


Inter-Organelle Crosstalk in Oxidative Distress: A Unified TRPM2-NOX2 Mediated Vicious Cycle Involving Ca2+, Zn2+, and ROS Amplification

Abstract
1. Introduction
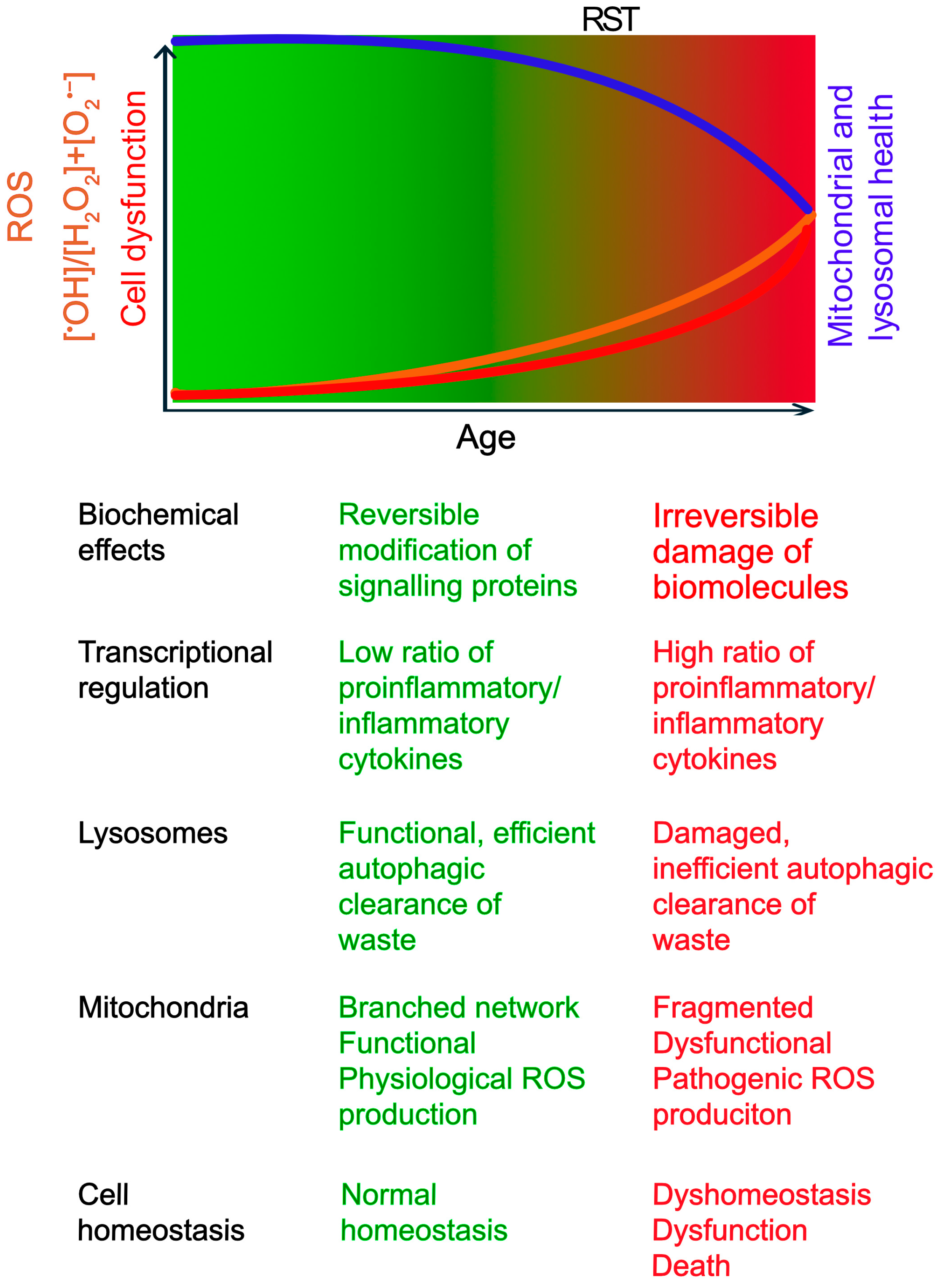
1.1. The Double-Edged Sword of ROS
1.2. Maintaining Redox Balance: Production vs. Defence
1.3. Mitochondrial and Lysosomal Contributions to Oxidative Distress
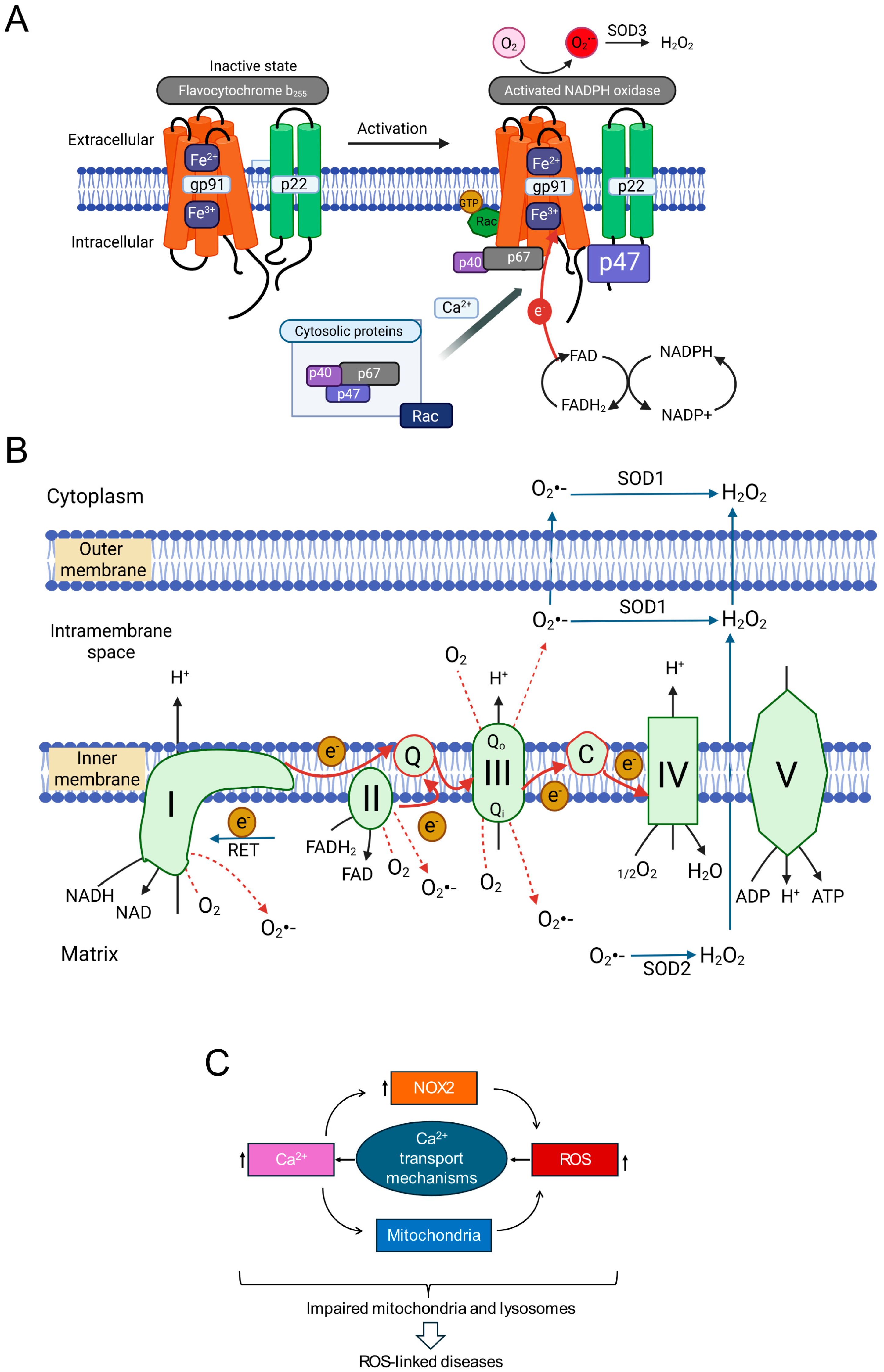
1.4. Cellular Sources of Reactive Oxygen Species
1.4.1. NADPH Oxidases (NOX Enzymes)
1.4.2. Mitochondrial Electron Transport Chain
1.4.3. Lysosomes: Indirect Contributors to ROS Generation
1.5. Amplifying the Damage: ROS-Induced ROS Production (RIRP)
2. Ionic Messengers in RIRP: The Roles of Ca2+ and Zn2+
2.1. The TRPM2 Channel: A Key Ca2+ Conduit in Oxidative Stress
2.2. Zinc Dyshomeostasis and Mitochondrial ROS
3. Integrating the Pieces: Towards a Unified Mechanism
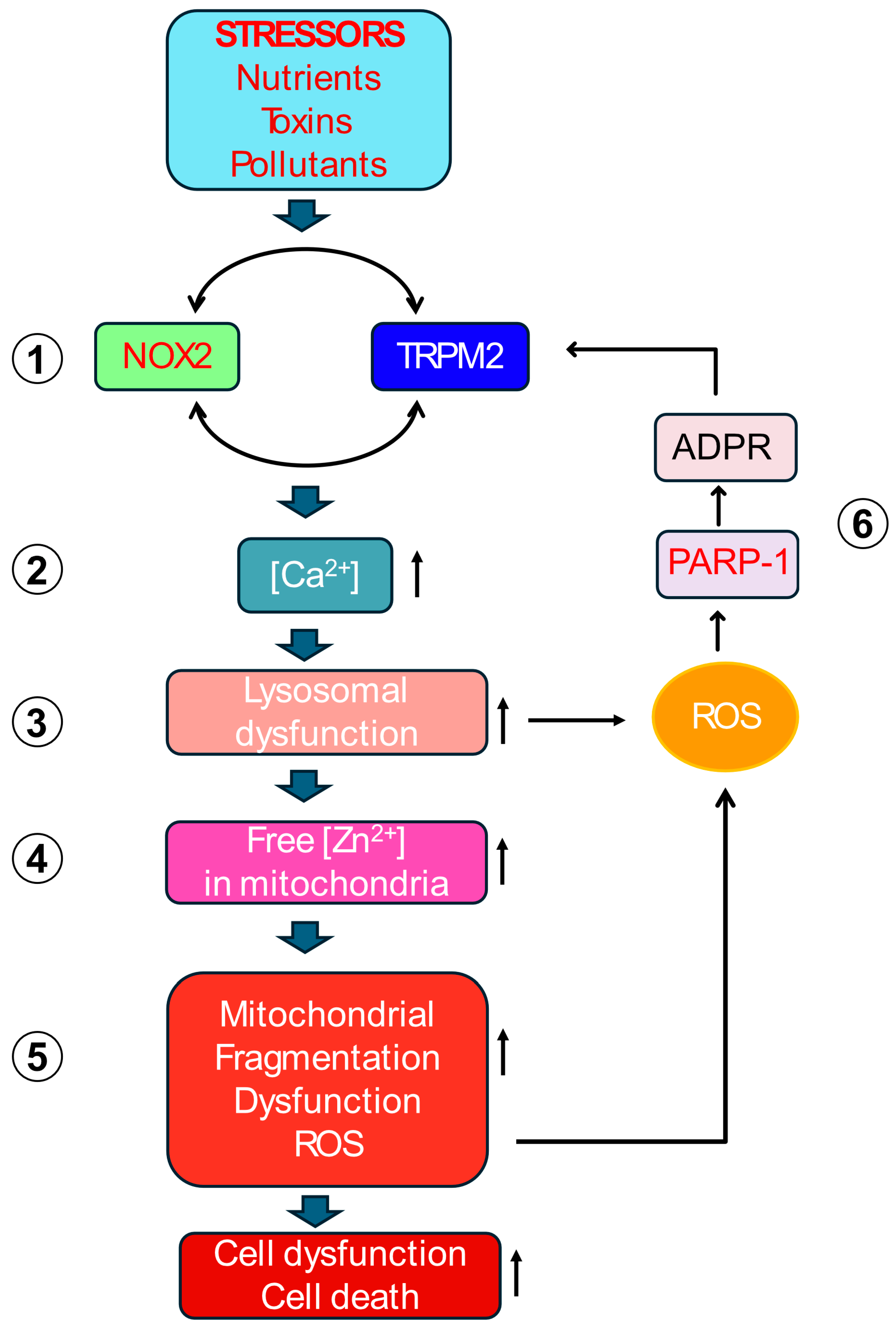
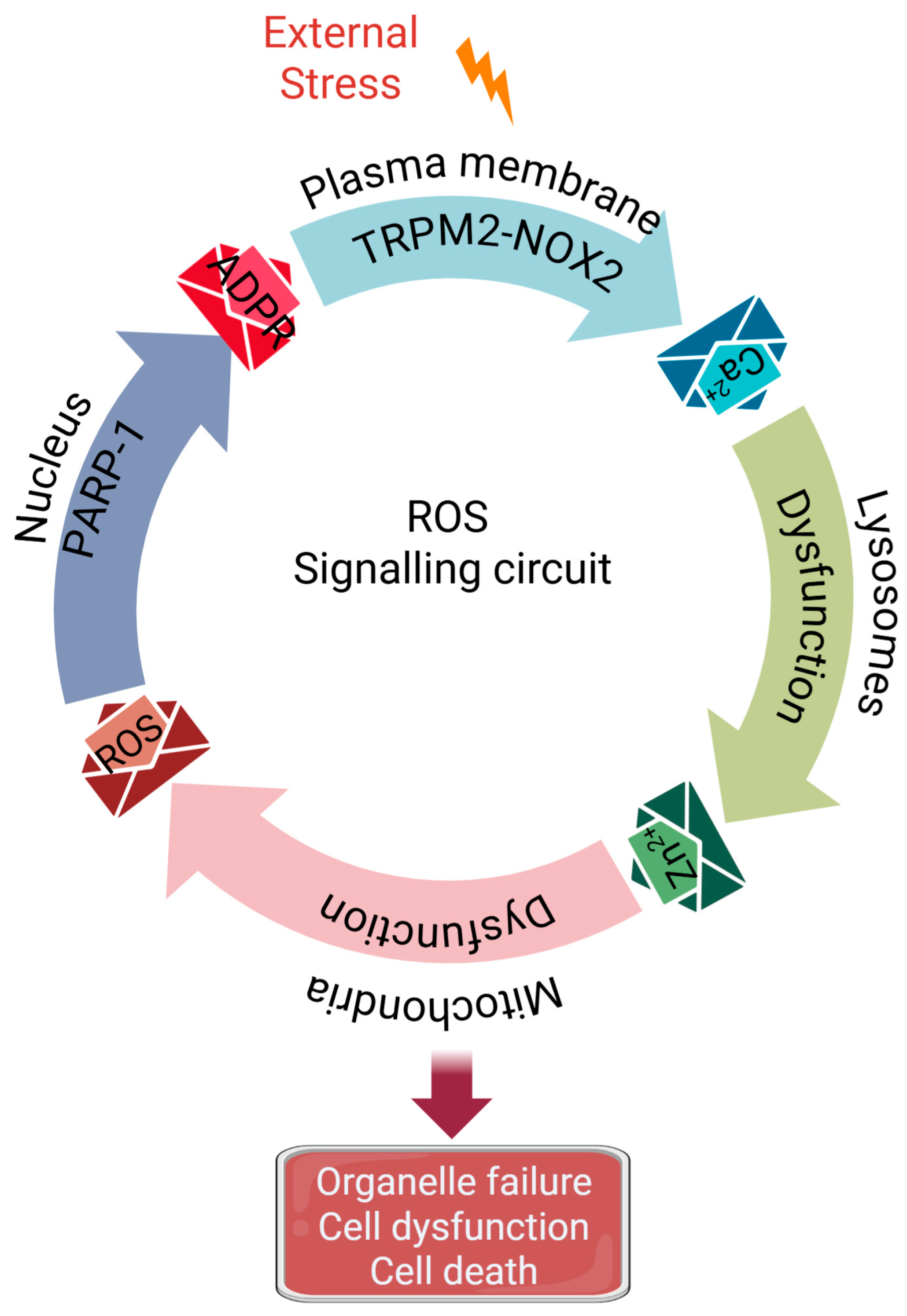
3.1. A Unified Vicious Cycle: Inter-Organelle Crosstalk Drives Pathological ROS Amplification
3.1.1. Initiating the Cycle: Synergistic Activation of TRPM2 and NOX2 at the Plasma Membrane
3.1.2. Calcium Overload Targets Lysosomes
3.1.3. From Damaged Lysosomes to Mitochondria: The Journey of Zinc
3.1.4. Zinc Disrupts Mitochondrial Function and Bolsters ROS Production
3.1.5. Closing the Loop: Mitochondrial ROS Stimulates ADPR Production to Perpetuate the Cycle
3.2. The Impact of Activating the Signalling Cycle
3.3. Evidence Supporting the Unified Mechanism Across Disease Models
4. Therapeutic Opportunities and Future Directions

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease | TRPM2 Involvement | NOX2 Involvement | Zn2+ Involvement | Mitochondrial ROS Involvement | PARP Involvement |
|---|---|---|---|---|---|
| Alzheimer’s Disease (AD) | TRPM2 inhibition (2-APB or ACA): ↓Aβ42-induced neuronal death in mouse hippocampus [154]. TRPM2 KO in mice: ↓Aβ-induced neurotoxicity ↓Ca2+ influx ↓TNF-α release [154]. TRPM2 KO (APP/PS1 mice): Improved spatial memory, ↓Microglial activation in hippocampus [155]. | NOX2 KO in mice: improved spatial memory [156]. Postmortem analyses: ↑NOX2 activity and expression in frontal and temporal cortices in patients with mild cognitive impairment [157]. | Zn2+ chelation (Clioquinol): Potential in reducing plaque load in AD models [158]. ZnT3-deficient mice: ↓Aβ oligomer accumulation [158]. | Scavenging mito-ROS with mitochondria targeted ROS scavengers in 3xTg-AD mice: ↓Oxidative stress ↓Aβ oligomer accumulation ↓Cell death ↓Cognitive impairment [159]. | PARP inhibition (pharmacological and genetic): ↓neuronal loss through parthanatos, neuroinflammation, cognitive impairment [153]. |
| Parkinson’s Disease (PD) | TRPM2 inhibition (2-APB, PJ34) and siRNA silencing in a cellular model: ↓MPP+- induced mtROS production and cell death [88,160]. Post-mortem brains of PD patients: ↑TRPM2 protein levels in SNpc [160]. | NOX2 inhibition (chemical) in a cellular model: ↓MPP+-induced Ca2+ rise ↓mtROS production ↓Cell death [88]. NOX2 inhibition (apocynin) in paraquat and 6-OHDA administered mice: ↓Cognitive deficits ↓Oxidative stress ↓Neuroinflammation [161,162]. NOX2 KO in 6-OHDA administered mice: ↓Dopaminergic neuron loss [162]. Post-mortem brains of PD patients: ↑gp91phox expression in midbrain [163]. | Zn2+ chelation (TPEN): ↓ROS levels ↓MPP+-induced cytotoxicity [88]. Zn2+ chelation (Clioquinol) in Lewy Bodies-injected mice: ↓α-synuclein-associated degeneration [164]. Post-mortem brains of PD patients: ↑Zn2+ levels observed in SNpc [165]. Genetic mutations in PARK9: ↑Mitochondrial Zn2+ in dopaminergic neurons ↑Mitochondrial damage [57,69]. | Scavenging mito-ROS with mitochondria- targeted ROS scavengers: ↓MPP+-induced cell death [88]. MitoQ in preclinical models: Neuroprotective [166]. | PARP-1 chemical inhibition or KO in mice: ↓α-synuclein-induced toxicity and neuronal cell death [167]. Post-mortem PD patient brains and CSF: ↑PAR levels [167]. |
| Cardiac ischemia | TRPM2 inhibition (chemical) or KO in mice subjected to IR injury: ↓Infarct size ↓Inflammation ↑Cardiac outcome [168]. | NOX2 KO in mice subjected to IR injury: ↓Infarct size [169]. | Zn2+ chelation (TPEN) in rat hearts during IR injury: ↓Infarct area [170]. | Scavenging mito-ROS with MitoQ in rats subjected to IR injury: ↓Mitochondrial damage ↓Cell death ↑Cardiac function [171]. | PARP1 inhibition (chemical) in mice subjected to IR injury: ↓Infarct size ↓Inflammation ↑Cardiac function [172]. |
| Stroke/Cerebral Ischemia | TRPM2 KO or inhibition (chemical) in male mice subjected to IR injury: ↓Neuronal cell death ↓Infarct size ↓Memory loss [173,174]. | NOX2 KO in mice subjected to IR injury: Delay infarct progression, but no protection from brain injury [175]. | Zn2+ chelation (TPEN): Protects mice from ischaemic brain damage [118]. | Mitochondrial ROS in IR injury mouse model: ↑Mitochondrial ROS in hippocampus in mice. MitoQ: ↓Hippocampal damage [176]. | PARP1 gene inactivation: Protection against ischemic insults [177]. |
| Various Cancers | TRPM2 Inhibition (chemical/genetic: SiRNA/KO): Breast cancer cells ↓Proliferation ↑DNA damage [178]. Neuroblastoma cells ↓Viability ↑ROS ↑DNA damage (sensitised to doxorubicin) [179]. Leukaemia ↓Proliferation ↑Chemo sensitivity [180]. Ovarian Cancer ↓Cell viability ↓Proliferation ↑Apoptosis [181]. PC3 and HeLa ↓Cell migration [115]. | NOX2 inhibition: Leukaemia cells ↑Cell death [182]. NOX2-KO and inhibition in mice: ↓Lung metastases [183]. | Zn2+ depletion: Breast cancer cells ZIP10 KO or zinc depletion: ↓Cell migration [184]. Zn2+ chelation (TPEN) in PC3 and HeLa: ↓Cell migration [115]. | Scavenging mito-ROS in mice: Mice lung carcinoma cells ↓Metastasis [185]. Scavenging mito-ROS in mouse melanoma cells ↓Cell growth ↓viability ↑Apoptosis [186]. | ↑PARP1 expression in breast, ovarian, and lung cancers. [143]. PARP1 inhibition in cervical cancer cell lines: ↓Proliferation ↑Cell death ↓Metastasis [187]. PARP1 inhibition in liver cancer cells: ↓Proliferation ↓Cell migration [188]. PARP inhibition in PC3 and HeLa: ↓Cell migration [115]. |
