Synthesis, DFT Calculations, and In Vitro Antioxidant Study on Novel Carba-Analogs of Vitamin E

 , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemical Synthesis
2.1.1. General Information
2.1.2. Synthesis of all-rac-1-carba-α-tocopherol (6)
(6-Methoxy-2,5,7,8-tetramethyl-1,2,3,4-tetrahydronaphthalen-2-yl)methanol (11)
6-Methoxy-2,5,7,8-tetramethyl-1,2,3,4-tetrahydronaphthalene-2-carbaldehyde (12)
6-Methoxy-2,5,7,8-tetramethyl-2-(4,8,12-trimethyltridec-1-enyl)-1,2,3,4-tetrahydronaphthalene (13)
6-Hydroxy-2,5,7,8-tetramethyl-2-(4,8,12-trimethyltridecyl)-1,2,3,4-tetrahydronaphthalene (6)
2.2. Autoxidation Procedure
2.2.1. Materials
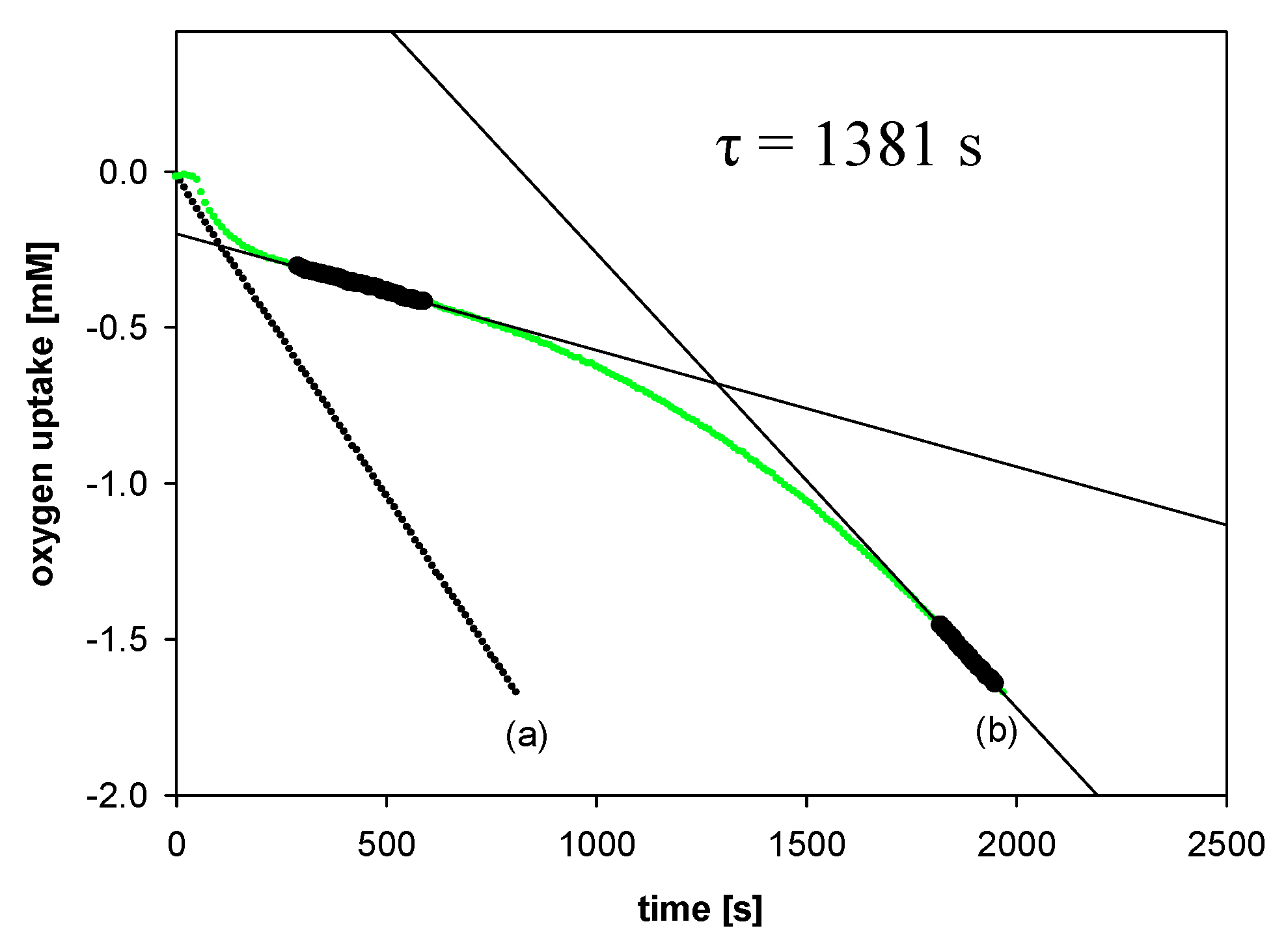
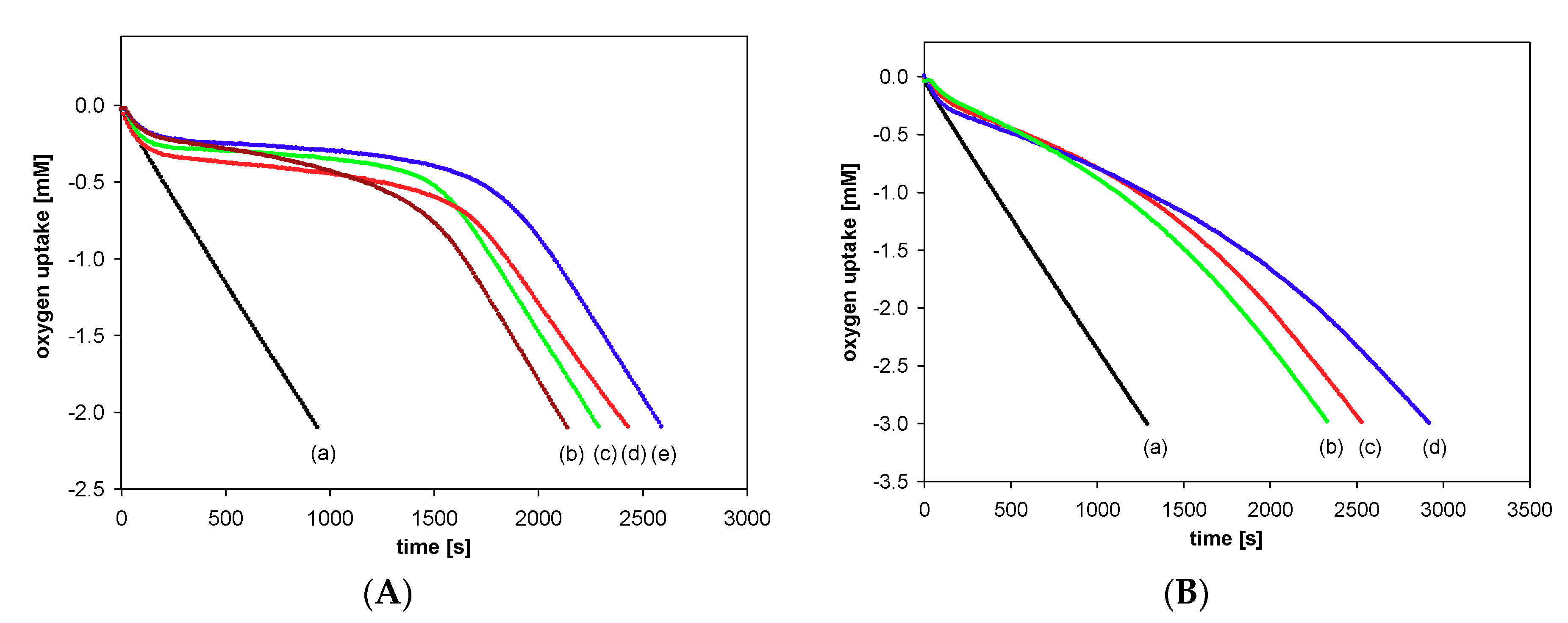
2.2.2. Measurements of Autoxidation Rate
2.2.3. Methodology of Autoxidation Measurements
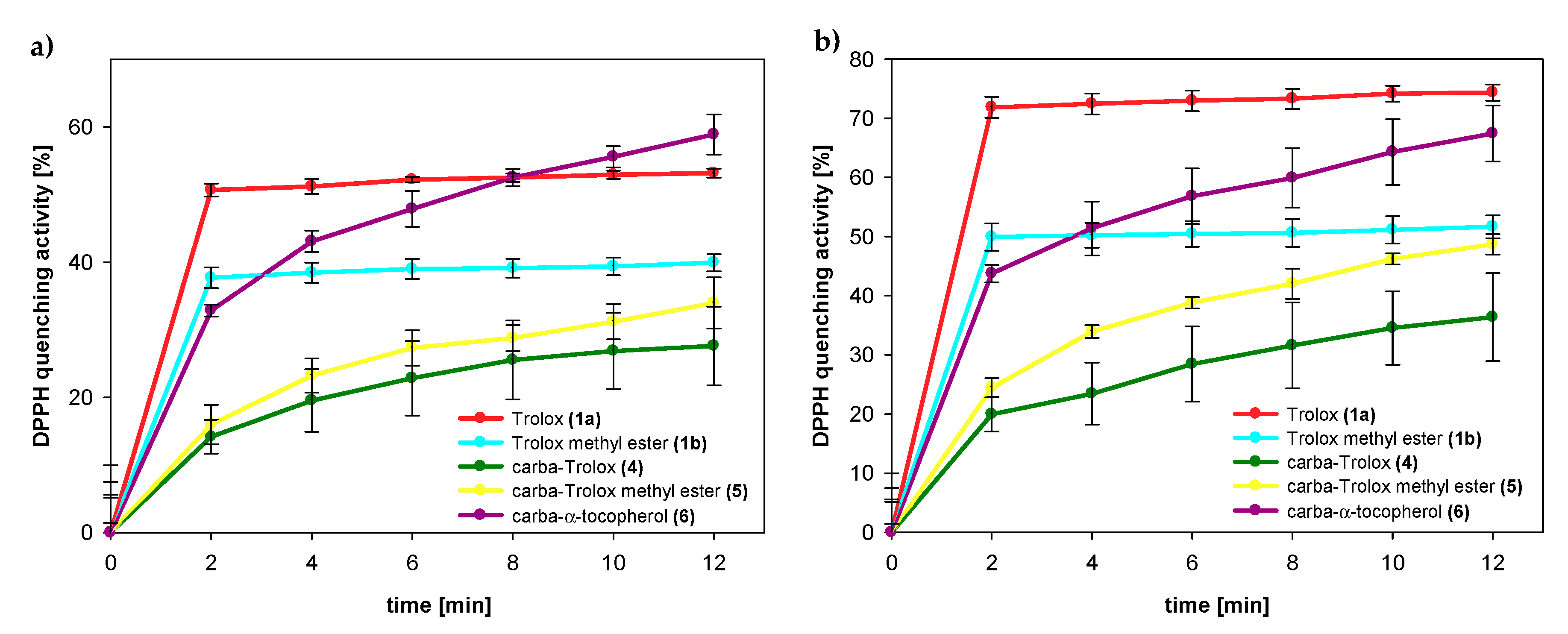
2.3. DPPH Radicals EPR Measurements
2.4. Computational Details
3. Results and Discussion
3.1. Synthesis of all-rac-1-carba-α-tocopherol (6)
3.2. Radical-Scavenging Ability
3.2.1. Measurements of Autoxidation Rate
3.2.2. DPPH Radical Scavenging Activity
3.3. DFT Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ARP | antiradical power |
| BDE | bond dissociation enthalpy |
| DFT | density functional theory |
| DPPH● | 2,2-diphenyl-1-picrylhydrazyl free radical |
| EC50 | the concentration required causing 50% of maximum effect |
| ETE | electron transfer enthalpy |
| HAT | hydrogen atom transfer |
| IP | ionization potential |
| LOO● | lipid peroxyl radical |
| PA | proton affinity |
| PDE | proton dissociation enthalpy |
| PMHC | 2,2,5,7,8-pentamethylchroman-6-ol |
| ROO● | alkyl peroxyl radical |
| SET-PT | sequential electron transfer—proton transfer |
| SPLET | sequential proton loss—electron transfer |
| Trolox | 6-hydroxy-2,5,7,8-tetramethylchromane-2-carboxylic acid |
References
