Luteolin 7-Sulfate Attenuates Melanin Synthesis through Inhibition of CREB- and MITF-Mediated Tyrosinase Expression

 ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Instrumental Analysis
2.3. Synthesis and Purification of Luteolin 7-Sulfate
2.4. Cell Culture
2.5. Melanin Content Assay
2.6. TYR Activity Assay
2.7. Western Blotting
2.8. Quantitative Reverse Transcription Polymerase Chain Reaction (qRT-PCR) Analysis
2.9. Statistical Analysis
3. Results
3.1. Luteolin 7-Sulfate
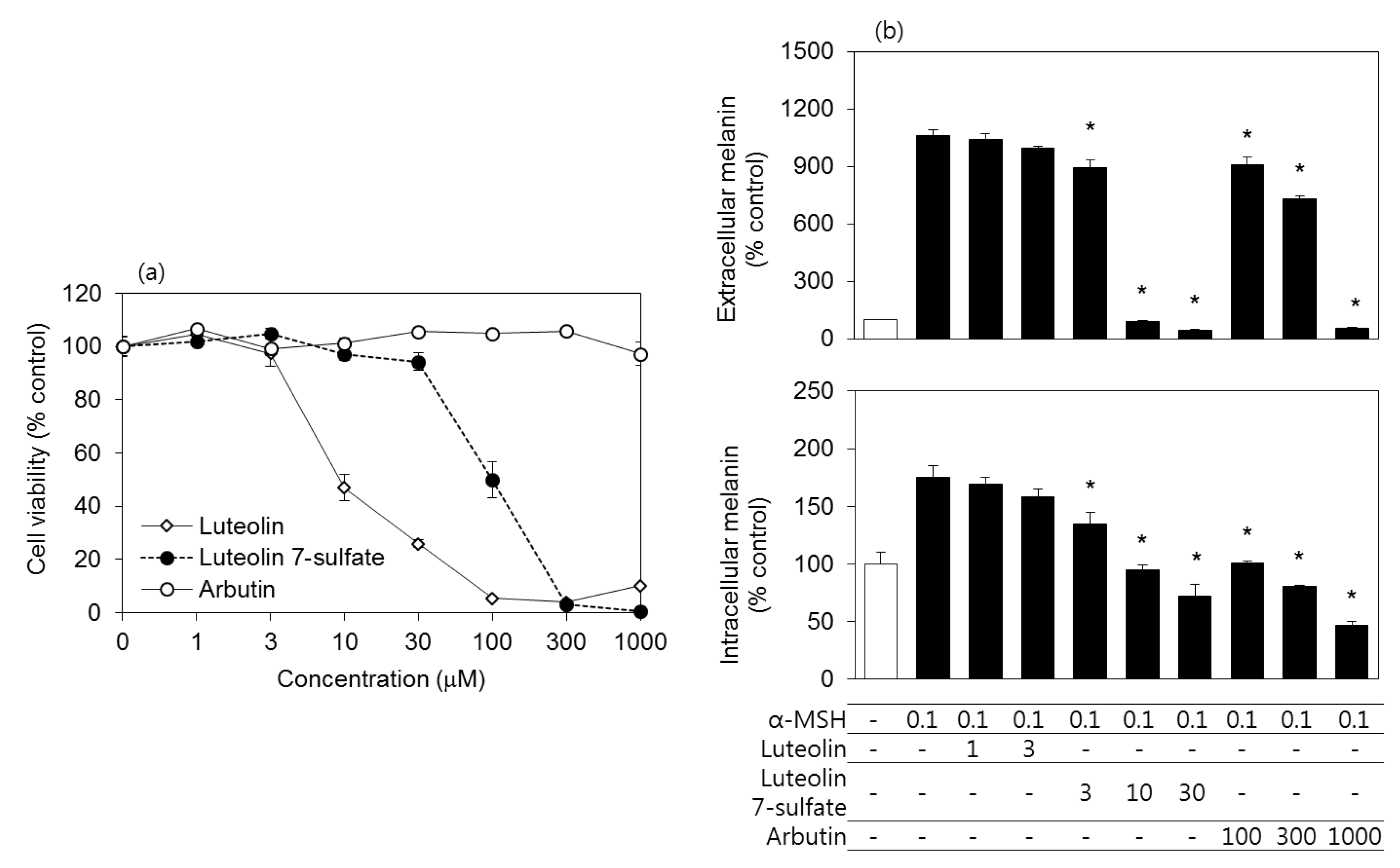
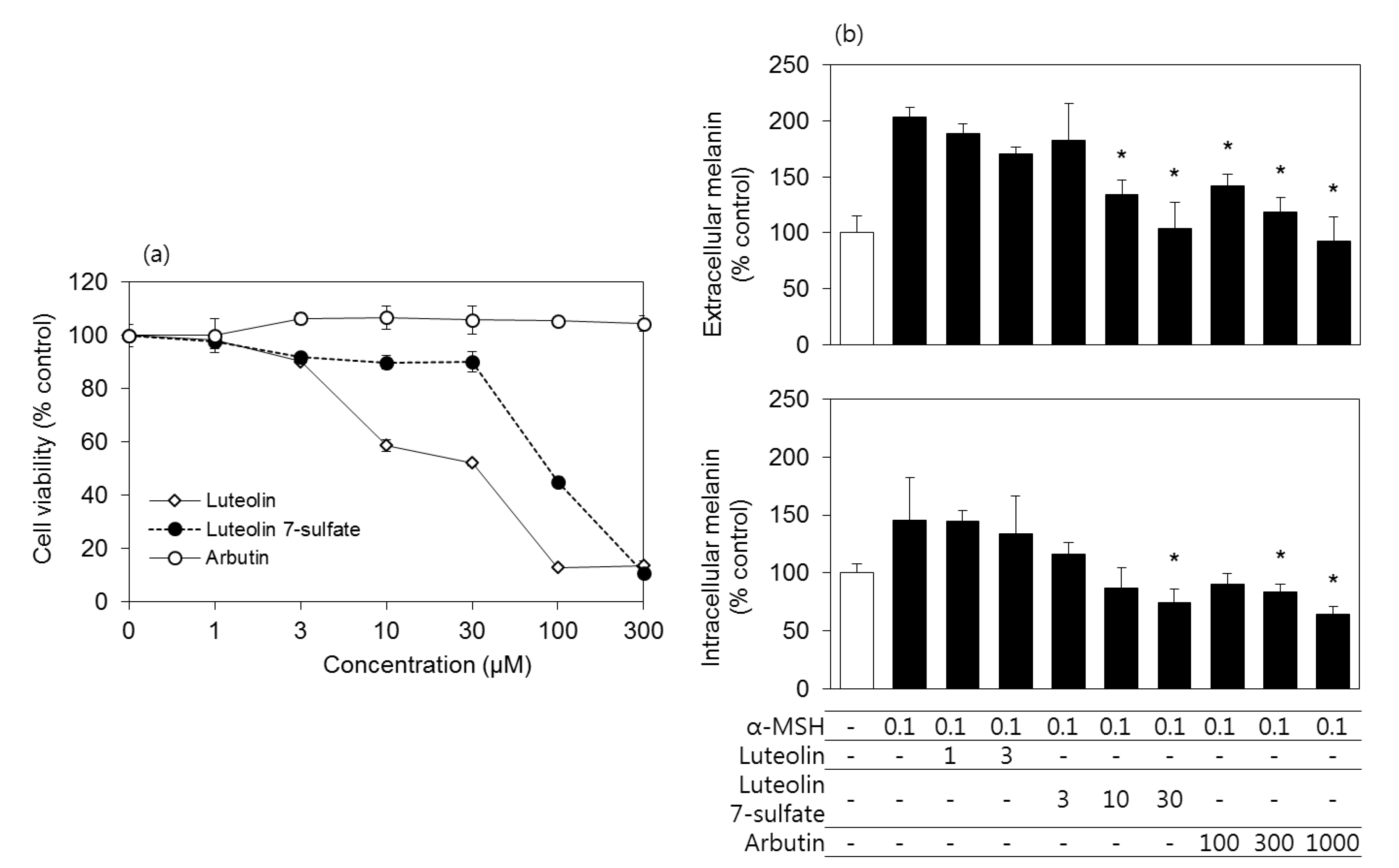
3.2. Effects of Luteolin, Luteolin 7-Sulfate, and Arbutin on the Viability and the Melanin Contents of B16-F10 Cells
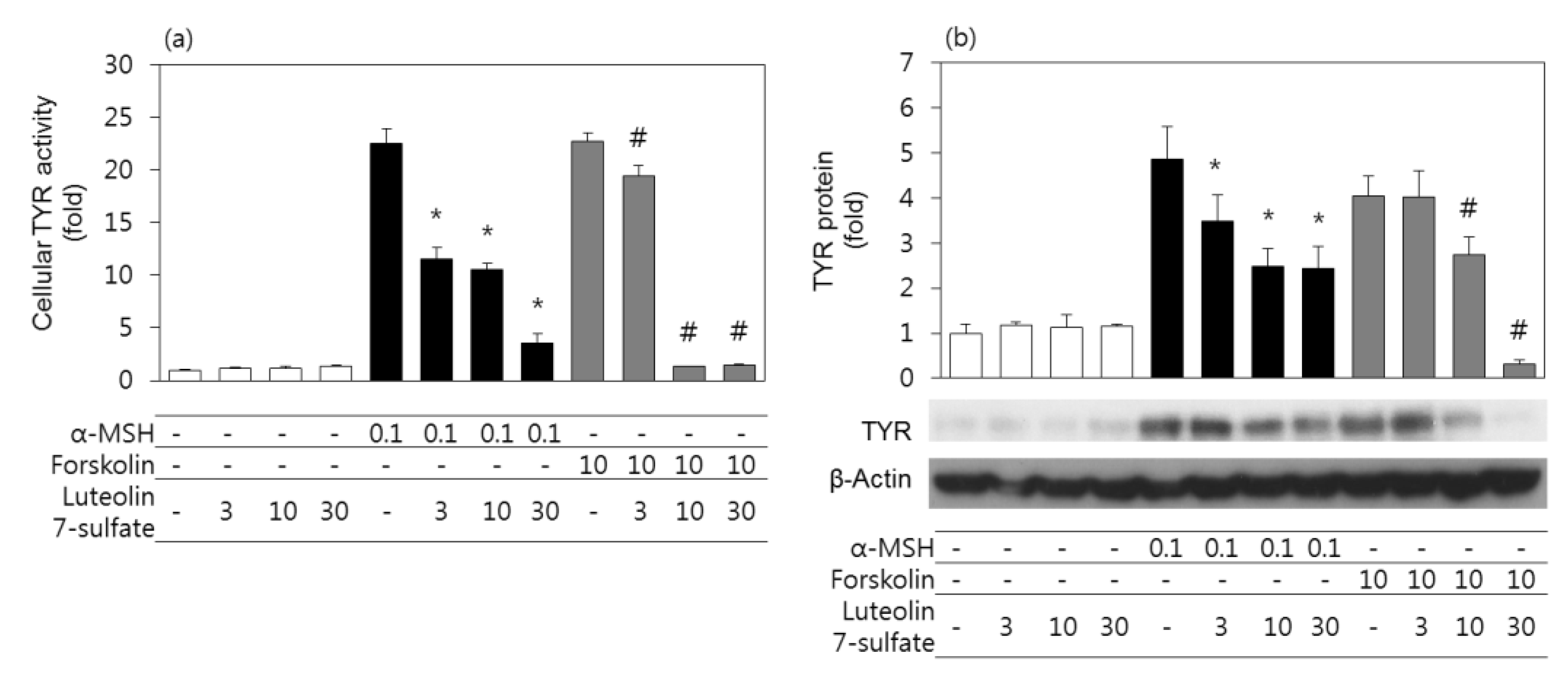
3.3. Effects of Luteolin 7-Sulfate on Cellular TYR Activities and TYR Protein Levels
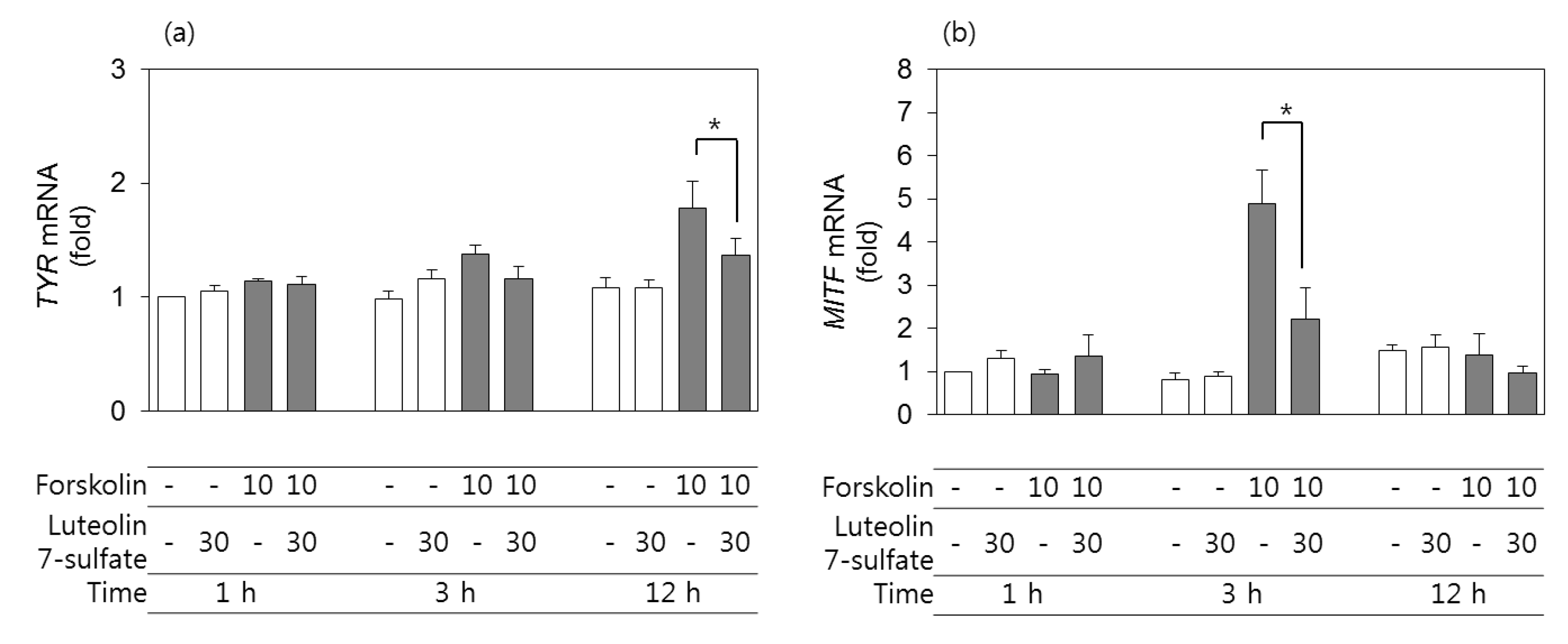
3.4. Effects of Luteolin 7-Sulfate on TYR and MITF mRNA Levels
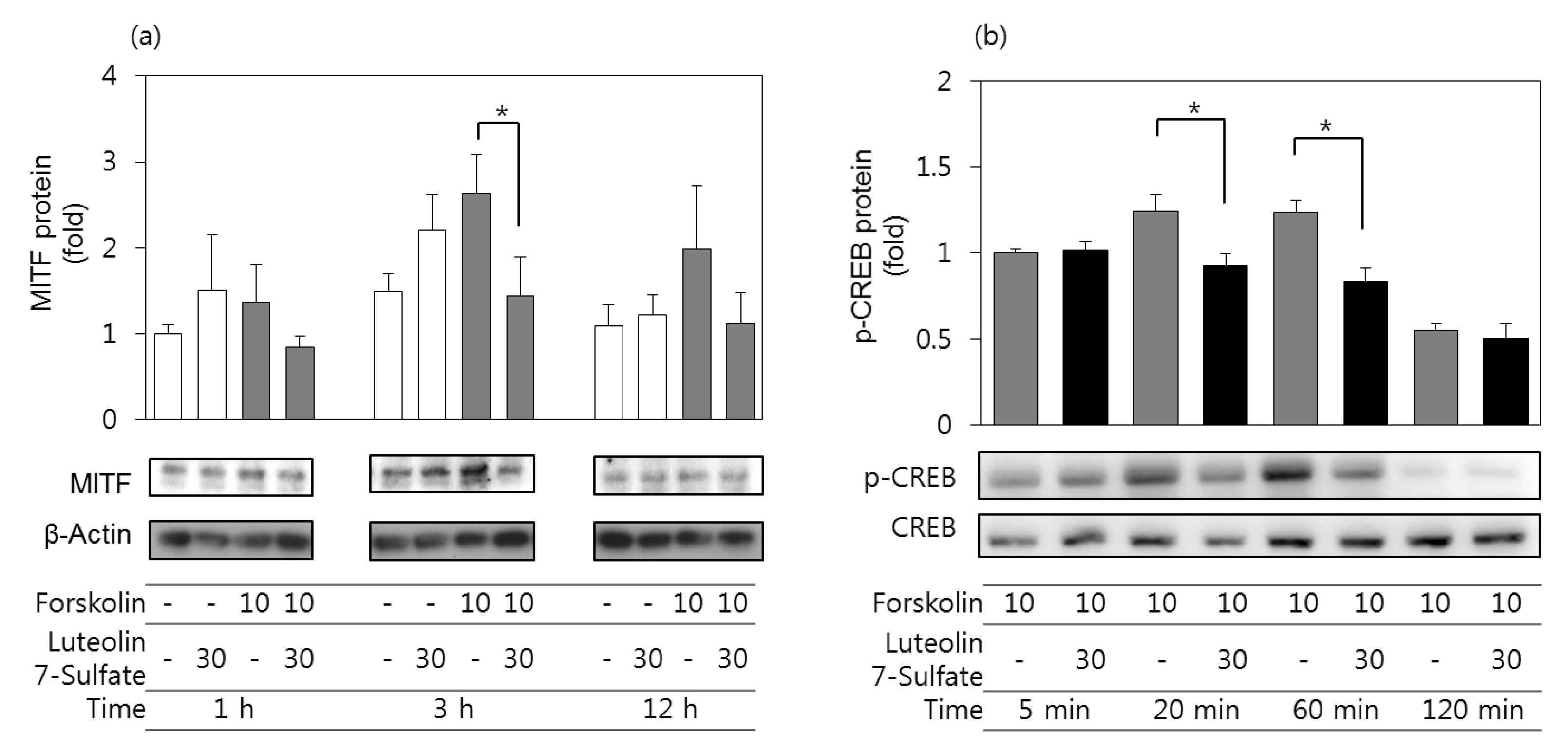
3.5. Effects of Luteolin 7-Sulfate on MITF Protein and Phospho-CREB Protein Levels
3.6. Effects of Luteolin, Luteolin 7-Sulfate, and Arbutin on the Viability and the Melanin Contents of HEMs
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Slominski, A.T.; Zmijewski, M.A.; Skobowiat, C.; Zbytek, B.; Slominski, R.M.; Steketee, J.D. Sensing the environment: Regulation of local and global homeostasis by the skin’s neuroendocrine system. Adv. Anat. Embryol. Cell Biol. 2012, 212, v-115. [Google Scholar] [PubMed]
- Fistarol, S.K.; Itin, P.H. Disorders of pigmentation. J. Dtsch. Dermatol. Ges. 2010, 8, 187–201. [Google Scholar] [CrossRef]