| Atherosclerosis (AS) | TRPM2 KO in Apoe-/- mice: ↓Progression of AS [189]. TRPM2 inhibition, KO and KD: ↓Mitochondrial damage in EC [112]. | NOX2 KO in Apoe/-e mice: ↓Plaque formation due to absence of NOX2 in macrophages and vessel wall cells [190]. | Data not available | MitoQ treatment HFD Apoe-/- mice: ↓Macrophages in plaques ↓Cell proliferation macrophages in plaques [191]. | PARP1 inhibition or KO Apoe/-e mice: ↓Plaque formation ↓Progression of atherosclerosis [192]. |
| Type 2 Diabetes | TRPM2 KO in HFD mice: ↑Insulin Sensitivity ↑Resistance to Diet-Induced Obesity ↑Glucose Metabolism ↓Inflammation [141]. Pancreatic β-cells (FFA treated): ↑NOX-dependent ROS ↑Mitochondrial damage ↑Cell death [113]. | NOX2 KO: ↑Insulin Sensitivity ↑Resistance to Diet-Induced Obesity [193]. NOX2 KD using SiRNA in pancreatic β-cell line exposed to high glucose and FFA: ↑ β-cell function ↑ β-cell survival [194,195] | Zn2+ chelation (TPEN): ↓FFA -induced β-cell death [113]. Loss of function mutations in hZnT8 in humans: ↑Risk of T2D ↑β-cell survival [196] Overexpression of LoF hZnT8 mutant in HFD-mice: ↑Glucose tolerance [197]. | Excess nutrition: ↑mtROS production ↑Insulin resistance ↑β-cell dysfunction [198]. | PARP-1 KO: ↓β-cell dysfunction ↓Insulin resistance ↓Vascular damage [199]. PARP-1 inhibition (PJ34) in pancreatic β-cells: ↓Cell death [113]. |
| Type 1 diabetes | TRPM2 KO in mice (STZ model): ↓β-cell death ↓Hyperglycaemia [200]. | NOX2 KO: ↓Glucose-induced superoxide in islets ↑Glucose-induced insulin secretion ↓β-cell apoptosis [201]. | Zn2+ chelation in STZ mouse model: ↓β-cell death ↓Hyperglycaemia [202] Pancreatic β-cells: Zn2+ chelation prevents oxidant- induced β-cell death [200]. | Mitochondrial ROS: ↑β-cell damage from cytokines and immune cells [23]. | PARP-1 KO in STZ mouse model: ↓β-cell death ↓Hyperglycaemia [203]. |
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chew, N.W.S.; Ng, C.H.; Tan, D.J.H.; Kong, G.; Lin, C.; Chin, Y.H.; Lim, W.H.; Huang, D.Q.; Quek, J.; Fu, C.E.; et al. The global burden of metabolic disease: Data from 2000 to 2019. Cell Metab. 2023, 35, 414–428.e3. [Google Scholar] [CrossRef] [PubMed]
- Nugent, R.; Fottrell, E. Non-communicable diseases and climate change: Linked global emergencies. Lancet 2019, 394, 622–623. [Google Scholar] [CrossRef] [PubMed]
- Kazibwe, J.; Tran, P.B.; Annerstedt, K.S. The household financial burden of non-communicable diseases in low- and middle-income countries: A systematic review. Health Res. Policy Syst. 2021, 19, 96. [Google Scholar] [CrossRef] [PubMed]
- Nunnari, J.; Suomalainen, A. Mitochondria: In sickness and in health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef]
- Sena, L.A.; Chandel, N.S. Physiological roles of mitochondrial reactive oxygen species. Mol. Cell 2012, 48, 158–167. [Google Scholar] [CrossRef]
- Dai, D.F.; Chiao, Y.A.; Marcinek, D.J.; Szeto, H.H.; Rabinovitch, P.S. Mitochondrial oxidative stress in aging and healthspan. Longev. Heal. 2014, 3, 6. [Google Scholar] [CrossRef]
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative Stress. Annu. Rev. Biochem. 2017, 86, 715–748. [Google Scholar] [CrossRef]
- Murphy, M.P.; Hartley, R.C. Mitochondria as a therapeutic target for common pathologies. Nat. Rev. Drug Discov. 2018, 17, 865–886. [Google Scholar] [CrossRef]
- Parvez, S.; Long, M.J.C.; Poganik, J.R.; Aye, Y. Redox Signaling by Reactive Electrophiles and Oxidants. Chem. Rev. 2018, 118, 8798–8888. [Google Scholar] [CrossRef]
- Brand, M.D. Riding the tiger—Physiological and pathological effects of superoxide and hydrogen peroxide generated in the mitochondrial matrix. Crit. Rev. Biochem. Mol. Biol. 2020, 55, 592–661. [Google Scholar] [CrossRef]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef] [PubMed]
- Forman, H.J.; Zhang, H. Targeting oxidative stress in disease: Promise and limitations of antioxidant therapy. Nat. Rev. Drug Discov. 2021, 20, 689–709. [Google Scholar] [CrossRef]
- Sies, H.; Belousov, V.V.; Chandel, N.S.; Davies, M.J.; Jones, D.P.; Mann, G.E.; Murphy, M.P.; Yamamoto, M.; Winterbourn, C. Defining roles of specific reactive oxygen species (ROS) in cell biology and physiology. Nat. Rev. Mol. Cell Biol. 2022, 23, 499–515. [Google Scholar] [CrossRef] [PubMed]
- Sies, H.; Mailloux, R.J.; Jakob, U. Fundamentals of redox regulation in biology. Nat. Rev. Mol. Cell Biol. 2024, 25, 701–719. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Li, Y.; Li, Y.; Ren, X.; Zhang, X.; Hu, D.; Gao, Y.; Xing, Y.; Shang, H. Oxidative Stress-Mediated Atherosclerosis: Mechanisms and Therapies. Front. Physiol. 2017, 8, 600. [Google Scholar] [CrossRef]
- Touyz, R.M.; Rios, F.J.; Alves-Lopes, R.; Neves, K.B.; Camargo, L.L.; Montezano, A.C. Oxidative Stress: A Unifying Paradigm in Hypertension. Can. J. Cardiol. 2020, 36, 659–670. [Google Scholar] [CrossRef]
- Chouchani, E.T.; Pell, V.R.; James, A.M.; Work, L.M.; Saeb-Parsy, K.; Frezza, C.; Krieg, T.; Murphy, M.P. A Unifying Mechanism for Mitochondrial Superoxide Production during Ischemia-Reperfusion Injury. Cell Metab. 2016, 23, 254–263. [Google Scholar] [CrossRef]
- Dias, V.; Junn, E.; Mouradian, M.M. The role of oxidative stress in Parkinson’s disease. J. Park. Dis. 2013, 3, 461–491. [Google Scholar] [CrossRef]
- Panicker, N.; Ge, P.; Dawson, V.L.; Dawson, T.M. The cell biology of Parkinson’s disease. J. Cell Biol. 2021, 220, e202012095. [Google Scholar] [CrossRef]
- Vázquez-Vélez, G.E.; Zoghbi, H.Y. Parkinson’s Disease Genetics and Pathophysiology. Annu. Rev. Neurosci. 2021, 44, 87–108. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Halliwell, B. Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat. Rev. Neurosci. 2019, 20, 148–160. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.L.; Goldfine, I.D.; Maddux, B.A.; Grodsky, G.M. Oxidative stress and stress-activated signaling pathways: A unifying hypothesis of type 2 diabetes. Endocr. Rev. 2002, 23, 599–622. [Google Scholar] [CrossRef]
- Chen, J.; Stimpson, S.E.; Fernandez-Bueno, G.A.; Mathews, C.E. Mitochondrial Reactive Oxygen Species and Type, 1 Diabetes. Antioxid. Redox Signal. 2018, 29, 1361–1372. [Google Scholar] [CrossRef]