- Falk, J.; Munné-Bosch, S. Tocochromanol functions in plants: Antioxidation and beyond. J. Exp. Bot. 2010, 61, 1549–1566. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Lee, Y.-K.; Kim, D.-G.; Son, M.-S.; Nam, T.; Jeong, B.-S. Tocopherol side chain synthesis via asymmetric organocatalytic transfer hydrogenation and convenient measurement of stereoselectivity. Tetrahedron Lett. 2014, 55, 5895–5897. [Google Scholar] [CrossRef]
- Cerecetto, H.; Lopez, G. Antioxidants Derived from Vitamin E: An Overview. Mini-Rev. Med. Chem. 2007, 7, 315–338. [Google Scholar] [CrossRef] [PubMed]
- Olcott, H.S.; Emerson, O.H. Antioxidants and the Autoxidation of Fats. IX. The Antioxidant Properties of the Tocopherols. J. Am. Chem. Soc. 1937, 59, 1008–1009. [Google Scholar] [CrossRef]
- Burton, G.W.; Doba, T.; Gabe, E.; Hughes, L.; Lee, F.L.; Prasad, L.; Ingold, K.U. Autoxidation of biological molecules. 4. Maximizing the antioxidant activity of phenols. J. Am. Chem. Soc. 1985, 107, 7053–7065. [Google Scholar] [CrossRef]
- Ingold, K.U.; Pratt, D.A. Advances in Radical-Trapping Antioxidant Chemistry in the 21st Century: A Kinetics and Mechanisms Perspective. Chem. Rev. 2014, 114, 9022–9046. [Google Scholar] [CrossRef]
- Marteau, C.; Favier, D.; Nardello-Rataj, V.; Aubry, J.-M. Dramatic solvent effect on the synergy between α-tocopherol and BHT antioxidants. Food Chem. 2014, 160, 190–195. [Google Scholar] [CrossRef]
- Burton, G.W. Vitamin E: Molecular and biological function. Proc. Nutr. Soc. 1994, 53, 251–262. [Google Scholar] [CrossRef]
- Mulder, P.; Korth, H.-G.; Ingold, K. Why Quantum-Thermochemical Calculations Must Be Used with Caution to Indicate “a Promising Lead Antioxidant”. Helv. Chim. Acta 2005, 88, 370–374. [Google Scholar] [CrossRef]
- Howard, J.A. Free Radicals; Kochi, J.K., Ed.; Wiley-Interscience, Wiley: New York, NY, USA, 1973; Volume 2, p. 906. [Google Scholar]
- Howard, J.A.; Ingold, K.U. Absolute constants for hydrocarbon autoxidation I. Styrene. Can. J. Chem. 1965, 43, 2729–2736. [Google Scholar] [CrossRef]
- Burton, G.W.; Ingold, K.U. Vitamin E: Application of the principles of physical organic chemistry to the exploration of its structure and function. Acc. Chem. Res. 1986, 19, 194–201. [Google Scholar] [CrossRef]
