Oxidative Stress and Arterial Dysfunction in Peripheral Artery Disease




Abstract
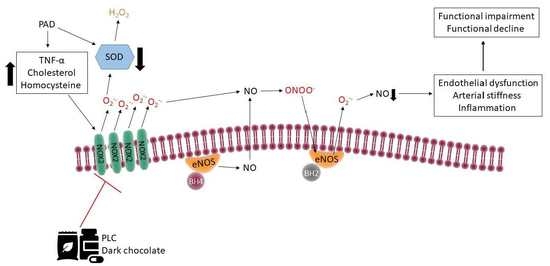
{kind=link}
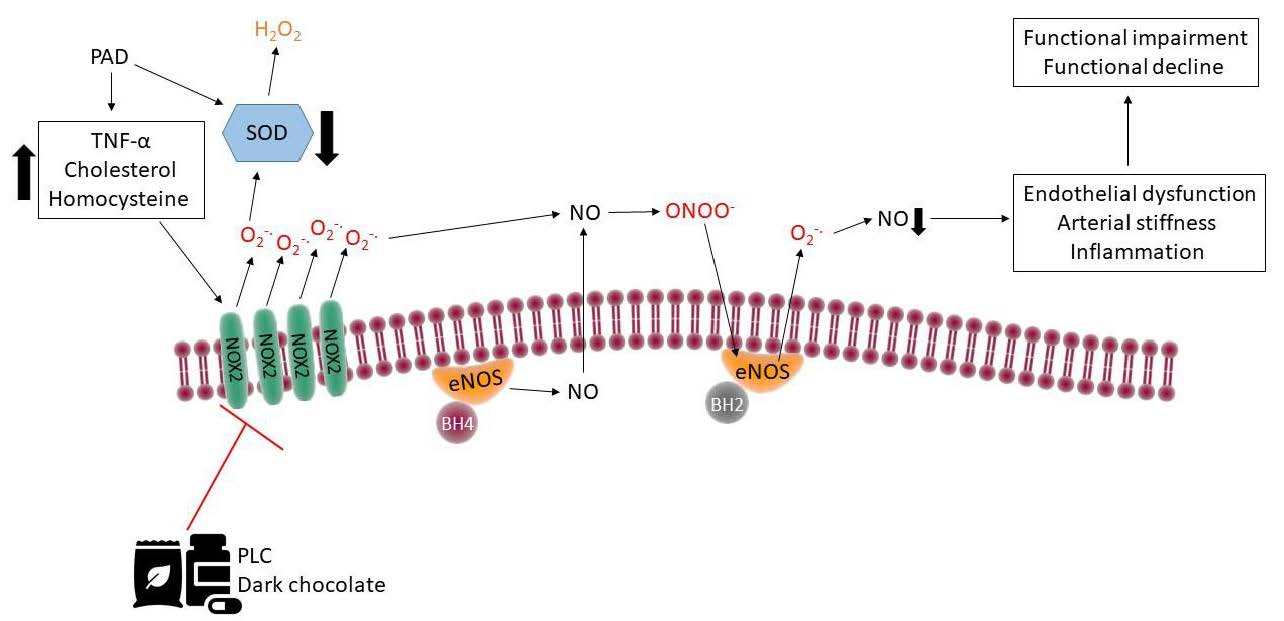
{kind=link}
1. Introduction
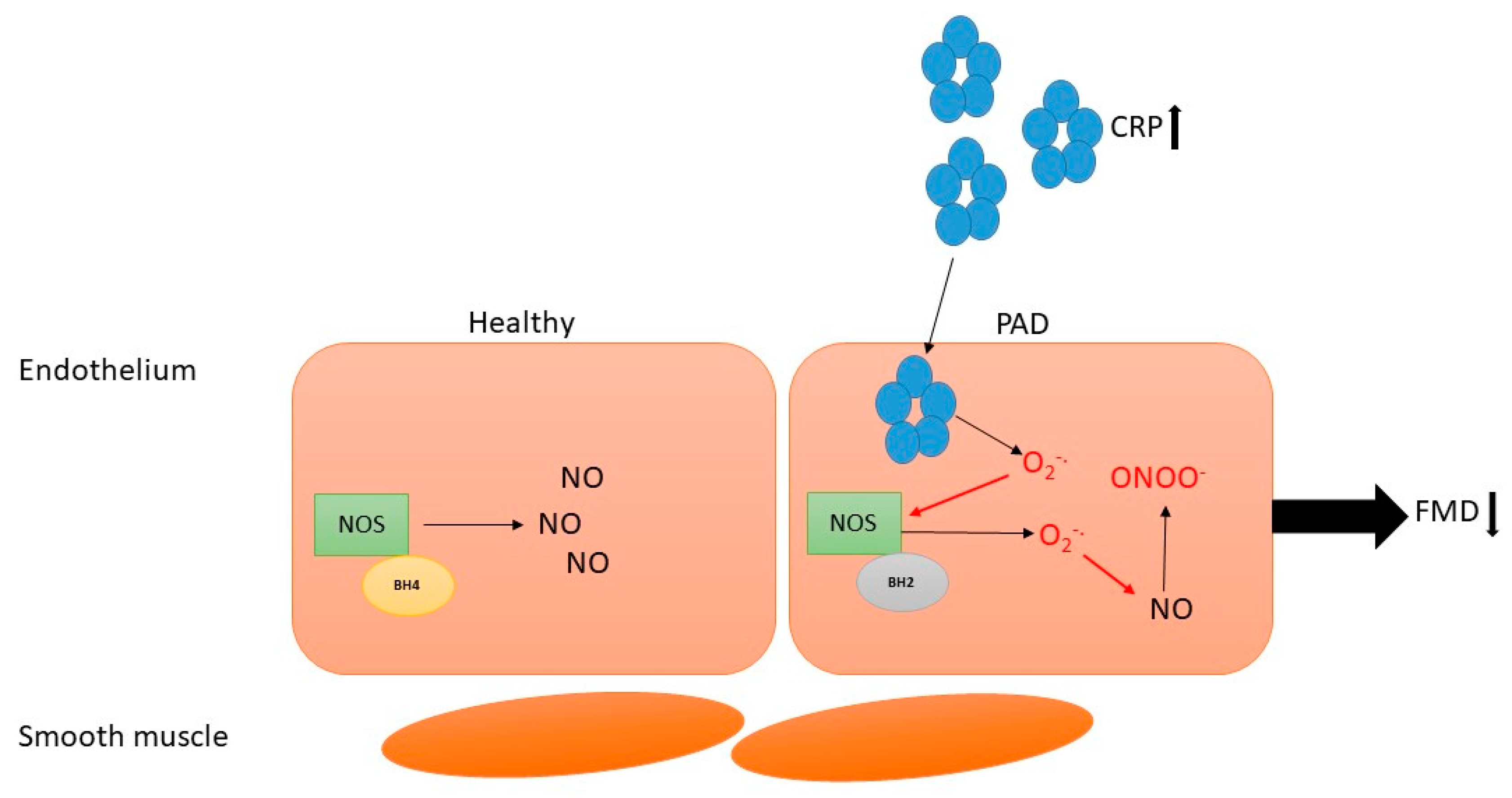
2. Endothelial Dysfunction
3. NADPH Oxidase 2 (NOX2)
4. Arterial Stiffness
5. Inflammation
6. Antioxidants
7. Discussion
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kullo, I.J.; Rooke, T.W. Peripheral Artery Disease. N. Engl. J. Med. 2016, 374, 861–871. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.L.; Halperin, J.L.; Albert, N.M.; Bozkurt, B.; Brindis, R.G.; Curtis, L.H.; DeMets, D.; Guyton, R.A.; Hochman, J.S.; Kovacs, R.J.; et al. Management of patients with peripheral artery disease (compilation of 2005 and 2011 ACCF/AHA guideline recommendations): A report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation 2013, 127, 1425–1443. [Google Scholar] [CrossRef] [PubMed]
- Pipinos, I.I.; Judge, A.R.; Selsby, J.T.; Zhu, Z.; Swanson, S.A.; Nella, A.A.; Dodd, S.L. The myopathy of peripheral arterial occlusive disease: Part 1. Functional and histomorphological changes and evidence for mitochondrial dysfunction. Vasc. Endovasc. Surg. 2007, 41, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Grenon, S.M.; Chong, K.; Alley, H.; Nosova, E.; Gasper, W.; Hiramoto, J.; Boscardin, W.J.; Owens, C.D. Walking disability in patients with peripheral artery disease is associated with arterial endothelial function. J. Vasc. Surg. 2014, 59, 1025–1034. [Google Scholar] [CrossRef] [PubMed]
- Hiatt, W.R. Medical treatment of peripheral arterial disease and claudication. N. Engl. J. Med. 2001, 344, 1608–1621. [Google Scholar] [CrossRef] [PubMed]
- Szuba, A.; Oka, R.K.; Harada, R.; Cooke, J.P. Limb hemodynamics are not predictive of functional capacity in patients with PAD. Vasc. Med. 2006, 11, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Parr, B.; Noakes, T.D.; Derman, E.W. Factors predicting walking intolerance in patients with peripheral arterial disease and intermittent claudication. S. Afr. Med. J. 2008, 98, 958–962. [Google Scholar] [PubMed]
- Coutinho, T.; Rooke, T.W.; Kullo, I.J. Arterial dysfunction and functional performance in patients with peripheral artery disease: A review. Vasc. Med. 2011, 16, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Steven, S.; Daiber, A.; Dopheide, J.F.; Munzel, T.; Espinola-Klein, C. Peripheral artery disease, redox signaling, oxidative stress-Basic and clinical aspects. Redox Biol. 2017, 12, 787–797. [Google Scholar] [CrossRef] [PubMed]
- Signorelli, S.S.; Katsiki, N. Oxidative stress and inflammation: Their role in the pathogenesis of peripheral artery disease with or without type 2 diabetes mellitus. Curr. Vasc. Pharmacol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Higashi, Y.; Noma, K.; Yoshizumi, M.; Kihara, Y. Endothelial function and oxidative stress in cardiovascular diseases. Circ. J. 2009, 73, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Hadi, H.A.; Carr, C.S.; Al Suwaidi, J. Endothelial dysfunction: Cardiovascular risk factors, therapy, and outcome. Vasc. Health Risk Manag. 2005, 1, 183–198. [Google Scholar] [PubMed]
- Green, D.J.; Dawson, E.A.; Groenewoud, H.M.; Jones, H.; Thijssen, D.H. Is flow-mediated dilation nitric oxide mediated? A meta-analysis. Hypertension 2014, 63, 376–382. [Google Scholar] [CrossRef] [PubMed]
- Brevetti, G.; Silvestro, A.; Schiano, V.; Chiariello, M. Endothelial dysfunction and cardiovascular risk prediction in peripheral arterial disease: Additive value of flow-mediated dilation to ankle-brachial pressure index. Circulation 2003, 108, 2093–2098. [Google Scholar] [CrossRef] [PubMed]
