Triple Combination of Ascorbate, Menadione and the Inhibition of Peroxiredoxin-1 Produces Synergistic Cytotoxic Effects in Triple-Negative Breast Cancer Cells

 , , ,
, , ,
Abstract
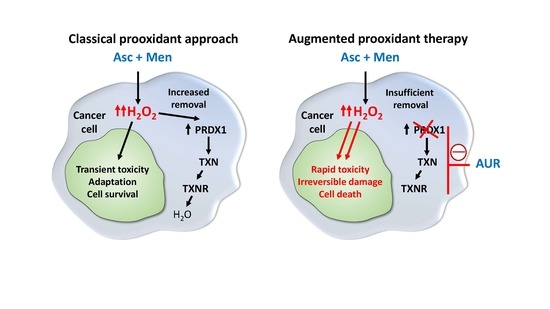
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials Methods
2.1. Analysis of RNA-seq Data
2.2. TMA Cohort
2.3. Immunohistochemistry
2.4. Digital Slide Scanning and Automated Image Analysis
2.5. Cell Line Culture
2.6. Western Blotting
2.7. Chemical Reagents
2.8. Stable shRNA-Mediated Knockdown of PRDX1
2.9. In Vitro Combinations with Prooxidant Agents
2.10. ROS Detection
2.11. Crystal Violet Assay
2.12. Colony Formation Assay
2.13. Statistical Analysis
3. Results
3.1. Expression of PRDX1 in TNBC
3.2. Effects of Downregulation of PRDX1 in TNBC Cell Lines
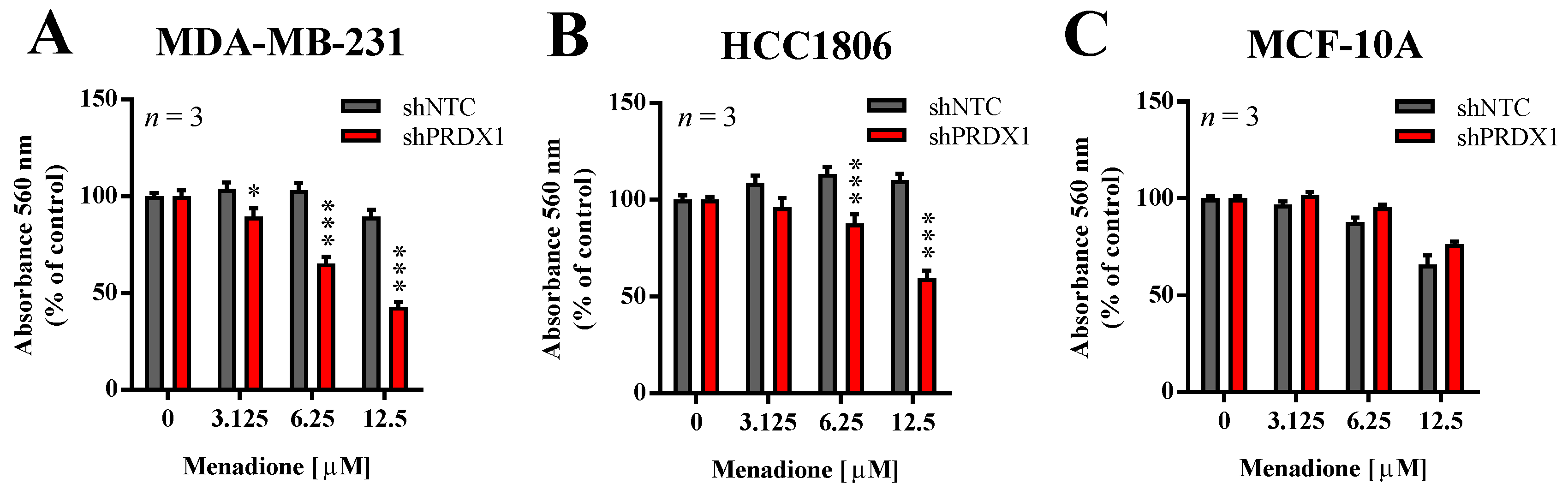
3.3. Effects of PRDX1 Knockdown on the Susceptibility TNBC Cells to Menadione
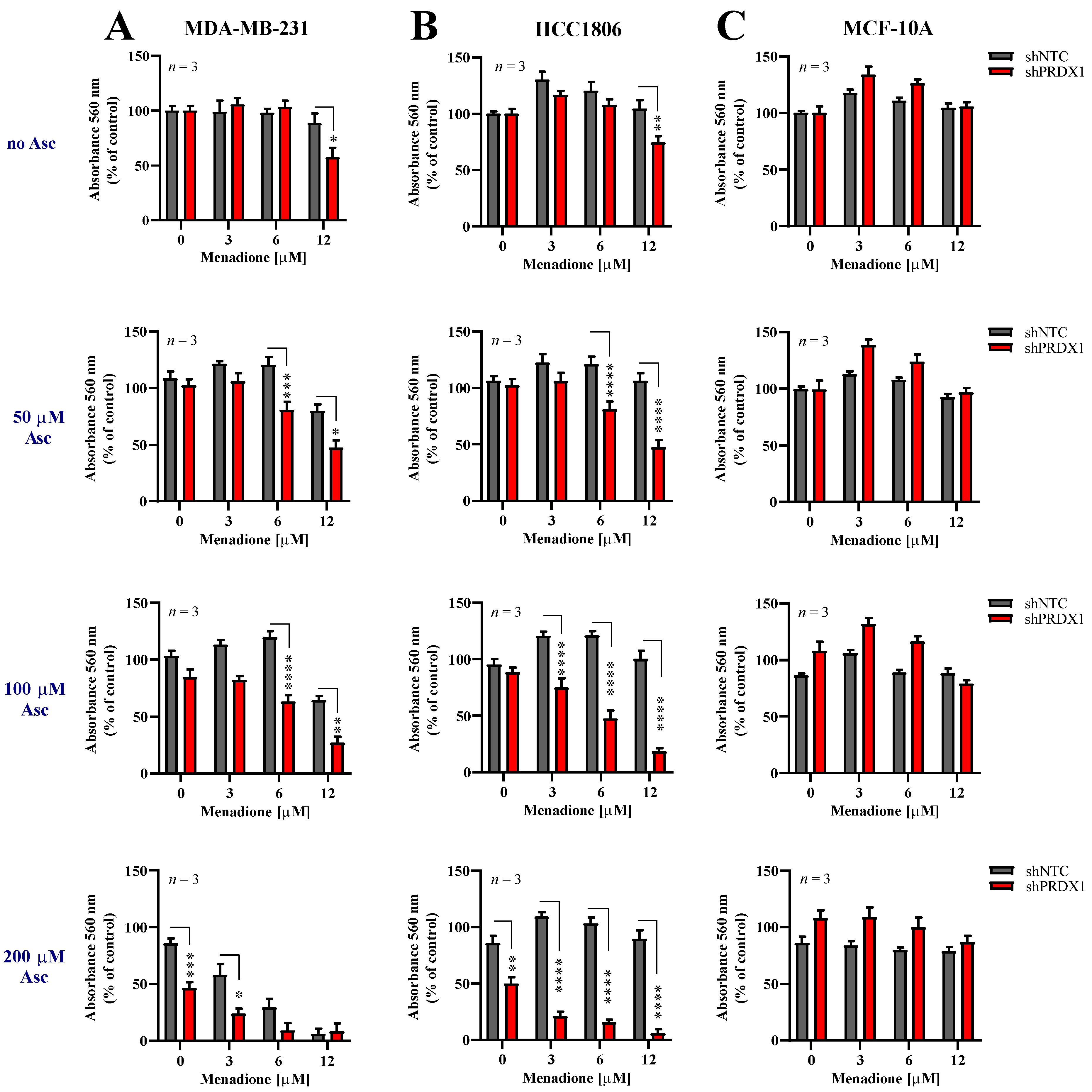
3.4. Effects of Asc/Men Combination in TNBC Cells with Downregulated PRDX1
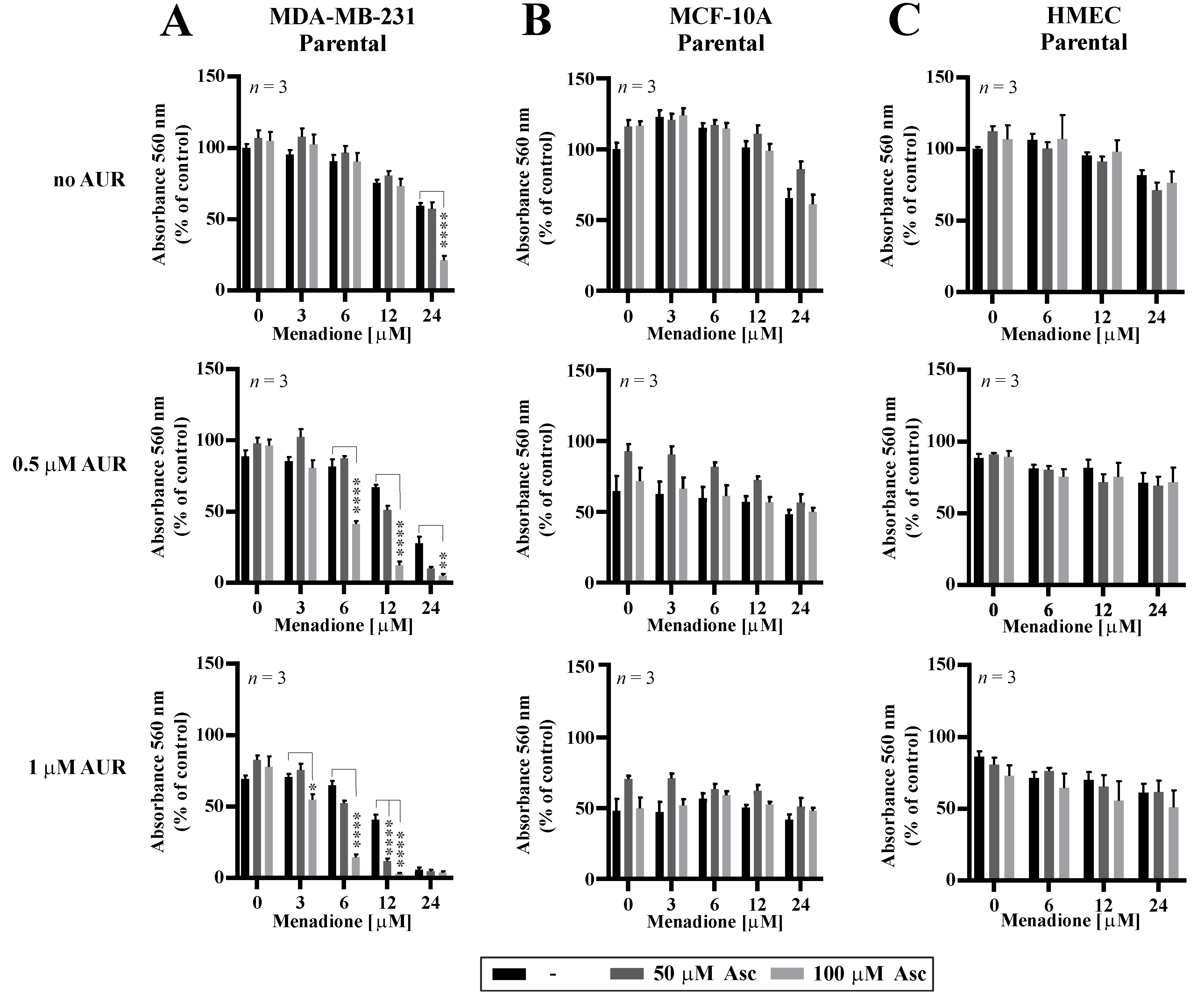
3.5. Effects of Auranofin on the Susceptibility of TNBC Cells to Asc/Men
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Marra, A.; Viale, G.; Curigliano, G. Recent advances in triple negative breast cancer: The immunotherapy era. BMC Med. 2019, 17, 90. [Google Scholar] [CrossRef] [PubMed]
- Khan, H.Y.; Zubair, H.; Ullah, M.F.; Ahmad, A.; Hadi, S.M. A prooxidant mechanism for the anticancer and chemopreventive properties of plant polyphenols. Curr. Drug Targets 2012, 13, 1738–1749. [Google Scholar] [CrossRef] [PubMed]
- Verrax, J.; Taper, H.; Buc Calderon, P. Targeting cancer cells by an oxidant-based therapy. Curr. Mol. Pharmacol. 2008, 1, 80–92. [Google Scholar] [PubMed]
- Beck, R.; Verrax, J.; Dejeans, N.; Taper, H.; Calderon, P.B. Menadione reduction by pharmacological doses of ascorbate induces an oxidative stress that kills breast cancer cells. Int. J. Toxicol. 2009, 28, 33–42. [Google Scholar] [CrossRef]