- Sun, N.; Youle, R.J.; Finkel, T. The Mitochondrial Basis of Aging. Mol. Cell 2016, 61, 654–666. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Huang, X.; Dou, L.; Yan, M.; Shen, T.; Tang, W.; Li, J. Aging and aging-related diseases: From molecular mechanisms to interventions and treatments. Signal Transduct. Target. Ther. 2022, 7, 391. [Google Scholar] [CrossRef]
- Hayes, J.D.; Dinkova-Kostova, A.T.; Tew, K.D. Oxidative Stress in Cancer. Cancer Cell 2020, 38, 167–197. [Google Scholar] [CrossRef]
- Liu, S.Z.; Chiao, Y.A.; Rabinovitch, P.S.; Marcinek, D.J. Mitochondrial Targeted Interventions for Aging. Cold Spring Harb. Perspect. Med. 2024, 14, a041199. [Google Scholar] [CrossRef]
- Ristow, M. Unraveling the truth about antioxidants: Mitohormesis explains ROS-induced health benefits. Nat. Med. 2014, 20, 709–711. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B. Understanding mechanisms of antioxidant action in health and disease. Nat. Rev. Mol. Cell Biol. 2024, 25, 13–33. [Google Scholar] [CrossRef]
- Padayatty, S.J.; Levine, M. Vitamin C: The known and the unknown and Goldilocks. Oral Dis. 2016, 22, 463–493. [Google Scholar] [CrossRef]
- Halliwell, B. Vitamin C: Poison, prophylactic or panacea? Trends Biochem. Sci. 1999, 24, 255–259. [Google Scholar] [CrossRef]
- Kumar, A.; Singh, A. A review on mitochondrial restorative mechanism of antioxidants in Alzheimer’s disease and other neurological conditions. Front. Pharmacol. 2015, 6, 206. [Google Scholar] [CrossRef] [PubMed]
- Meng, J.; Lv, Z.; Zhang, Y.; Wang, Y.; Qiao, X.; Sun, C.; Chen, Y.; Guo, M.; Han, W.; Ye, A.; et al. Precision Redox: The Key for Antioxidant Pharmacology. Antioxid. Redox Signal. 2021, 34, 1069–1082. [Google Scholar] [CrossRef]
- Meng, J.; Lv, Z.; Wang, Y.; Chen, C. Identification of the redox-stress signaling threshold (RST): Increased RST helps to delay aging in C. elegans. Free Radic. Biol. Med. 2022, 178, 54–58. [Google Scholar] [CrossRef]
- Archer, S.L. Mitochondrial dynamics—Mitochondrial fission and fusion in human diseases. N. Engl. J. Med. 2013, 369, 2236–2251. [Google Scholar] [CrossRef]
- Bourdenx, M.; Dehay, B. What lysosomes actually tell us about Parkinson’s disease? Ageing Res. Rev. 2016, 32, 140–149. [Google Scholar] [CrossRef]
- Carmona-Gutierrez, D.; Hughes, A.L.; Madeo, F.; Ruckenstuhl, C. The crucial impact of lysosomes in aging and longevity. Ageing Res. Rev. 2016, 32, 2–12. [Google Scholar] [CrossRef]
- Friedman, J.R.; Nunnari, J. Mitochondrial form and function. Nature 2014, 505, 335–343. [Google Scholar] [CrossRef]
- Jheng, H.F.; Tsai, P.J.; Guo, S.M.; Kuo, L.H.; Chang, C.S.; Su, I.J.; Chang, C.R.; Tsai, Y.S. Mitochondrial fission contributes to mitochondrial dysfunction and insulin resistance in skeletal muscle. Mol. Cell Biol. 2012, 32, 309–319. [Google Scholar] [CrossRef]
- Macdonald, R.; Barnes, K.; Hastings, C.; Mortiboys, H. Mitochondrial abnormalities in Parkinson’s disease and Alzheimer’s disease: Can mitochondria be targeted therapeutically? Biochem. Soc. Trans. 2018, 46, 891–909. [Google Scholar] [CrossRef]
- Mao, K.; Zhang, G. The role of PARP1 in neurodegenerative diseases and aging. FEBS J. 2022, 289, 2013–2024. [Google Scholar] [CrossRef] [PubMed]
- Perera, R.M.; Zoncu, R. The Lysosome as a Regulatory Hub. Annu. Rev. Cell Dev. Biol. 2016, 32, 223–253. [Google Scholar] [CrossRef] [PubMed]
- Shenouda, S.M.; Widlansky, M.E.; Chen, K.; Xu, G.; Holbrook, M.; Tabit, C.E.; Hamburg, N.M.; Frame, A.A.; Caiano, T.L.; Kluge, M.A.; et al. Altered mitochondrial dynamics contributes to endothelial dysfunction in diabetes mellitus. Circulation 2011, 124, 444–453. [Google Scholar] [CrossRef]
- Tabara, L.C.; Segawa, M.; Prudent, J. Molecular mechanisms of mitochondrial dynamics. Nat. Rev. Mol. Cell Biol. 2025, 26, 123–146. [Google Scholar] [CrossRef]
- Udayar, V.; Chen, Y.; Sidransky, E.; Jagasia, R. Lysosomal dysfunction in neurodegeneration: Emerging concepts and methods. Trends Neurosci. 2022, 45, 184–199. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.X.; Finkel, T. Lysosomes in senescence and aging. EMBO Rep. 2023, 24, e57265. [Google Scholar] [CrossRef]
- Qian, L.; Zhu, Y.; Deng, C.; Liang, Z.; Chen, J.; Chen, Y.; Wang, X.; Liu, Y.; Tian, Y.; Yang, Y. Peroxisome proliferator-activated receptor gamma coactivator-1 (PGC-1) family in physiological and pathophysiological process and diseases. Signal Transduct. Target. Ther. 2024, 9, 50. [Google Scholar] [CrossRef]
- Dorn, G.W., 2nd; Vega, R.B.; Kelly, D.P. Mitochondrial biogenesis and dynamics in the developing and diseased heart. Genes Dev. 2015, 29, 1981–1991. [Google Scholar] [CrossRef]
- Luzio, J.P.; Pryor, P.R.; Bright, N.A. Lysosomes: Fusion and function. Nat. Rev. Mol. Cell Biol. 2007, 8, 622–632. [Google Scholar] [CrossRef]
- Saffi, G.T.; Botelho, R.J. Lysosome Fission: Planning for an Exit. Trends Cell Biol. 2019, 29, 635–646. [Google Scholar] [CrossRef]
- Levine, B.; Kroemer, G. Biological Functions of Autophagy Genes: A Disease Perspective. Cell 2019, 176, 11–42. [Google Scholar] [CrossRef] [PubMed]
- Meyer, H.; Kravic, B. The Endo-Lysosomal Damage Response. Annu. Rev. Biochem. 2024, 93, 367–387. [Google Scholar] [CrossRef] [PubMed]
- Bajaj, L.; Lotfi, P.; Pal, R.; Ronza, A.D.; Sharma, J.; Sardiello, M. Lysosome biogenesis in health and disease. J. Neurochem. 2019, 148, 573–589. [Google Scholar] [CrossRef]
- Settembre, C.; Di Malta, C.; Polito, V.A.; Garcia Arencibia, M.; Vetrini, F.; Erdin, S.; Erdin, S.U.; Huynh, T.; Medina, D.; Colella, P.; et al. TFEB links autophagy to lysosomal biogenesis. Science 2011, 332, 1429–1433. [Google Scholar] [CrossRef] [PubMed]
- Todkar, K.; Ilamathi, H.S.; Germain, M. Mitochondria and Lysosomes: Discovering Bonds. Front. Cell Dev. Biol. 2017, 5, 106. [Google Scholar] [CrossRef]
- Mutvei, A.P.; Nagiec, M.J.; Blenis, J. Balancing lysosome abundance in health and disease. Nat. Cell Biol. 2023, 25, 1254–1264. [Google Scholar] [CrossRef]