- Burton, G.W.; Ingold, K.U. Autoxidation of biological molecules. 1. Antioxidant activity of vitamin E and related chain-breaking phenolic antioxidants in vitro. J. Am. Chem. Soc. 1981, 103, 6472–6477. [Google Scholar] [CrossRef]
- Upston, J.M.; Terentis, A.C.; Stocker, R. Tocopherol-mediated peroxidation of lipoproteins: Implications for vitamin E as a potential antiatherogenic supplement. Faseb J. 1999, 13, 977–994. [Google Scholar] [CrossRef] [PubMed]
- Poljšak, B.; Bučar-Miklavčič, M.; Glavan, U.; Butinar, B. Pro-oxidant effects of vitamin E. In Tocopherol: Sources, Uses and Health Benefits; Nova Science Publishers Inc.: New York, NY, USA, 2012; pp. 117–139. [Google Scholar]
- Bowry, V.W.; Ingold, K.U.; Stocker, R. Vitamin E in human low-density lipoprotein. When and how this antioxidant becomes a pro-oxidant. Biochem. J. 1992, 288, 341–344. [Google Scholar] [CrossRef]
- Ricciarelli, R.; Zingg, J.-M.; Azzi, A. Vitamin E: Protective role of a Janus molecule. Faseb J. 2001, 15, 2314–2325. [Google Scholar] [CrossRef]
- Ingold, K.U.; Burton, G.W.; Foster, D.O.; Zuker, M.; Hughes, L.; Lacelle, S.; Lusztyk, E.; Slaby, M. A new vitamin E analogue more active than α-tocopherol in the rat curative myopathy bioassay. Febs Lett. 1986, 205, 117–120. [Google Scholar] [CrossRef]
- Burton, G.W.; Hughes, L.; Ingold, K.U. Antioxidant activity of phenols related to vitamin E. Are there chain-breaking antioxidants better than α-tocopherol? J. Am. Chem. Soc. 1983, 105, 5950–5951. [Google Scholar] [CrossRef]
- Doba, T.; Burton, G.W.; Ingold, K.U. EPR spectra of some α-tocopherol model compounds. Polar and conformational effects and their relation to antioxidant activities. J. Am. Chem. Soc. 1983, 105, 6505–6506. [Google Scholar] [CrossRef]
- Baj, A.; Wałejko, P.; Witkowski, S. Synthesis of new carbacyclic analogs of trolox. Monatshefte Chem.-Chem. Mon. 2014, 146, 375–382. [Google Scholar] [CrossRef]
