Inhibition of LPS-Induced Oxidative Damages and Potential Anti-Inflammatory Effects of Phyllanthus emblica Extract via Down-Regulating NF-κB, COX-2, and iNOS in RAW 264.7 Cells

 ,
,  and
and
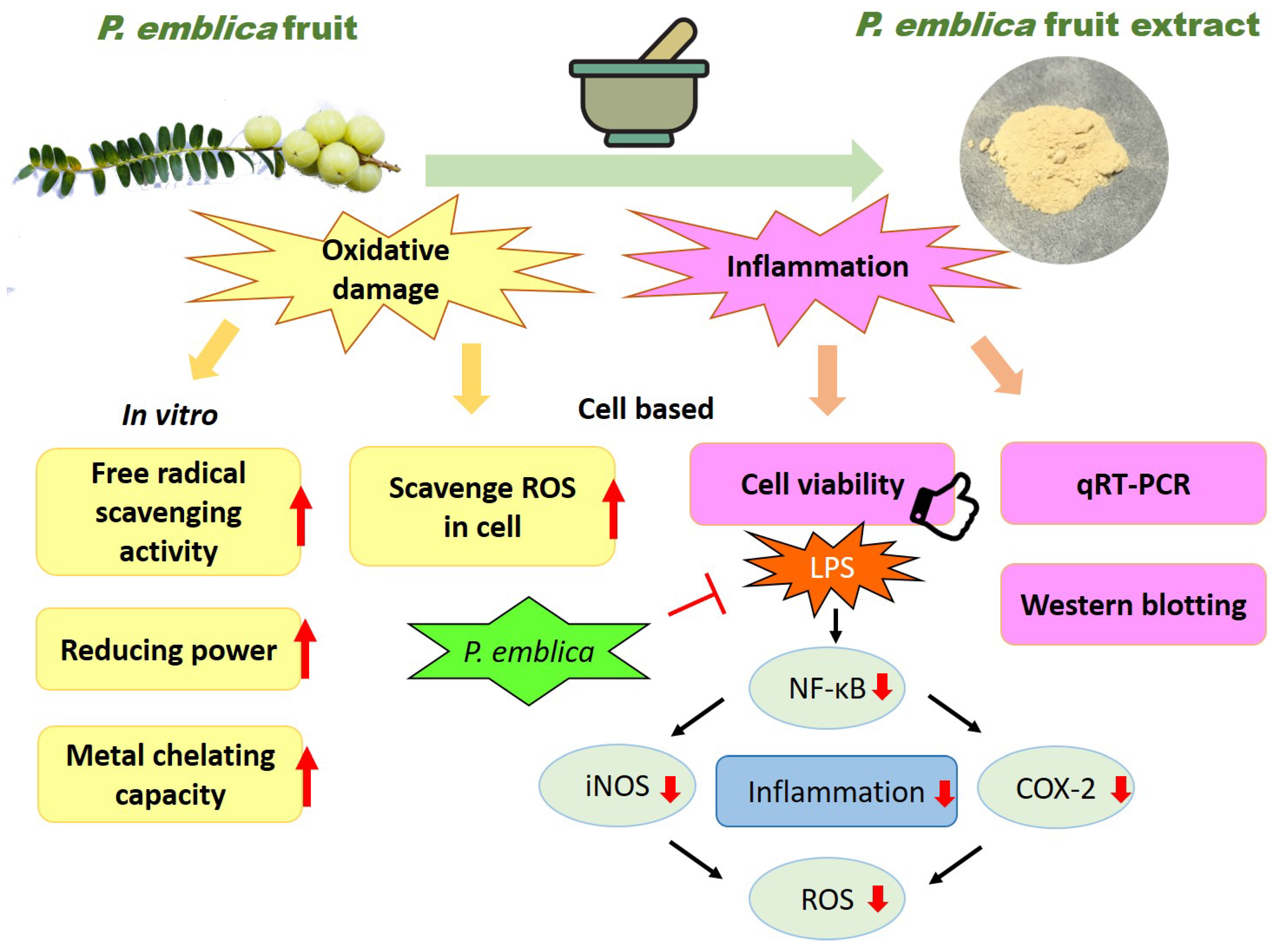
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. P. emblica Fruit Powder Extracts Preparation
2.3. Free Radical Scavenging Activity
2.4. Ferric Reducing Antioxidant Power (FRAP) Assay
2.5. Metal Chelating Ability Test
2.6. Cell Culture and Treatment
2.7. Cell Viability Assay
2.8. Measurement of Intracellular ROS Level
2.9. Quantitative Reverse Transcription Polymerase Chain Reaction (qRT-PCR)
2.10. Western Blotting
2.11. Statistical Analysis
3. Results
3.1. Antioxidant Activity of P. emblica Fruit Extracts Powder
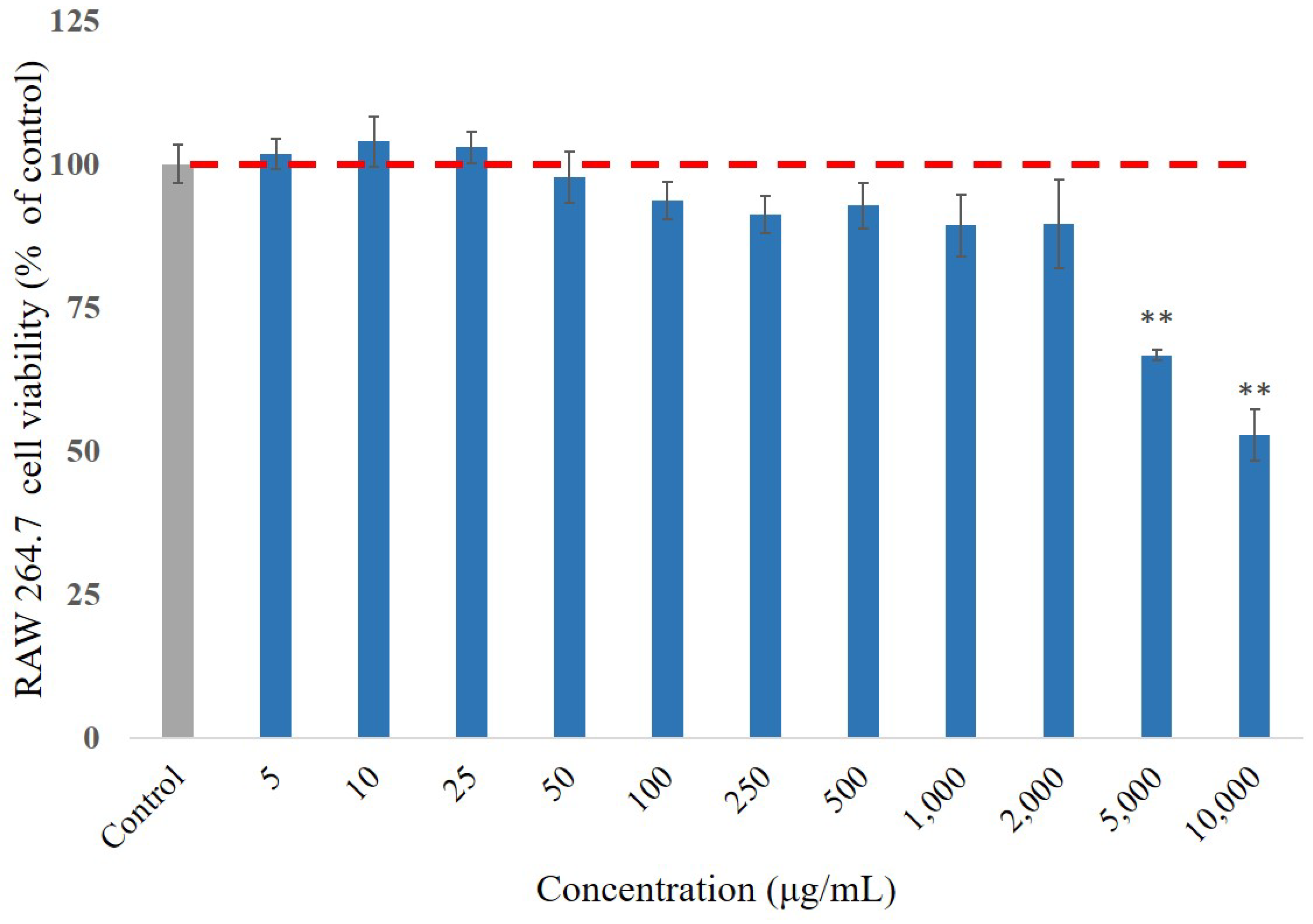
3.2. Cell Viability Effect of P. emblica Fruit Extract Treatment
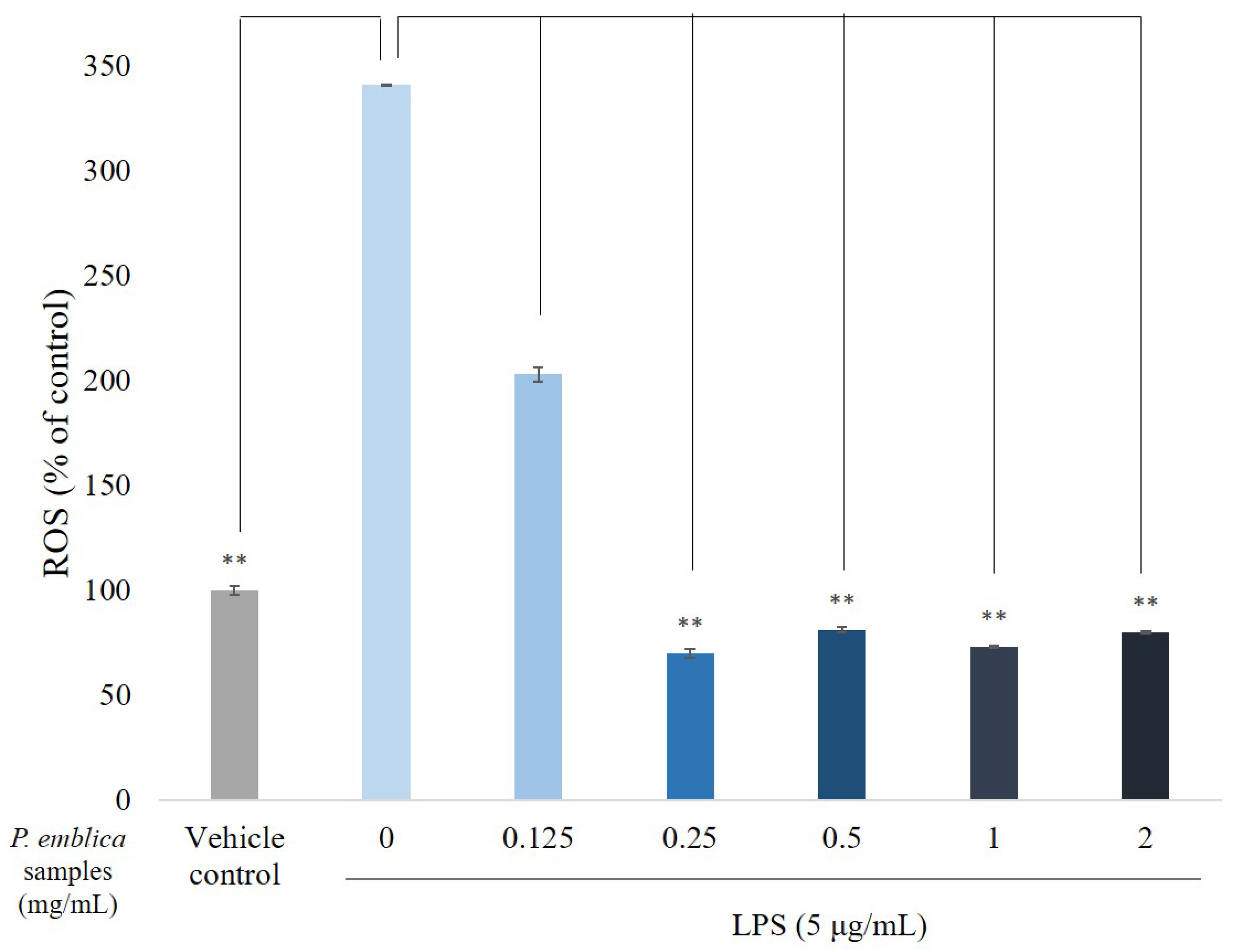
3.3. ROS Scavenge by P. emblica Fruit Extract Powder
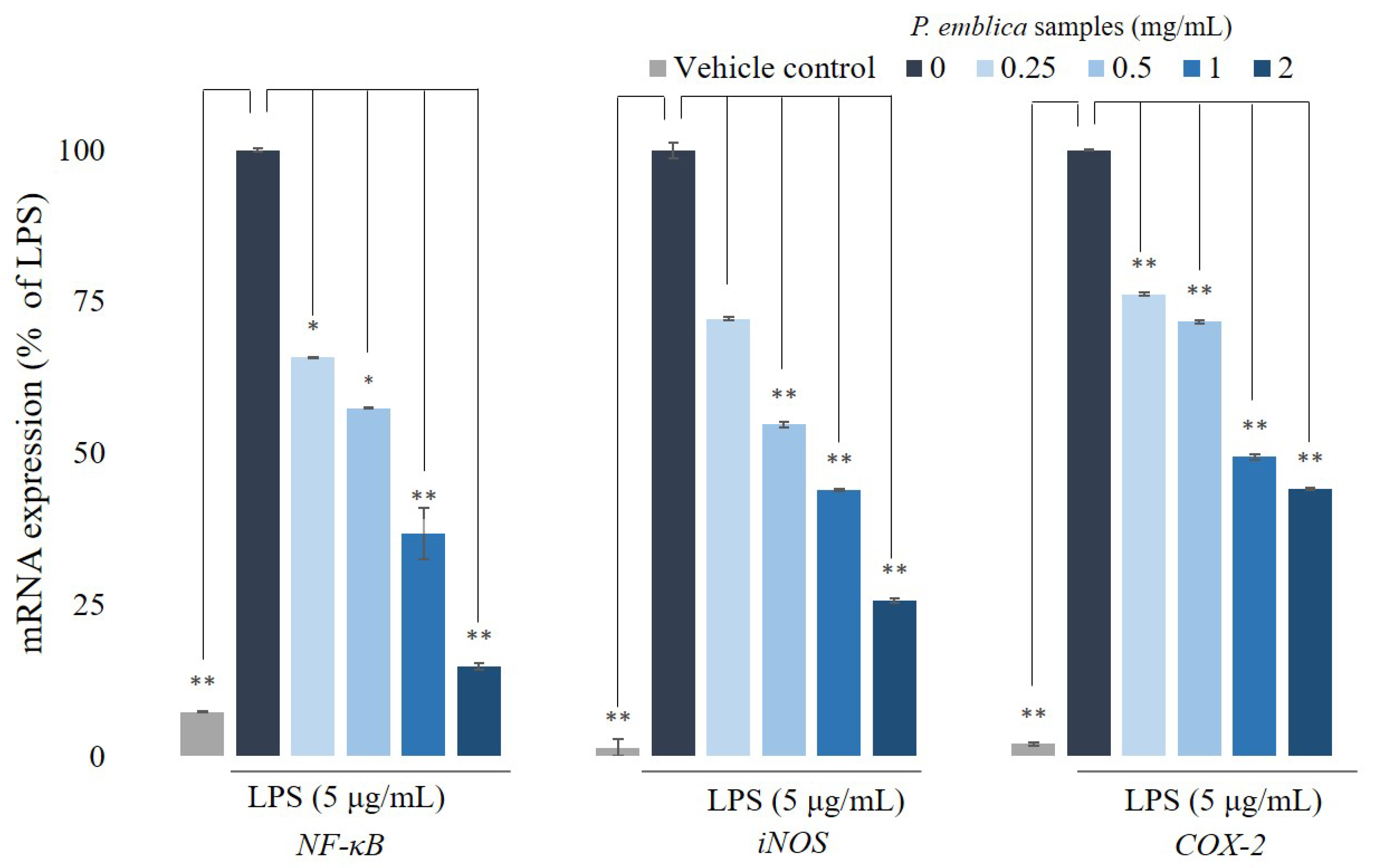
3.4. Quantitative Reverse Transcription Polymerase Chain Reaction Analysis for NF-κB, iNOS, and COX-2
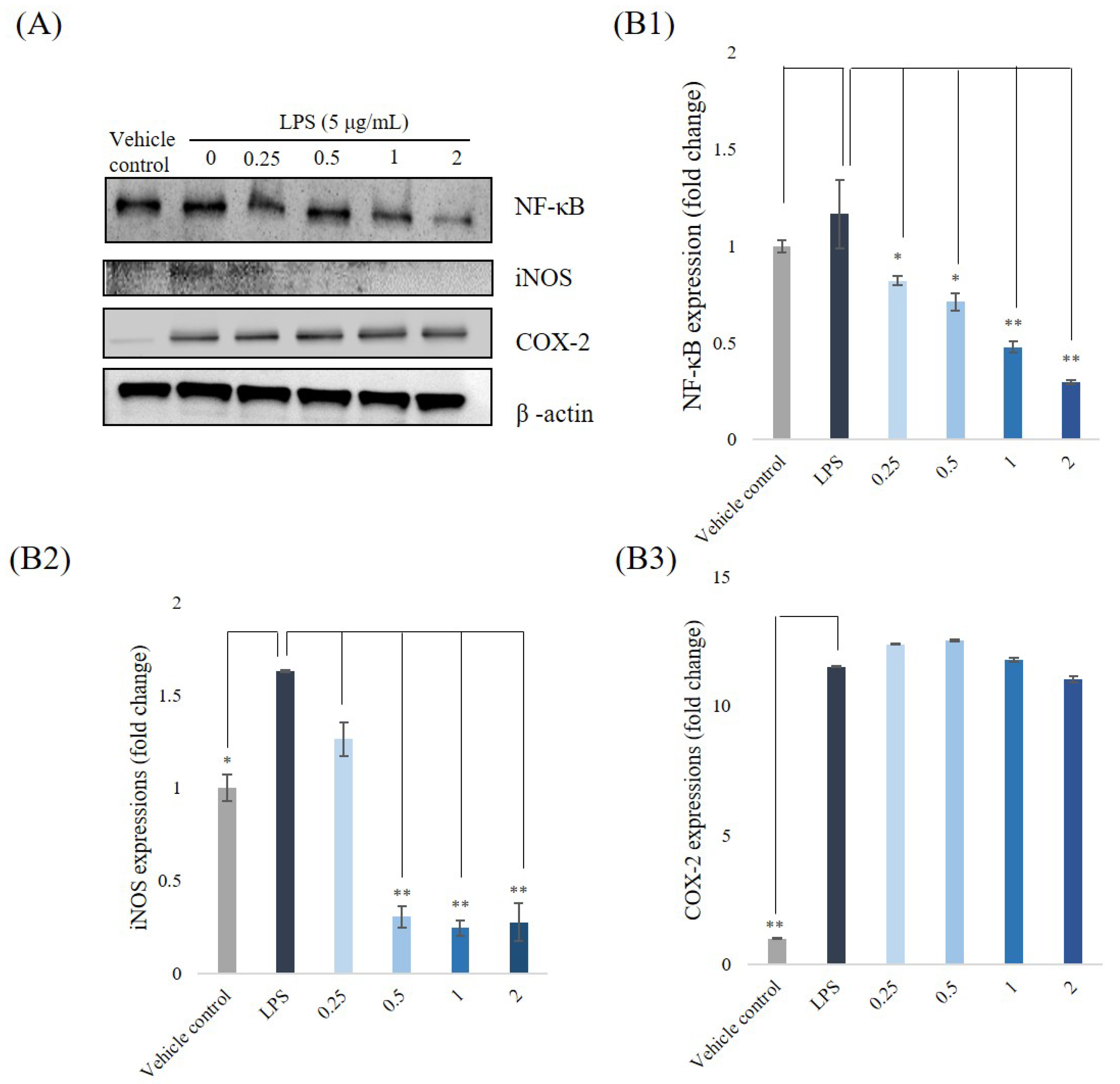
3.5. Western Blotting Analyses for NF-κB, iNOS, and COX-2
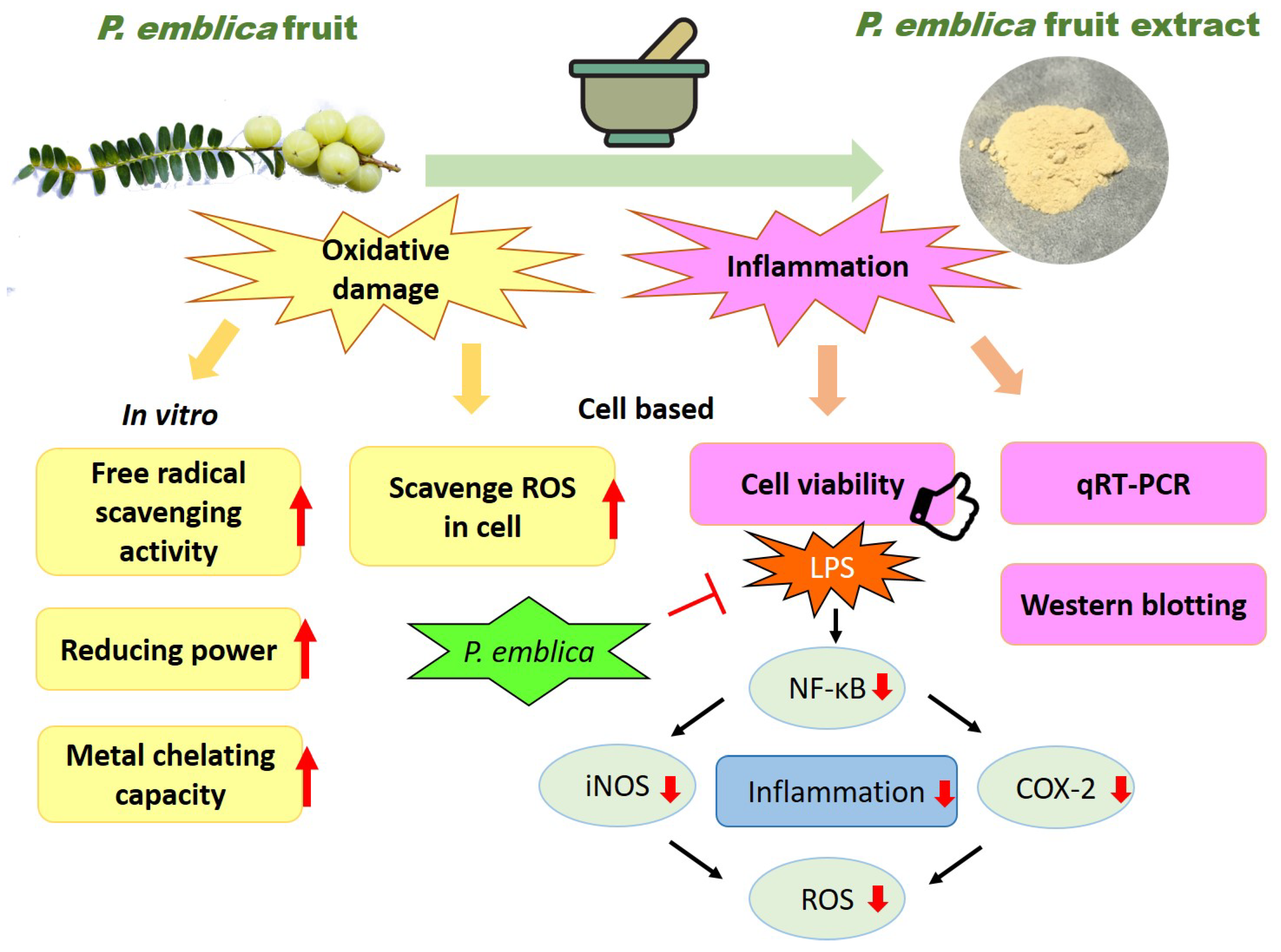
4. Discussion
5. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Huang, S.-Y.; Feng, C.-W.; Hung, H.-C.; Chakraborty, C.; Chen, C.-H.; Chen, W.-F.; Jean, Y.-H.; Wang, H.-M.D.; Sung, C.-S.; Sun, Y.-M.; et al. A novel zebrafish model to provide mechanistic insights into the inflammatory events in carrageenan-induced abdominal edema. PLoS ONE 2014, 9, e104414. [Google Scholar] [CrossRef]