- Gokce, N.; Keaney, J.F., Jr.; Hunter, L.M.; Watkins, M.T.; Nedeljkovic, Z.S.; Menzoian, J.O.; Vita, J.A. Predictive value of noninvasively determined endothelial dysfunction for long-term cardiovascular events in patients with peripheral vascular disease. J. Am. Coll. Cardiol. 2003, 41, 1769–1775. [Google Scholar] [CrossRef]
- Maldonado, F.J.; Miralles, J.D.H.; Aguilar, E.M.; Gonzalez, A.F.; Garcia, J.R.; Garcia, F.A. Relationship between noninvasively measured endothelial function and peripheral arterial disease. Angiology 2009, 60, 725–731. [Google Scholar] [CrossRef] [PubMed]
- Igari, K.; Kudo, T.; Toyofuku, T.; Inoue, Y. Endothelial Dysfunction of Patients with Peripheral Arterial Disease Measured by Peripheral Arterial Tonometry. Int. J. Vasc. Med. 2016, 2016, 3805380. [Google Scholar] [CrossRef] [PubMed]
- Marti, C.N.; Gheorghiade, M.; Kalogeropoulos, A.P.; Georgiopoulou, V.V.; Quyyumi, A.A.; Butler, J. Endothelial dysfunction, arterial stiffness, and heart failure. J. Am. Coll. Cardiol. 2012, 60, 1455–1469. [Google Scholar] [CrossRef] [PubMed]
- Koutakis, P.; Ismaeel, A.; Farmer, P.; Purcell, S.; Smith, R.S.; Eidson, J.L.; Bohannon, W.T. Oxidative stress and antioxidant treatment in patients with peripheral artery disease. Physiol. Rep. 2018, 6, e13650. [Google Scholar] [CrossRef] [PubMed]
- Forstermann, U. Nitric oxide and oxidative stress in vascular disease. Pflugers Arch. 2010, 459, 923–939. [Google Scholar] [CrossRef] [PubMed]
- Loffredo, L.; Marcoccia, A.; Pignatelli, P.; Andreozzi, P.; Borgia, M.C.; Cangemi, R.; Chiarotti, F.; Violi, F. Oxidative-stress-mediated arterial dysfunction in patients with peripheral arterial disease. Eur. Heart J. 2007, 28, 608–612. [Google Scholar] [CrossRef] [PubMed]
- Lauer, T.; Preik, M.; Rassaf, T.; Strauer, B.E.; Deussen, A.; Feelisch, M.; Kelm, M. Plasma nitrite rather than nitrate reflects regional endothelial nitric oxide synthase activity but lacks intrinsic vasodilator action. Proc. Natl. Acad. Sci. USA 2001, 98, 12814–12819. [Google Scholar] [CrossRef] [PubMed]
- Drummond, G.R.; Sobey, C.G. Endothelial NADPH oxidases: Which NOX to target in vascular disease? Trends Endocrinol. Metab. 2014, 25, 452–463. [Google Scholar] [CrossRef] [PubMed]
- Drummond, G.R.; Selemidis, S.; Griendling, K.K.; Sobey, C.G. Combating oxidative stress in vascular disease: NADPH oxidases as therapeutic targets. Nat. Rev. Drug Discov. 2011, 10, 453–471. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Ma, X.; Xie, X.; Shen, G.X. Involvement of NADPH oxidase in oxidized LDL-induced upregulation of heat shock factor-1 and plasminogen activator inhibitor-1 in vascular endothelial cells. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E104–E111. [Google Scholar] [CrossRef] [PubMed]
- Bendall, J.K.; Rinze, R.; Adlam, D.; Tatham, A.L.; de Bono, J.; Wilson, N.; Volpi, E.; Channon, K.M. Endothelial Nox2 overexpression potentiates vascular oxidative stress and hemodynamic response to angiotensin II: Studies in endothelial-targeted Nox2 transgenic mice. Circ. Res. 2007, 100, 1016–1025. [Google Scholar] [CrossRef] [PubMed]
- Douglas, G.; Bendall, J.K.; Crabtree, M.J.; Tatham, A.L.; Carter, E.E.; Hale, A.B.; Channon, K.M. Endothelial-specific Nox2 overexpression increases vascular superoxide and macrophage recruitment in ApoE(/) mice. Cardiovasc. Res. 2012, 94, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Touyz, R.M.; Mercure, C.; He, Y.; Javeshghani, D.; Yao, G.; Callera, G.E.; Yogi, A.; Lochard, N.; Reudelhuber, T.L. Angiotensin II-dependent chronic hypertension and cardiac hypertrophy are unaffected by gp91phox-containing NADPH oxidase. Hypertension 2005, 45, 530–537. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.D.; Xu, S.; Johns, D.G.; Du, Y.; Quinn, M.T.; Cayatte, A.J.; Cohen, R.A. Role of NADPH oxidase in the vascular hypertrophic and oxidative stress response to angiotensin II in mice. Circ. Res. 2001, 88, 947–953. [Google Scholar] [CrossRef] [PubMed]