- Wałejko, P.; Paradowska, K.; Szeleszczuk, Ł.; Wojtulewski, S.; Baj, A. Racemic crystals of trolox derivatives compared to their chiral counterparts: Structural studies using solid-state NMR, DFT calculations and X-ray diffraction. J. Mol. Struct. 2018, 1156, 290–300. [Google Scholar] [CrossRef]
- Smith, L.I.; Opie, J.W.; Wawzonek, S.; Prichard, W.W. The chemistry of vitamin E. VII. Preparation of quinones from methylphenols. J. Org. Chem. 1939, 4, 318–322. [Google Scholar] [CrossRef]
- Jodko-Piorecka, K.; Cedrowski, J.; Litwinienko, G. Measurement of Antioxidant Activity and Capacity: Recent Trends and Applications; Apak, R., Çapanoğlu, E., Shahidi, F., Eds.; Wiley: Hoboken, NJ, USA, 2018. [Google Scholar]
- Lucarini, M.; Pedulli, G.F. Free radical intermediates in the inhibition of the autoxidation reaction. Chem. Soc. Rev. 2010, 39, 2106. [Google Scholar] [CrossRef] [PubMed]
- Boozer, C.E.; Hammond, G.S.; Hamilton, C.E.; Peterson, C. Air Oxidation of Hydrocarbons. IV. The Effects of Varying Solvent and the Mechanism of Uninhibited Chain Termination. J. Am. Chem. Soc. 1955, 77, 3380–3383. [Google Scholar] [CrossRef]
- Olchowik-Grabarek, E.; Mavlyanov, S.; Abdullajanova, N.; Gieniusz, R.; Zamaraeva, M. Specificity of Hydrolysable Tannins from Rhus typhina L. to Oxidants in Cell and Cell-Free Models. Appl. Biochem. Biotechnol. 2017, 181, 495–510. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Wałejko, P.; Dobrzycki, Ł.; Ratkiewicz, A.; Socha, P.; Witkowski, S.; Cyrański, M.K. An X-ray and Natural Bond Orbital (NBO) structural study of α-tocopheryl and 2,2,5,7,8-pentamethylchroman-6-yl succinates. J. Saudi Chem. Soc. 2019, 23, 365–377. [Google Scholar] [CrossRef]
- Barone, V.; Cossi, M.; Tomasi, J. Geometry optimization of molecular structures in solution by the polarizable continuum model. J. Comput. Chem. 1998, 19, 404–417. [Google Scholar] [CrossRef]
- Wojtkielewicz, A.; Uścinowicz, P.; Siergiejczyk, L.; Kiełczewska, U.; Ratkiewicz, A.; Morzycki, J.W. A study on the reaction of 16-dehydropregnenolone acetate with 2-aminobenzimidazole. Steroids 2017, 117, 71–76. [Google Scholar] [CrossRef]