- Du, C.; Bhatia, M.; Tang, S.C.W.; Zhang, M.; Steiner, T. Mediators of inflammation: Inflammation in cancer, chronic diseases, and wound healing. Mediat. Inflamm. 2015, 2015, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Tamura, Y.; Harada, Y.; Nishikawa, S.-I.; Yamano, K.; Kamiya, M.; Shiota, T.; Kuroda, T.; Kuge, O.; Sesaki, H.; Imai, K.; et al. Tam41 is a CDP-diacylglycerol synthase required for cardiolipin biosynthesis in mitochondria. Cell Metab. 2013, 17, 709–718. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-κB signaling in inflammation. Signal Transduct. Target Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.-L.; Chong, I.-W.; Lee, Y.-C.; Tsai, J.-R.; Wang, H.-M.; Hsieh, C.-C.; Kuo, H.-F.; Liu, W.-L.; Chen, Y.-H.; Chen, H.-L. Anti-inflammatory effects of resveratrol on hypoxia/reoxygenation-induced alveolar epithelial cell dysfunction. J. Agric. Food Chem. 2015, 63, 9480–9487. [Google Scholar] [CrossRef] [PubMed]
- Frattaruolo, L.; Carullo, G.; Brindisi, M.; Mazzotta, S.; Bellissimo, L.; Rago, V.; Curcio, R.; Dolce, V.; Aiello, F.; Cappello, A.R. Antioxidant and anti-inflammatory activities of flavanones from Glycyrrhiza glabra L. (licorice) leaf phytocomplexes: Identification of Licoflavanone as a modulator of NF-κB/MAPK pathway. Antioxidants 2019, 8, 186. [Google Scholar] [CrossRef]