- Schiaffino, M.V. Signaling pathways in melanosome biogenesis and pathology. Int. J. Biochem. Cell Biol. 2010, 42, 1094–1104. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Park, J.; Song, K.; Kim, H.G.; Koh, J.S.; Boo, Y.C. Screening of plant extracts for human tyrosinase inhibiting effects. Int. J. Cosmet. Sci. 2012, 34, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; An, S.M.; Koh, J.S.; Jang, D.I.; Boo, Y.C. Use of non-melanocytic HEK293 cells stably expressing human tyrosinase for the screening of anti-melanogenic agents. J. Cosmet. Sci. 2011, 62, 515–523. [Google Scholar] [PubMed]
- An, S.M.; Koh, J.S.; Boo, Y.C. p-Coumaric acid not only inhibits human tyrosinase activity in vitro but also melanogenesis in cells exposed to UVB. Phytother. Res. 2010, 24, 1175–1180. [Google Scholar] [CrossRef] [PubMed]
- An, S.M.; Lee, S.I.; Choi, S.W.; Moon, S.W.; Boo, Y.C. p-Coumaric acid, a constituent of Sasa quelpaertensis Nakai, inhibits cellular melanogenesis stimulated by alpha-melanocyte stimulating hormone. Br. J. Dermatol. 2008, 159, 292–299. [Google Scholar] [CrossRef] [PubMed]
- Seo, Y.K.; Kim, S.J.; Boo, Y.C.; Baek, J.H.; Lee, S.H.; Koh, J.S. Effects of p-coumaric acid on erythema and pigmentation of human skin exposed to ultraviolet radiation. Clin. Exp. Dermatol. 2011, 36, 260–266. [Google Scholar] [CrossRef]
- Park, J.; Park, J.H.; Suh, H.J.; Lee, I.C.; Koh, J.; Boo, Y.C. Effects of resveratrol, oxyresveratrol, and their acetylated derivatives on cellular melanogenesis. Arch. Dermatol. Res. 2014, 306, 475–487. [Google Scholar] [CrossRef]
- Park, J.; Boo, Y.C. Isolation of resveratrol from vitis viniferae caulis and its potent inhibition of human tyrosinase. Evid. Based Complement. Alternat. Med. 2013, 2013, 645257. [Google Scholar] [CrossRef]