- Tsunemi, T.; Krainc, D. Zn2+ dyshomeostasis caused by loss of ATP13A2/PARK9 leads to lysosomal dysfunction and alpha-synuclein accumulation. Hum. Mol. Genet. 2014, 23, 2791–2801. [Google Scholar] [CrossRef]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef]
- Cao, M.; Luo, X.; Wu, K.; He, X. Targeting lysosomes in human disease: From basic research to clinical applications. Signal Transduct. Target. Ther. 2021, 6, 379. [Google Scholar] [CrossRef]
- Kurz, T.; Terman, A.; Gustafsson, B.; Brunk, U.T. Lysosomes in iron metabolism, ageing and apoptosis. Histochem. Cell Biol. 2008, 129, 389–406. [Google Scholar] [CrossRef]
- Nixon, R.A. New perspectives on lysosomes in ageing and neurodegenerative disease. Ageing Res. Rev. 2016, 32, 1. [Google Scholar] [CrossRef] [PubMed]
- Deus, C.M.; Yambire, K.F.; Oliveira, P.J.; Raimundo, N. Mitochondria-Lysosome Crosstalk: From Physiology to Neurodegeneration. Trends Mol. Med. 2020, 26, 71–88. [Google Scholar] [CrossRef] [PubMed]
- Kiraly, S.; Stanley, J.; Eden, E.R. Lysosome-Mitochondrial Crosstalk in Cellular Stress and Disease. Antioxidants 2025, 14, 125. [Google Scholar] [CrossRef]
- Dan, X.; Croteau, D.L.; Liu, W.; Chu, X.; Robbins, P.D.; Bohr, V.A. Mitochondrial accumulation and lysosomal dysfunction result in mitochondrial plaques in Alzheimer’s disease. bioRxiv 2025. [Google Scholar] [CrossRef]
- Demers-Lamarche, J.; Guillebaud, G.; Tlili, M.; Todkar, K.; Belanger, N.; Grondin, M.; Nguyen, A.P.; Michel, J.; Germain, M. Loss of Mitochondrial Function Impairs Lysosomes. J. Biol. Chem. 2016, 291, 10263–10276. [Google Scholar] [CrossRef]
- Plotegher, N.; Duchen, M.R. Crosstalk between Lysosomes and Mitochondria in Parkinson’s Disease. Front. Cell Dev. Biol. 2017, 5, 110. [Google Scholar] [CrossRef]
- Ballabio, A.; Bonifacino, J.S. Lysosomes as dynamic regulators of cell and organismal homeostasis. Nat. Rev. Mol. Cell Biol. 2020, 21, 101–118. [Google Scholar] [CrossRef]
- Do, J.; McKinney, C.; Sharma, P.; Sidransky, E. Glucocerebrosidase and its relevance to Parkinson disease. Mol. Neurodegener. 2019, 14, 36. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Koentjoro, B.; Veivers, D.; Mackay-Sim, A.; Sue, C.M. Parkinson’s disease-associated human ATP13A2 (PARK9) deficiency causes zinc dyshomeostasis and mitochondrial dysfunction. Hum. Mol. Genet. 2014, 23, 2802–2815. [Google Scholar] [CrossRef] [PubMed]
- Trist, B.G.; Hare, D.J.; Double, K.L. Oxidative stress in the aging substantia nigra and the etiology of Parkinson’s disease. Aging Cell 2019, 18, e13031. [Google Scholar] [CrossRef]
- Malpartida, A.B.; Williamson, M.; Narendra, D.P.; Wade-Martins, R.; Ryan, B.J. Mitochondrial Dysfunction and Mitophagy in Parkinson’s Disease: From Mechanism to Therapy. Trends Biochem. Sci. 2021, 46, 329–343. [Google Scholar] [CrossRef] [PubMed]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Belarbi, K.; Cuvelier, E.; Destée, A.; Gressier, B.; Chartier-Harlin, M.C. NADPH oxidases in Parkinson’s disease: A systematic review. Mol. Neurodegener. 2017, 12, 84. [Google Scholar] [CrossRef]
- Vermot, A.; Petit-Hartlein, I.; Smith, S.M.E.; Fieschi, F. NADPH Oxidases (NOX): An Overview from Discovery, Molecular Mechanisms to Physiology and Pathology. Antioxidants 2021, 10, 890. [Google Scholar] [CrossRef]
- Noreng, S.; Ota, N.; Sun, Y.; Ho, H.; Johnson, M.; Arthur, C.P.; Schneider, K.; Lehoux, I.; Davies, C.W.; Mortara, K.; et al. Structure of the core human NADPH oxidase NOX2. Nat. Commun. 2022, 13, 6079. [Google Scholar] [CrossRef] [PubMed]
- Lambeth, J.D.; Neish, A.S. Nox enzymes and new thinking on reactive oxygen: A double-edged sword revisited. Annu. Rev. Pathol. 2014, 9, 119–145. [Google Scholar] [CrossRef]
- Fan, L.M.; Geng, L.; Cahill-Smith, S.; Liu, F.; Douglas, G.; McKenzie, C.A.; Smith, C.; Brooks, G.; Channon, K.M.; Li, J.M. Nox2 contributes to age-related oxidative damage to neurons and the cerebral vasculature. J. Clin. Investig. 2019, 129, 3374–3386. [Google Scholar] [CrossRef]
- Keeney, M.T.; Hoffman, E.K.; Farmer, K.; Bodle, C.R.; Fazzari, M.; Zharikov, A.; Castro, S.L.; Hu, X.; Mortimer, A.; Kofler, J.K.; et al. NADPH oxidase 2 activity in Parkinson’s disease. Neurobiol. Dis. 2022, 170, 105754. [Google Scholar] [CrossRef]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef]
- Quinlan, C.L.; Perevoshchikova, I.V.; Hey-Mogensen, M.; Orr, A.L.; Brand, M.D. Sites of reactive oxygen species generation by mitochondria oxidizing different substrates. Redox Biol. 2013, 1, 304–312. [Google Scholar] [CrossRef]
- Scialo, F.; Fernandez-Ayala, D.J.; Sanz, A. Role of Mitochondrial Reverse Electron Transport in ROS Signaling: Potential Roles in Health and Disease. Front. Physiol. 2017, 8, 428. [Google Scholar] [CrossRef]
- Bennett, N.K.; Lee, M.; Orr, A.L.; Nakamura, K. Systems-level analyses dissociate genetic regulators of reactive oxygen species and energy production. Proc. Natl. Acad. Sci. USA 2024, 121, e2307904121. [Google Scholar] [CrossRef] [PubMed]
- Protasoni, M.; Perez-Perez, R.; Lobo-Jarne, T.; Harbour, M.E.; Ding, S.; Penas, A.; Diaz, F.; Moraes, C.T.; Fearnley, I.M.; Zeviani, M.; et al. Respiratory supercomplexes act as a platform for complex III-mediated maturation of human mitochondrial complexes I and IV. EMBO J. 2020, 39, e102817. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.H.; Tsai, P.I.; Lin, H.Y.; Hattori, N.; Funayama, M.; Jeon, B.; Sato, K.; Abe, K.; Mukai, Y.; Takahashi, Y.; et al. Mitochondrial UQCRC1 mutations cause autosomal dominant parkinsonism with polyneuropathy. Brain 2020, 143, 3352–3373. [Google Scholar] [CrossRef] [PubMed]
- Diaz, F.; Garcia, S.; Padgett, K.R.; Moraes, C.T. A defect in the mitochondrial complex III, but not complex IV, triggers early ROS-dependent damage in defined brain regions. Hum. Mol. Genet. 2012, 21, 5066–5077. [Google Scholar] [CrossRef] [PubMed]
- Lang, A.L.; Nissanka, N.; Louzada, R.A.; Tamayo, A.; Pereira, E.; Moraes, C.T.; Caicedo, A. A Defect in Mitochondrial Complex III but Not in Complexes I or IV Causes Early beta-Cell Dysfunction and Hyperglycemia in Mice. Diabetes 2023, 72, 1262–1276. [Google Scholar] [CrossRef]