- Rao, T.P.; Okamoto, T.; Akita, N.; Hayashi, T.; Kato-Yasuda, N.; Suzuki, K. Amla (Emblica officinalis Gaertn.) extract inhibits lipopolysaccharide-induced procoagulant and pro-inflammatory factors in cultured vascular endothelial cells. Br. J. Nutr. 2013, 110, 2201–2206. [Google Scholar]
- Shih, C.-C.; Hwang, H.-R.; Chang, C.-I.; Su, H.-M.; Chen, P.-C.; Kuo, H.-M.; Li, P.-J.; Wang, H.-M.D.; Tsui, K.-H.; Lin, Y.-C.; et al. Anti-inflammatory and antinociceptive effects of ethyl acetate fraction of an edible red macroalgae Sarcodia ceylanica. Int. J. Mol. Sci. 2017, 18, 2437. [Google Scholar] [CrossRef]
- Gaire, B.P.; Subedi, L. Phytochemistry, pharmacology and medicinal properties of Phyllanthus emblica Linn. Chin. J. Integr. Med. 2014, 1–8. [Google Scholar] [CrossRef]
- Khanna, S.; Das, A.; Spieldenner, J.; Rink, C.; Roy, S. Supplementation of a standardized Extract from Phyllanthus emblica improves cardiovascular risk factors and platelet aggregation in overweight/class-1 obese adults. J. Med. Food 2015, 18, 415–420. [Google Scholar] [CrossRef]
- Wang, F.; Pan, T.; Yuan, R.; Li, C.; Li, K. Optimization of extraction process of flavonoids in Phyllanthus emblica L. by response surface methodology and content determination. Indian J. Tradit. knowl. 2015, 14, 213–219. [Google Scholar]