- Violi, F.; Sanguigni, V.; Carnevale, R.; Plebani, A.; Rossi, P.; Finocchi, A.; Pignata, C.; De Mattia, D.; Martire, B.; Pietrogrande, M.C.; et al. Hereditary deficiency of gp91(phox) is associated with enhanced arterial dilatation: Results of a multicenter study. Circulation 2009, 120, 1616–1622. [Google Scholar] [CrossRef] [PubMed]
- Zalba, G.; San Jose, G.; Moreno, M.U.; Fortuno, M.A.; Fortuno, A.; Beaumont, F.J.; Diez, J. Oxidative stress in arterial hypertension: Role of NAD(P)H oxidase. Hypertension 2001, 38, 1395–1399. [Google Scholar] [CrossRef] [PubMed]
- Loffredo, L.; Carnevale, R.; Cangemi, R.; Angelico, F.; Augelletti, T.; Di Santo, S.; Calabrese, C.M.; Della Volpe, L.; Pignatelli, P.; Perri, L.; et al. NOX2 up-regulation is associated with artery dysfunction in patients with peripheral artery disease. Int. J. Cardiol. 2013, 165, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Anderson, T.J. Arterial stiffness or endothelial dysfunction as a surrogate marker of vascular risk. Can. J. Cardiol. 2006, 22, 72B–80B. [Google Scholar] [CrossRef]
- Laurent, S.; Cockcroft, J.; Van Bortel, L.; Boutouyrie, P.; Giannattasio, C.; Hayoz, D.; Pannier, B.; Vlachopoulos, C.; Wilkinson, I.; Struijker-Boudier, H.; et al. Expert consensus document on arterial stiffness: Methodological issues and clinical applications. Eur. Heart J. 2006, 27, 2588–2605. [Google Scholar] [CrossRef] [PubMed]
- Catalano, M.; Scandale, G.; Carzaniga, G.; Cinquini, M.; Minola, M.; Dimitrov, G.; Carotta, M. Increased aortic stiffness and related factors in patients with peripheral arterial disease. J. Clin. Hypertens. (Greenwich) 2013, 15, 712–716. [Google Scholar] [CrossRef] [PubMed]
- Janner, J.H.; Godtfredsen, N.S.; Ladelund, S.; Vestbo, J.; Prescott, E. Aortic Augmentation Index: Reference Values in a Large Unselected Population by Means of the SphygmoCor Device. Am. J. Hypertens. 2010, 23, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, M.; Yamakado, T.; Kurachi, H.; Kato, T.; Kuroda, K.; Ishisu, R.; Okamoto, S.; Isaka, N.; Nakano, J.; Ito, M. The relationship between aortic augmentation index and pulse wave velocity: An invasive study. J. Hypertens. 2007, 25, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, I.B.; MacCallum, H.; Flint, L.; Cockcroft, J.R.; Newby, D.E.; Webb, D.J. The influence of heart rate on augmentation index and central arterial pressure in humans. J. Physiol. 2000, 525 Pt 1, 263–270. [Google Scholar] [CrossRef]
- Zahner, G.J.; Gruendl, M.A.; Spaulding, K.A.; Schaller, M.S.; Hills, N.K.; Gasper, W.J.; Grenon, S.M. Association between arterial stiffness and peripheral artery disease as measured by radial artery tonometry. J. Vasc. Surg. 2017, 66, 1518–1526. [Google Scholar] [CrossRef] [PubMed]
- Vlachopoulos, C.; Aznaouridis, K.; Stefanadis, C. Prediction of cardiovascular events and all-cause mortality with arterial stiffness: A systematic review and meta-analysis. J. Am. Coll. Cardiol. 2010, 55, 1318–1327. [Google Scholar] [CrossRef] [PubMed]
- Janic, M.; Lunder, M.; Sabovic, M. Arterial stiffness and cardiovascular therapy. Bio. Med. Res. Int. 2014, 2014, 621437. [Google Scholar] [CrossRef]
- Mozos, I.; Luca, C.T. Crosstalk between Oxidative and Nitrosative Stress and Arterial Stiffness. Curr. Vasc. Pharmacol. 2017, 15, 446–456. [Google Scholar] [CrossRef] [PubMed]
