2 proteins. Genes Dev. 2001, 15, 3278–3285. [Google Scholar] [CrossRef]
- Gunasekar, P.G.; Kanthasamy, A.G.; Borowitz, J.L.; Isom, G.E. Monitoring intracellular nitric oxide formation by dichlorofluorescin in neuronal cells. J. Neurosci. Methods 1995, 61, 15–21. [Google Scholar] [CrossRef]
- Kooy, N.W.; Royall, J.A.; Ischiropoulos, H. Oxidation of 2′,7′-dichlorofluorescin by peroxynitrite. Free Radic. Res. 1997, 27, 245–254. [Google Scholar] [CrossRef]
- Rhee, S.G.; Chang, T.S.; Jeong, W.; Kang, D. Methods for detection and measurement of hydrogen peroxide inside and outside of cells. Mol. Cells 2010, 29, 539–549. [Google Scholar] [CrossRef]
- Marsh, B.J.; Howell, K.E. The mammalian Golgi--complex debates. Nat. Rev. Mol. Cell Biol. 2002, 3, 789–795. [Google Scholar] [CrossRef]
- Sutterlin, C.; Hsu, P.; Mallabiabarrena, A.; Malhotra, V. Fragmentation and dispersal of the pericentriolar Golgi complex is required for entry into mitosis in mammalian cells. Cell 2002, 109, 359–369. [Google Scholar] [CrossRef]
- Sutterlin, C.; Lin, C.Y.; Feng, Y.; Ferris, D.K.; Erikson, R.L.; Malhotra, V. Polo-like kinase is required for the fragmentation of pericentriolar Golgi stacks during mitosis. Proc. Natl. Acad. Sci. USA 2001, 98, 9128–9132. [Google Scholar] [CrossRef] [PubMed]
- Acharya, U.; Mallabiabarrena, A.; Acharya, J.K.; Malhotra, V. Signaling via mitogen-activated protein kinase kinase (MEK1) is required for Golgi fragmentation during mitosis. Cell 1998, 92, 183–192. [Google Scholar] [CrossRef]
- Lowe, M.; Rabouille, C.; Nakamura, N.; Watson, R.; Jackman, M.; Jamsa, E.; Rahman, D.; Pappin, D.J.; Warren, G. Cdc2 kinase directly phosphorylates the cis-Golgi matrix protein GM130 and is required for Golgi fragmentation in mitosis. Cell 1998, 94, 783–793. [Google Scholar] [CrossRef]
- Park, S.; Lim, J.M.; Park, S.H.; Kim, S.; Heo, S.; Balla, T.; Jeong, W.; Rhee, S.G.; Kang, D. Inactivation of the PtdIns(4)P phosphatase Sac1 at the Golgi by H2O2 produced via Ca(2+)-dependent Duox in EGF-stimulated cells. Free Radic. Biol. Med. 2019, 131, 40–49. [Google Scholar] [CrossRef]
- Keith, C.H.; Ratan, R.; Maxfield, F.R.; Bajer, A.; Shelanski, M.L. Local cytoplasmic calcium gradients in living mitotic cells. Nature 1985, 316, 848–850. [Google Scholar] [CrossRef]
- Poenie, M.; Alderton, J.; Steinhardt, R.; Tsien, R. Calcium rises abruptly and briefly throughout the cell at the onset of anaphase. Science 1986, 233, 886–889. [Google Scholar] [CrossRef]
- Ratan, R.R.; Shelanski, M.L.; Maxfield, F.R. Transition from metaphase to anaphase is accompanied by local changes in cytoplasmic free calcium in Pt K2 kidney epithelial cells. Proc. Natl. Acad. Sci. USA 1986, 83, 5136–5140. [Google Scholar] [CrossRef]
- Vafai, S.B.; Mootha, V.K. Mitochondrial disorders as windows into an ancient organelle. Nature 2012, 491, 374–383. [Google Scholar] [CrossRef]