- Verrax, J.; Cadrobbi, J.; Marques, C.; Taper, H.; Habraken, Y.; Piette, J.; Calderon, P.B. Ascorbate potentiates the cytotoxicity of menadione leading to an oxidative stress that kills cancer cells by a non-apoptotic caspase-3 independent form of cell death. Apoptosis 2004, 9, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Verrax, J.; Stockis, J.; Tison, A.; Taper, H.S.; Calderon, P.B. Oxidative stress by ascorbate/menadione association kills K562 human chronic myelogenous leukaemia cells and inhibits its tumour growth in nude mice. Biochem. Pharmacol. 2006, 72, 671–680. [Google Scholar] [CrossRef]
- Zhang, W.; Negoro, T.; Satoh, K.; Jiang, Y.; Hashimoto, K.; Kikuchi, H.; Nishikawa, H.; Miyata, T.; Yamamoto, Y.; Nakano, K.; et al. Synergistic cytotoxic action of vitamin C and vitamin K3. Anticancer Res. 2001, 21, 3439–3444. [Google Scholar]
- Tareen, B.; Summers, J.L.; Jamison, J.M.; Neal, D.R.; McGuire, K.; Gerson, L.; Diokno, A. A 12 week, open label, phase I/IIa study using apatone for the treatment of prostate cancer patients who have failed standard therapy. Int. J. Med. Sci. 2008, 5, 62–67. [Google Scholar] [CrossRef]
- Duconge, J.; Miranda-Massari, J.R.; Gonzalez, M.J.; Jackson, J.A.; Warnock, W.; Riordan, N.H. Pharmacokinetics of vitamin C: Insights into the oral and intravenous administration of ascorbate. Proc. R. Health Sci. J. 2008, 27, 7–19. [Google Scholar]
- Hirota, Y.; Tsugawa, N.; Nakagawa, K.; Suhara, Y.; Tanaka, K.; Uchino, Y.; Takeuchi, A.; Sawada, N.; Kamao, M.; Wada, A.; et al. Menadione (vitamin K3) is a catabolic product of oral phylloquinone (vitamin K1) in the intestine and a circulating precursor of tissue menaquinone-4 (vitamin K2) in rats. J. Biol. Chem. 2013, 288, 33071–33080. [Google Scholar] [CrossRef]
- Nauman, G.; Gray, J.C.; Parkinson, R.; Levine, M.; Paller, C.J. Systematic Review of Intravenous Ascorbate in Cancer Clinical Trials. Antioxidants (Basel) 2018, 7, 89. [Google Scholar] [CrossRef]
- Lim, D.; Morgan, R.J., Jr.; Akman, S.; Margolin, K.; Carr, B.I.; Leong, L.; Odujinrin, O.; Doroshow, J.H. Phase I trial of menadiol diphosphate (vitamin K3) in advanced malignancy. Investig. New Drugs 2005, 23, 235–239. [Google Scholar] [CrossRef] [PubMed]