- Zanoli, L.; Rastelli, S.; Inserra, G.; Castellino, P. Arterial structure and function in inflammatory bowel disease. World J. Gastroenterol. 2015, 21, 11304–11311. [Google Scholar] [CrossRef] [PubMed]
- Kals, J.; Kampus, P.; Kals, M.; Pulges, A.; Teesalu, R.; Zilmer, K.; Kullisaar, T.; Salum, T.; Eha, J.; Zilmer, M. Inflammation and oxidative stress are associated differently with endothelial function and arterial stiffness in healthy subjects and in patients with atherosclerosis. Scand. J. Clin. Lab. Investig. 2008, 68, 594–601. [Google Scholar] [CrossRef] [PubMed]
- Zagura, M.; Kals, J.; Kilk, K.; Serg, M.; Kampus, P.; Eha, J.; Soomets, U.; Zilmer, M. Metabolomic signature of arterial stiffness in male patients with peripheral arterial disease. Hypertens. Res. 2015, 38, 840–846. [Google Scholar] [CrossRef] [PubMed]
- Zimlichman, R.; Shargorodsky, M.; Boaz, M.; Duprez, D.; Rahn, K.H.; Rizzoni, D.; Payeras, A.C.; Hamm, C.; McVeigh, G. Determination of arterial compliance using blood pressure waveform analysis with the CR-2000 system: Reliability, repeatability, and establishment of normal values for healthy European population--the seven European sites study (SESS). Am. Hypertens. 2005, 18, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Kals, J.; Lieberg, J.; Kampus, P.; Zagura, M.; Eha, J.; Zilmer, M. Prognostic impact of arterial stiffness in patients with symptomatic peripheral arterial disease. Eur. J. Vasc. Endovasc. Surg. 2014, 48, 308–315. [Google Scholar] [CrossRef] [PubMed]
- Kals, J.; Kampus, P.; Kals, M.; Zilmer, K.; Kullisaar, T.; Teesalu, R.; Pulges, A.; Zilmer, M. Impact of oxidative stress on arterial elasticity in patients with atherosclerosis. Am. J. Hypertens. 2006, 19, 902–908. [Google Scholar] [CrossRef] [PubMed]
- Ross, R. Atherosclerosis-an inflammatory disease. N. Engl. J. Med. 1999, 340, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Brevetti, G.; Schiano, V.; Chiariello, M. Endothelial dysfunction: A key to the pathophysiology and natural history of peripheral arterial disease? Atherosclerosis 2008, 197, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Cushman, M.; Stampfer, M.J.; Tracy, R.P.; Hennekens, C.H. Plasma concentration of C-reactive protein and risk of developing peripheral vascular disease. Circulation 1998, 97, 425–428. [Google Scholar] [CrossRef] [PubMed]
- Tzoulaki, I.; Murray, G.D.; Lee, A.J.; Rumley, A.; Lowe, G.D.; Fowkes, F.G. Inflammatory, haemostatic, and rheological markers for incident peripheral arterial disease: Edinburgh Artery Study. Eur. Heart J. 2007, 28, 354–362. [Google Scholar] [CrossRef] [PubMed]
- Folsom, A.R.; Pankow, J.S.; Tracy, R.P.; Arnett, D.K.; Peacock, J.M.; Hong, Y.; Djousse, L.; Eckfeldt, J.H. Investigators of the National Heart, Lung, and Blood Institute (NHLBI) Family Heart Study. Association of C-reactive protein with markers of prevalent atherosclerotic disease. Am. J. Cardiol. 2001, 88, 112–117. [Google Scholar] [CrossRef]
- Lin, C.W.; Hsu, L.A.; Chen, C.C.; Yeh, J.T.; Sun, J.H.; Lin, C.H.; Chen, S.T.; Hsu, B.R.; Huang, Y.Y. C-reactive protein as an outcome predictor for percutaneous transluminal angioplasty in diabetic patients with peripheral arterial disease and infected foot ulcers. Diabetes Res. Clin. Pract. 2010, 90, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Du Clos, T.W. Function of C-reactive protein. Ann. Med. 2000, 32, 274–278. [Google Scholar] [CrossRef] [PubMed]
- Jialal, I.; Verma, S.; Devaraj, S. Inhibition of endothelial nitric oxide synthase by C-reactive protein: Clinical relevance. Clin. Chem. 2009, 55, 206–208. [Google Scholar] [CrossRef] [PubMed]