- Pinto, M.; Diaz, F.; Nissanka, N.; Guastucci, C.S.; Illiano, P.; Brambilla, R.; Moraes, C.T. Adult-Onset Deficiency of Mitochondrial Complex III in a Mouse Model of Alzheimer’s Disease Decreases Amyloid Beta Plaque Formation. Mol. Neurobiol. 2022, 59, 6552–6566. [Google Scholar] [CrossRef]
- AlAhmad, M.; Isbea, H.; Shitaw, E.; Li, F.; Sivaprasadarao, A. NOX2-TRPM2 coupling promotes Zn(2+) inhibition of complex III to exacerbate ROS production in a cellular model of Parkinson’s disease. Sci. Rep. 2024, 14, 18431. [Google Scholar] [CrossRef]
- Bleier, L.; Drose, S. Superoxide generation by complex III: From mechanistic rationales to functional consequences. Biochim. Biophys. Acta. 2013, 1827, 1320–1331. [Google Scholar] [CrossRef]
- Orr, A.L.; Vargas, L.; Turk, C.N.; Baaten, J.E.; Matzen, J.T.; Dardov, V.J.; Attle, S.J.; Li, J.; Quackenbush, D.C.; Goncalves, R.L.; et al. Suppressors of superoxide production from mitochondrial complex III. Nat. Chem. Biol. 2015, 11, 834–836. [Google Scholar] [CrossRef]
- Han, D.; Antunes, F.; Canali, R.; Rettori, D.; Cadenas, E. Voltage-dependent anion channels control the release of the superoxide anion from mitochondria to cytosol. J. Biol. Chem. 2003, 278, 5557–5563. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Williams, E.; Cadenas, E. Mitochondrial respiratory chain-dependent generation of superoxide anion and its release into the intermembrane space. Biochem. J. 2001, 353, 411–416. [Google Scholar] [CrossRef]
- Palmer, J.E.; Wilson, N.; Son, S.M.; Obrocki, P.; Wrobel, L.; Rob, M.; Takla, M.; Korolchuk, V.I.; Rubinsztein, D.C. Autophagy, aging, and age-related neurodegeneration. Neuron 2025, 113, 29–48. [Google Scholar] [CrossRef]
- Samie, M.; Wang, X.; Zhang, X.; Goschka, A.; Li, X.; Cheng, X.; Gregg, E.; Azar, M.; Zhuo, Y.; Garrity, A.G.; et al. A TRP channel in the lysosome regulates large particle phagocytosis via focal exocytosis. Dev. Cell 2013, 26, 511–524. [Google Scholar] [CrossRef] [PubMed]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef]
- Dikalov, S. Cross talk between mitochondria and NADPH oxidases. Free Radic. Biol. Med. 2011, 51, 1289–1301. [Google Scholar] [CrossRef]
- Jiang, Q.; Yin, J.; Chen, J.; Ma, X.; Wu, M.; Liu, G.; Yao, K.; Tan, B.; Yin, Y. Mitochondria-Targeted Antioxidants: A Step towards Disease Treatment. Oxid. Med. Cell Longev. 2020, 2020, 8837893. [Google Scholar] [CrossRef]
- Berridge, M.J. Calcium signalling remodelling and disease. Biochem. Soc. T. 2012, 40, 297–309. [Google Scholar] [CrossRef] [PubMed]
- Gorlach, A.; Bertram, K.; Hudecova, S.; Krizanova, O. Calcium and ROS: A mutual interplay. Redox Biol. 2015, 6, 260–271. [Google Scholar] [CrossRef]
- Belrose, J.C.; Jackson, M.F. TRPM2: A candidate therapeutic target for treating neurological diseases. Acta Pharmacol. Sin. 2018, 39, 722–732. [Google Scholar] [CrossRef]
- Jiang, L.H.; Gamper, N.; Beech, D.J. Properties and therapeutic potential of transient receptor potential channels with putative roles in adversity: Focus on TRPC5, TRPM2 and TRPA1. Curr. Drug Targets 2011, 12, 724–736. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.S.; Ren, H.C.; Li, H.; Xing, M.; Cao, J.H. From oxidative stress to metabolic dysfunction: The role of TRPM2. Int. J. Biol. Macromol. 2025, 284, 138081. [Google Scholar] [CrossRef] [PubMed]
- Sumoza-Toledo, A.; Penner, R. TRPM2: A multifunctional ion channel for calcium signalling. J. Physiol. 2011, 589, 1515–1525. [Google Scholar] [CrossRef]
- Takahashi, N.; Kozai, D.; Kobayashi, R.; Ebert, M.; Mori, Y. Roles of TRPM2 in oxidative stress. Cell Calcium 2011, 50, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Shimizu, S. Targeting TRPM2 in ROS-Coupled Diseases. Pharmaceuticals 2016, 9, 57. [Google Scholar] [CrossRef]
- Yamamoto, S.; Shimizu, S.; Mori, Y. Involvement of TRPM2 channel in amplification of reactive oxygen species-induced signaling and chronic inflammation. Nihon yakurigaku zasshi. Folia Pharmacol. Jpn. 2009, 134, 122–126. [Google Scholar] [CrossRef]
- Zhong, Z.; Zhai, Y.; Liang, S.; Mori, Y.; Han, R.; Sutterwala, F.S.; Qiao, L. TRPM2 links oxidative stress to NLRP3 inflammasome activation. Nat. Commun. 2013, 4, 1611. [Google Scholar] [CrossRef]
- Hara, Y.; Wakamori, M.; Ishii, M.; Maeno, E.; Nishida, M.; Yoshida, T.; Yamada, H.; Shimizu, S.; Mori, E.; Kudoh, J.; et al. LTRPC2 Ca2+-permeable channel activated by changes in redox status confers susceptibility to cell death. Mol. Cell 2002, 9, 163–173. [Google Scholar] [CrossRef]
- Wang, L.; Fu, T.M.; Zhou, Y.; Xia, S.; Greka, A.; Wu, H. Structures and gating mechanism of human TRPM2. Science 2018, 362, eaav4809. [Google Scholar] [CrossRef]
- McHugh, D.; Flemming, R.; Xu, S.Z.; Perraud, A.L.; Beech, D.J. Critical intracellular Ca2+ dependence of transient receptor potential melastatin 2 (TRPM2) cation channel activation. J. Biol. Chem. 2003, 278, 11002–11006. [Google Scholar] [CrossRef]
- Tan, C.H.; McNaughton, P.A. The TRPM2 ion channel is required for sensitivity to warmth. Nature 2016, 536, 460–463. [Google Scholar] [CrossRef] [PubMed]
- Abuarab, N.; Munsey, T.; Jiang, L.; Li, J.; Sivaprasadarao, A. High glucose-induced ROS activates TRPM2 to trigger lysosomal membrane permeabilization and Zn(2+)-mediated mitochondrial fission. Sci. Signal. 2017, 10, eaal4161. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Munsey, T.S.; Sivaprasadarao, A. TRPM2-mediated rise in mitochondrial Zn(2+) promotes palmitate-induced mitochondrial fission and pancreatic beta-cell death in rodents. Cell Death Differ. 2017, 24, 1999–2012. [Google Scholar] [CrossRef]
- Miller, B.A. TRPM2 in Cancer. Cell Calcium 2019, 80, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Abuarab, N.; Sivaprasadarao, A. Reciprocal regulation of actin cytoskeleton remodelling and cell migration by Ca2+ and Zn2+: Role of TRPM2 channels. J. Cell Sci. 2016, 129, 2016–2029. [Google Scholar] [CrossRef]
- Sivaprasadarao, A.; Abuarab, N.; Li, F. TRPM2 channels in mitochondrial dynamics and cancer. Oncotarget 2017, 8, 84620–84621. [Google Scholar] [CrossRef]
