Bardoxolone-Methyl (CDDO-Me) Suppresses Androgen Receptor and Its Splice-Variant AR-V7 and Enhances Efficacy of Enzalutamide in Prostate Cancer Cells

 , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Reagents
2.3. MTT Assay
2.4. Western Blot Analysis
2.5. ROS Assay
2.6. Wound-Heal Assay
2.7. Colony Forming Units Assay
2.8. Immunofluorescence Microscopy
2.9. Quantitative RT-PCR
2.10. Statistical Analysis
3. Results
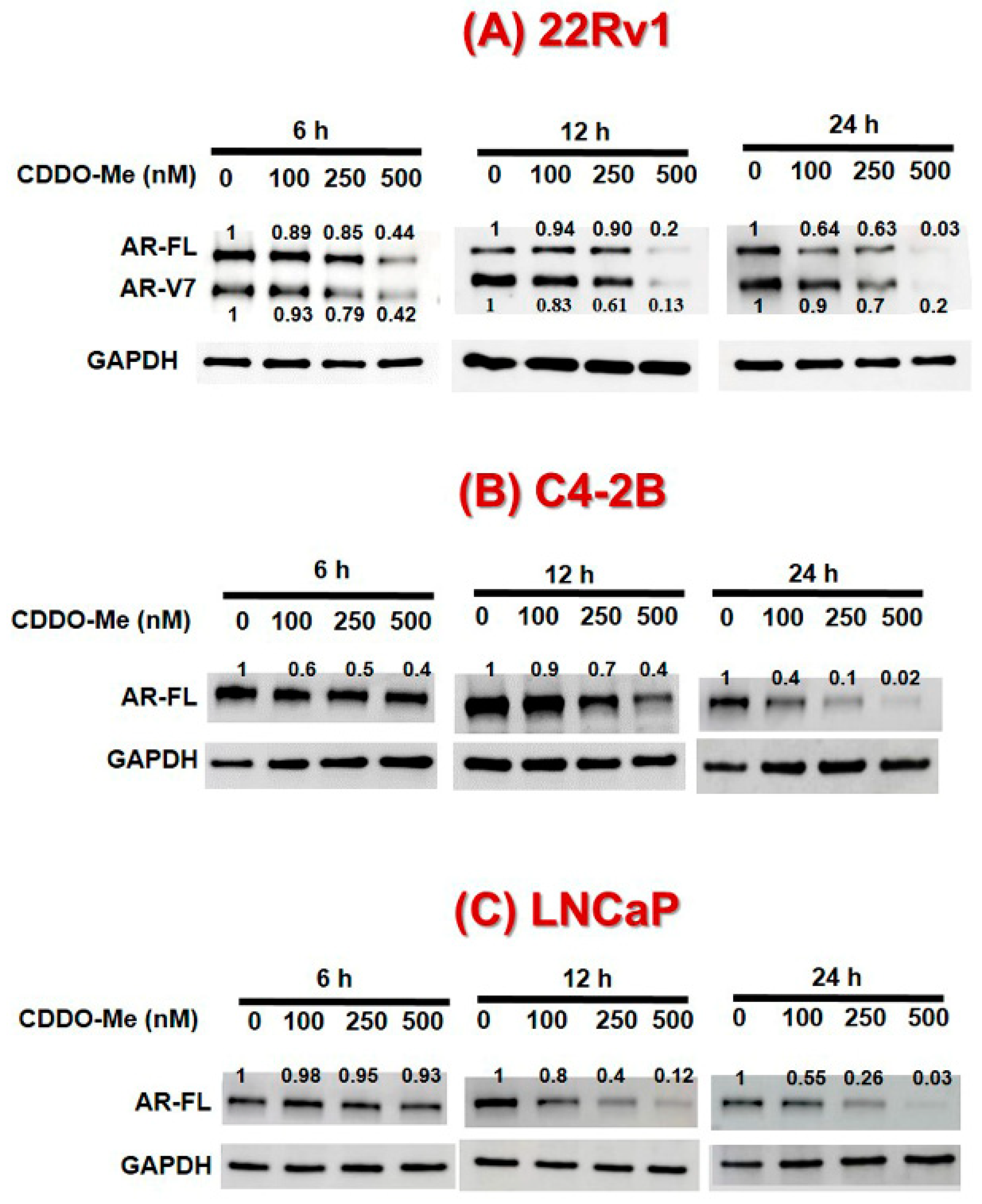
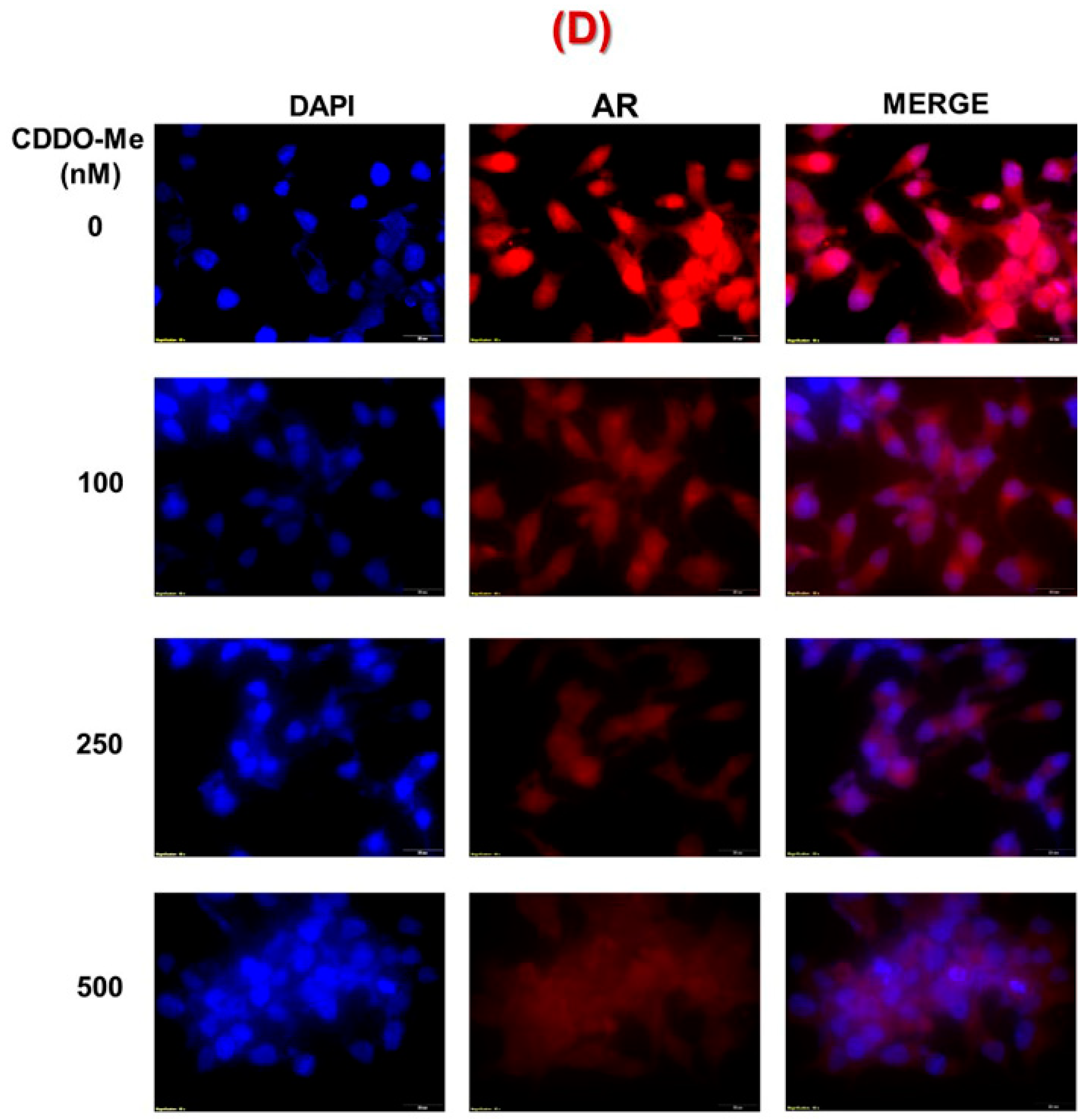
3.1. Exposure to Low-Dose CDDO-Me Decreases AR-FL and AR-V7 Protein Levels in PC Cells, in a Time- and Dose-Dependent Manner
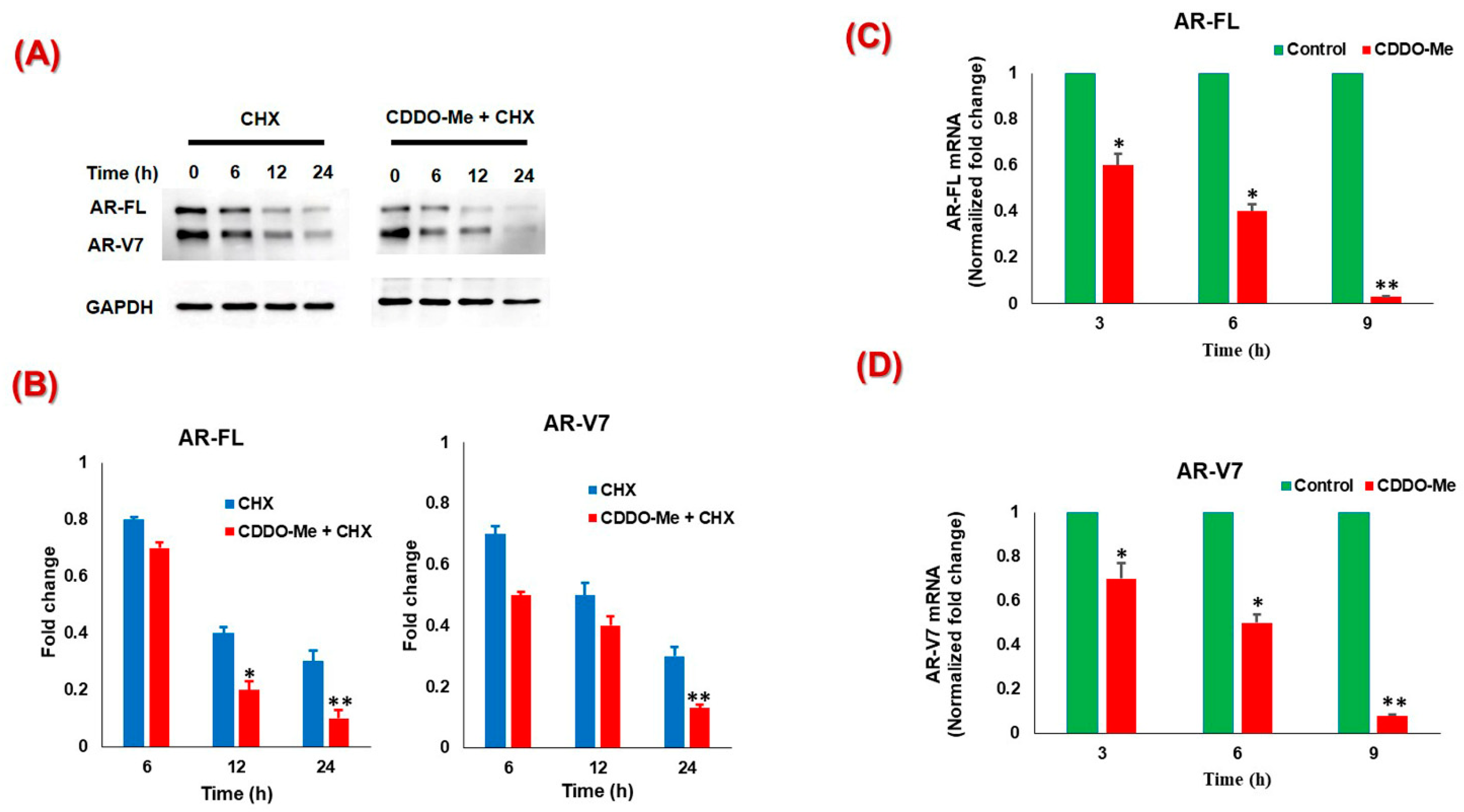
3.2. CDDO-Me-Mediated Suppression of AR-FL and AR-V7 is Regulated at both mRNA and Protein Levels
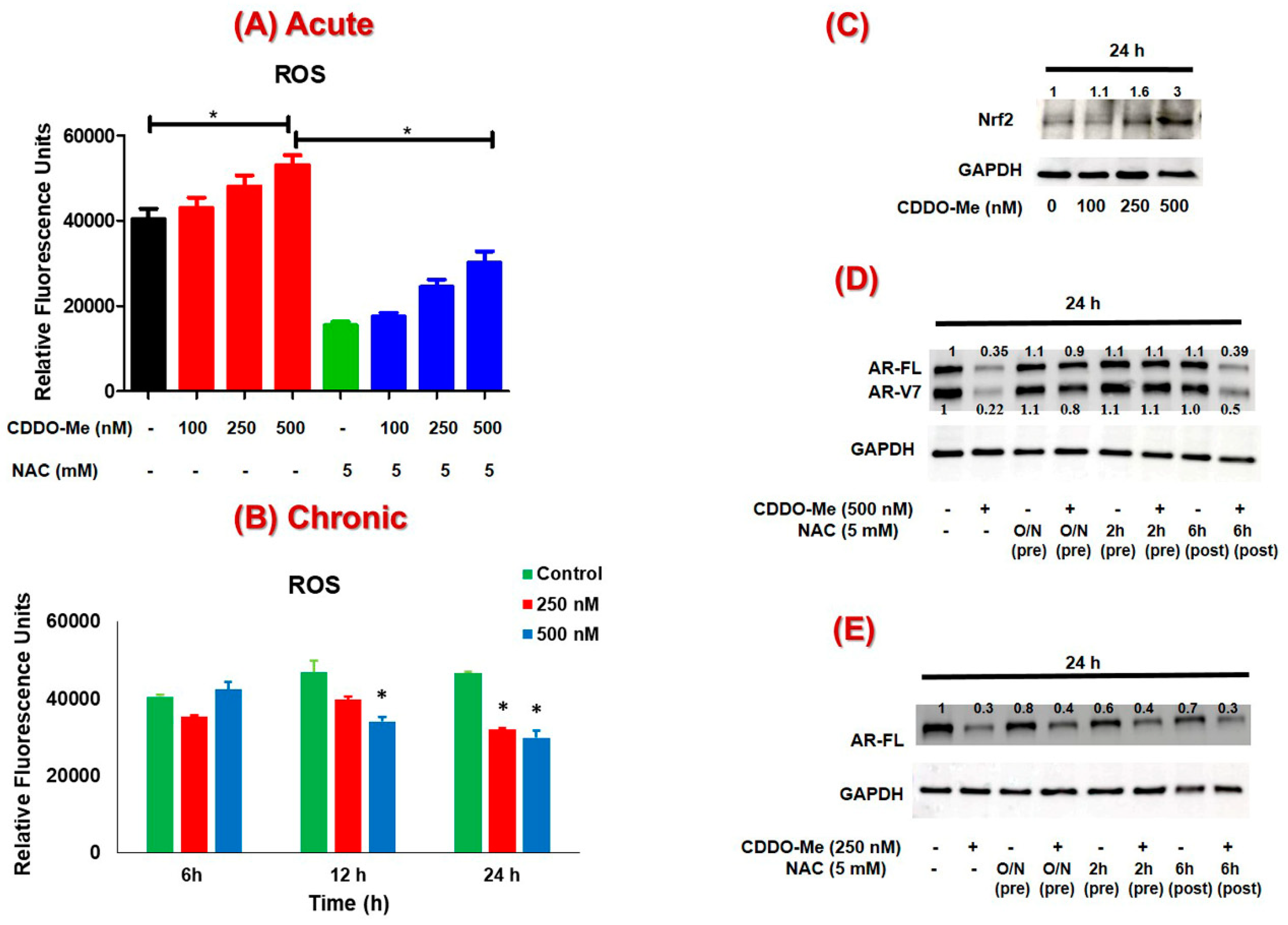
3.3. The Suppression of AR-FL and AR-V7 by CDDO-Me is Primarily Mediated via Oxidative Stress in both C4-2B and 22Rv1 Cells
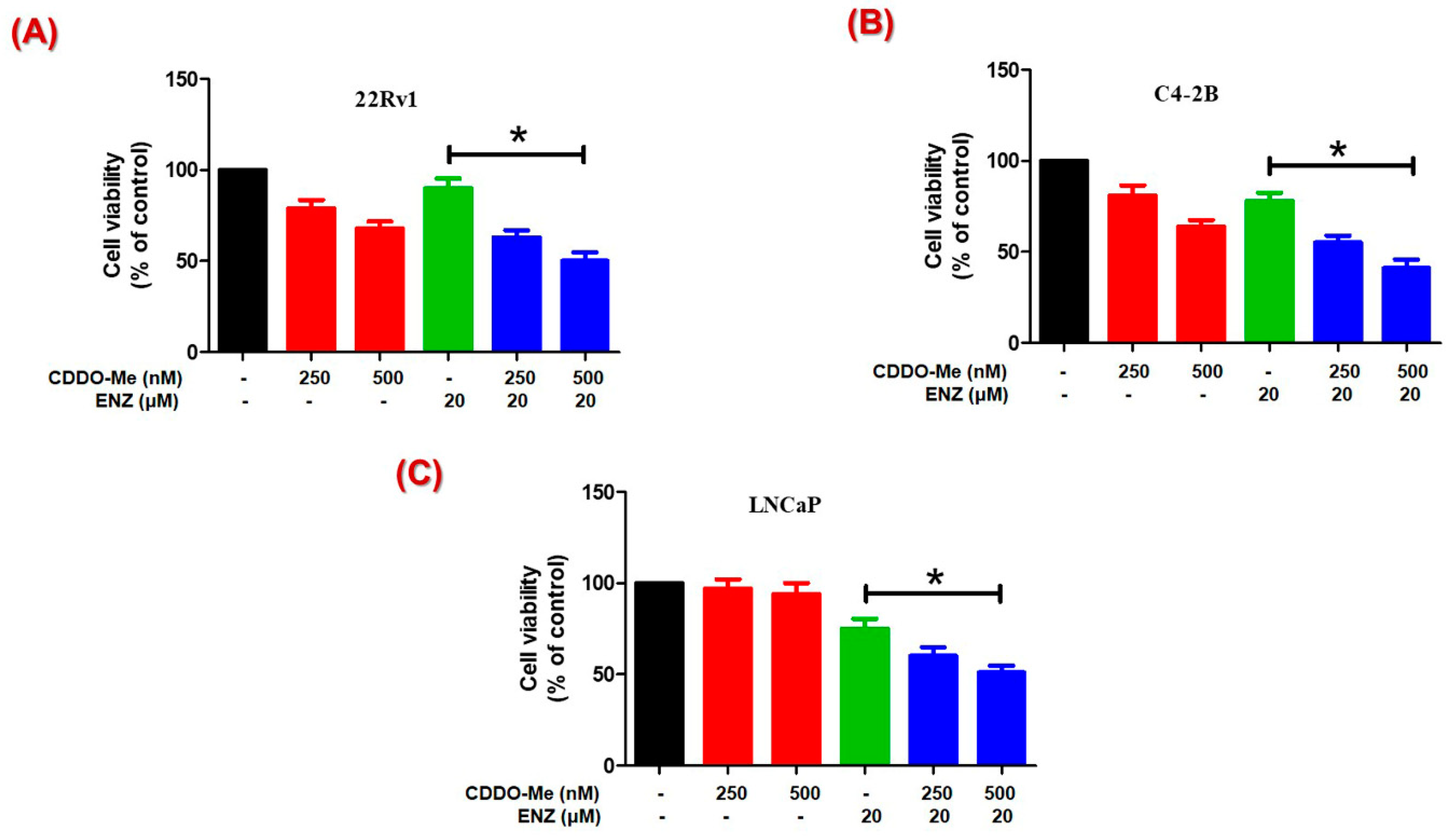
3.4. Co-Exposure to CDDO-Me Increases the Anticancer Efficacy of ENZ
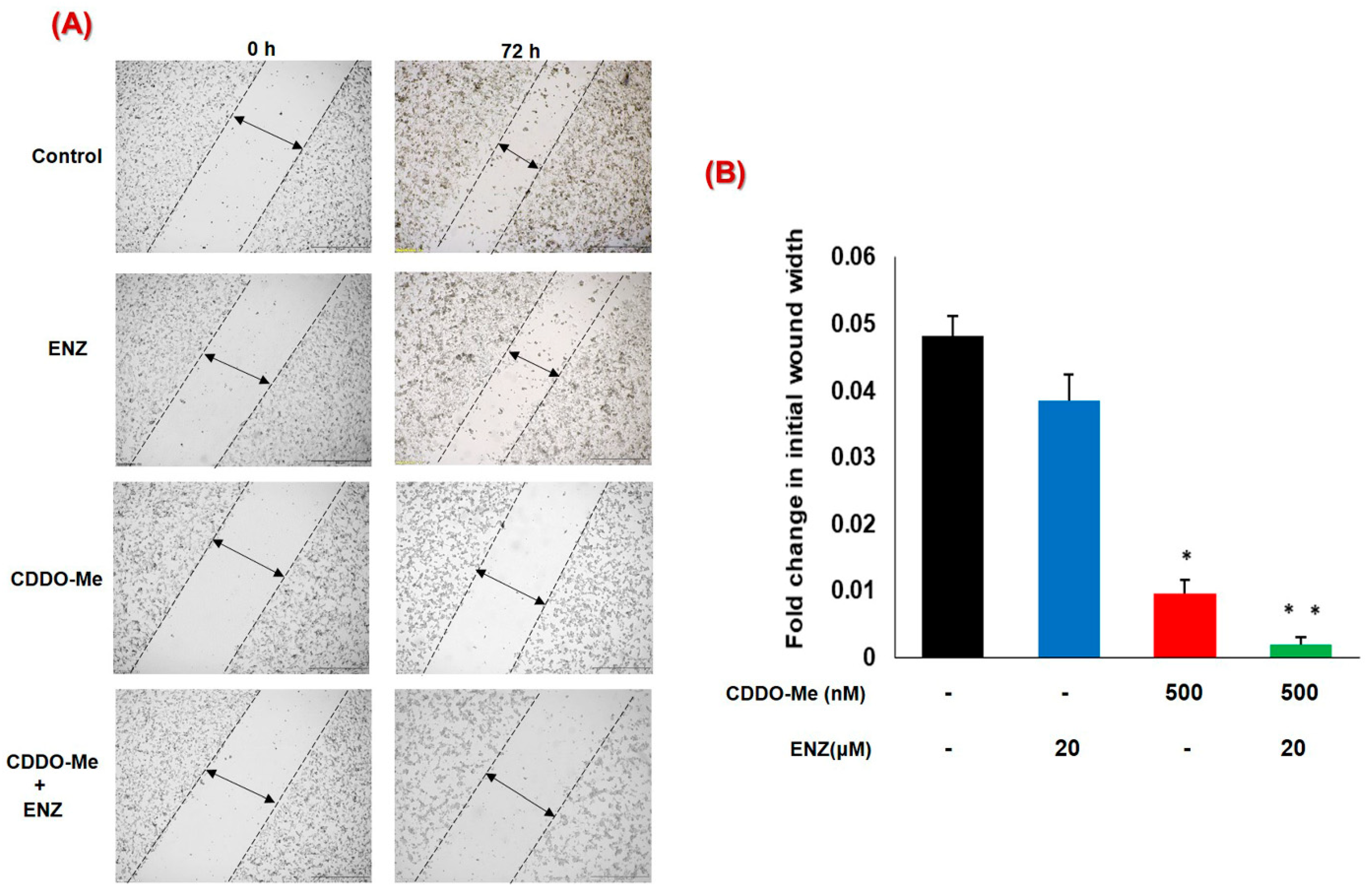
3.5. Co-Exposure to CDDO-Me and ENZ Abrogates the Migratory Potential of CRPC Cells
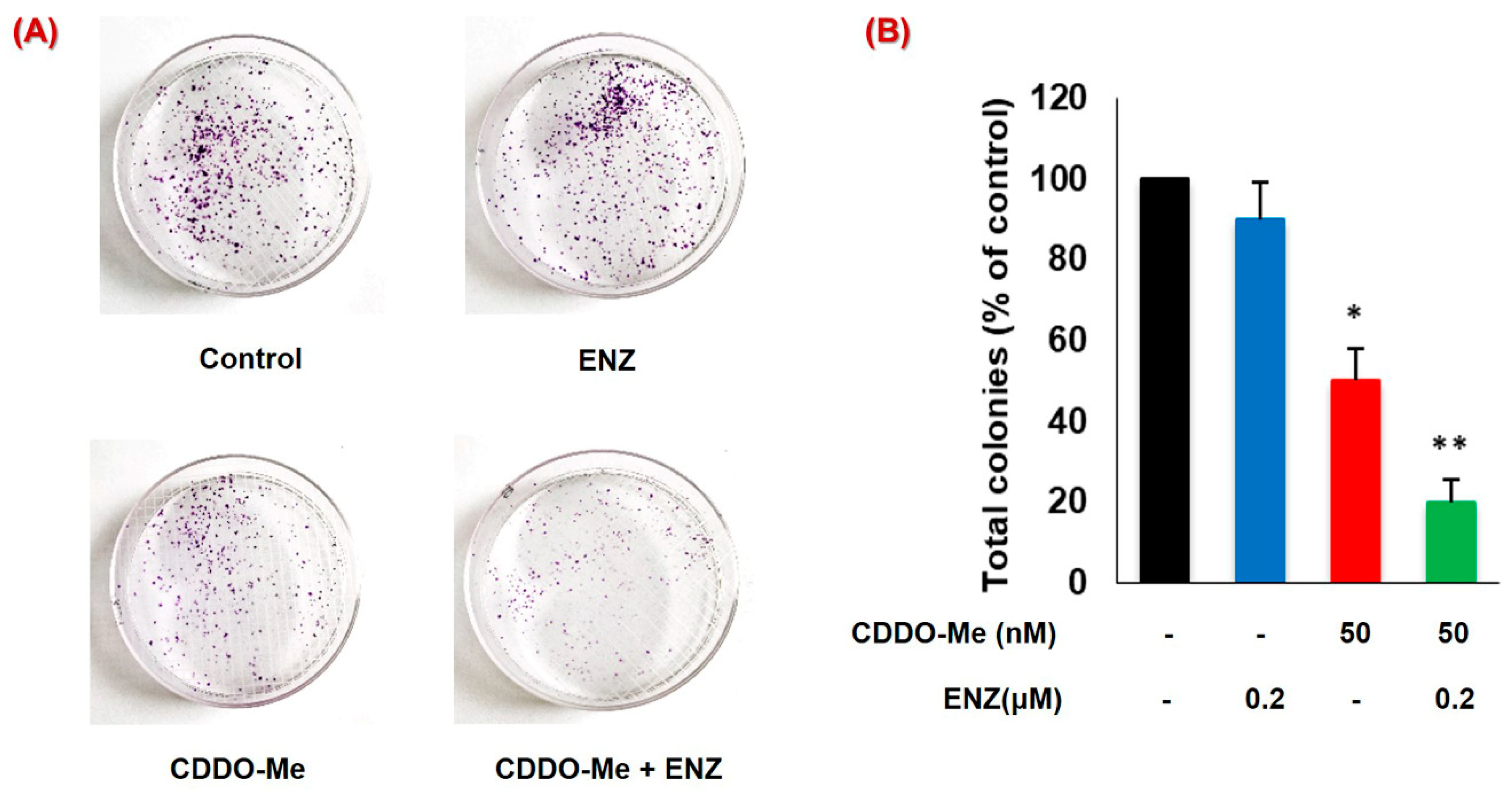
3.6. CDDO-Me Increases ENZ Efficacy by Inhibiting the Clonogenic Ability of PC Cells
4. Discussion
5. Conlusions
Author Contributions
Funding
Conflicts of Interest
References
- Yap, T.A.; Zivi, A.; Omlin, A.; de Bono, J.S. The changing therapeutic landscape of castration-resistant prostate cancer. Nat. Rev. Clin. Oncol. 2011, 8, 597–610. [Google Scholar] [CrossRef] [PubMed]
- Hodgson, M.C.; Bowden, W.A.; Agoulnik, I.U. Androgen receptor footprint on the way to prostate cancer progression. World, J. Urol. 2012, 30, 279–285. [Google Scholar] [CrossRef] [PubMed]
- Harris, W.P.; Mostaghel, E.A.; Nelson, P.S.; Montgomery, B. Androgen deprivation therapy: Progress in understanding mechanisms of resistance and optimizing androgen depletion. Nat. Clin. Pract. Urol. 2009, 6, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Ryan, C.J. Androgen Receptor Directed Therapies in Castration-Resistant Metastatic Prostate Cancer. Curr. Treat. Options Oncol. 2012, 13, 189–200. [Google Scholar] [CrossRef]
- Godbole, A.M.; Njar, V.C.O. New insights into the androgen-targeted therapies and epigenetic therapies in prostate cancer. Prostate Cancer 2011, 2011, 918707. [Google Scholar] [CrossRef]
- Scher, H.I.; Buchanan, G.; Gerald, W.; Butler, L.M.; Tilley, W.D. Targeting the androgen receptor: Improving outcomes for castration-resistant prostate cancer. Endocr. Relat. Cancer 2004, 11, 459–476. [Google Scholar] [CrossRef]
- Chang, K.H.; Ercole, C.E.; Sharifi, N. Androgen metabolism in prostate cancer: From molecular mechanisms to clinical consequences. Br. J. Cancer 2014, 111, 1249–1254. [Google Scholar] [CrossRef]