- Sinisalo, J.; Paronen, J.; Mattila, K.J.; Syrjala, M.; Alfthan, G.; Palosuo, T.; Nieminen, M.S.; Vaarala, O. Relation of inflammation to vascular function in patients with coronary heart disease. Atherosclerosis 2000, 149, 403–411. [Google Scholar] [CrossRef]
- Gardner, A.W.; Parker, D.E.; Montgomery, P.S.; Sosnowska, D.; Casanegra, A.I.; Ungvari, Z.; Csiszar, A.; Sonntag, W.E. Greater endothelial apoptosis and oxidative stress in patients with peripheral artery disease. Int. J. Vasc. Med. 2014, 2014, 160534. [Google Scholar] [CrossRef] [PubMed]
- Brevetti, G.; Silvestro, A.; Di Giacomo, S.; Bucur, R.; Di Donato, A.; Schiano, V.; Scopacasa, F. Endothelial dysfunction in peripheral arterial disease is related to increase in plasma markers of inflammation and severity of peripheral circulatory impairment but not to classic risk factors and atherosclerotic burden. J. Vasc. Surg. 2003, 38, 374–379. [Google Scholar] [CrossRef]
- De Haro, J.; Acin, F.; Lopez-Quintana, A.; Medina, F.J.; Martinez-Aguilar, E.; Florez, A.; March, J.R. Direct association between C-reactive protein serum levels and endothelial dysfunction in patients with claudication. Eur. J. Vasc. Endovasc. Surg. 2008, 35, 480–486. [Google Scholar] [CrossRef] [PubMed]
- Silvestro, A.; Scopacasa, F.; Ruocco, A.; Oliva, G.; Schiano, V.; Zincarelli, C.; Brevetti, G. Inflammatory status and endothelial function in asymptomatic and symptomatic peripheral arterial disease. Vasc. Med. 2003, 8, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Brevetti, G.; Piscione, F.; Cirillo, P.; Galasso, G.; Schiano, V.; Barbato, E.; Scopacasa, F.; Chiariello, M. In concomitant coronary and peripheral arterial disease, inflammation of the affected limbs predicts coronary artery endothelial dysfunction. Atherosclerosis 2008, 201, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Shammas, N.W. Epidemiology, classification, and modifiable risk factors of peripheral arterial disease. Vasc. Health Risk Manag. 2007, 3, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive oxygen species in inflammation and tissue injury. Antioxid. Redox Signal. 2014, 20, 1126–1167. [Google Scholar] [CrossRef] [PubMed]
- Touyz, R.M. Reactive oxygen species and angiotensin II signaling in vascular cells-implications in cardiovascular disease. Braz. J. Med. Biol. Res. 2004, 37, 1263–1273. [Google Scholar] [CrossRef] [PubMed]
- Rull, A.; Garcia, R.; Fernandez-Sender, L.; Beltran-Debon, R.; Aragones, G.; Alegret, J.M.; Alonso-Villaverde, C.; Mackness, B.; Mackness, M.; Camps, J.; et al. The role of combined assessment of defense against oxidative stress and inflammation in the evaluation of peripheral arterial disease. Curr. Mol. Med. 2011, 11, 453–464. [Google Scholar] [CrossRef] [PubMed]
- de Haro Miralles, J.; Martinez-Aguilar, E.; Florez, A.; Varela, C.; Bleda, S.; Acin, F. Nitric oxide: Link between endothelial dysfunction and inflammation in patients with peripheral arterial disease of the lower limbs. Interact. Cardiovasc. Thorac. Surg. 2009, 9, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Barbato, J.E.; Tzeng, E. Nitric oxide and arterial disease. J. Vasc. Surg. 2004, 40, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Weber, C.; Zernecke, A.; Libby, P. The multifaceted contributions of leukocyte subsets to atherosclerosis: Lessons from mouse models. Nat. Rev. Immunol. 2008, 8, 802–815. [Google Scholar] [CrossRef] [PubMed]
- Haselmayer, P.; Grosse-Hovest, L.; von Landenberg, P.; Schild, H.; Radsak, M.P. TREM-1 ligand expression on platelets enhances neutrophil activation. Blood 2007, 110, 1029–1035. [Google Scholar] [CrossRef] [PubMed]