- Chen, B.; Yu, P.; Chan, W.N.; Xie, F.; Zhang, Y.; Liang, L.; Leung, K.T.; Lo, K.W.; Yu, J.; Tse, G.M.K.; et al. Cellular zinc metabolism and zinc signaling: From biological functions to diseases and therapeutic targets. Signal Transduct. Target. Ther. 2024, 9, 6. [Google Scholar] [CrossRef]
- Clausen, A.; McClanahan, T.; Ji, S.G.; Weiss, J.H. Mechanisms of rapid reactive oxygen species generation in response to cytosolic Ca2+ or Zn2+ loads in cortical neurons. PLoS ONE 2013, 8, e83347. [Google Scholar] [CrossRef]
- Liu, H.Y.; Gale, J.R.; Reynolds, I.J.; Weiss, J.H.; Aizenman, E. The Multifaceted Roles of Zinc in Neuronal Mitochondrial Dysfunction. Biomedicines 2021, 9, 489. [Google Scholar] [CrossRef]
- Maret, W. The redox biology of redox-inert zinc ions. Free Radic. Biol. Med. 2019, 134, 311–326. [Google Scholar] [CrossRef]
- Portbury, S.D.; Adlard, P.A. Zinc Signal in Brain Diseases. Int. J. Mol. Sci. 2017, 18, 2506. [Google Scholar] [CrossRef] [PubMed]
- Sensi, S.L.; Paoletti, P.; Bush, A.I.; Sekler, I. Zinc in the physiology and pathology of the CNS. Nat. Rev. Neurosci. 2009, 10, 780–791. [Google Scholar] [CrossRef] [PubMed]
- Sensi, S.L.; Paoletti, P.; Koh, J.Y.; Aizenman, E.; Bush, A.I.; Hershfinkel, M. The neurophysiology and pathology of brain zinc. J. Neurosci. Off. J. Soc. Neurosci. 2011, 31, 16076–16085. [Google Scholar] [CrossRef]
- Shuttleworth, C.W.; Weiss, J.H. Zinc: New clues to diverse roles in brain ischemia. Trends Pharmacol. Sci. 2011, 32, 480–486. [Google Scholar] [CrossRef]
- Kambe, T.; Taylor, K.M.; Fu, D. Zinc transporters and their functional integration in mammalian cells. J. Biol. Chem. 2021, 296, 100320. [Google Scholar] [CrossRef] [PubMed]
- Bush, A.I.; Tanzi, R.E. Therapeutics for Alzheimer’s disease based on the metal hypothesis. Neurotherapeutics 2008, 5, 421–432. [Google Scholar] [CrossRef]
- Kambe, T.; Tsuji, T.; Hashimoto, A.; Itsumura, N. The Physiological, Biochemical, and Molecular Roles of Zinc Transporters in Zinc Homeostasis and Metabolism. Physiol. Rev. 2015, 95, 749–784. [Google Scholar] [CrossRef] [PubMed]
- Dai, D.F.; Rabinovitch, P.S.; Ungvari, Z. Mitochondria and cardiovascular aging. Circ. Res. 2012, 110, 1109–1124. [Google Scholar] [CrossRef]
- Yoon, Y.; Galloway, C.A.; Jhun, B.S.; Yu, T. Mitochondrial dynamics in diabetes. Antioxid. Redox Signal. 2011, 14, 439–457. [Google Scholar] [CrossRef]
- Boya, P.; Kroemer, G. Lysosomal membrane permeabilization in cell death. Oncogene 2008, 27, 6434–6451. [Google Scholar] [CrossRef]
- Feno, S.; Butera, G.; Vecellio Reane, D.; Rizzuto, R.; Raffaello, A. Crosstalk between Calcium and ROS in Pathophysiological Conditions. Oxid. Med. Cell Longev. 2019, 2019, 9324018. [Google Scholar] [CrossRef] [PubMed]
- Harraz, O.F.; Jensen, L.J. Vascular calcium signalling and ageing. J. Physiol. 2021, 599, 5361–5377. [Google Scholar] [CrossRef]
- Daiber, A. Redox signaling (cross-talk) from and to mitochondria involves mitochondrial pores and reactive oxygen species. Biochim. Biophys. Acta. 2010, 1797, 897–906. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Otin, C.; Kroemer, G. Hallmarks of Health. Cell 2021, 184, 33–63. [Google Scholar] [CrossRef] [PubMed]
- Bon, R.S.; Wright, D.J.; Beech, D.J.; Sukumar, P. Pharmacology of TRPC Channels and Its Potential in Cardiovascular and Metabolic Medicine. Annu. Rev. Pharmacol. Toxicol. 2022, 62, 427–446. [Google Scholar] [CrossRef]
- Giorgi, C.; Marchi, S.; Pinton, P. The machineries, regulation and cellular functions of mitochondrial calcium. Nat. Rev. Mol. Cell Biol. 2018, 19, 713–730. [Google Scholar] [CrossRef]
- Link, T.A.; von Jagow, G. Zinc ions inhibit the QP center of bovine heart mitochondrial bc1 complex by blocking a protonatable group. J. Biol. Chem. 1995, 270, 25001–25006. [Google Scholar] [CrossRef]
- Sharpley, M.S.; Hirst, J. The inhibition of mitochondrial complex I (NADH:ubiquinone oxidoreductase) by Zn2+. J. Biol. Chem. 2006, 281, 34803–34809. [Google Scholar] [CrossRef]
- Liu, R.; Kowada, T.; Du, Y.; Amagai, Y.; Matsui, T.; Inaba, K.; Mizukami, S. Organelle-Level Labile Zn. ACS Sens. 2022, 7, 748–757. [Google Scholar] [CrossRef]
- Morris, G.; Walker, A.J.; Berk, M.; Maes, M.; Puri, B.K. Cell Death Pathways: A Novel Therapeutic Approach for Neuroscientists. Mol. Neurobiol. 2018, 55, 5767–5786. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, W.; Jung, D.Y.; Ko, H.J.; Lee, Y.; Friedline, R.H.; Lee, E.; Jun, J.; Ma, Z.; Kim, F.; et al. TRPM2 Ca2+ channel regulates energy balance and glucose metabolism. Am. J. Physiol. Endocrinol. Metab. 2012, 302, E807–E816. [Google Scholar] [CrossRef]
- Conceicao, C.J.F.; Moe, E.; Ribeiro, P.A.; Raposo, M. PARP1: A comprehensive review of its mechanisms, therapeutic implications and emerging cancer treatments. Biochim. Biophys. Acta. Rev. Cancer 2025, 1880, 189282. [Google Scholar] [CrossRef]
- Wang, L.; Liang, C.; Li, F.; Guan, D.; Wu, X.; Fu, X.; Lu, A.; Zhang, G. PARP1 in Carcinomas and PARP1 Inhibitors as Antineoplastic Drugs. Int. J. Mol. Sci. 2017, 18, 2111. [Google Scholar] [CrossRef] [PubMed]
- Block, K.; Gorin, Y. Aiding and abetting roles of NOX oxidases in cellular transformation. Nat. Rev. Cancer 2012, 12, 627–637. [Google Scholar] [CrossRef]
- Westermann, B. Mitochondrial fusion and fission in cell life and death. Nat. Rev. Mol. Cell Biol. 2010, 11, 872–884. [Google Scholar] [CrossRef]
- Colpman, P.; Dasgupta, A.; Archer, S.L. The Role of Mitochondrial Dynamics and Mitotic Fission in Regulating the Cell Cycle in Cancer and Pulmonary Arterial Hypertension: Implications for Dynamin-Related Protein 1 and Mitofusin2 in Hyperproliferative Diseases. Cells 2023, 12, 1897. [Google Scholar] [CrossRef] [PubMed]
- Qian, W.; Choi, S.; Gibson, G.A.; Watkins, S.C.; Bakkenist, C.J.; Van Houten, B. Mitochondrial hyperfusion induced by loss of the fission protein Drp1 causes ATM-dependent G2/M arrest and aneuploidy through DNA replication stress. J. Cell Sci. 2012, 125, 5745–5757. [Google Scholar] [CrossRef]
- Martner, A.; Aydin, E.; Hellstrand, K. NOX2 in autoimmunity, tumor growth and metastasis. J. Pathol. 2019, 247, 151–154. [Google Scholar] [CrossRef] [PubMed]