- Mitra, K.; Wunder, C.; Roysam, B.; Lin, G.; Lippincott-Schwartz, J. A hyperfused mitochondrial state achieved at G1-S regulates cyclin E buildup and entry into S phase. Proc. Natl. Acad. Sci. USA 2009, 106, 11960–11965. [Google Scholar] [CrossRef]
- Salazar-Roa, M.; Malumbres, M. Fueling the Cell Division Cycle. Trends Cell Biol. 2017, 27, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, oxidants, and aging. Cell 2005, 120, 483–495. [Google Scholar] [CrossRef]
- Chance, B.; Sies, H.; Boveris, A. Hydroperoxide metabolism in mammalian organs. Physiol. Rev. 1979, 59, 527–605. [Google Scholar] [CrossRef]
- Owusu-Ansah, E.; Yavari, A.; Mandal, S.; Banerjee, U. Distinct mitochondrial retrograde signals control the G1-S cell cycle checkpoint. Nat. Genet. 2008, 40, 356–361. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, N.; Ishihara, N.; Jofuku, A.; Oka, T.; Mihara, K. Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J. Biol. Chem. 2007, 282, 11521–11529. [Google Scholar] [CrossRef] [PubMed]
- Horn, S.R.; Thomenius, M.J.; Johnson, E.S.; Freel, C.D.; Wu, J.Q.; Coloff, J.L.; Yang, C.S.; Tang, W.; An, J.; Ilkayeva, O.R.; et al. Regulation of mitochondrial morphology by APC/CCdh1-mediated control of Drp1 stability. Mol. Biol. Cell 2011, 22, 1207–1216. [Google Scholar] [CrossRef]
- Rhee, S.G.; Woo, H.A.; Kil, I.S.; Bae, S.H. Peroxiredoxin functions as a peroxidase and a regulator and sensor of local peroxides. J. Biol. Chem. 2012, 287, 4403–4410. [Google Scholar] [CrossRef]
- Hanschmann, E.M.; Godoy, J.R.; Berndt, C.; Hudemann, C.; Lillig, C.H. Thioredoxins, Glutaredoxins, and Peroxiredoxins-Molecular Mechanisms and Health Significance: From Cofactors to Antioxidants to Redox Signaling. Antioxid. Redox Signal. 2013, 19, 1539–1605. [Google Scholar] [CrossRef]
- Oberley, T.D.; Verwiebe, E.; Zhong, W.; Kang, S.W.; Rhee, S.G. Localization of the thioredoxin system in normal rat kidney. Free Radic. Biol. Med. 2001, 30, 412–424. [Google Scholar] [CrossRef]
- Woo, H.A.; Yim, S.H.; Shin, D.H.; Kang, D.; Yu, D.Y.; Rhee, S.G. Inactivation of peroxiredoxin I by phosphorylation allows localized H(2)O(2) accumulation for cell signaling. Cell 2010, 140, 517–528. [Google Scholar] [CrossRef]
- Kang, S.W.; Chae, H.Z.; Seo, M.S.; Kim, K.H.; Baines, I.C.; Rhee, S.G. Mammalian peroxiredoxin isoforms can reduce hydrogen peroxide generated in response to growth factors and tumor necrosis factor-alpha. J. Biol. Chem. 1998, 273, 6297–6302. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.W.; Lee, D.J.; Lee, J.Y.; Kang, D.H.; Kwon, J.; Kang, S.W. Peroxiredoxin II restrains DNA damage-induced death in cancer cells by positively regulating JNK-dependent DNA repair. J. Biol. Chem. 2011, 286, 8394–8404. [Google Scholar] [CrossRef] [PubMed]
- Cox, A.G.; Winterbourn, C.C.; Hampton, M.B. Mitochondrial peroxiredoxin involvement in antioxidant defence and redox signalling. Biochem. J. 2009, 425, 313–325. [Google Scholar] [CrossRef] [PubMed]