- Newton, R.A.; Cook, A.L.; Roberts, D.W.; Leonard, J.H.; Sturm, R.A. Post-transcriptional regulation of melanin biosynthetic enzymes by cAMP and resveratrol in human melanocytes. J. Investig. Dermatol. 2007, 127, 2216–2227. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Seok, J.K.; Kwak, J.Y.; Choi, Y.H.; Hong, S.S.; Suh, H.J.; Park, W.; Boo, Y.C. Anti-melanogenic effects of resveratryl triglycolate, a novel hybrid compound derived by esterification of resveratrol with glycolic acid. Arch. Dermatol. Res. 2016, 308, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Jo, D.J.; Seok, J.K.; Kim, S.Y.; Park, W.; Baek, J.H.; Kim, Y.M.; Boo, Y.C. Human skin-depigmenting effects of resveratryl triglycolate, a hybrid compound of resveratrol and glycolic acid. Int. J. Cosmet. Sci. 2018. [Google Scholar] [CrossRef] [PubMed]
- Choi, G.W.; Jeong, H.J.; Sek, J.K.; Baek, J.H.; Kim, Y.M.; Boo, Y.C. Skin Anti-aging Effects of a Cream Containing Resveratryl Triacetate (RTA). J. Soc. Cosmet. Sci. Korea 2018, 44, 161–170. [Google Scholar]
- Ryu, J.H.; Seok, J.K.; An, S.M.; Baek, J.H.; Koh, J.S.; Boo, Y.C. A study of the human skin-whitening effects of resveratryl triacetate. Arch. Dermatol. Res. 2015, 307, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Mustapha, N.; Bzeouich, I.M.; Ghedira, K.; Hennebelle, T.; Chekir-Ghedira, L. Compounds isolated from the aerial part of Crataegus azarolus inhibit growth of B16F10 melanoma cells and exert a potent inhibition of the melanin synthesis. Biomed. Pharmacother. 2015, 69, 139–144. [Google Scholar] [CrossRef]
