Impact of Thermal Degradation of Cyanidin-3-O-Glucoside of Haskap Berry on Cytotoxicity of Hepatocellular Carcinoma HepG2 and Breast Cancer MDA-MB-231 Cells



Abstract
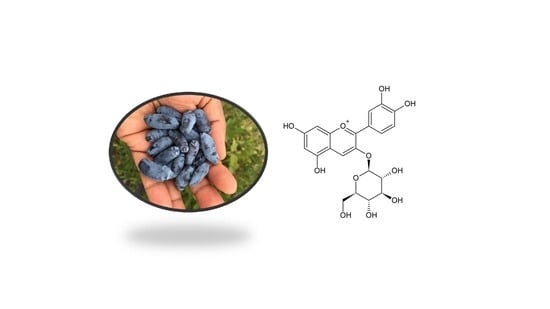
1. Introduction
2. Materials and Methods
2.1. Plant Material
2.2. Chemicals and Reagents
2.3. Preparation of Extracts
2.3.1. Preparation of Crude Extract
2.3.2. Preparation of C3G-Rich Fraction
2.4. Degradation Kinetics
2.4.1. Sample Preparation
2.4.2. Quantification of Total Monomeric Anthocyanins, C3G, PCA, and PGA
2.4.3. Statistical Analysis
2.5. Antiproliferative Activity
2.5.1. Preparation of Extracts
2.5.2. Cell Culture
2.5.3. Cell Viability MTS Assay
2.5.4. Acid Phosphatase Assay
2.5.5. Adenosine Triphosphate (ATP) Assay
2.5.6. Statistical Analysis
3. Results
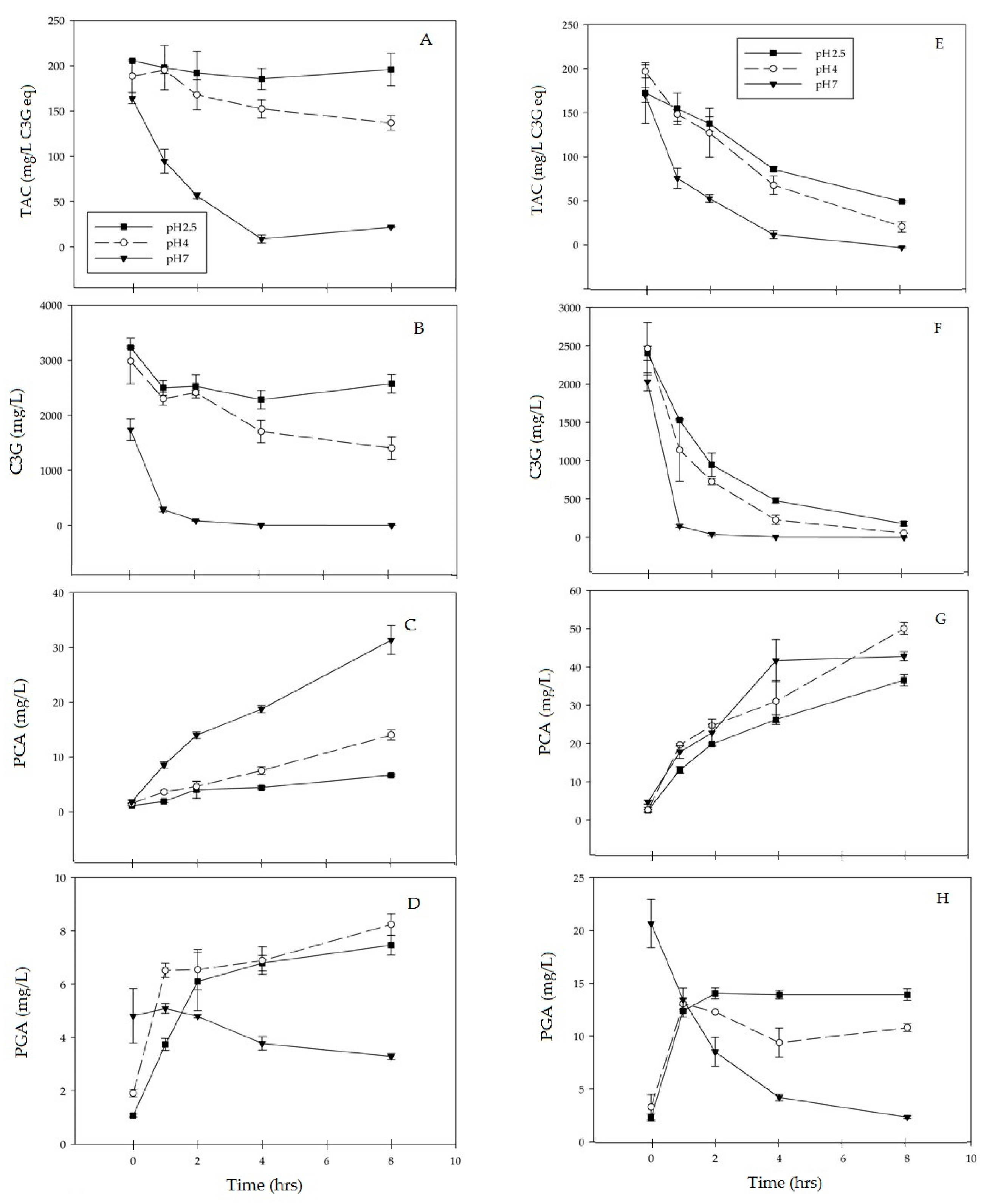
3.1. C3G Degradation
3.2. Formation of PCA
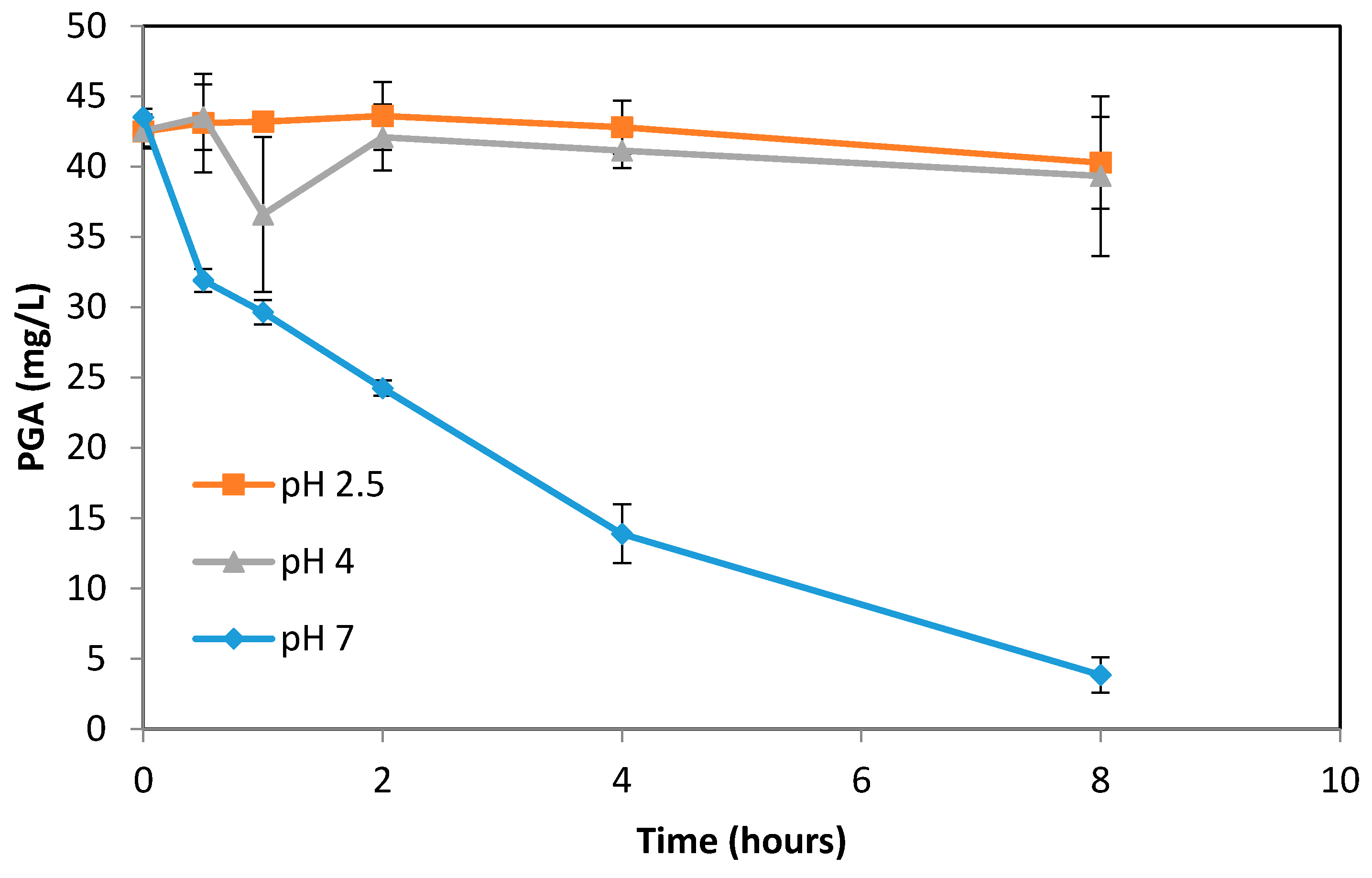
3.3. Formation of PGA
3.4. Breast Adenocarcinoma MDA-MB-231 Viability
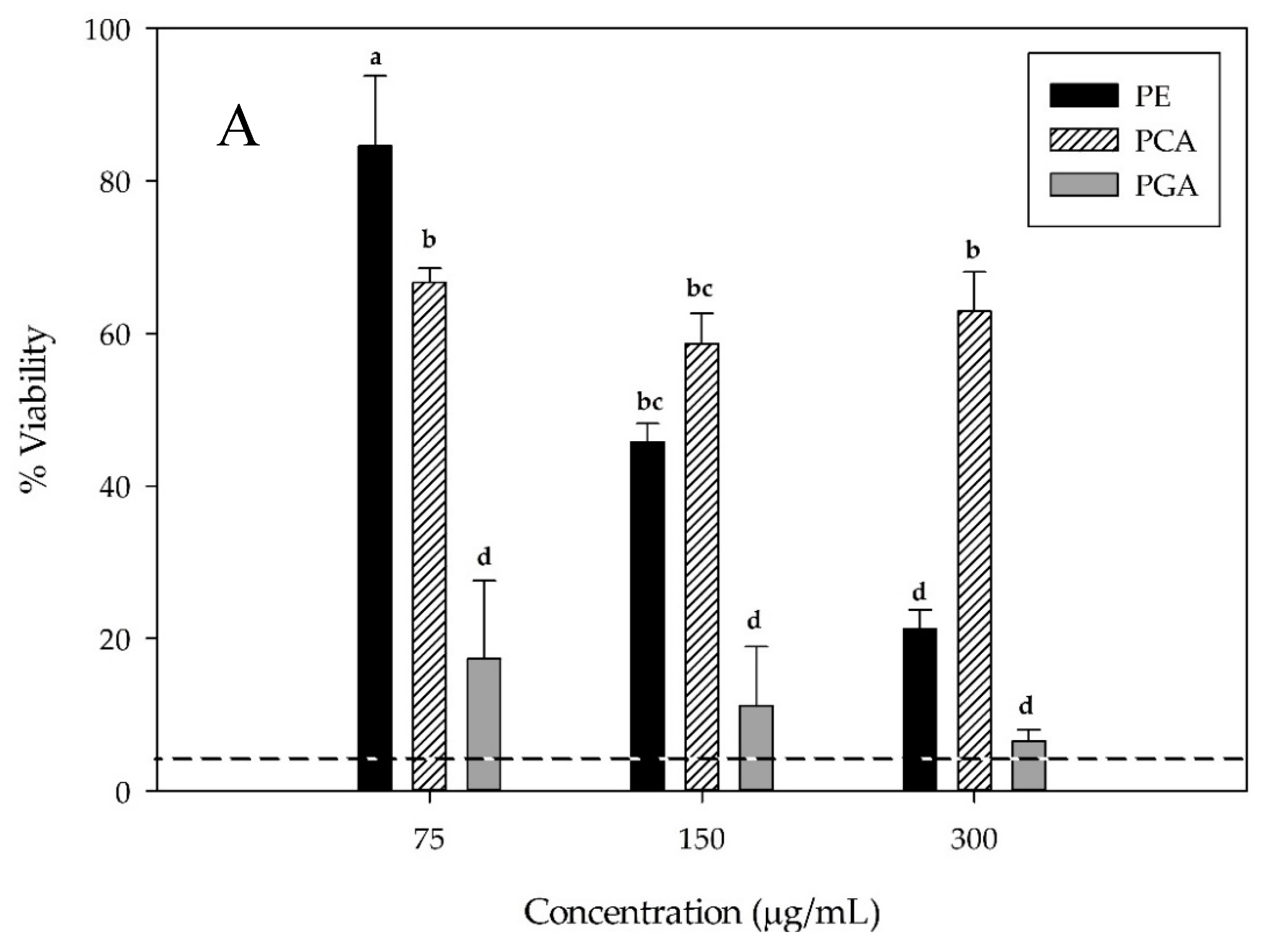
3.5. Hepatocellular Carcinoma HepG2 Cell Viability
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Rupasinghe, H.P.V.; Yu, L.J.; Bhullar, K.S.; Bors, B. Haskap (Lonicera caerulea): A new berry crop with high antioxidant capacity. Can. J. Plant Sci. 2012, 92, 1311–1317. [Google Scholar] [CrossRef]
- Celli, G.B.; Ghanem, A.; Brooks, M.S.L. Haskap berries (Lonicera caerulea L.)—A critical review of antioxidant capacity and health-related studies for potential value-added products. Food Bioprocess Technol. 2014, 7, 1541–1554. [Google Scholar] [CrossRef]
- Khattab, R.; Brooks, M.S.; Ghanem, A. Phenolic analyses of haskap berries (Lonicera caerulea L.): Spectrophotometry versus high performance liquid chromatography. Int. J. Food Prop. 2016, 19, 1708–1725. [Google Scholar] [CrossRef]