- Zhong, H.; Xiao, M.; Zarkovic, K.; Zhu, M.; Sa, R.; Lu, J.; Tao, Y.; Chen, Q.; Xia, L.; Cheng, S.; et al. Mitochondrial control of apoptosis through modulation of cardiolipin oxidation in hepatocellular carcinoma: A novel link between oxidative stress and cancer. Free Radic. Biol. Med. 2017, 102, 67–76. [Google Scholar] [CrossRef]
- Matsumoto, A.; Okado, A.; Fujii, T.; Fujii, J.; Egashira, M.; Niikawa, N.; Taniguchi, N. Cloning of the peroxiredoxin gene family in rats and characterization of the fourth member. FEBS Lett. 1999, 443, 246–250. [Google Scholar] [CrossRef]
- Okado-Matsumoto, A.; Matsumoto, A.; Fujii, J.; Taniguchi, N. Peroxiredoxin IV is a secretable protein with heparin-binding properties under reduced conditions. J. Biochem. 2000, 127, 493–501. [Google Scholar] [CrossRef]
- Tavender, T.J.; Springate, J.J.; Bulleid, N.J. Recycling of peroxiredoxin IV provides a novel pathway for disulphide formation in the endoplasmic reticulum. EMBO J. 2010, 29, 4185–4197. [Google Scholar] [CrossRef]
- Zito, E.; Melo, E.P.; Yang, Y.; Wahlander, A.; Neubert, T.A.; Ron, D. Oxidative protein folding by an endoplasmic reticulum-localized peroxiredoxin. Mol. Cell 2010, 40, 787–797. [Google Scholar] [CrossRef]
- Tavender, T.J.; Sheppard, A.M.; Bulleid, N.J. Peroxiredoxin IV is an endoplasmic reticulum-localized enzyme forming oligomeric complexes in human cells. Biochem. J. 2008, 411, 191–199. [Google Scholar] [CrossRef]
- Knoops, B.; Clippe, A.; Bogard, C.; Arsalane, K.; Wattiez, R.; Hermans, C.; Duconseille, E.; Falmagne, P.; Bernard, A. Cloning and characterization of AOEB166, a novel mammalian antioxidant enzyme of the peroxiredoxin family. J. Biol. Chem. 1999, 274, 30451–30458. [Google Scholar] [CrossRef]
- Nguyen-Nhu, N.T.; Berck, J.; Clippe, A.; Duconseille, E.; Cherif, H.; Boone, C.; Van der Eecken, V.; Bernard, A.; Banmeyer, I.; Knoops, B. Human peroxiredoxin 5 gene organization, initial characterization of its promoter and identification of alternative forms of mRNA. Biochim. Biophys. Acta 2007, 1769, 472–483. [Google Scholar] [CrossRef] [PubMed]
- Seo, M.S.; Kang, S.W.; Kim, K.; Baines, I.C.; Lee, T.H.; Rhee, S.G. Identification of a new type of mammalian peroxiredoxin that forms an intramolecular disulfide as a reaction intermediate. J. Biol. Chem. 2000, 275, 20346–20354. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Kok, K.H.; Chun, A.C.; Wong, C.M.; Wu, H.W.; Lin, M.C.; Fung, P.C.; Kung, H.; Jin, D.Y. Mouse peroxiredoxin V is a thioredoxin peroxidase that inhibits p53-induced apoptosis. Biochem. Biophys. Res. Commun. 2000, 268, 921–927. [Google Scholar] [CrossRef]
- Fisher, A.B. Peroxiredoxin 6: A bifunctional enzyme with glutathione peroxidase and phospholipase A(2) activities. Antioxid. Redox Signal. 2011, 15, 831–844. [Google Scholar] [CrossRef]