- Wu, P.-F.; Wang, H.-M.D.; Chen, C.-Y. Isophilippinolide A arrests cell cycle progression and induces apoptosis for anticancer inhibitory agents in human melanoma cells. J. Agric. Food Chem. 2014, 62, 1057–1065. [Google Scholar]
- Li, P.-H.; Chiu, Y.-P.; Shih, C.-C.; Wen, Z.-H.; Ibeto, L.K.; Huang, S.-H.; Chiu, C.-C.; Ma, D.-L.; Leung, C.-H.; Chang, Y.-N.; et al. Biofunctional activities of Equisetum ramosissimum extract: Protective effects against oxidation, melanoma, and melanogenesis. Oxidative Med. Cell. Longev. 2016, 2016, 1–9. [Google Scholar]
- Chen, Y.; Huang, J.-Y.; Lin, Y.; Lin, I.-F.; Lu, Y.-R.; Liu, L.-H.; Wang, H.-M.D. Antioxidative and antimelanoma effects of various tea extracts via a green extraction method. J. Food Qual. 2018, 2018, 1–6. [Google Scholar] [CrossRef]
- Zhao, C.-N.; Tang, G.-Y.; Cao, S.-Y.; Xu, X.-Y.; Gan, R.-Y.; Liu, Q.; Mao, Q.-Q.; Shang, A.; Li, H.-B. Phenolic profiles and antioxidant activities of 30 tea infusions from green, black, oolong, white, yellow and dark Teas. Antioxidants 2019, 8, 215. [Google Scholar] [CrossRef]
- Rossin, D.; Barbosa-Pereira, L.; Iaia, N.; Testa, G.; Sottero, B.; Poli, G.; Zeppa, G.; Biasi, F. A dietary mixture of oxysterols induces in vitro intestinal inflammation through TLR2/4 activation: The protective effect of cocoa bean shells. Antioxidants 2019, 8, 151. [Google Scholar] [CrossRef]
- Wang, H.-M.; Chiu, C.-C.; Wu, P.-F.; Chen, C.-Y. Subamolide E from Cinnamomum subavenium induces sub-G1 cell-cycle arrest and caspase-dependent apoptosis and reduces the migration ability of human melanoma cells. J. Agric. Food Chem. 2011, 59, 8187–8192. [Google Scholar] [CrossRef]