- Chen, L.; Xin, X.; Yuan, Q.; Su, D.; Liu, W. Phytochemical properties and antioxidant capacities of various colored berries. J. Sci. Food Agric. 2014, 94, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Fan, Z.L.; Wang, Z.Y.; Liu, J.R. Cold-field fruit extracts exert different antioxidant and antiproliferative activities in vitro. Food Chem. 2011, 129, 402–407. [Google Scholar] [CrossRef]
- Sun, C.; Zheng, Y.; Chen, Q.; Tang, X.; Jiang, M.; Zhang, J.; Li, X.; Chen, K. Purification and anti-tumour activity of cyanidin-3-O-glucoside from Chinese bayberry fruit. Food Chem. 2012, 131, 1287–1294. [Google Scholar] [CrossRef]
- Liu, W.; Xu, J.; Wu, S.; Liu, Y.; Yu, X.; Chen, J.; Tang, X.; Wang, Z.; Zhu, X.; Li, X. Selective anti-proliferation of HER2-positive breast cancer cells by anthocyanins identified by high-throughput screening. PLoS ONE 2013, 8, e81586. [Google Scholar] [CrossRef] [PubMed]
- Serra, D.; Paixão, J.; Nunes, C.; Dinis, T.C.P.; Almeida, L.M. Cyanidin-3-glucoside suppresses cytokine-induced inflammatory response in human intestinal cells: Comparison with 5-aminosalicylic acid. PLoS ONE 2013, 8, e73001. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.G.; Yan, Q.-Q.; Lu, L.-Z.; Zhang, Y.-Q. In vivo antioxidant, hypoglycemic, and anti-tumor activities of anthocyanin extracts from purple sweet potato. Nutr. Res. Pract. 2013, 7, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Aboonabi, A.; Singh, I. Chemopreventive role of anthocyanins in atherosclerosis via activation of Nrf2–ARE as an indicator and modulator of redox. Biomed. Pharmacother. 2015, 72, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Rupasinghe, H.P.V.; Boehm, M.M.; Sekhon-Loodu, S.; Parmar, I.; Bors, B.; Jamieson, A.R. Anti-inflammatory activity of haskap cultivars is polyphenols-dependent. Biomolecules 2015, 5, 1079–1098. [Google Scholar] [CrossRef] [PubMed]