- Ford, J.W.; McVicar, D.W. TREM and TREM-like receptors in inflammation and disease. Curr. Opin. Immunol. 2009, 21, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Dopheide, J.F.; Doppler, C.; Scheer, M.; Obst, V.; Radmacher, M.C.; Radsak, M.P.; Gori, T.; Warnholtz, A.; Fottner, C.; Munzel, T.; et al. Critical limb ischaemia is characterised by an increased production of whole blood reactive oxygen species and expression of TREM-1 on neutrophils. Atherosclerosis 2013, 229, 396–403. [Google Scholar] [CrossRef] [PubMed]
- Dubois, R.N.; Abramson, S.B.; Crofford, L.; Gupta, R.A.; Simon, L.S.; Van De Putte, L.B.; Lipsky, P.E. Cyclooxygenase in biology and disease. FASEB J. 1998, 12, 1063–1073. [Google Scholar] [CrossRef] [PubMed]
- Schonbeck, U.; Sukhova, G.K.; Graber, P.; Coulter, S.; Libby, P. Augmented expression of cyclooxygenase-2 in human atherosclerotic lesions. Am. J. Pathol. 1999, 155, 1281–1291. [Google Scholar] [CrossRef]
- Florez, A.; de Haro, J.; Martinez, E.; Varela, C.; Bleda, S.; Acin, F. Selective cyclooxygenase-2 inhibition reduces endothelial dysfunction and improves inflammatory status in patients with intermittent claudication. Rev. Esp. Cardiol. 2009, 62, 851–857. [Google Scholar] [CrossRef]
- Giannopoulos, G.; Angelidis, C.; Vogiatzi, G.; Cleman, M.W.; Deftereos, S. Antioxidant treatment in peripheral artery disease: The rationale is there, but what about clinical results? Curr. Opin. Pharmacol. 2018, 39, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Myung, S.K.; Ju, W.; Cho, B.; Oh, S.W.; Park, S.M.; Koo, B.K.; Park, B.J. Efficacy of vitamin and antioxidant supplements in prevention of cardiovascular disease: Systematic review and meta-analysis of randomised controlled trials. BMJ 2013, 346, f10. [Google Scholar] [CrossRef] [PubMed]
- Lonn, E.; Yusuf, S.; Hoogwerf, B.; Pogue, J.; Yi, Q.; Zinman, B.; Bosch, J.; Dagenais, G.; Mann, J.F.; Gerstein, H.C.; et al. Effects of vitamin E on cardiovascular and microvascular outcomes in high-risk patients with diabetes: Results of the HOPE study and MICRO-HOPE substudy. Diabetes Care 2002, 25, 1919–1927. [Google Scholar] [CrossRef] [PubMed]
- Belch, J.; MacCuish, A.; Campbell, I.; Cobbe, S.; Taylor, R.; Prescott, R.; Lee, R.; Bancroft, J.; MacEwan, S.; Shepherd, J.; et al. The prevention of progression of arterial disease and diabetes (POPADAD) trial: Factorial randomised placebo controlled trial of aspirin and antioxidants in patients with diabetes and asymptomatic peripheral arterial disease. BMJ 2008, 337, a1840. [Google Scholar] [CrossRef] [PubMed]
- Wijnen, M.H.; Coolen, S.A.; Vader, H.L.; Reijenga, J.C.; Huf, F.A.; Roumen, R.M. Antioxidants reduce oxidative stress in claudicants. J. Surg. Res. 2001, 96, 183–187. [Google Scholar] [CrossRef] [PubMed]
- Arosio, E.; De Marchi, S.; Zannoni, M.; Prior, M.; Lechi, A. Effect of glutathione infusion on leg arterial circulation, cutaneous microcirculation, and pain-free walking distance in patients with peripheral obstructive arterial disease: A randomized, double-blind, placebo-controlled trial. Mayo Clin. Proc. 2002, 77, 754–759. [Google Scholar] [CrossRef] [PubMed]
- Muller, M.D.; Drew, R.C.; Blaha, C.A.; Mast, J.L.; Cui, J.; Reed, A.B.; Sinoway, L.I. Oxidative stress contributes to the augmented exercise pressor reflex in peripheral arterial disease patients. J. Physiol. 2012, 590, 6237–6246. [Google Scholar] [CrossRef] [PubMed]
- Rey, F.E.; Cifuentes, M.E.; Kiarash, A.; Quinn, M.T.; Pagano, P.J. Novel competitive inhibitor of NAD(P)H oxidase assembly attenuates vascular O-2(-) and systolic blood pressure in mice. Circ. Res. 2001, 89, 408–414. [Google Scholar] [CrossRef] [PubMed]