- Wang, S.; Suh, J.H.; Hung, W.-L.; Zheng, X.; Wang, Y.; Ho, C.-T. Use of UHPLC-TripleQ with synthetic standards to profile anti-inflammatory hydroxycinnamic acid amides in root barks and leaves of Lycium barbarum. J. Food Drug Anal. 2018, 26, 572–582. [Google Scholar] [CrossRef]
- Lin, C.Y.; Lee, C.H.; Chang, Y.W.; Wang, H.M.; Chen, C.Y.; Chen, Y.H. Pheophytin a inhibits inflammation via suppression of LPS-induced nitric oxide synthase-2, prostaglandin E2, and interleukin-1beta of macrophages. Int. J. Mol. Sci. 2014, 15, 22819–22834. [Google Scholar] [CrossRef]
- Wang, C.C.; Huang, S.Y.; Huang, S.H.; Wen, Z.H.; Huang, J.Y.; Liu, W.S.; Wang, H.M.D. A synthetic biological secondary metabolite, LycogenTM, produced and extracted from Rhodobacter sphaeroides WL-APD911 in an optimizatioal scale-up strategy. Food Sci. Hum. Wellness 2017, 6, 195–201. [Google Scholar] [CrossRef]
- Li, P.-H.; Liu, L.-H.; Chang, C.-C.; Gao, R.; Leung, C.-H.; Ma, D.-L.; Wang, H.-M.D. Silencing stem cell factor gene in fibroblasts to regulate paracrine factor productions and enhance c-Kit expression in melanocytes on melanogenesis. Int. J. Mol. Sci. 2018, 19, 1475. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-T.; Kao, C.-J.; Huang, H.-Y.; Huang, S.-Y.; Chen, C.-Y.; Lin, Y.-S.; Wen, Z.-H.; Wang, H.-M.D. Astaxanthin reduces MMP expressions, suppresses cancer cell migrations, and triggers apoptotic caspases of in vitro and in vivo models in melanoma. J. Funct. Foods 2017, 31, 20–31. [Google Scholar] [CrossRef]