- Roleira, F.M.F.; Tavares-da-Silva, E.J.; Varela, C.L.; Costa, S.C.; Silva, T.; Garrido, J.; Borges, F. Plant derived and dietary phenolic antioxidants: Anticancer properties. Food Chem. 2015, 183, 235–258. [Google Scholar] [CrossRef] [PubMed]
- Duthie, S.J.; Gardner, P.T.; Morrice, P.C.; Wood, S.G.; Pirie, L.; Bestwick, C.C.; Milne, L.; Duthie, G.G. DNA stability and lipid peroxidation in vitamin E-deficient rats in vivo and colon cells in vitro: Modulation by the dietary anthocyanin, cyanidin-3-glycoside. Eur. J. Nutr. 2005, 44, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Fantini, M.; Benvenuto, M.; Masuelli, L.; Frajese, G.; Tresoldi, I.; Modesti, A.; Bei, R. In vitro and in vivo antitumoral effects of combinations of polyphenols, or polyphenols and anticancer drugs: Perspectives on cancer treatment. Int. J. Mol. Sci. 2015, 16, 9236–9282. [Google Scholar] [CrossRef] [PubMed]
- Hui, C.; Bin, Y.; Xiaoping, Y.; Long, Y.; Chunye, C.; Mantian, M. Anticancer activities of an anthocyanin-rich extract from black rice against breast cancer cells in vitro and in vivo. Nutr. Cancer 2010, 62, 1128–1136. [Google Scholar] [CrossRef] [PubMed]
- Kopjar, M.; Piližota, V. Prevention of thermal degradation of anthocyanins in blackberry juice with addition of different sugars. CyTA-J. Food 2011, 9, 237–242. [Google Scholar] [CrossRef]
- Kamiloglu, S.; Capanoglu, E.; Grootaert, C.; Van Camp, J. Anthocyanin absorption and metabolism by human intestinal caco-2 cells—A review. Int. J. Mol. Sci. 2015, 16, 21555–21574. [Google Scholar] [CrossRef] [PubMed]