- Altenhofer, S.; Radermacher, K.A.; Kleikers, P.W.; Wingler, K.; Schmidt, H.H. Evolution of NADPH Oxidase Inhibitors: Selectivity and Mechanisms for Target Engagement. Antioxid. Redox Signal. 2015, 23, 406–427. [Google Scholar] [CrossRef] [PubMed]
- Chandasana, H.; Chhonker, Y.S.; Bala, V.; Prasad, Y.D.; Chaitanya, T.K.; Sharma, V.L.; Bhatta, R.S. Pharmacokinetic, bioavailability, metabolism and plasma protein binding evaluation of NADPH-oxidase inhibitor apocynin using LC-MS/MS. J. Chromatogr. B 2015, 985, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Heumuller, S.; Wind, S.; Barbosa-Sicard, E.; Schmidt, H.H.; Busse, R.; Schroder, K.; Brandes, R.P. Apocynin is not an inhibitor of vascular NADPH oxidases but an antioxidant. Hypertension 2008, 51, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Stefanska, J.; Sokolowska, M.; Sarniak, A.; Wlodarczyk, A.; Doniec, Z.; Nowak, D.; Pawliczak, R. Apocynin decreases hydrogen peroxide and nirtate concentrations in exhaled breath in healthy subjects. Pulm. Pharmacol. Ther. 2010, 23, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Stefanska, J.; Sarniak, A.; Wlodarczyk, A.; Sokolowska, M.; Pniewska, E.; Doniec, Z.; Nowak, D.; Pawliczak, R. Apocynin reduces reactive oxygen species concentrations in exhaled breath condensate in asthmatics. Exp. Lung Res. 2012, 38, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Stefanska, J.; Sarniak, A.; Wlodarczyk, A.; Sokolowska, M.; Doniec, Z.; Bialasiewicz, P.; Nowak, D.; Pawliczak, R. Hydrogen peroxide and nitrite reduction in exhaled breath condensate of COPD patients. Pulm. Pharmacol. Ther. 2012, 25, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Pignatelli, P.; Lenti, L.; Sanguigni, V.; Frati, G.; Simeoni, I.; Gazzaniga, P.P.; Pulcinelli, F.M.; Violi, F. Carnitine inhibits arachidonic acid turnover, platelet function, and oxidative stress. Am. J. Physiol. Heart Circ. Physiol. 2003, 284, H41–H48. [Google Scholar] [CrossRef] [PubMed]
- Stasi, M.A.; Scioli, M.G.; Arcuri, G.; Mattera, G.G.; Lombardo, K.; Marcellini, M.; Riccioni, T.; De Falco, S.; Pisano, C.; Spagnoli, L.G.; et al. Propionyl-L-carnitine improves postischemic blood flow recovery and arteriogenetic revascularization and reduces endothelial NADPH-oxidase 4-mediated superoxide production. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 426–435. [Google Scholar] [CrossRef] [PubMed]
- Scioli, M.G.; Stasi, M.A.; Passeri, D.; Doldo, E.; Costanza, G.; Camerini, R.; Fociani, P.; Arcuri, G.; Lombardo, K.; Pace, S.; et al. Propionyl-L-Carnitine is Efficacious in Ulcerative Colitis Through its Action on the Immune Function and Microvasculature. Clin. Transl. Gastroenterol. 2014, 5, e55. [Google Scholar] [CrossRef] [PubMed]
- Loffredo, L.; Pignatelli, P.; Cangemi, R.; Andreozzi, P.; Panico, M.A.; Meloni, V.; Violi, F. Imbalance between nitric oxide generation and oxidative stress in patients with peripheral arterial disease: Effect of an antioxidant treatment. J. Vasc. Surg. 2006, 44, 525–530. [Google Scholar] [CrossRef] [PubMed]
- El-Benna, J.; Dang, P.M.; Gougerot-Pocidalo, M.A.; Marie, J.C.; Braut-Boucher, F. p47phox, the phagocyte NADPH oxidase/NOX2 organizer: Structure, phosphorylation and implication in diseases. Exp. Mol. Med. 2009, 41, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Pandey, K.B.; Rizvi, S.I. Plant polyphenols as dietary antioxidants in human health and disease. Oxid. Med. Cell Longev. 2009, 2, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Carnevale, R.; Loffredo, L.; Nocella, C.; Bartimoccia, S.; Bucci, T.; De Falco, E.; Peruzzi, M.; Chimenti, I.; Biondi-Zoccai, G.; Pignatelli, P.; et al. Epicatechin and catechin modulate endothelial activation induced by platelets of patients with peripheral artery disease. Oxid. Med. Cell Longev. 2014, 2014, 691015. [Google Scholar] [CrossRef] [PubMed]