- Lazny, R.; Ratkiewicz, A.; Brzezinski, K.; Nodzewska, A.; Sidorowicz, K. An Investigation of the Enolization and Isomeric Products Distribution in the Water Promoted Aldol Reaction of Tropinone and Granatanone. J. Chem. 2016, 2016, 4674901. [Google Scholar] [CrossRef]
- Klein, E.; Lukeš, V.; Ilčin, M. DFT/B3LYP study of tocopherols and chromans antioxidant action energetics. Chem. Phys. 2007, 336, 51–57. [Google Scholar] [CrossRef]
- Fifen, J.J. Thermodynamics of the Electron Revisited and Generalized. J. Chem. Theory Comput. 2013, 9, 3165–3169. [Google Scholar] [CrossRef]
- Fifen, J.J.; Dhaouadi, Z.; Nsangou, M. Revision of the Thermodynamics of the Proton in Gas Phase. J. Phys. Chem. A 2014, 118, 11090–11097. [Google Scholar] [CrossRef] [PubMed]
- Mulder, P.; Korth, H.-G.; Pratt, D.A.; DiLabio, G.A.; Valgimigli, L.; Pedulli, G.F.; Ingold, K.U. Critical Re-evaluation of the O-H Bond Dissociation Enthalpy in Phenol. J. Phys. Chem. A 2005, 109, 2647–2655. [Google Scholar] [CrossRef]
- Valgimigli, L.; Banks, J.T.; Lusztyk, J.; Ingold, K.U. Solvent Effects on the Antioxidant Activity of Vitamin E. J. Org. Chem. 1999, 64, 3381–3383. [Google Scholar] [CrossRef] [PubMed]
- Culbertson, S.M.; Antunes, F.; Havrilla, C.M.; Milne, G.L.; Porter, N.A. Determination of the α-Tocopherol Inhibition Rate Constant for Peroxidation in Low-Density Lipoprotein. Chem. Res. Toxicol. 2002, 15, 870–876. [Google Scholar] [CrossRef] [PubMed]
- Müller, L.; Theile, K.; Böhm, V. In vitro antioxidant activity of tocopherols and tocotrienols and comparison of vitamin E concentration and lipophilic antioxidant capacity in human plasma. Mol. Nutr. Food Res. 2010, 54, 731–742. [Google Scholar] [CrossRef]
- Musialik, M.; Litwinienko, G. Scavenging of dpph∙ Radicals by Vitamin E Is Accelerated by Its Partial Ionization: The Role of Sequential Proton Loss Electron Transfer. Org. Lett. 2005, 7, 4951–4954. [Google Scholar] [CrossRef]