- Nanni, V.; Canuti, L.; Gismondi, A.; Canini, A. Hydroalcoholic extract of Spartium junceum L. flowers inhibits growth and melanogenesis in B16-F10 cells by inducing senescence. Phytomedicine 2018, 46, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Meng, T.X.; Irino, N.; Kondo, R. Melanin biosynthesis inhibitory activity of a compound isolated from young green barley (Hordeum vulgare L.) in B16 melanoma cells. J. Nat. Med. 2015, 69, 427–431. [Google Scholar] [CrossRef]
- Kwak, J.Y.; Seok, J.K.; Suh, H.J.; Choi, Y.H.; Hong, S.S.; Kim, D.S.; Boo, Y.C. Antimelanogenic effects of luteolin 7-sulfate isolated from Phyllospadix iwatensis Makino. Br. J. Dermatol. 2016, 175, 501–511. [Google Scholar] [CrossRef]
- Barron, D.; Ibrahim, K. Synthesis of flavonoid sulfates: 1. Stepwise fulfation of positions 3, 7, and 4‘ using N,N‘-dicyclohexylcarbodiimide and tetrabutylammonium hydrogen sulfate. Tetrahedron 1987, 43, 5197–5202. [Google Scholar] [CrossRef]
- Mitani, K.; Takano, F.; Kawabata, T.; Allam, A.E.; Ota, M.; Takahashi, T.; Yahagi, N.; Sakurada, C.; Fushiya, S.; Ohta, T. Suppression of melanin synthesis by the phenolic constituents of sappanwood (Caesalpinia sappan). Planta Med. 2013, 79, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Jimenez, A.; Teruel-Puche, J.A.; Berna, J.; Rodriguez-Lopez, J.N.; Tudela, J.; Garcia-Canovas, F. Action of tyrosinase on alpha and beta-arbutin: A kinetic study. PLoS ONE 2017, 12, e0177330. [Google Scholar] [CrossRef] [PubMed]