- Graczyk-Jarzynka, A.; Goral, A.; Muchowicz, A.; Zagozdzon, R.; Winiarska, M.; Bajor, M.; Trzeciecka, A.; Fidyt, K.; Krupka, J.A.; Cyran, J.; et al. Inhibition of thioredoxin-dependent H2O2 removal sensitizes malignant B-cells to pharmacological ascorbate. Redox Biol. 2019, 21, 101062. [Google Scholar] [CrossRef] [PubMed]
- Trzeciecka, A.; Klossowski, S.; Bajor, M.; Zagozdzon, R.; Gaj, P.; Muchowicz, A.; Malinowska, A.; Czerwoniec, A.; Barankiewicz, J.; Domagala, A.; et al. Dimeric peroxiredoxins are druggable targets in human Burkitt lymphoma. Oncotarget 2016, 7, 1717–1731. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.G. Overview on Peroxiredoxin. Mol. Cells 2016, 39, 1. [Google Scholar] [CrossRef]
- Argyropoulou, V.; Goemaere, J.; Clippe, A.; Lefort, C.; Tissir, F.; Schakman, O.; Philippe, G.; Ahn, M.T.; Guiot, Y.; Galant, C.; et al. 188—Peroxiredoxin-5 as a Novel Actor in Inflammation and Tumor Suppression. Free Radic. Biol. Med. 2016, 100, S92. [Google Scholar] [CrossRef]
- Hwang, I.; Uddin, M.J.; Lee, G.; Jiang, S.; Pak, E.S.; Ha, H. Peroxiredoxin 3 deficiency accelerates chronic kidney injury in mice through interactions between macrophages and tubular epithelial cells. Free Radic. Biol. Med. 2019, 131, 162–172. [Google Scholar] [CrossRef]
- Iuchi, Y.; Okada, F.; Tsunoda, S.; Kibe, N.; Shirasawa, N.; Ikawa, M.; Okabe, M.; Ikeda, Y.; Fujii, J. Peroxiredoxin 4 knockout results in elevated spermatogenic cell death via oxidative stress. Biochem. J. 2009, 419, 149–158. [Google Scholar] [CrossRef]
- Lee, T.H.; Kim, S.U.; Yu, S.L.; Kim, S.H.; Park, D.S.; Moon, H.B.; Dho, S.H.; Kwon, K.S.; Kwon, H.J.; Han, Y.H.; et al. Peroxiredoxin II is essential for sustaining life span of erythrocytes in mice. Blood 2003, 101, 5033–5038. [Google Scholar] [CrossRef]
- Neumann, C.A.; Krause, D.S.; Carman, C.V.; Das, S.; Dubey, D.P.; Abraham, J.L.; Bronson, R.T.; Fujiwara, Y.; Orkin, S.H.; Van Etten, R.A. Essential role for the peroxiredoxin Prdx1 in erythrocyte antioxidant defence and tumour suppression. Nature 2003, 424, 561–565. [Google Scholar] [CrossRef]