- Chaovanalikit, A.; Thompson, M.M.; Wrolstad, R.E. Characterization and quantification of anthocyanins and polyphenolics in blue honeysuckle (Lonicera caerulea L.). J. Agric. Food Chem. 2004, 52, 848–852. [Google Scholar] [CrossRef] [PubMed]
- Kusznierewicz, B.; Piekarska, A.; Mrugalska, B.; Konieczka, P.; Namies, J. Phenolic composition and antioxidant properties of polish blue-berried honeysuckle genotypes by HPLC-DAD-MS, HPLC postcolumn derivatization with ABTS or FC, and TLC with DPPH visualization. J. Agric. Food Chem. 2012, 60, 1755–1763. [Google Scholar] [CrossRef] [PubMed]
- Sadilova, E.; Stintzing, F.C.; Carle, R. Thermal degradation of acylated and nonacylated anthocyanins. J. Food Sci. 2006, 71, 504–512. [Google Scholar] [CrossRef]
- Liu, C.L.; Wang, J.M.; Chu, C.Y.; Cheng, M.T.; Tseng, T.H. In vivo protective effect of protocatechuic acid on tert-butyl hydroperoxide-induced rat hepatotoxicity. Food Chem. Toxicol. 2002, 40, 635–641. [Google Scholar] [CrossRef]
- Babich, H.; Sedletcaia, A.; Kenigsberg, B. In vitro cytotoxicity of protocatechuic acid to cultured human cells from oral tissue: Involvement in oxidative stress. Pharmacol. Toxicol. 2002, 91, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Torikai, K.; Ohto, Y.; Murakami, A.; Tanaka, T.; Ohigashi, H. A simple phenolic antioxidant protocatechuic acid enhances tumor promotion and oxidative stress in female ICR mouse skin: Dose-and timing-dependent enhancement and involvement of bioactivation by tyrosinase. Carcinogenesis 2000, 21, 1899–1907. [Google Scholar] [CrossRef] [PubMed]
- Forester, S.C.; Waterhouse, A.L. Gut metabolites of anthocyanins, gallic acid, 3-O-methylgallic proliferation of caco-2 cells. J. Agric. Food Chem. 2010, 58, 5320–5327. [Google Scholar] [CrossRef] [PubMed]