- An, S.M.; Koh, J.S.; Boo, Y.C. Inhibition of melanogenesis by tyrosinase siRNA in human melanocytes. BMB Rep. 2009, 42, 178–183. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Zheng, Y.; Li, L.; Liu, S.; Burrows, M.; Wei, Z.; Nace, A.; Herlyn, M.; Cui, R.; Guo, W.; et al. Direct conversion of mouse and human fibroblasts to functional melanocytes by defined factors. Nat. Commun. 2014, 5, 5807. [Google Scholar] [CrossRef] [PubMed]
- Han, M.S.; Che, X.; Cho, G.H.; Park, H.R.; Lim, K.E.; Park, N.R.; Jin, J.S.; Jung, Y.K.; Jeong, J.H.; Lee, I.K.; et al. Functional Cooperation between Vitamin D Receptor and Runx2 in Vitamin D-Induced Vascular Calcification. PLoS ONE 2013, 8, e83584. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.S.; Jeong, W.; Lee, J.Y.; Ahn, E.K.; Oh, J.S. Inhibition of Antigen-Induced Degranulation by Flavonoids Isolated from the Leaves of Quercus Acuta in RBL-2H3 Cells. Chem. Nat. Compd. 2016, 52, 1089–1092. [Google Scholar] [CrossRef]
- Zhang, C.F.; Sun, Z.H.; Zhang, D.; Zhang, M.A. Sulphur compounds from the aerial parts of Eclipta prostrata. Biochem. Syst. Ecol. 2010, 38, 1253–1256. [Google Scholar] [CrossRef]
- Maeda, K.; Fukuda, M. Arbutin: Mechanism of its depigmenting action in human melanocyte culture. J. Pharmacol. Exp. Ther. 1996, 276, 765–769. [Google Scholar]
- Lopez-Lazaro, M. Distribution and biological activities of the flavonoid luteolin. Mini Rev. Med. Chem. 2009, 9, 31–59. [Google Scholar] [CrossRef]
- An, S.M.; Kim, H.J.; Kim, J.E.; Boo, Y.C. Flavonoids, taxifolin and luteolin attenuate cellular melanogenesis despite increasing tyrosinase protein levels. Phytother. Res. 2008, 22, 1200–1207. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.Y.; Song, H.S.; Hur, H.S.; Sim, S.S. Whitening activity of luteolin related to the inhibition of cAMP pathway in alpha-MSH-stimulated B16 melanoma cells. Arch. Pharm. Res. 2008, 31, 1166–1171. [Google Scholar] [CrossRef] [PubMed]