- Navarrete, M.; Rangel, C.; Corchado, J.C.; Espinosa-García, J. Trapping of the OH Radical by α-Tocopherol: A Theoretical Study. J. Phys. Chem. A 2005, 109, 4777–4784. [Google Scholar] [CrossRef]
- Navarrete, M.; Rangel, C.; Espinosa-García, J.; Corchado, J.C. Theoretical Study of the Antioxidant Activity of Vitamin E: Reactions of α-Tocopherol with the Hydroperoxy Radical. J. Chem. Theory Comput. 2005, 1, 337–344. [Google Scholar] [CrossRef]
- Bakhouche, K.; Dhaouadi, Z.; Jaidane, N.; Hammoutène, D. Comparative antioxidant potency and solvent polarity effects on HAT mechanisms of tocopherols. Comput. Theor. Chem. 2015, 1060, 58–65. [Google Scholar] [CrossRef]
- Guitard, R.; Nardello-Rataj, V.; Aubry, J.-M. Theoretical and Kinetic Tools for Selecting Effective Antioxidants: Application to the Protection of Omega-3 Oils with Natural and Synthetic Phenols. Int. J. Mol. Sci. 2016, 17, 1220. [Google Scholar] [CrossRef]
- Farmanzadeh, D.; Najafi, M. Novel Trolox derivatives as antioxidant: A DFT investigation. J. Serb. Chem. Soc. 2016, 81, 277–290. [Google Scholar] [CrossRef]
- Foti, M.C. Use and Abuse of the DPPH∙ Radical. J. Agric. Food Chem. 2015, 63, 8765–8776. [Google Scholar] [CrossRef] [PubMed]
- Amorati, R.; Fumo, M.G.; Menichetti, S.; Mugnaini, V.; Pedulli, G.F. Electronic and Hydrogen Bonding Effects on the Chain-Breaking Activity of Sulfur-Containing Phenolic Antioxidants. J. Org. Chem. 2006, 71, 6325–6332. [Google Scholar] [CrossRef] [PubMed]
- Barclay, L.R.C.; Vinqvist, M.R.; Mukai, K.; Itoh, S.; Morimoto, H. Chain-Breaking Phenolic Antioxidants: Steric and Electronic Effects in Polyalkylchromanols, Tocopherol Analogs, Hydroquinones, and Superior Antioxidants of the Polyalkylbenzochromanol and Naphthofuran. J. Org. Chem. 1993, 58, 7416–7420. [Google Scholar] [CrossRef]
- Amorati, R.; Valgimigli, L. Advantages and limitations of common testing methods for antioxidants. Free Radic. Res. 2015, 49, 633–649. [Google Scholar] [CrossRef] [PubMed]