- Forester, S.C.; Oteiza, P.; Waterhouse, A.L.; Forester, S.C.; Oteiza, P.; Waterhouse, A.L. The anthocyanin metabolites gallic acid, 3-O-methylgallic acid, and 2,4,6-trihydroxybenzaldehyde decrease human colon cancer cell viability by regulating pro-oncogenic signals. Mol. Carcinog. 2014, 53, 432–439. [Google Scholar] [CrossRef] [PubMed]
- De Ferrars, R.M.; Czank, C.; Zhang, Q.; Botting, N.P.; Kroon, P.A.; Cassidy, A.; Kay, C.D. The pharmacokinetics of anthocyanins and their metabolites in humans. Br. J. Pharmacol. 2014, 171, 3268–3282. [Google Scholar] [CrossRef] [PubMed]
- Czank, C.; Cassidy, A.; Zhang, Q.; Morrison, D.J.; Preston, T.; Kroon, P.A.; Botting, N.P.; Kay, C.D. Human metabolism and elimination of the anthocyanin, cyanidin-3-glucoside: A 13C-Tracer study. Am. J. Clin. Nutr. 2013, 97, 995–1003. [Google Scholar] [CrossRef] [PubMed]
- Bhullar, K.S.; Rupasinghe, H.P.V. Antioxidant and cytoprotective properties of partridgeberry polyphenols. Food Chem. 2015, 168, 595–605. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Durst, R.W.; Wrolstad, R.E. Determination of total monomeric anthocyanin pigment content of fruit juices, beverages, natural colorants, and wines by the pH differential method: Collaborative study. J. AOAC Int. 2005, 88, 1269–1278. [Google Scholar] [PubMed]
- Liu, L.; Cao, S.Q.; Pan, S.Y. Thermal degradation kinetics of three kinds of representative anthocyanins obtained from blood orange. Agric. Sci. China 2011, 10, 642–649. [Google Scholar] [CrossRef]
- Nayak, B.; Liu, R.H.; Tang, J. Effect of processing on phenolic antioxidants of effect of processing on phenolic antioxidants of fruits, vegetables, and grains—A review. Crit. Rev. Food Sci. Nutr. 2015, 55, 887–918. [Google Scholar] [CrossRef] [PubMed]