- Takagi, M.; Funahashi, S.; Ohta, K.; Nakabayashi, T. Phyllospadine, a New Flavonoidal Alkaloid from the Sea-Grass Phyllosphadix iwatensis. Agric. Biol. Chem. 1980, 44, 3019–3020. [Google Scholar] [CrossRef]
- Enerstvedt, K.H.; Jordheim, M.; Andersen, O.M. Isolation and Identification of Flavonoids Found in Zostera marina Collected in Norwegian Coastal Waters. Am. J. Plant Sci. 2016, 2016, 1163–1172. [Google Scholar] [CrossRef]
- Slominski, A.; Tobin, D.J.; Shibahara, S.; Wortsman, J. Melanin pigmentation in mammalian skin and its hormonal regulation. Physiol. Rev. 2004, 84, 1155–1228. [Google Scholar] [CrossRef] [PubMed]
- Simon, J.D.; Peles, D.; Wakamatsu, K.; Ito, S. Current challenges in understanding melanogenesis: Bridging chemistry, biological control, morphology, and function. Pigment Cell Melanoma Res. 2009, 22, 563–579. [Google Scholar] [CrossRef] [PubMed]
- Bracalente, C.; Salguero, N.; Notcovich, C.; Muller, C.B.; da Motta, L.L.; Klamt, F.; Ibanez, I.L.; Duran, H. Reprogramming human A375 amelanotic melanoma cells by catalase overexpression: Reversion or promotion of malignancy by inducing melanogenesis or metastasis. Oncotarget 2016, 7, 41142–41153. [Google Scholar] [CrossRef] [PubMed]
- Liu-Smith, F.; Meyskens, F.L. Molecular mechanisms of flavonoids in melanin synthesis and the potential for the prevention and treatment of melanoma. Mol. Nutr. Food Res. 2016, 60, 1264–1274. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Chou, G.X.; Wang, H.; Chu, J.H.; Yu, Z.L. Flavonoids, apigenin and icariin exert potent melanogenic activities in murine B16 melanoma cells. Phytomedicine 2010, 18, 32–35. [Google Scholar] [CrossRef] [PubMed]