- Karihtala, P.; Mantyniemi, A.; Kang, S.W.; Kinnula, V.L.; Soini, Y. Peroxiredoxins in breast carcinoma. Clin. Cancer Res. 2003, 9, 3418–3424. [Google Scholar]
- O’Leary, P.C.; Terrile, M.; Bajor, M.; Gaj, P.; Hennessy, B.T.; Mills, G.B.; Zagozdzon, A.; O’Connor, D.P.; Brennan, D.J.; Connor, K.; et al. Peroxiredoxin-1 protects estrogen receptor alpha from oxidative stress-induced suppression and is a protein biomarker of favorable prognosis in breast cancer. Breast Cancer Res. 2014, 16, R79. [Google Scholar] [CrossRef]
- Kalinina, E.V.; Berezov, T.T.; Shtil, A.A.; Chernov, N.N.; Glazunova, V.A.; Novichkova, M.D.; Nurmuradov, N.K. Expression of peroxiredoxin 1, 2, 3, and 6 genes in cancer cells during drug resistance formation. Bull. Exp. Biol. Med. 2012, 153, 878–881. [Google Scholar] [CrossRef]
- McDonald, C.; Muhlbauer, J.; Perlmutter, G.; Taparra, K.; Phelan, S.A. Peroxiredoxin proteins protect MCF-7 breast cancer cells from doxorubicin-induced toxicity. Int. J. Oncol. 2014, 45, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Varley, K.E.; Gertz, J.; Roberts, B.S.; Davis, N.S.; Bowling, K.M.; Kirby, M.K.; Nesmith, A.S.; Oliver, P.G.; Grizzle, W.E.; Forero, A.; et al. Recurrent read-through fusion transcripts in breast cancer. Breast Cancer Res. Treat. 2014, 146, 287–297. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Ni Chonghaile, T.; Fan, Y.; Madden, S.F.; Klinger, R.; O’Connor, A.E.; Walsh, L.; O’Hurley, G.; Mallya Udupi, G.; Joseph, J.; et al. Therapeutic Rationale to Target Highly Expressed CDK7 Conferring Poor Outcomes in Triple-Negative Breast Cancer. Cancer Res. 2017, 77, 3834–3845. [Google Scholar] [CrossRef] [PubMed]
- Gutta, C.; Rahman, A.; Aura, C.; Dynoodt, P.; Charles, E.M.; Hirschenhahn, E.; Joseph, J.; Wouters, J.; de Chaumont, C.; Rafferty, M.; et al. Low expression of pro-apoptotic proteins Bax, Bak and Smac indicates prolonged progression-free survival in chemotherapy-treated metastatic melanoma. Cell Death Dis. 2020, 11, 124. [Google Scholar] [CrossRef] [PubMed]