- Olivas-Aguirre, F.J.; Rodrigo-García, J.; Martínez-Ruiz, N.R.; Cárdenas-Robles, A.I.; Mendoza-Díaz, S.O.; Álvarez-Parrilla, E.; González-Aguilar, G.A.; de la Rosa, L.A.; Ramos-Jiménez, A.; Wall-Medrano, A. Cyanidin-3-O-glucoside: Physical-Chemistry, foodomics and health effects. Molecules 2016, 21, 1264. [Google Scholar] [CrossRef] [PubMed]
- Patras, A.; Brunton, N.P.; Donnell, C.O.; Tiwari, B.K. Effect of thermal processing on anthocyanin stability in foods; mechanisms and kinetics of degradation. Trends Food Sci. Technol. 2010, 21, 3–11. [Google Scholar] [CrossRef]
- Brenes, C.H.; Del Pozo-Insfran, D.; Talcott, S.T. Stability of copigmented anthocyanins and ascorbic acid in a grape juice model system. J. Agric. Food Chem. 2005, 53, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Zorić, Z.; Dragović-Uzelac, V.; Pedisić, S.; Kurtanjek, Ž.; Garofulić, I.E. Kinetics of the degradation of anthocyanins, phenolic acids and flavonols during heat treatments of freeze-dried sour cherry Marasca paste. Food Technol. Biotechnol. 2014, 52, 101–108. [Google Scholar]
- Khattab, R.; Ghanem, A.; Brooks, M.S.-L. Stability of haskap berry (Lonicera caerulea L.) anthocyanins at different storage and processing conditions. J. Food Res. 2016, 5, 67. [Google Scholar] [CrossRef]
- Sadilova, E.; Carle, R.; Stintzing, F.C. Thermal degradation of anthocyanins and its impact on color and in vitro antioxidant capacity. Mol. Nutr. Food Res. 2007, 51, 1461–1471. [Google Scholar] [CrossRef] [PubMed]
- Arslan, D. Effects of degradation preventive agents on storage stability of anthocyanins in sour cherry concentrate. Agron. Res. 2015, 13, 892–899. [Google Scholar]
- Yin, M.; Lin, C.-C.; Wu, H.; Tsao, S.; Hsu, C.-K. Apoptotic effects of protocatechuic acid in human breast, lung, liver, cervix, and prostate cancer cells: Potential mechanisms of action. J. Agric. Food Chem. 2009, 57, 6468–6473. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment | C3G (µg/mL) | PCA (µg/mL) | PGA (µg/mL) |