- Frederick, M.I.; Abdesselam, D.; Clouvel, A.; Croteau, L.; Hassan, S. Leveraging PARP-1/2 to Target Distant Metastasis. Int. J. Mol. Sci. 2024, 25, 9032. [Google Scholar] [CrossRef]
- Nurse, P.M. Nobel Lecture. Cyclin dependent kinases and cell cycle control. Biosci. Rep. 2002, 22, 487–499. [Google Scholar] [CrossRef]
- Patke, A.; Young, M.W.; Axelrod, S. Molecular mechanisms and physiological importance of circadian rhythms. Nat. Rev. Mol. Cell Biol. 2020, 21, 67–84. [Google Scholar] [CrossRef]
- Rose, M.; Burgess, J.T.; O’Byrne, K.; Richard, D.J.; Bolderson, E. PARP Inhibitors: Clinical Relevance, Mechanisms of Action and Tumor Resistance. Front. Cell Dev. Biol. 2020, 8, 564601. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Sun, B.; Zhao, C. Poly (ADP-Ribose) polymerase 1 and parthanatos in neurological diseases: From pathogenesis to therapeutic opportunities. Neurobiol. Dis. 2023, 187, 106314. [Google Scholar] [CrossRef]
- Li, X.; Jiang, L.H. Multiple molecular mechanisms form a positive feedback loop driving amyloid beta42 peptide-induced neurotoxicity via activation of the TRPM2 channel in hippocampal neurons. Cell Death Dis. 2018, 9, 195. [Google Scholar] [CrossRef] [PubMed]
- Ostapchenko, V.G.; Chen, M.; Guzman, M.S.; Xie, Y.F.; Lavine, N.; Fan, J.; Beraldo, F.H.; Martyn, A.C.; Belrose, J.C.; Mori, Y.; et al. The Transient Receptor Potential Melastatin 2 (TRPM2) Channel Contributes to β-Amyloid Oligomer-Related Neurotoxicity and Memory Impairment. J. Neurosci. Off. J. Soc. Neurosci. 2015, 35, 15157–15169. [Google Scholar] [CrossRef] [PubMed]
- Park, L.; Zhou, P.; Pitstick, R.; Capone, C.; Anrather, J.; Norris, E.H.; Younkin, L.; Younkin, S.; Carlson, G.; McEwen, B.S.; et al. Nox2-derived radicals contribute to neurovascular and behavioral dysfunction in mice overexpressing the amyloid precursor protein. Proc. Natl. Acad. Sci. USA 2008, 105, 1347–1352. [Google Scholar] [CrossRef]
- Shimohama, S.; Tanino, H.; Kawakami, N.; Okamura, N.; Kodama, H.; Yamaguchi, T.; Hayakawa, T.; Nunomura, A.; Chiba, S.; Perry, G.; et al. Activation of NADPH oxidase in Alzheimer’s disease brains. Biochem. Biophys. Res. Commun. 2000, 273, 5–9. [Google Scholar] [CrossRef]
- Deshpande, A.; Kawai, H.; Metherate, R.; Glabe, C.G.; Busciglio, J. A role for synaptic zinc in activity-dependent Abeta oligomer formation and accumulation at excitatory synapses. J. Neurosci. Off. J. Soc. Neurosci. 2009, 29, 4004–4015. [Google Scholar] [CrossRef]
- McManus, M.J.; Murphy, M.P.; Franklin, J.L. The mitochondria-targeted antioxidant MitoQ prevents loss of spatial memory retention and early neuropathology in a transgenic mouse model of Alzheimer’s disease. J. Neurosci. Off. J. Soc. Neurosci. 2011, 31, 15703–15715. [Google Scholar] [CrossRef]
- Sun, Y.; Sukumaran, P.; Selvaraj, S.; Cilz, N.I.; Schaar, A.; Lei, S.; Singh, B.B. TRPM2 Promotes Neurotoxin MPP(+)/MPTP-Induced Cell Death. Mol. Neurobiol. 2018, 55, 409–420. [Google Scholar] [CrossRef]
- Hou, L.; Sun, F.; Huang, R.; Sun, W.; Zhang, D.; Wang, Q. Inhibition of NADPH oxidase by apocynin prevents learning and memory deficits in a mouse Parkinson’s disease model. Redox Biol. 2019, 22, 101134. [Google Scholar] [CrossRef] [PubMed]
- Hernandes, M.S.; Cafe-Mendes, C.C.; Britto, L.R. NADPH oxidase and the degeneration of dopaminergic neurons in parkinsonian mice. Oxid. Med. Cell Longev. 2013, 2013, 157857. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.C.; Teismann, P.; Tieu, K.; Vila, M.; Jackson-Lewis, V.; Ischiropoulos, H.; Przedborski, S. NADPH oxidase mediates oxidative stress in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2003, 100, 6145–6150. [Google Scholar] [CrossRef] [PubMed]
- Teil, M.; Doudnikoff, E.; Thiolat, M.L.; Bohic, S.; Bezard, E.; Dehay, B. The Zinc Ionophore Clioquinol Reduces Parkinson’s Disease Patient-Derived Brain Extracts-Induced Neurodegeneration. Mol. Neurobiol. 2022, 59, 6245–6259. [Google Scholar] [CrossRef]
- Dexter, D.T.; Carayon, A.; Javoy-Agid, F.; Agid, Y.; Wells, F.R.; Daniel, S.E.; Lees, A.J.; Jenner, P.; Marsden, C.D. Alterations in the levels of iron, ferritin and other trace metals in Parkinson’s disease and other neurodegenerative diseases affecting the basal ganglia. Brain 1991, 114, 1953–1975. [Google Scholar] [CrossRef]
- Murphy, M.P.; Smith, R.A. Targeting antioxidants to mitochondria by conjugation to lipophilic cations. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 629–656. [Google Scholar] [CrossRef]
- Kam, T.I.; Mao, X.; Park, H.; Chou, S.C.; Karuppagounder, S.S.; Umanah, G.E.; Yun, S.P.; Brahmachari, S.; Panicker, N.; Chen, R.; et al. Poly(ADP-ribose) drives pathologic alpha-synuclein neurodegeneration in Parkinson’s disease. Science 2018, 362, eaat8407. [Google Scholar] [CrossRef]
- Hiroi, T.; Wajima, T.; Negoro, T.; Ishii, M.; Nakano, Y.; Kiuchi, Y.; Mori, Y.; Shimizu, S. Neutrophil TRPM2 channels are implicated in the exacerbation of myocardial ischaemia/reperfusion injury. Cardiovasc. Res. 2013, 97, 271–281. [Google Scholar] [CrossRef]
- Matsushima, S.; Kuroda, J.; Ago, T.; Zhai, P.; Ikeda, Y.; Oka, S.; Fong, G.H.; Tian, R.; Sadoshima, J. Broad suppression of NADPH oxidase activity exacerbates ischemia/reperfusion injury through inadvertent downregulation of hypoxia-inducible factor-1alpha and upregulation of peroxisome proliferator-activated receptor-alpha. Circ. Res. 2013, 112, 1135–1149. [Google Scholar] [CrossRef]
- Lin, C.L.; Tseng, H.C.; Chen, R.F.; Chen, W.P.; Su, M.J.; Fang, K.M.; Wu, M.L. Intracellular zinc release-activated ERK-dependent GSK-3beta-p53 and Noxa-Mcl-1 signaling are both involved in cardiac ischemic-reperfusion injury. Cell Death Differ. 2011, 18, 1651–1663. [Google Scholar] [CrossRef]
- Adlam, V.J.; Harrison, J.C.; Porteous, C.M.; James, A.M.; Smith, R.A.; Murphy, M.P.; Sammut, I.A. Targeting an antioxidant to mitochondria decreases cardiac ischemia-reperfusion injury. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2005, 19, 1088–1095. [Google Scholar] [CrossRef]