- Lamont, K.R.; Tindall, D.J. Minireview: Alternative activation pathways for the androgen receptor in prostate cancer. Mol. Endocrinol. 2011, 25, 897–907. [Google Scholar] [CrossRef]
- Brooke, G.; Bevan, C. The Role of Androgen Receptor Mutations in Prostate Cancer Progression. Curr. Genom. 2009, 10, 18–25. [Google Scholar] [CrossRef]
- Armstrong, C.M.; Gao, A.C. Drug resistance in castration resistant prostate cancer: Resistance mechanisms and emerging treatment strategies. Am. J. Clin. Exp. Urol. 2015, 3, 64–76. [Google Scholar]
- Guo, Z.; Yang, X.; Sun, F.; Jiang, R.; Linn, D.E.; Chen, H.; Chen, H.; Kong, X.; Melamed, J.; Tepper, C.G.; et al. A novel androgen receptor splice variant is up-regulated during prostate cancer progression and promotes androgen depletion-resistant growth. Cancer Res. 2009, 69, 2305–2313. [Google Scholar] [CrossRef] [PubMed]
- Dehm, S.M.; Tindall, D.J. Alternatively spliced androgen receptor variants. Endocr. Relat. Cancer 2011, 18, R183–R196. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Morrissey, C.; Sun, S.; Ketchandji, M.; Nelson, P.S.; True, L.D.; Vakar-Lopez, F.; Vessella, R.L.; Plymate, S.R. Androgen receptor variants occur frequently in castration resistant prostate cancer metastases. PLoS ONE 2011, 6. [Google Scholar] [CrossRef]
- Sun, S.; Sprenger, C.C.T.; Vessella, R.L.; Haugk, K.; Soriano, K.; Mostaghel, E.A.; Page, S.T.; Coleman, I.M.; Nguyen, H.M.; Sun, H.; et al. Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. J. Clin. Invest. 2010, 120, 2715–2730. [Google Scholar] [CrossRef] [PubMed]
- Hu, R.; Dunn, T.A.; Wei, S.; Isharwal, S.; Veltri, R.W.; Humphreys, E.; Han, M.; Partin, A.W.; Vessella, R.L.; Isaacs, W.B.; et al. Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Res. 2009, 69, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Dehm, S.M.; Schmidt, L.J.; Heemers, H.V.; Vessella, R.L.; Tindall, D.J. Splicing of a novel androgen receptor exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance. Cancer Res. 2008, 68, 5469–5477. [Google Scholar] [CrossRef] [PubMed]
- Del Re, M.; Biasco, E.; Crucitta, S.; Derosa, L.; Rofi, E.; Orlandini, C.; Miccoli, M.; Galli, L.; Falcone, A.; Jenster, G.W.; et al. The Detection of Androgen Receptor Splice Variant 7 in Plasma-derived Exosomal RNA Strongly Predicts Resistance to Hormonal Therapy in Metastatic Prostate Cancer Patients. Eur. Urol. 2017, 71, 680–687. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Armstrong, A.J.; Dehm, S.M.; Luo, J. Androgen receptor variant-driven prostate cancer: Clinical implications and therapeutic targeting. Prostate Cancer Prostatic Dis. 2016, 19, 231–241. [Google Scholar] [CrossRef]