|---|---|---|---|
| PE | 59.92 ± 1.6 | 0.28 ± 0.001 | 0.023 ± 0.0002 |
| HPE2 | 34.23 ± 3.3 | 0.77 ± 0.04 | 0.29 ± 0.01 |
| HPE8 | 5.76 ± 0.5 | 2.02 ± 0.2 | 0.467 ± 0.01 |
| CE | 11.35 ± 0.1 | N/A | N/A |
| Treatment | Concentration | 24 h | 48 h |
|---|---|---|---|
| (µg/mL) | (% Viability ± SD) | (% Viability ± SD) | |
| MDA-MB-231 | |||
| PE | 50 | 97.43 ± 2.04 ab | 103.10 ± 10.17 a |
| 100 | 107.02 ± 6.71 ab | 105.15 ± 5.29 a | |
| 200 | 109.86 ± 1.50 ab | 106.52 ± 6.50 a | |
| HPE2 | 50 | 100.22 ± 2.14 cd | 98.34 ± 0.91 a |
| 100 | 104.96 ± 1.42 ab | 106.43 ± 4.20 a | |
| 200 | 108.38 ± 4.16 ab | 94.13 ± 8.44 a | |
| HPE8 | 50 | 103.82 ± 2.48 ab | 107.68 ± 6.24 a |
| 100 | 104.19 ± 4.13 ab | 104.78 ± 4.45 a | |
| 200 | 100.55 ± 3.11 ab | 85.10 ± 11.13 a | |
| CE | 50 | 103.31 ± 4.98 ab | 105.37 ± 3.46 a |
| 100 | 99.33 ± 3.24 ab | 98.99 ± 12.10 a | |
| 200 | 98.73 ± 2.18 ab | 99.16 ± 3.86 a | |
| PCA | 50 | 101.18 ± 11.40 ab | 94.43 ± 6.65 a |
| 100 | 101.08 ± 8.12 ab | 81.96 ± 13.33 a | |
| 200 | 107.89 ± 10.21 ab | 86.96 ± 29.52 a | |
| PGA | 50 | 78.22 ± 2.37 bc | 80.35 ± 5.54 a |
| 100 | 35.30 ± 26.66 d | 36.25 ± 5.11 bc | |
| 200 | 7.09 ± 8.93 e | 17.71 ± 13.55 c | |
| Dox | 27.2 | 53.30 ± 13.56 cd | 7.89 ± 7.29 c |
| HepG2 | |||
| PE | 75 | 89.26 ± 11.01 bc | 72.9 ± 3.07 cd |
| 150 | 88.39 ± 2.43 bc | 70.97 ± 7.32 d | |
| 300 | 64.32 ± 14.33 cd | 35.57 ± 11.55 e | |
| HPE2 | 75 | 105.92 ± 7.78 ab | 92.09 ± 4.61 abc |
| 150 | 104.28 ± 9.19 ab | 91.26 ± 5.18 abcd | |
| 300 | 97.34 ± 8.48 ab | 81.58 ± 6.69 bcd | |
| HPE8 | 75 | 105.64 ± 8.35 ab | 90.15 ± 7.96 abcd |
| 150 | 102.85 ± 5.86 ab | 81.65 ± 10.15 bcd | |