- Sorokina, E.M.; Feinstein, S.I.; Milovanova, T.N.; Fisher, A.B. Identification of the amino acid sequence that targets peroxiredoxin 6 to lysosome-like structures of lung epithelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2009, 297, L871–L880. [Google Scholar] [CrossRef]
- Sorokina, E.M.; Feinstein, S.I.; Zhou, S.; Fisher, A.B. Intracellular targeting of peroxiredoxin 6 to lysosomal organelles requires MAPK activity and binding to 14-3-3epsilon. Am. J. Physiol. Cell Physiol. 2011, 300, C1430–C1441. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Feinstein, S.I.; Manevich, Y.; Chowdhury, I.; Pak, J.H.; Kazi, A.; Dodia, C.; Speicher, D.W.; Fisher, A.B. Mitogen-activated protein kinase-mediated phosphorylation of peroxiredoxin 6 regulates its phospholipase A(2) activity. Biochem. J. 2009, 419, 669–679. [Google Scholar] [CrossRef] [PubMed]
- Belousov, V.V.; Fradkov, A.F.; Lukyanov, K.A.; Staroverov, D.B.; Shakhbazov, K.S.; Terskikh, A.V.; Lukyanov, S. Genetically encoded fluorescent indicator for intracellular hydrogen peroxide. Nat. Methods 2006, 3, 281–286. [Google Scholar] [CrossRef]
- van Rossum, G.S.; Vlug, A.S.; van den Bosch, H.; Verkleij, A.J.; Boonstra, J. Cytosolic phospholipase A(2) activity during the ongoing cell cycle. J. Cell. Physiol. 2001, 188, 321–328. [Google Scholar] [CrossRef]
- Cho, K.J.; Seo, J.M.; Kim, J.H. Bioactive lipoxygenase metabolites stimulation of NADPH oxidases and reactive oxygen species. Mol. Cells 2011, 32, 1–5. [Google Scholar] [CrossRef]
- Zaal, K.J.; Smith, C.L.; Polishchuk, R.S.; Altan, N.; Cole, N.B.; Ellenberg, J.; Hirschberg, K.; Presley, J.F.; Roberts, T.H.; Siggia, E.; et al. Golgi membranes are absorbed into and reemerge from the ER during mitosis. Cell 1999, 99, 589–601. [Google Scholar] [CrossRef]
- Jezek, J.; Cooper, K.F.; Strich, R. Reactive Oxygen Species and Mitochondrial Dynamics: The Yin and Yang of Mitochondrial Dysfunction and Cancer Progression. Antioxidants 2018, 7, 13. [Google Scholar] [CrossRef]
- Hung, C.H.; Cheng, S.S.; Cheung, Y.T.; Wuwongse, S.; Zhang, N.Q.; Ho, Y.S.; Lee, S.M.; Chang, R.C. A reciprocal relationship between reactive oxygen species and mitochondrial dynamics in neurodegeneration. Redox Biol. 2018, 14, 7–19. [Google Scholar] [CrossRef] [PubMed]
- Luders, J.; Stearns, T. Microtubule-organizing centres: A re-evaluation. Nat. Rev. Mol. Cell Biol. 2007, 8, 161–167. [Google Scholar] [CrossRef]
- Bonnet, J.; Coopman, P.; Morris, M.C. Characterization of centrosomal localization and dynamics of Cdc25C phosphatase in mitosis. Cell Cycle 2008, 7, 1991–1998. [Google Scholar] [CrossRef][Green Version]
- Cho, H.P.; Liu, Y.; Gomez, M.; Dunlap, J.; Tyers, M.; Wang, Y. The dual-specificity phosphatase CDC14B bundles and stabilizes microtubules. Mol. Cell Biol. 2005, 25, 4541–4551. [Google Scholar] [CrossRef]
- Clute, P.; Pines, J. Temporal and spatial control of cyclin B1 destruction in metaphase. Nat. Cell Biol. 1999, 1, 82–87. [Google Scholar] [CrossRef]