- Marković, Z.; Tošović, J.; Milenković, D.; Marković, S. Revisiting the solvation enthalpies and free energies of the proton and electron in various solvents. Comput. Theor. Chem. 2016, 1077, 11–17. [Google Scholar] [CrossRef]
- Wright, J.S.; Johnson, E.R.; DiLabio, G.A. Predicting the Activity of Phenolic Antioxidants: Theoretical Method, Analysis of Substituent Effects, and Application to Major Families of Antioxidants. J. Am. Chem. Soc. 2001, 123, 1173–1183. [Google Scholar] [CrossRef]
- Litwinienko, G.; Megiel, E.; Wojnicz, M. Hydrogen Bonding between Phenols and Fatty Acid Esters: 1H NMR Study and ab Initio Calculations. Org. Lett. 2002, 4, 2425–2428. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structure | Phenol | 105 kinh [M−1 s−1] |
|---|---|---|
 | α-T: R1 = CH3, R2 = CH3 | 32.0 |
| β-T: R1 = CH3, R2 = H | 13.0 | |
| γ-T: R1 = H, R2 = CH3 | 14.0 | |
| δ-T: R1 = H, R2 = H | 4.4 | |
 | 1a: R = COOH (Trolox) | 11.0 |
| 1b: R = COOCH3 (Trolox methyl ester) | 18.0 | |
| 1c: R = CH3 (PMHC) | 38.0 | |
| 1d: R = (CH2)2COOH | 37.0 | |
 | 2a: R = H | 54.0 |
| 2b: R = CH3 | 57.0 | |
| 2c: R = COOH | 16.0 | |
| 2d: R = C16H33 [18] | 47.0 | |
 | 3a: R1 = H, R2 = OCH3, R3 = CH3 | 13.0 |
| 3b: R1 = H, R2 = OCH3, R3 = H | 9.4 | |
| 3c: R1 = CH3, R2 = OCH3, R3 = CH3 | 3.9 | |
| 3d: R1 = CH3, R2 = CH3, R3 = CH3 | 3.6 |
| Phenol | kinh [M−1s−1] | n ** |
|---|---|---|
| Autoxidation of Styrene | ||
| 1a | (9.0 ± 0.4) × 105 | 1.6 |
| 1b | (1.5 ± 0.1) × 106 | 1.7 |
| α-T | (2.0 ± 0.2) × 106 | 1.9 |
| 1c | (2.2 ± 0.1) × 106 | 1.9 |
| 4 | (2.8 ± 0.1) × 105 | 1.6 |
| 5 | (3.1 ± 0.2) × 105 | 1.4 |
| 6 | (2.3 ± 0.1) × 105 | 1.5 |
| Autoxidation of Cumene | ||
| 4 | (2.6 ± 0.4) × 105 | 1.5 |
| 5 | (3.1 ± 0.2) × 105 | 1.7 |
| 6 | (2.3 ± 0.5) × 105 | 1.5 |
| Compound | EC50 [µM] | ARP [1/EC50 × 10−3] |
|---|---|---|
| 1a | 48.0 ± 0.1 | 20.8 ± 0.1 |
| 1b | 68.9 ± 2.3 | 14.5 ± 0.5 |
| 4 | 106.8 ± 2.1 | 9.4 ± 0.2 |
| 5 | 75.1 ± 3.3 | 13.3 ± 0.6 |
| 6 | 46.8 ± 3.7 | 21.5 ± 1.7 |