- Giandomenico, A.R.; Cerniglia, G.E.; Biaglow, J.E.; Stevens, C.W.; Koch, C.J. The importance of sodium pyruvate in assessing damage produced by hydrogen peroxide. Free Radic. Biol. Med. 1997, 23, 426–434. [Google Scholar] [CrossRef]
- Bajor, M.; Zych, A.O.; Graczyk-Jarzynka, A.; Muchowicz, A.; Firczuk, M.; Trzeciak, L.; Gaj, P.; Domagala, A.; Siernicka, M.; Zagozdzon, A.; et al. Targeting peroxiredoxin 1 impairs growth of breast cancer cells and potently sensitises these cells to prooxidant agents. Br. J. Cancer 2018, 119, 873–884. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Chattopadhyay, N.; Liu, W.J.; Chan, C.; Pignol, J.P.; Reilly, R.M. Optimized digital counting colonies of clonogenic assays using ImageJ software and customized macros: Comparison with manual counting. Int. J. Radiat. Biol. 2011, 87, 1135–1146. [Google Scholar] [CrossRef] [PubMed]
- Brzozowska, B.; Galecki, M.; Tartas, A.; Ginter, J.; Kazmierczak, U.; Lundholm, L. Freeware tool for analysing numbers and sizes of cell colonies. Radiat. Environ. Biophys. 2019, 58, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Muchowicz, A.; Firczuk, M.; Chlebowska, J.; Nowis, D.; Stachura, J.; Barankiewicz, J.; Trzeciecka, A.; Klossowski, S.; Ostaszewski, R.; Zagozdzon, R.; et al. Adenanthin targets proteins involved in the regulation of disulphide bonds. Biochem. Pharmacol. 2014, 89, 210–216. [Google Scholar] [CrossRef]
- Hatem, E.; Azzi, S.; El Banna, N.; He, T.; Heneman-Masurel, A.; Vernis, L.; Baille, D.; Masson, V.; Dingli, F.; Loew, D.; et al. Auranofin/Vitamin C: A Novel Drug Combination Targeting Triple-Negative Breast Cancer. J. Natl. Cancer Inst. 2019, 111, 597–608. [Google Scholar] [CrossRef]
- Montero, A.J.; Jassem, J. Cellular redox pathways as a therapeutic target in the treatment of cancer. Drugs 2011, 71, 1385–1396. [Google Scholar] [CrossRef]
- Kim, S.J.; Kim, H.S.; Seo, Y.R. Understanding of ROS-Inducing Strategy in Anticancer Therapy. Oxid. Med. Cell. Longev. 2019, 2019, 5381692. [Google Scholar] [CrossRef]