- Lokhandwala, P.M.; Riel, S.L.; Haley, L.; Lu, C.; Chen, Y.; Silberstein, J.; Zhu, Y.; Zheng, G.; Lin, M.T.; Gocke, C.D.; et al. Analytical Validation of Androgen Receptor Splice Variant 7 Detection in a Clinical Laboratory Improvement Amendments (CLIA) Laboratory Setting. J. Mol. Diagnostics 2017, 19, 115–125. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Lu, C.; Wang, H.; Luber, B.; Nakazawa, M.; Roeser, J.C.; Chen, Y.; Mohammad, T.A.; Chen, Y.; Fedor, H.L.; et al. AR-V7 and Resistance to Enzalutamide and Abiraterone in Prostate Cancer. N. Engl. J. Med. 2014, 371, 1028–1038. [Google Scholar] [CrossRef]
- Sarwar, M.; Semenas, J.; Miftakhova, R.; Simoulis, A.; Robinson, B.; Gjörloff Wingren, A.; Mongan, N.P.; Heery, D.M.; Johnsson, H.; Abrahamsson, P.-A.; et al. Targeted suppression of AR-V7 using PIP5K1α inhibitor overcomes enzalutamide resistance in prostate cancer cells. Oncotarget 2016, 7, 63065–63081. [Google Scholar] [CrossRef] [PubMed]
- Watson, P.A.; Chen, Y.F.; Balbas, M.D.; Wongvipat, J.; Socci, N.D.; Viale, A.; Kim, K.; Sawyers, C.L. Constitutively active androgen receptor splice variants expressed in castration-resistant prostate cancer require full-length androgen receptor. Proc. Natl. Acad. Sci. USA 2010, 107, 16759–16765. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Zhan, Y.; Qi, Y.; Cao, B.; Bai, S.; Xu, W.; Gambhir, S.S.; Lee, P.; Sartor, O.; Flemington, E.K.; et al. Androgen receptor splice variants dimerize to transactivate target genes. Cancer Res. 2015, 75, 3663–3671. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Y.; Yang, Y.X.; Zhe, H.; He, Z.X.; Zhou, S.F. Bardoxolone methyl (CDDO-Me) as a therapeutic agent: An update on its pharmacokinetic and pharmacodynamic properties. Drug Des. Devel. Ther. 2014, 8, 2075–2088. [Google Scholar] [CrossRef]
- Wang, Y.Y.; Zhe, H.; Zhao, R. Preclinical evidences toward the use of triterpenoid CDDO-Me for solid cancer prevention and treatment. Mol. Cancer 2014, 13, 30. [Google Scholar] [CrossRef]
- Deeb, D.; Gao, X.; Jiang, H.; Dulchavsky, S.A.; Gautam, S.C. Oleanane triterpenoid CDDO-Me inhibits growth and induces apoptosis in prostate cancer cells by independently targeting pro-survival Akt and mTOR. Prostate 2009, 69, 851–860. [Google Scholar] [CrossRef]
- Kim, E.H.; Deng, C.; Sporn, M.B.; Royce, D.B.; Risingsong, R.; Williams, C.R.; Liby, K.T. CDDO-methyl ester delays breast cancer development in Brca1-mutated mice. Cancer Prev. Res. 2012, 5, 89–97. [Google Scholar] [CrossRef]
- Gao, X.; Liu, Y.; Deeb, D.; Arbab, A.S.; Guo, A.M.; Dulchavsky, S.A.; Gautam, S.C. Synthetic oleanane triterpenoid, CDDO-Me, induces apoptosis in ovarian cancer cells by inhibiting prosurvival AKT/NF-κB/mTOR signaling. Anticancer Res. 2011, 31, 3673–3681. [Google Scholar]
- Liby, K.; Royce, D.B.; Williams, C.R.; Risingsong, R.; Yore, M.M.; Honda, T.; Gribble, G.W.; Dmitrovsky, E.; Sporn, T.A.; Sporn, M.B. The synthetic triterpenoids CDDO-methyl ester and CDDO-ethyl amide prevent lung cancer induced by vinyl carbamate in A/J mice. Cancer Res. 2007, 67, 2414–2419. [Google Scholar] [CrossRef]