| Environment | Compound | BDE | IP | PDE | PA | ETE |
|---|---|---|---|---|---|---|
| Gas phase | 1a | 78.3 | 161.6 | 231.2 | 350.2 | 39.9 |
| 1b | 78.1 | 161.2 | 231.4 | 350.9 | 40.0 | |
| α-T | 77.3* | 157.2 | 234.5 | 352.2 | 38.2 | |
| 4 | 81.2 | 171.2 | 224.6 | 348.5 | 45.3 | |
| 5 | 81.3 | 171.6 | 224.2 | 348.2 | 45.4 | |
| 6 | 81.0 | 168.6 | 226.9 | 350.1 | 43.4 | |
| Chlorobenzene | 1a | 77.2 | 132.2 | 259.4 | 311.6 | 77.3 |
| 1b | 77.1 | 132.0 | 259.6 | 311.6 | 78.1 | |
| α-T | 75.9 | 128.6 | 261.8 | 312.8 | 75.8 | |
| 4 | 80.3 | 139.8 | 255.0 | 310.7 | 81.9 | |
| 5 | 80.4 | 139.9 | 255.0 | 310.7 | 82.0 | |
| 6 | 79.9 | 138.1 | 256.3 | 311.6 | 80.7 | |
| Ethanol | 1a | 76.9 | 127.4 | 263.9 | 304.6 | 84.0 |
| 1b | 76.8 | 127.4 | 263.9 | 304.6 | 84.7 | |
| α-T | 75.4 | 124.6 | 265.3 | 305.5 | 82.7 | |
| 4 | 80.1 | 134.6 | 260.0 | 303.9 | 88.4 | |
| 5 | 80.1 | 135.0 | 259.7 | 303.9 | 88.5 | |
| 6 | 79.6 | 133.6 | 260.4 | 304.5 | 87.4 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baj, A.; Cedrowski, J.; Olchowik-Grabarek, E.; Ratkiewicz, A.; Witkowski, S. Synthesis, DFT Calculations, and In Vitro Antioxidant Study on Novel Carba-Analogs of Vitamin E. Antioxidants 2019, 8, 589. https://doi.org/10.3390/antiox8120589
Baj A, Cedrowski J, Olchowik-Grabarek E, Ratkiewicz A, Witkowski S. Synthesis, DFT Calculations, and In Vitro Antioxidant Study on Novel Carba-Analogs of Vitamin E. Antioxidants. 2019; 8(12):589. https://doi.org/10.3390/antiox8120589
Chicago/Turabian StyleBaj, Aneta, Jakub Cedrowski, Ewa Olchowik-Grabarek, Artur Ratkiewicz, and Stanislaw Witkowski. 2019. "Synthesis, DFT Calculations, and In Vitro Antioxidant Study on Novel Carba-Analogs of Vitamin E" Antioxidants 8, no. 12: 589. https://doi.org/10.3390/antiox8120589
APA StyleBaj, A., Cedrowski, J., Olchowik-Grabarek, E., Ratkiewicz, A., & Witkowski, S. (2019). Synthesis, DFT Calculations, and In Vitro Antioxidant Study on Novel Carba-Analogs of Vitamin E. Antioxidants, 8(12), 589. https://doi.org/10.3390/antiox8120589