- Nagata, H.; Takekoshi, S.; Takeyama, R.; Homma, T.; Yoshiyuki Osamura, R. Quercetin enhances melanogenesis by increasing the activity and synthesis of tyrosinase in human melanoma cells and in normal human melanocytes. Pigment Cell Res. 2004, 17, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.M.; Son, Y.O.; Lee, S.A.; Jeon, Y.M.; Lee, J.C. Quercetin Inhibits alpha-MSH-stimulated Melanogenesis in B16F10 Melanoma Cells. Phytother. Res. 2011, 25, 1166–1173. [Google Scholar] [CrossRef] [PubMed]
- Ko, W.C.; Shih, C.M.; Lai, Y.H.; Chen, J.H.; Huang, H.L. Inhibitory effects of flavonoids on phosphodiesterase isozymes from guinea pig and their structure-activity relationships. Biochem. Pharmacol. 2004, 68, 2087–2094. [Google Scholar] [CrossRef] [PubMed]
- Roubalova, L.; Purchartova, K.; Papouskova, B.; Vacek, J.; Kren, V.; Ulrichova, J.; Vrba, J. Sulfation modulates the cell uptake, antiradical activity and biological effects of flavonoids in vitro: An examination of quercetin, isoquercitrin and taxifolin. Bioorg. Med. Chem. 2015, 23, 5402–5409. [Google Scholar] [CrossRef] [PubMed]
- Thilakarathna, S.H.; Rupasinghe, H.P. Flavonoid bioavailability and attempts for bioavailability enhancement. Nutrients 2013, 5, 3367–3387. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Luteolin 7-Sulfate | Luteolin | Luteolin 7-Sulfate | Luteolin |
|---|---|---|---|---|
| δH Multiplicity (J = Hz) | δC | |||
| 2 | 164.3 | 163.9 | ||