- Konopleva, M.; Tsao, T.; Ruvolo, P.; Stiouf, I.; Estrov, Z.; Leysath, C.E.; Zhao, S.; Harris, D.; Chang, S.; Jackson, C.E.; et al. Novel triterpenoid CDDO-Me is a potent inducer of apoptosis and differentiation in acute myelogenous leukemia. Blood 2002, 99, 326–335. [Google Scholar] [CrossRef]
- Liby, K.T.; Royce, D.B.; Risingsong, R.; Williams, C.R.; Maitra, A.; Hruban, R.H.; Sporn, M.B. Synthetic triterpenoids prolong survival in a transgenic mouse model of pancreatic cancer. Cancer Prev. Res. 2010, 3, 1427–1434. [Google Scholar] [CrossRef] [PubMed]
- Ryu, K.; Susa, M.; Choy, E.; Yang, C.; Hornicek, F.J.; Mankin, H.J.; Duan, Z. Oleanane triterpenoid CDDO-Me induces apoptosis in multidrug resistant osteosarcoma cells through inhibition of Stat3 pathway. BMC Cancer 2010, 10. [Google Scholar] [CrossRef] [PubMed]
- Dinkova-Kostova, A.T.; Liby, K.T.; Stephenson, K.K.; Holtzclaw, W.D.; Gao, X.; Suh, N.; Williams, C.; Risingsong, R.; Honda, T.; Gribble, G.W.; et al. Extremely potent triterpenoid inducers of the phase 2 response: Correlations of protection against oxidant and inflammatory stress. Proc. Natl. Acad. Sci. USA 2005, 102, 4584–4589. [Google Scholar] [CrossRef] [PubMed]
- Yates, M.S.; Tauchi, M.; Katsuoka, F.; Flanders, K.C.; Liby, K.T.; Honda, T.; Gribble, G.W.; Johnson, D.A.; Johnson, J.A.; Burton, N.C.; et al. Pharmacodynamic characterization of chemopreventive triterpenoids as exceptionally potent inducers of Nrf2-regulated genes. Mol. Cancer Ther. 2007, 6, 154–162. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, R.; Raina, D.; Meyer, C.; Kharbanda, S.; Kufe, D. Triterpenoid CDDO-Me blocks the NF-κB pathway by direct inhibition of IKKβ on Cys-179. J. Biol. Chem. 2006, 281, 35764–35769. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, R.; Raina, D.; Meyer, C.; Kufe, D. Triterpenoid CDDO-methyl ester inhibits the Janus-activated kinase-1 (JAK1)→signal transducer and activator of transcription-3 (STAT3) pathway by direct inhibition of JAK1 and STAT3. Cancer Res. 2008, 68, 2920–2926. [Google Scholar] [CrossRef]
- Hyer, M.L.; Shi, R.; Krajewska, M.; Meyer, C.; Lebedeva, I.V.; Fisher, P.B.; Reed, J.C. Apoptotic activity and mechanism of 2-cyano-3,12-dioxoolean-1,9-dien-28- oic-acid and related synthetic triterpenoids in prostate cancer. Cancer Res. 2008, 68, 2927–2933. [Google Scholar] [CrossRef]
- Ikeda, T.; Sporn, M.; Honda, T.; Gribble, G.W.; Kufe, D. The novel triterpenoid CDDO and its derivatives induce apoptosis by disruption of intracellular redox balance. Cancer Res. 2003, 63, 5551–5558. [Google Scholar]