- Loffredo, L.; Perri, L.; Catasca, E.; Pignatelli, P.; Brancorsini, M.; Nocella, C.; De Falco, E.; Bartimoccia, S.; Frati, G.; Carnevale, R.; et al. Dark chocolate acutely improves walking autonomy in patients with peripheral artery disease. J. Am. Heart Assoc. 2014, 3, e001072. [Google Scholar] [CrossRef] [PubMed]
- Silvestro, A.; Scopacasa, F.; Oliva, G.; de Cristofaro, T.; Iuliano, L.; Brevetti, G. Vitamin C prevents endothelial dysfunction induced by acute exercise in patients with intermittent claudication. Atherosclerosis 2002, 165, 277–283. [Google Scholar] [CrossRef]
- McDermott, M.M. Functional impairment in peripheral artery disease and how to improve it in 2013. Curr. Cardiol. Rep. 2013, 15, 347. [Google Scholar] [CrossRef] [PubMed]
- Karim, A.S.; Reese, S.R.; Wilson, N.A.; Jacobson, L.M.; Zhong, W.; Djamali, A. Nox2 is a mediator of ischemia reperfusion injury. Am. J. Transplant. 2015, 15, 2888–2899. [Google Scholar] [CrossRef] [PubMed]
- Pipinos, I.I.; Judge, A.R.; Selsby, J.T.; Zhu, Z.; Swanson, S.A.; Nella, A.A.; Dodd, S.L. The myopathy of peripheral arterial occlusive disease: Part 2. Oxidative stress, neuropathy, and shift in muscle fiber type. Vasc. Endovascular Surg. 2008, 42, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Cauley, J.A.; Kassem, A.M.; Lane, N.E.; Thorson, S.; Osteoporotic Fractures in Men Study Research, G. Prevalent peripheral arterial disease and inflammatory burden. BMC Geriatr. 2016, 16, 213. [Google Scholar] [CrossRef] [PubMed]
- Khandanpour, N.; Loke, Y.K.; Meyer, F.J.; Jennings, B.; Armon, M.P. Homocysteine and peripheral arterial disease: systematic review and meta-analysis. Eur. J. Vasc. Endovasc. Surg. 2009, 38, 316–322. [Google Scholar] [CrossRef] [PubMed]
- Andreozzi, G.M. Propionyl l-carnitine: Intermittent claudication and peripheral arterial disease. Expert Opin. Pharmacother. 2009, 10, 2697–2707. [Google Scholar] [CrossRef] [PubMed]
- Brass, E.P.; Koster, D.; Hiatt, W.R.; Amato, A. A systematic review and meta-analysis of propionyl-L-carnitine effects on exercise performance in patients with claudication. Vasc. Med. 2013, 18, 3–12. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ismaeel, A.; Brumberg, R.S.; Kirk, J.S.; Papoutsi, E.; Farmer, P.J.; Bohannon, W.T.; Smith, R.S.; Eidson, J.L.; Sawicki, I.; Koutakis, P. Oxidative Stress and Arterial Dysfunction in Peripheral Artery Disease. Antioxidants 2018, 7, 145. https://doi.org/10.3390/antiox7100145
Ismaeel A, Brumberg RS, Kirk JS, Papoutsi E, Farmer PJ, Bohannon WT, Smith RS, Eidson JL, Sawicki I, Koutakis P. Oxidative Stress and Arterial Dysfunction in Peripheral Artery Disease. Antioxidants. 2018; 7(10):145. https://doi.org/10.3390/antiox7100145
Chicago/Turabian StyleIsmaeel, Ahmed, Robert S. Brumberg, Jeffrey S. Kirk, Evlampia Papoutsi, Patrick J. Farmer, William T. Bohannon, Robert S. Smith, Jack L. Eidson, Ian Sawicki, and Panagiotis Koutakis. 2018. "Oxidative Stress and Arterial Dysfunction in Peripheral Artery Disease" Antioxidants 7, no. 10: 145. https://doi.org/10.3390/antiox7100145
APA StyleIsmaeel, A., Brumberg, R. S., Kirk, J. S., Papoutsi, E., Farmer, P. J., Bohannon, W. T., Smith, R. S., Eidson, J. L., Sawicki, I., & Koutakis, P. (2018). Oxidative Stress and Arterial Dysfunction in Peripheral Artery Disease. Antioxidants, 7(10), 145. https://doi.org/10.3390/antiox7100145