| 3 | 6.76 s | 6.67 s | 103.0 | 102.9 |
| 4 | 182.0 | 181.7 | ||
| 5 | 160.5 | 161.5 | ||
| 6 | 6.52 d (2.1) | 6.19 d (2.1) | 102.0 | 98.9 |
| 7 | 159.5 | 164.2 | ||
| 8 | 7.03 d (2.1) | 6.45 d (2.1) | 97.5 | 93.9 |
| 9 | 156.3 | 157.3 | ||
| 10 | 105.6 | 103.7 | ||
| 1’ | 121.3 | 121.6 | ||
| 2’ | 7.47 d (2.1) | 7.42 2 | 113.3 | 113.4 |
| 3’ | 145.7 | 145.8 | ||
| 4’ | 149.8 | 149.7 | ||
| 5’ | 6.89 d (8.4) | 6.90 d (8.4) | 116.1 | 116.1 |
| 6’ | 7.46 dd (8.4, 2.1) | 7.42 2 | 119.1 | 119.0 |
| 5-OH | 12.87 s | 12.98 s | ||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, S.W.; Kim, J.H.; Song, H.; Seok, J.K.; Hong, S.S.; Boo, Y.C. Luteolin 7-Sulfate Attenuates Melanin Synthesis through Inhibition of CREB- and MITF-Mediated Tyrosinase Expression. Antioxidants 2019, 8, 87. https://doi.org/10.3390/antiox8040087
Lee SW, Kim JH, Song H, Seok JK, Hong SS, Boo YC. Luteolin 7-Sulfate Attenuates Melanin Synthesis through Inhibition of CREB- and MITF-Mediated Tyrosinase Expression. Antioxidants. 2019; 8(4):87. https://doi.org/10.3390/antiox8040087
Chicago/Turabian StyleLee, Seok Won, Jae Heon Kim, Hyerim Song, Jin Kyung Seok, Seong Su Hong, and Yong Chool Boo. 2019. "Luteolin 7-Sulfate Attenuates Melanin Synthesis through Inhibition of CREB- and MITF-Mediated Tyrosinase Expression" Antioxidants 8, no. 4: 87. https://doi.org/10.3390/antiox8040087
APA StyleLee, S. W., Kim, J. H., Song, H., Seok, J. K., Hong, S. S., & Boo, Y. C. (2019). Luteolin 7-Sulfate Attenuates Melanin Synthesis through Inhibition of CREB- and MITF-Mediated Tyrosinase Expression. Antioxidants, 8(4), 87. https://doi.org/10.3390/antiox8040087