- Chintharlapalli, S.; Papineni, S.; Konopleva, M.; Andreef, M.; Samudio, I.; Safe, S. 2-Cyano-3,12-dioxoolean-1,9-dien-28-oic acid and related compounds inhibit growth of colon cancer cells through peroxisome proliferator-activated receptor γ-dependent and -independent pathways. Mol. Pharmacol. 2005, 68, 119–128. [Google Scholar] [CrossRef]
- Deeb, D.; Gao, X.; Liu, Y.; Jiang, D.; Divine, G.W.; Arbab, A.S.; Dulchavsky, S.A.; Gautam, S.C. Synthetic triterpenoid CDDO prevents the progression and metastasis of prostate cancer in TRAMP mice by inhibiting survival signaling. Carcinogenesis 2011, 32, 757–764. [Google Scholar] [CrossRef]
- Gao, X.; Deeb, D.; Liu, Y.; Arbab, A.S.; Divine, G.W.; Dulchavsky, S.A.; Gautam, S.C. Prevention of prostate cancer with oleanane synthetic triterpenoid CDDO-Me in the TRAMP mouse model of prostate cancer. Cancers 2011, 3, 3353–3369. [Google Scholar] [CrossRef]
- Hong, D.S.; Kurzrock, R.; Supko, J.G.; He, X.; Naing, A.; Wheler, J.; Lawrence, D.; Eder, J.P.; Meyer, C.J.; Ferguson, D.A.; et al. A phase I first-in-human trial of bardoxolone methyl in patients with advanced solid tumors and lymphomas. Clin. Cancer Res. 2012, 18, 3396–3406. [Google Scholar] [CrossRef] [PubMed]
- Deeb, D.; Gao, X.; Dulchavsky, S.A.; Gautam, S.C. CDDO-me induces apoptosis and inhibits Akt, mTOR and NF-kappaB signaling proteins in prostate cancer cells. Anticancer Res. 2007, 27, 3035–3044. [Google Scholar] [PubMed]
- Liu, Y.; Gao, X.; Deeb, D.; Arbab, A.S.; Gautam, S.C. Telomerase reverse transcriptase (TERT) is a therapeutic target of oleanane triterpenoid cddo-me in prostate cancer. Molecules 2012, 17, 14795–14809. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.-C.; Hsieh, J.-T.; Gleave, M.E.; Brown, N.M.; Pathak, S.; Chung, L.W.K. Derivation of androgen-independent human LNCaP prostatic cancer cell sublines: Role of bone stromal cells. Int. J. Cancer 1994, 57, 406–412. [Google Scholar] [CrossRef]
- Uygur, B.; Wu, W.S. SLUG promotes prostate cancer cell migration and invasion via CXCR4/CXCL12 axis. Mol. Cancer 2011, 10, 139. [Google Scholar] [CrossRef]
- Chou, T.C. Drug combination studies and their synergy quantification using the chou-talalay method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef]
- Shiota, M.; Yokomizo, A.; Naito, S. Oxidative stress and androgen receptor signaling in the development and progression of castration-resistant prostate cancer. Free Radic. Biol. Med. 2011, 51, 1320–1328. [Google Scholar] [CrossRef]
- Schultz, M.A.; Abdel-Mageed, A.B.; Mondal, D. The NrF1 and NrF2 balance in oxidative stress regulation and androgen signaling in prostate cancer cells. Cancers 2010, 2, 1354–1378. [Google Scholar] [CrossRef]
- Jutooru, I.; Guthrie, A.S.; Chadalapaka, G.; Pathi, S.; Kim, K.; Burghardt, R.; Jin, U.-H.; Safe, S. Mechanism of Action of Phenethylisothiocyanate and Other Reactive Oxygen Species-Inducing Anticancer Agents. Mol. Cell. Biol. 2014, 34, 2382–2395. [Google Scholar] [CrossRef]