| 300 | 103.35 ± 5.08 ab | 80.07 ± 6.22 bcd | |
| CE | 75 | 102.92 ± 3.59 ab | 95.2 ± 5.03 ab |
| 150 | 102.59 ± 7.62 ab | 94.67 ± 3.6 ab | |
| 300 | 104.76 ± 4.90 ab | 93.35 ± 3.29 abc | |
| PCA | 75 | 84.98 ± 4.04 bc | 90.943 ± 1.6 abcd |
| 150 | 92.41 ± 8.27 ab | 89.99 ± 7.21 abcd | |
| 300 | 114.06 ± 7.53 ab | 108.66 ± 7.77 a | |
| PGA | 75 | 52.85 ± 3.82 de | 25.71 ± 7.07 ef |
| 150 | 18.34 ± 10.46 f | 15.16 ± 7.36 efg | |
| 300 | 20.23 ± 8.25 f | 1.80 ± 1.70 g | |
| Sorafenib | 10 | 39.38 ± 5.48 ef | 11.40 ± 9.22 fg |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pace, E.; Jiang, Y.; Clemens, A.; Crossman, T.; Rupasinghe, H.P.V. Impact of Thermal Degradation of Cyanidin-3-O-Glucoside of Haskap Berry on Cytotoxicity of Hepatocellular Carcinoma HepG2 and Breast Cancer MDA-MB-231 Cells. Antioxidants 2018, 7, 24. https://doi.org/10.3390/antiox7020024
Pace E, Jiang Y, Clemens A, Crossman T, Rupasinghe HPV. Impact of Thermal Degradation of Cyanidin-3-O-Glucoside of Haskap Berry on Cytotoxicity of Hepatocellular Carcinoma HepG2 and Breast Cancer MDA-MB-231 Cells. Antioxidants. 2018; 7(2):24. https://doi.org/10.3390/antiox7020024
Chicago/Turabian StylePace, Eric, Yuanyuan Jiang, Amy Clemens, Tennille Crossman, and H.P. Vasantha Rupasinghe. 2018. "Impact of Thermal Degradation of Cyanidin-3-O-Glucoside of Haskap Berry on Cytotoxicity of Hepatocellular Carcinoma HepG2 and Breast Cancer MDA-MB-231 Cells" Antioxidants 7, no. 2: 24. https://doi.org/10.3390/antiox7020024
APA StylePace, E., Jiang, Y., Clemens, A., Crossman, T., & Rupasinghe, H. P. V. (2018). Impact of Thermal Degradation of Cyanidin-3-O-Glucoside of Haskap Berry on Cytotoxicity of Hepatocellular Carcinoma HepG2 and Breast Cancer MDA-MB-231 Cells. Antioxidants, 7(2), 24. https://doi.org/10.3390/antiox7020024