- Galletti, E.; Bonilauri, P.; Bardasi, L.; Fontana, M.C.; Ramini, M.; Renzi, M.; Dosa, G.; Merialdi, G. Development of a minor groove binding probe based real-time PCR for the diagnosis and quantification of Leishmania infantum in dog specimens. Res. Vet. Sci. 2011, 91, 243–245. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.-C.; Chen, C.-Y.; Kuo, C.-H.; Lin, Y.-S.; Hwang, B.H.; Wang, T.K.; Kuo, Y.-H.; Wang, H.-M.D. 36H: A novel potent inhibitor for antimelanogenesis. Oxidative Med. Cell. Longev. 2018, 2018, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Parveen, R.; Shamsi, T.N.; Singh, G.; Athar, T.; Fatima, S. Phytochemical analysis and in-vitro biochemical characterization of aqueous and methanolic extract of Triphala, a conventional herbal remedy. Biotechnol. Rep. 2018, 17, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R. Origin and physiological roles of inflammation. Nat. 2008, 454, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.-H.; Wu, S.-H.; Lee, S.-S.; Chang, K.-P.; Chai, C.-Y.; Yeh, J.-L.; Lin, S.-D.; Kwan, A.-L.; Wang, H.-M.D.; Lai, C.-S. Fat grafting in burn scar alleviates neuropathic pain via anti-inflammation effect in scar and spinal cord. PLoS ONE 2015, 10, e0137563. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Yang, Z.; Zhang, J.; Mu, J.; Zhou, X.; Zhao, X. Liver injury induced by carbon tetrachloride in mice is prevented by the antioxidant capacity of Anji white tea polyphenols. Antioxidants 2019, 8, 64. [Google Scholar] [CrossRef] [PubMed]