- Jin, U.H.; Cheng, Y.; Zhou, B.; Safe, S. Bardoxolone methyl and a related triterpenoid downregulate cMyc expression in leukemia cells. Mol. Pharmacol. 2017, 91, 438–450. [Google Scholar] [CrossRef] [PubMed]
- Matuszak, E.A.; Kyprianou, N. Androgen regulation of epithelial-mesenchymal transition in prostate tumorigenesis. Expert Rev. Endocrinol. Metab. 2011, 6, 469–482. [Google Scholar] [CrossRef] [PubMed]
- Nomura, Y.; Tashiro, H.; Hisamatsu, K. In Vitro Clonogenic Growth and Metastatic Potential of Human Operable Breast Cancer. Cancer Res. 1989, 49, 5288–5293. [Google Scholar] [PubMed]
- Khurana, N.; Sikka, S. Targeting Crosstalk between Nrf-2, NF-κB and Androgen Receptor Signaling in Prostate Cancer. Cancers 2018, 10, 352. [Google Scholar] [CrossRef] [PubMed]
- Khurana, N.; Sikka, S.C. Interplay Between SOX9, Wnt/β-Catenin and Androgen Receptor Signaling in Castration-Resistant Prostate Cancer. Int. J. Mol. Sci. 2019, 20, 2066. [Google Scholar] [CrossRef] [PubMed]
- Khurana, N.; Talwar, S.; Chandra, P.K.; Sharma, P.; Abdel-Mageed, A.B.; Mondal, D.; Sikka, S.C. Sulforaphane increases the efficacy of anti-androgens by rapidly decreasing androgen receptor levels in prostate cancer cells. Int. J. Oncol. 2016, 49, 1609–1619. [Google Scholar] [CrossRef]
- Khurana, N.; Kim, H.; Chandra, P.K.; Talwar, S.; Sharma, P.; Abdel-Mageed, A.B.; Sikka, S.C.; Mondal, D. Multimodal actions of the phytochemical sulforaphane suppress both AR and AR-V7 in 22Rv1 cells: Advocating a potent pharmaceutical combination against castration-resistant prostate cancer. Oncol. Rep. 2017, 38, 2774–2786. [Google Scholar] [CrossRef]
- Horoszewicz, J.S.; Leong, S.S.; Chu, T.M.; Wajsman, Z.L.; Friedman, M.; Papsidero, L.; Kim, U.; Chai, L.S.; Kakati, S.; Arya, S.K.; et al. The LNCaP cell line--a new model for studies on human prostatic carcinoma. Prog. Clin. Biol. Res. 1980, 37, 115–132. [Google Scholar]
- Cunningham, D.; You, Z. In vitro and in vivo model systems used in prostate cancer research. J. Biol. Methods 2015, 2, 17. [Google Scholar] [CrossRef]
- Sporn, M.B.; Liby, K.T.; Yore, M.M.; Fu, L.; Lopchuk, J.M.; Gribble, G.W. New synthetic triterpenoids: Potent agents for prevention and treatment of tissue injury caused by inflammatory and oxidative stress. J. Nat. Prod. 2011, 74, 537–545. [Google Scholar] [CrossRef]
- Yore, M.M.; Kettenbach, A.N.; Sporn, M.B.; Gerber, S.A.; Liby, K.T. Proteomic analysis shows synthetic oleanane triterpenoid binds to mTOR. PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.A.; Kim, I.Y.; Lee, A.R.; Yoon, M.J.; Cho, H.; Lee, J.S.; Choi, K.S. Ca 2+ influx-mediated dilation of the endoplasmic reticulum and c-FLIP L downregulation trigger CDDO-Me-induced apoptosis in breast cancer cells. Oncotarget 2015, 6, 21173–21192. [Google Scholar] [CrossRef] [PubMed]