- Tomasetti, M.; Santarelli, L.; Alleva, R.; Dong, L.F.; Neuzil, J. Redox-active and redox-silent compounds: Synergistic therapeutics in cancer. Curr. Med. Chem. 2015, 22, 552–568. [Google Scholar] [CrossRef]
- Firczuk, M.; Bajor, M.; Graczyk-Jarzynka, A.; Fidyt, K.; Goral, A.; Zagozdzon, R. Harnessing altered oxidative metabolism in cancer by augmented prooxidant therapy. Cancer Lett. 2020, 471, 1–11. [Google Scholar] [CrossRef]
- Mei, J.; Hao, L.; Liu, X.; Sun, G.; Xu, R.; Wang, H.; Liu, C. Comprehensive analysis of peroxiredoxins expression profiles and prognostic values in breast cancer. Biomark. Res. 2019, 7, 16. [Google Scholar] [CrossRef]
- Lunetti, P.; Di Giacomo, M.; Vergara, D.; De Domenico, S.; Maffia, M.; Zara, V.; Capobianco, L.; Ferramosca, A. Metabolic reprogramming in breast cancer results in distinct mitochondrial bioenergetics between luminal and basal subtypes. FEBS J. 2019, 286, 688–709. [Google Scholar] [CrossRef]
- Bae, J.Y.; Ahn, S.J.; Han, W.; Noh, D.Y. Peroxiredoxin I and II inhibit H2O2-induced cell death in MCF-7 cell lines. J. Cell. Biochem. 2007, 101, 1038–1045. [Google Scholar] [CrossRef] [PubMed]
- Siernicka, M.; Winiarska, M.; Bajor, M.; Firczuk, M.; Muchowicz, A.; Bobrowicz, M.; Fauriat, C.; Golab, J.; Olive, D.; Zagozdzon, R. Adenanthin, a new inhibitor of thiol-dependent antioxidant enzymes, impairs the effector functions of human natural killer cells. Immunology 2015, 146, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Guo, W.; Zhu, Y.; Peng, S.; Zheng, W.; Zhang, C.; Shao, F.; Zhu, Y.; Hang, N.; Kong, L.; et al. Targeting Peroxiredoxin 1 by a Curcumin Analogue, AI-44, Inhibits NLRP3 Inflammasome Activation and Attenuates Lipopolysaccharide-Induced Sepsis in Mice. J. Immunol. 2018, 201, 2403–2413. [Google Scholar] [CrossRef]
- Ye, Q.; Zhang, Y.; Cao, Y.; Wang, X.; Guo, Y.; Chen, J.; Horn, J.; Ponomareva, L.V.; Chaiswing, L.; Shaaban, K.A.; et al. Frenolicin B Targets Peroxiredoxin 1 and Glutaredoxin 3 to Trigger ROS/4E-BP1-Mediated Antitumor Effects. Cell Chem. Biol. 2019, 26, 366–377/e312. [Google Scholar] [CrossRef] [PubMed]
- Roder, C.; Thomson, M.J. Auranofin: Repurposing an old drug for a golden new age. Drugs R D 2015, 15, 13–20. [Google Scholar] [CrossRef]