- Ning, C.; Wang, H.-M.D.; Gao, R.; Chang, Y.-C.; Hu, F.; Meng, X.; Huang, S.-Y. Marine-derived protein kinase inhibitors for neuroinflammatory diseases. Biomed. Eng. Online 2018, 17, 46. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NF-κB |
| Forward: 5′-TATGTGTGTGAAGGCCCATCA-3′ |
| Reverse: 5′-ACCAACTGAACGATAACCTTTGC-3′ |
| iNOS |
| Forward: 5′-CGAGACGGATAGGCAGAGATTG-3′ |
| Reverse: 5′-CTCTTCAAGCACCTCCAGGAA-3′ |
| COX-2 |
| Forward: 5′-CCAGCACTTCACCCATCAGTTT-3′ |
| Reverse: 5′-TCTGTCCAGAGTTTCACCATAAATG-3′ |
| Concentration (mg/mL) | DPPH Free Radical Scavenging Activity (%) | Reducing Power (OD700) | Metal Chelating Activity (%) |
|---|---|---|---|
| 0 | 0 ± 0 | 0.105 ± 0.001 | 0 ± 0 |
| 0.5 | 3.92 ± 0.07 | 0.151 ± 0.001 | 5.66 ± 0.20 |
| 1 | 7.04 ± 0.10 | 0.180 ± 0.002 | 7.72 ± 0.69 |
| 2 | 16.43 ± 0.25 | 0.205 ± 0.002 | 11.63 ± 0.66 |
| 5 | 42.43 ± 0.51 | 0.436 ± 0.006 | 15.06 ± 0.13 |
| 10 | 67.03 ± 0.07 | 0.796 ± 0.023 | 16.12 ± 0.25 |
| 50 | 88.71 ± 0.30 | 2.311 ± 0.054 | 16.92 ± 0.11 |
| Vitamin C a | 89.97 ± 0.17 | - | - |
| BHA b | - | 0.604 ± 0.002 | - |