- Dutertre, S.; Cazales, M.; Quaranta, M.; Froment, C.; Trabut, V.; Dozier, C.; Mirey, G.; Bouche, J.P.; Theis-Febvre, N.; Schmitt, E.; et al. Phosphorylation of CDC25B by Aurora-A at the centrosome contributes to the G2-M transition. J. Cell Sci. 2004, 117, 2523–2531. [Google Scholar] [CrossRef]
- Kramer, A.; Mailand, N.; Lukas, C.; Syljuasen, R.G.; Wilkinson, C.J.; Nigg, E.A.; Bartek, J.; Lukas, J. Centrosome-associated Chk1 prevents premature activation of cyclin-B-Cdk1 kinase. Nat. Cell Biol. 2004, 6, 884–891. [Google Scholar] [CrossRef]
- Wu, J.; Cho, H.P.; Rhee, D.B.; Johnson, D.K.; Dunlap, J.; Liu, Y.; Wang, Y. Cdc14B depletion leads to centriole amplification, and its overexpression prevents unscheduled centriole duplication. J. Cell Biol. 2008, 181, 475–483. [Google Scholar] [CrossRef]
- Raff, J.W.; Jeffers, K.; Huang, J.Y. The roles of Fzy/Cdc20 and Fzr/Cdh1 in regulating the destruction of cyclin B in space and time. J. Cell Biol. 2002, 157, 1139–1149. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.S.; Jeong, W.; Choi, S.Y.; Yu, S.; Kang, S.W.; Rhee, S.G. Regulation of peroxiredoxin I activity by Cdc2-mediated phosphorylation. J. Biol. Chem. 2002, 277, 25370–25376. [Google Scholar] [CrossRef]
- Wurzenberger, C.; Gerlich, D.W. Phosphatases: Providing safe passage through mitotic exit. Nat. Rev. Mol. Cell Biol. 2011, 12, 469–482. [Google Scholar] [CrossRef]
- Andersen, J.S.; Wilkinson, C.J.; Mayor, T.; Mortensen, P.; Nigg, E.A.; Mann, M. Proteomic characterization of the human centrosome by protein correlation profiling. Nature 2003, 426, 570–574. [Google Scholar] [CrossRef]
- Jeong, A.L.; Lee, S.; Park, J.S.; Han, S.; Jang, C.Y.; Lim, J.S.; Lee, M.S.; Yang, Y. Cancerous Inhibitor of Protein Phosphatase 2A (CIP2A) Protein Is Involved in Centrosome Separation through the Regulation of NIMA (Never In Mitosis Gene A)-related Kinase 2 (NEK2) Protein Activity. J. Biol. Chem. 2014, 289, 28–40. [Google Scholar] [CrossRef] [PubMed]
- Meraldi, P.; Nigg, E.A. Centrosome cohesion is regulated by a balance of kinase and phosphatase activities. J. Cell Sci. 2001, 114, 3749–3757. [Google Scholar] [PubMed]
- Schlaitz, A.L.; Srayko, M.; Dammermann, A.; Quintin, S.; Wielsch, N.; MacLeod, I.; de Robillard, Q.; Zinke, A.; Yates, J.R., 3rd; Muller-Reichert, T.; et al. The C. elegans RSA complex localizes protein phosphatase 2A to centrosomes and regulates mitotic spindle assembly. Cell 2007, 128, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Domingo-Sananes, M.R.; Kapuy, O.; Hunt, T.; Novak, B. Switches and latches: A biochemical tug-of-war between the kinases and phosphatases that control mitosis. Philos. Trans. R. Soc. Lond B Biol. Sci. 2011, 366, 3584–3594. [Google Scholar] [CrossRef]
- Garcia-Higuera, I.; Manchado, E.; Dubus, P.; Canamero, M.; Mendez, J.; Moreno, S.; Malumbres, M. Genomic stability and tumour suppression by the APC/C cofactor Cdh1. Nat. Cell Biol. 2008, 10, 802–811. [Google Scholar] [CrossRef]