- Zhang, X.; Selvaraju, K.; Saei, A.A.; D’Arcy, P.; Zubarev, R.A.; Arner, E.S.; Linder, S. Repurposing of auranofin: Thioredoxin reductase remains a primary target of the drug. Biochimie 2019, 162, 46–54. [Google Scholar] [CrossRef]
- Onodera, T.; Momose, I.; Kawada, M. Potential Anticancer Activity of Auranofin. Chem. Pharm. Bull. (Tokyo) 2019, 67, 186–191. [Google Scholar] [CrossRef]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bajor, M.; Graczyk-Jarzynka, A.; Marhelava, K.; Kurkowiak, M.; Rahman, A.; Aura, C.; Russell, N.; Zych, A.O.; Firczuk, M.; Winiarska, M.; et al. Triple Combination of Ascorbate, Menadione and the Inhibition of Peroxiredoxin-1 Produces Synergistic Cytotoxic Effects in Triple-Negative Breast Cancer Cells. Antioxidants 2020, 9, 320. https://doi.org/10.3390/antiox9040320
Bajor M, Graczyk-Jarzynka A, Marhelava K, Kurkowiak M, Rahman A, Aura C, Russell N, Zych AO, Firczuk M, Winiarska M, et al. Triple Combination of Ascorbate, Menadione and the Inhibition of Peroxiredoxin-1 Produces Synergistic Cytotoxic Effects in Triple-Negative Breast Cancer Cells. Antioxidants. 2020; 9(4):320. https://doi.org/10.3390/antiox9040320
Chicago/Turabian StyleBajor, Malgorzata, Agnieszka Graczyk-Jarzynka, Katsiaryna Marhelava, Malgorzata Kurkowiak, Arman Rahman, Claudia Aura, Niamh Russell, Agata O. Zych, Malgorzata Firczuk, Magdalena Winiarska, and et al. 2020. "Triple Combination of Ascorbate, Menadione and the Inhibition of Peroxiredoxin-1 Produces Synergistic Cytotoxic Effects in Triple-Negative Breast Cancer Cells" Antioxidants 9, no. 4: 320. https://doi.org/10.3390/antiox9040320
APA StyleBajor, M., Graczyk-Jarzynka, A., Marhelava, K., Kurkowiak, M., Rahman, A., Aura, C., Russell, N., Zych, A. O., Firczuk, M., Winiarska, M., Gallagher, W. M., & Zagozdzon, R. (2020). Triple Combination of Ascorbate, Menadione and the Inhibition of Peroxiredoxin-1 Produces Synergistic Cytotoxic Effects in Triple-Negative Breast Cancer Cells. Antioxidants, 9(4), 320. https://doi.org/10.3390/antiox9040320