| EDTA c | - | - | 94.43 ± 0.21 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, H.M.-D.; Fu, L.; Cheng, C.C.; Gao, R.; Lin, M.Y.; Su, H.L.; Belinda, N.E.; Nguyen, T.H.; Lin, W.-H.; Lee, P.C.; et al. Inhibition of LPS-Induced Oxidative Damages and Potential Anti-Inflammatory Effects of Phyllanthus emblica Extract via Down-Regulating NF-κB, COX-2, and iNOS in RAW 264.7 Cells. Antioxidants 2019, 8, 270. https://doi.org/10.3390/antiox8080270
Wang HM-D, Fu L, Cheng CC, Gao R, Lin MY, Su HL, Belinda NE, Nguyen TH, Lin W-H, Lee PC, et al. Inhibition of LPS-Induced Oxidative Damages and Potential Anti-Inflammatory Effects of Phyllanthus emblica Extract via Down-Regulating NF-κB, COX-2, and iNOS in RAW 264.7 Cells. Antioxidants. 2019; 8(8):270. https://doi.org/10.3390/antiox8080270
Chicago/Turabian StyleWang, Hui Min-David, Ling Fu, Chia Chi Cheng, Rong Gao, Meng Yi Lin, Hong Lin Su, Nathania Earlene Belinda, Thi Hiep Nguyen, Wen-Hung Lin, Po Chun Lee, and et al. 2019. "Inhibition of LPS-Induced Oxidative Damages and Potential Anti-Inflammatory Effects of Phyllanthus emblica Extract via Down-Regulating NF-κB, COX-2, and iNOS in RAW 264.7 Cells" Antioxidants 8, no. 8: 270. https://doi.org/10.3390/antiox8080270
APA StyleWang, H. M.-D., Fu, L., Cheng, C. C., Gao, R., Lin, M. Y., Su, H. L., Belinda, N. E., Nguyen, T. H., Lin, W.-H., Lee, P. C., & Hsieh, L. P. (2019). Inhibition of LPS-Induced Oxidative Damages and Potential Anti-Inflammatory Effects of Phyllanthus emblica Extract via Down-Regulating NF-κB, COX-2, and iNOS in RAW 264.7 Cells. Antioxidants, 8(8), 270. https://doi.org/10.3390/antiox8080270