- Qin, D.-J.; Tang, C.-X.; Yang, L.; Lei, H.; Wei, W.; Wang, Y.-Y.; Ma, C.-M.; Gao, F.-H.; Xu, H.-Z.; Wu, Y.-L. Hsp90 Is a Novel Target Molecule of CDDO-Me in Inhibiting Proliferation of Ovarian Cancer Cells. PLoS ONE 2015, 10, e0132337. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.E.; Li, J.; Xu, H.E.; Melcher, K.; Yong, E.L. Androgen receptor: Structure, role in prostate cancer and drug discovery. Acta Pharmacol. Sin. 2015, 36, 3–23. [Google Scholar] [CrossRef] [PubMed]
- Schultz, M.A.; Hagan, S.S.; Datta, A.; Zhang, Y.; Freeman, M.L.; Sikka, S.C.; Abdel-Mageed, A.B.; Mondal, D. Nrf1 and Nrf2 Transcription Factors Regulate Androgen Receptor Transactivation in Prostate Cancer Cells. PLoS ONE 2014, 9, e87204. [Google Scholar] [CrossRef]
- Park, H.S.; Han, M.H.; Kim, G.-Y.; Moon, S.-K.; Kim, W.-J.; Hwang, H.J.; Park, K.Y.; Choi, Y.H. Sulforaphane induces reactive oxygen species-mediated mitotic arrest and subsequent apoptosis in human bladder cancer 5637 cells. Food Chem. Toxicol. 2014, 64, 157–165. [Google Scholar] [CrossRef]
- Kocyigit, A.; Guler, E.M. Curcumin induce DNA damage and apoptosis through generation of reactive oxygen species and reducing mitochondrial membrane potential in melanoma cancer cells. Cell. Mol. Biol. (Noisy-le-grand). 2017, 63, 97–105. [Google Scholar] [CrossRef]
- Gersey, Z.C.; Rodriguez, G.A.; Barbarite, E.; Sanchez, A.; Walters, W.M.; Ohaeto, K.C.; Komotar, R.J.; Graham, R.M. Curcumin decreases malignant characteristics of glioblastoma stem cells via induction of reactive oxygen species. BMC Cancer 2017, 17, 99. [Google Scholar] [CrossRef] [PubMed]
- Choi, W.Y.; Choi, B.T.; Lee, W.H.; Choi, Y.H. Sulforaphane generates reactive oxygen species leading to mitochondrial perturbation for apoptosis in human leukemia U937 cells. Biomed. Pharmacother. 2008, 62, 637–644. [Google Scholar] [CrossRef]
- Helness, A.; Gaudreau, M.-C.; Grapton, D.; Vadnais, C.; Fraszczak, J.; Wilhelm, B.; Robert, F.; Heyd, F.; Moroy, T. Loss of heterogeneous nuclear ribonucleoprotein L (HNRNP L) leads to mitochondrial dysfunction, DNA damage response and caspase-dependent cell death in hematopoietic stem cells. Exp. Hematol. 2016, 44, S78–S79. [Google Scholar] [CrossRef]
- Takayama, K.I. Splicing factors have an essential role in prostate cancer progression and androgen receptor signaling. Biomolecules 2019, 9, 131. [Google Scholar] [CrossRef] [PubMed]
- Huo, C.; Kao, Y.H.; Chuu, C.P. Androgen receptor inhibits epithelial-mesenchymal transition, migration, and invasion of PC-3 prostate cancer cells. Cancer Lett. 2015, 369, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Demir, A.; Cecen, K.; Karadag, M.A.; Kocaaslan, R.; Turkeri, L. The course of metastatic prostate cancer under treatment. Springerplus 2014, 3, 725. [Google Scholar] [CrossRef] [PubMed]