- Neumann, C.A.; Krause, D.S.; Carman, C.V.; Das, S.; Dubey, D.P.; Abraham, J.L.; Bronson, R.T.; Fujiwara, Y.; Orkin, S.H.; Van Etten, R.A. Essential role for the peroxiredoxin Prdx1 in erythrocyte antioxidant defence and tumour suppression. Nature 2003, 424, 561–565. [Google Scholar] [CrossRef]
- Wood, Z.A.; Poole, L.B.; Karplus, P.A. Peroxiredoxin evolution and the regulation of hydrogen peroxide signaling. Science 2003, 300, 650–653. [Google Scholar] [CrossRef] [PubMed]
- Kwon, J.; Lee, S.R.; Yang, K.S.; Ahn, Y.; Kim, Y.J.; Stadtman, E.R.; Rhee, S.G. Reversible oxidation and inactivation of the tumor suppressor PTEN in cells stimulated with peptide growth factors. Proc. Natl. Acad. Sci. USA 2004, 101, 16419–16424. [Google Scholar] [CrossRef] [PubMed]
- Tonks, N.K. Redox redux: Revisiting PTPs and the control of cell signaling. Cell 2005, 121, 667–670. [Google Scholar] [CrossRef]
- Deshpande, N.N.; Sorescu, D.; Seshiah, P.; Ushio-Fukai, M.; Akers, M.; Yin, Q.; Griendling, K.K. Mechanism of hydrogen peroxide-induced cell cycle arrest in vascular smooth muscle. Antioxid. Redox Signal. 2002, 4, 845–854. [Google Scholar] [CrossRef]
- Bae, S.H.; Sung, S.H.; Cho, E.J.; Lee, S.K.; Lee, H.E.; Woo, H.A.; Yu, D.Y.; Kil, I.S.; Rhee, S.G. Concerted action of sulfiredoxin and peroxiredoxin I protects against alcohol-induced oxidative injury in mouse liver. Hepatology 2011, 53, 945–953. [Google Scholar] [CrossRef]
- Smith-Pearson, P.S.; Kooshki, M.; Spitz, D.R.; Poole, L.B.; Zhao, W.; Robbins, M.E. Decreasing peroxiredoxin II expression decreases glutathione, alters cell cycle distribution, and sensitizes glioma cells to ionizing radiation and H(2)O(2). Free Radic. Biol. Med. 2008, 45, 1178–1189. [Google Scholar] [CrossRef]
- Chua, P.J.; Lee, E.H.; Yu, Y.; Yip, G.W.; Tan, P.H.; Bay, B.H. Silencing the Peroxiredoxin III gene inhibits cell proliferation in breast cancer. Int. J. Oncol. 2010, 36, 359–364. [Google Scholar]
- Bassermann, F.; Frescas, D.; Guardavaccaro, D.; Busino, L.; Peschiaroli, A.; Pagano, M. The Cdc14B-Cdh1-Plk1 axis controls the G2 DNA-damage-response checkpoint. Cell 2008, 134, 256–267. [Google Scholar] [CrossRef]
- Mocciaro, A.; Schiebel, E. Cdc14: A highly conserved family of phosphatases with non-conserved functions? J. Cell Sci. 2010, 123, 2867–2876. [Google Scholar] [CrossRef]
- Mochida, S.; Ikeo, S.; Gannon, J.; Hunt, T. Regulated activity of PP2A-B55 delta is crucial for controlling entry into and exit from mitosis in Xenopus egg extracts. EMBO J. 2009, 28, 2777–2785. [Google Scholar] [CrossRef]
- Schmitz, M.H.; Held, M.; Janssens, V.; Hutchins, J.R.; Hudecz, O.; Ivanova, E.; Goris, J.; Trinkle-Mulcahy, L.; Lamond, A.I.; Poser, I.; et al. Live-cell imaging RNAi screen identifies PP2A-B55alpha and importin-beta1 as key mitotic exit regulators in human cells. Nat. Cell Biol. 2010, 12, 886–893. [Google Scholar] [CrossRef]