- Aapro, M.S.; Eliason, J.F.; Krauer, F.; Alberto, P. Colony formation in vitro as a prognostic indicator for primary breast cancer. J. Clin. Oncol. 1987, 5, 890–896. [Google Scholar] [CrossRef]
- Sekhar, K.R.; Wang, J.; Freeman, M.L.; Kirschner, A.N. Radiosensitization by enzalutamide for human prostate cancer is mediated through the DNA damage repair pathway. PLoS ONE 2019, 14. [Google Scholar] [CrossRef]
- Ardiani, A.; Farsaci, B.; Rogers, C.J.; Protter, A.; Guo, Z.; King, T.H.; Apelian, D.; Hodge, J.W. Combination therapy with a second-generation androgen receptor antagonist and a metastasis vaccine improves survival in a spontaneous prostate cancer model. Clin. Cancer Res. 2013, 19, 6205–6218. [Google Scholar] [CrossRef]
- Luk, I.S.U.; Shrestha, R.; Xue, H.; Wang, Y.; Zhang, F.; Lin, D.; Haegert, A.; Wu, R.; Dong, X.; Collins, C.C.; et al. BIRC6 Targeting as Potential Therapy for Advanced, Enzalutamide-Resistant Prostate Cancer. Clin. Cancer Res. 2017, 23, 1542–1551. [Google Scholar] [CrossRef]
- Cheng, J.; Moore, S.; Gomez-Galeno, J.; Lee, D.-H.; Okolotowicz, K.J.; Cashman, J.R. A Novel Small Molecule Inhibits Tumor Growth and Synergizes Effects of Enzalutamide on Prostate Cancer. J. Pharmacol. Exp. Ther. 2019, 371, 703–712. [Google Scholar] [CrossRef]
- Toren, P.; Kim, S.; Johnson, F.; Zoubeidi, A. Combined AKT and MEK Pathway Blockade in Pre-Clinical Models of Enzalutamide-Resistant Prostate Cancer. PLoS ONE 2016, 11, e0152861. [Google Scholar] [CrossRef]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khurana, N.; Chandra, P.K.; Kim, H.; Abdel-Mageed, A.B.; Mondal, D.; Sikka, S.C. Bardoxolone-Methyl (CDDO-Me) Suppresses Androgen Receptor and Its Splice-Variant AR-V7 and Enhances Efficacy of Enzalutamide in Prostate Cancer Cells. Antioxidants 2020, 9, 68. https://doi.org/10.3390/antiox9010068
Khurana N, Chandra PK, Kim H, Abdel-Mageed AB, Mondal D, Sikka SC. Bardoxolone-Methyl (CDDO-Me) Suppresses Androgen Receptor and Its Splice-Variant AR-V7 and Enhances Efficacy of Enzalutamide in Prostate Cancer Cells. Antioxidants. 2020; 9(1):68. https://doi.org/10.3390/antiox9010068
Chicago/Turabian StyleKhurana, Namrata, Partha K. Chandra, Hogyoung Kim, Asim B. Abdel-Mageed, Debasis Mondal, and Suresh C. Sikka. 2020. "Bardoxolone-Methyl (CDDO-Me) Suppresses Androgen Receptor and Its Splice-Variant AR-V7 and Enhances Efficacy of Enzalutamide in Prostate Cancer Cells" Antioxidants 9, no. 1: 68. https://doi.org/10.3390/antiox9010068
APA StyleKhurana, N., Chandra, P. K., Kim, H., Abdel-Mageed, A. B., Mondal, D., & Sikka, S. C. (2020). Bardoxolone-Methyl (CDDO-Me) Suppresses Androgen Receptor and Its Splice-Variant AR-V7 and Enhances Efficacy of Enzalutamide in Prostate Cancer Cells. Antioxidants, 9(1), 68. https://doi.org/10.3390/antiox9010068