- Wu, J.Q.; Guo, J.Y.; Tang, W.; Yang, C.S.; Freel, C.D.; Chen, C.; Nairn, A.C.; Kornbluth, S. PP1-mediated dephosphorylation of phosphoproteins at mitotic exit is controlled by inhibitor-1 and PP1 phosphorylation. Nat. Cell Biol. 2009, 11, 644–651. [Google Scholar] [CrossRef]
- Pieri, L.; Dominici, S.; Del Bello, B.; Maellaro, E.; Comporti, M.; Paolicchi, A.; Pompella, A. Redox modulation of protein kinase/phosphatase balance in melanoma cells: The role of endogenous and gamma-glutamyltransferase-dependent H2O2 production. Biochim. Biophys. Acta 2003, 1621, 76–83. [Google Scholar] [CrossRef]
- Rusnak, F.; Reiter, T. Sensing electrons: Protein phosphatase redox regulation. Trends Biochem. Sci. 2000, 25, 527–529. [Google Scholar] [CrossRef]
- Wright, V.P.; Reiser, P.J.; Clanton, T.L. Redox modulation of global phosphatase activity and protein phosphorylation in intact skeletal muscle. J. Physiol. 2009, 587, 5767–5781. [Google Scholar] [CrossRef]
- Qu, D.; Rashidian, J.; Mount, M.P.; Aleyasin, H.; Parsanejad, M.; Lira, A.; Haque, E.; Zhang, Y.; Callaghan, S.; Daigle, M.; et al. Role of Cdk5-mediated phosphorylation of Prx2 in MPTP toxicity and Parkinson’s disease. Neuron 2007, 55, 37–52. [Google Scholar] [CrossRef]
- Przedborski, S. Pathogenesis of nigral cell death in Parkinson’s disease. Parkinsonism Relat. Disord. 2005, 11, S3–S7. [Google Scholar] [CrossRef]
- Maestre, C.; Delgado-Esteban, M.; Gomez-Sanchez, J.C.; Bolanos, J.P.; Almeida, A. Cdk5 phosphorylates Cdh1 and modulates cyclin B1 stability in excitotoxicity. EMBO J. 2008, 27, 2736–2745. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Prx Isoform | Subfamily | Subcellular Localization | Reference |
|---|---|---|---|
| PrxⅠ | 2-Cys Prx | Cytosol, nucleus, centrosome, plasma membrane | [13,17,47,48,49,50,51] |
| PrxⅡ | 2-Cys Prx | Cytosol, nucleus, plasma membrane | [13,47,48,49,52] |
| PrxⅢ | 2-Cys Prx | Mitochondria | [13,47,48,49,53,54] |
| PrxⅣ | 2-Cys Prx | Cytosol, ER, lysosome, extracellular localization | [13,47,48,49,55,56,57,58,59] |
| PrxⅤ | Atypical 2-Cys Prx | Cytosol, mitochondria, peroxisome, nucleus | [13,47,48,49,53,60,61,62,63] |
| PrxⅥ | 1-Cys Prx | Cytosol, lysosome, extracellular localization | [13,47,48,49,64,65,66,67] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heo, S.; Kim, S.; Kang, D. The Role of Hydrogen Peroxide and Peroxiredoxins throughout the Cell Cycle. Antioxidants 2020, 9, 280. https://doi.org/10.3390/antiox9040280
Heo S, Kim S, Kang D. The Role of Hydrogen Peroxide and Peroxiredoxins throughout the Cell Cycle. Antioxidants. 2020; 9(4):280. https://doi.org/10.3390/antiox9040280
Chicago/Turabian StyleHeo, Sukyeong, Suree Kim, and Dongmin Kang. 2020. "The Role of Hydrogen Peroxide and Peroxiredoxins throughout the Cell Cycle" Antioxidants 9, no. 4: 280. https://doi.org/10.3390/antiox9040280
APA StyleHeo, S., Kim, S., & Kang, D. (2020). The Role of Hydrogen Peroxide and Peroxiredoxins throughout the Cell Cycle. Antioxidants, 9(4), 280. https://doi.org/10.3390/antiox9040280