- Luo, J.M.; Lin, H.B.; Weng, Y.Q.; Lin, Y.H.; Lai, L.Y.; Li, J.; Li, F.X.; Xu, S.Y.; Zhang, H.F.; Zhao, W. Inhibition of PARP1 improves cardiac function after myocardial infarction via up-regulated NLRC5. Chem. Biol. Interact. 2024, 395, 111010. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.; Verma, S.; Nakayama, S.; Quillinan, N.; Grafe, M.R.; Hurn, P.D.; Herson, P.S. Sex differences in neuroprotection provided by inhibition of TRPM2 channels following experimental stroke. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2011, 31, 2160–2168. [Google Scholar] [CrossRef] [PubMed]
- Ye, M.; Yang, W.; Ainscough, J.F.; Hu, X.P.; Li, X.; Sedo, A.; Zhang, X.H.; Zhang, X.; Chen, Z.; Li, X.M.; et al. TRPM2 channel deficiency prevents delayed cytosolic Zn2+ accumulation and CA1 pyramidal neuronal death after transient global ischemia. Cell Death Dis. 2014, 5, e1541. [Google Scholar] [CrossRef]
- McCann, S.K.; Dusting, G.J.; Roulston, C.L. Nox2 knockout delays infarct progression and increases vascular recovery through angiogenesis in mice following ischaemic stroke with reperfusion. PLoS ONE 2014, 9, e110602. [Google Scholar] [CrossRef]
- Ibrahim, A.A.; Abdel Mageed, S.S.; Safar, M.M.; El-Yamany, M.F.; Oraby, M.A. MitoQ alleviates hippocampal damage after cerebral ischemia: The potential role of SIRT6 in regulating mitochondrial dysfunction and neuroinflammation. Life Sci. 2023, 328, 121895. [Google Scholar] [CrossRef] [PubMed]
- Eliasson, M.J.; Sampei, K.; Mandir, A.S.; Hurn, P.D.; Traystman, R.J.; Bao, J.; Pieper, A.; Wang, Z.Q.; Dawson, T.M.; Snyder, S.H.; et al. Poly(ADP-ribose) polymerase gene disruption renders mice resistant to cerebral ischemia. Nat. Med. 1997, 3, 1089–1095. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, M.M.; Feng, X.; Liu, M.; Parker, L.P.; Koh, D.W. Inhibition of the transient receptor potential melastatin-2 channel causes increased DNA damage and decreased proliferation in breast adenocarcinoma cells. Int. J. Oncol. 2015, 46, 2267–2276. [Google Scholar] [CrossRef]
- Hirschler-Laszkiewicz, I.; Festa, F.; Huang, S.; Moldovan, G.L.; Nicolae, C.; Dhoonmoon, A.; Bao, L.; Keefer, K.; Chen, S.J.; Wang, H.G.; et al. The human ion channel TRPM2 modulates cell survival in neuroblastoma through E2F1 and FOXM1. Sci. Rep. 2022, 12, 6311. [Google Scholar] [CrossRef]
- Chen, S.J.; Bao, L.; Keefer, K.; Shanmughapriya, S.; Chen, L.; Lee, J.; Wang, J.; Zhang, X.Q.; Hirschler-Laszkiewicz, I.; Merali, S.; et al. Transient receptor potential ion channel TRPM2 promotes AML proliferation and survival through modulation of mitochondrial function, ROS, and autophagy. Cell Death Dis. 2020, 11, 247. [Google Scholar] [CrossRef]
- Zhu, W.; Mao, S.; Jiang, J. TRPM2-AS promotes ovarian cancer cell proliferation and inhibits cell apoptosis by upregulating the nearby gene TRPM2 via miR-6764-5p. Cell Div. 2024, 19, 26. [Google Scholar] [CrossRef]
- Marlein, C.R.; Zaitseva, L.; Piddock, R.E.; Robinson, S.D.; Edwards, D.R.; Shafat, M.S.; Zhou, Z.; Lawes, M.; Bowles, K.M.; Rushworth, S.A. NADPH oxidase-2 derived superoxide drives mitochondrial transfer from bone marrow stromal cells to leukemic blasts. Blood 2017, 130, 1649–1660. [Google Scholar] [CrossRef] [PubMed]
- van der Weyden, L.; Speak, A.O.; Swiatkowska, A.; Clare, S.; Schejtman, A.; Santilli, G.; Arends, M.J.; Adams, D.J. Pulmonary metastatic colonisation and granulomas in NOX2-deficient mice. J. Pathol. 2018, 246, 300–310. [Google Scholar] [CrossRef] [PubMed]
- Kagara, N.; Tanaka, N.; Noguchi, S.; Hirano, T. Zinc and its transporter ZIP10 are involved in invasive behavior of breast cancer cells. Cancer Sci. 2007, 98, 692–697. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, K.; Takenaga, K.; Akimoto, M.; Koshikawa, N.; Yamaguchi, A.; Imanishi, H.; Nakada, K.; Honma, Y.; Hayashi, J. ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science 2008, 320, 661–664. [Google Scholar] [CrossRef]
- Nazarewicz, R.R.; Dikalova, A.; Bikineyeva, A.; Ivanov, S.; Kirilyuk, I.A.; Grigor’ev, I.A.; Dikalov, S.I. Does scavenging of mitochondrial superoxide attenuate cancer prosurvival signaling pathways? Antioxid. Redox Signal. 2013, 19, 344–349. [Google Scholar] [CrossRef]
- Mann, M.; Kumar, S.; Sharma, A.; Chauhan, S.S.; Bhatla, N.; Kumar, S.; Bakhshi, S.; Gupta, R.; Kumar, L. PARP-1 inhibitor modulate beta-catenin signaling to enhance cisplatin sensitivity in cancer cervix. Oncotarget 2019, 10, 4262–4275. [Google Scholar] [CrossRef]
- Mao, X.; Du, S.; Yang, Z.; Zhang, L.; Peng, X.; Jiang, N.; Zhou, H. Inhibitors of PARP-1 exert inhibitory effects on the biological characteristics of hepatocellular carcinoma cells in vitro. Mol. Med. Rep. 2017, 16, 208–214. [Google Scholar] [CrossRef]
- Zong, P.; Feng, J.; Yue, Z.; Yu, A.S.; Vacher, J.; Jellison, E.R.; Miller, B.; Mori, Y.; Yue, L. TRPM2 deficiency in mice protects against atherosclerosis by inhibiting TRPM2-CD36 inflammatory axis in macrophages. Nat. Cardiovasc. Res. 2022, 1, 344–360. [Google Scholar] [CrossRef]
- Vendrov, A.E.; Hakim, Z.S.; Madamanchi, N.R.; Rojas, M.; Madamanchi, C.; Runge, M.S. Atherosclerosis is attenuated by limiting superoxide generation in both macrophages and vessel wall cells. Arter. Thromb. Vasc. Biol. 2007, 27, 2714–2721. [Google Scholar] [CrossRef]
- Mercer, J.R.; Yu, E.; Figg, N.; Cheng, K.K.; Prime, T.A.; Griffin, J.L.; Masoodi, M.; Vidal-Puig, A.; Murphy, M.P.; Bennett, M.R. The mitochondria-targeted antioxidant MitoQ decreases features of the metabolic syndrome in ATM+/-/ApoE-/- mice. Free Radic. Biol. Med. 2012, 52, 841–849. [Google Scholar] [CrossRef]
- Oumouna-Benachour, K.; Hans, C.P.; Suzuki, Y.; Naura, A.; Datta, R.; Belmadani, S.; Fallon, K.; Woods, C.; Boulares, A.H. Poly(ADP-ribose) polymerase inhibition reduces atherosclerotic plaque size and promotes factors of plaque stability in apolipoprotein E-deficient mice: Effects on macrophage recruitment, nuclear factor-kappaB nuclear translocation, and foam cell death. Circulation 2007, 115, 2442–2450. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Li, B.; Brun, T.; Deffert-Delbouille, C.; Mahiout, Z.; Daali, Y.; Ma, X.J.; Krause, K.H.; Maechler, P. NADPH oxidase NOX2 defines a new antagonistic role for reactive oxygen species and cAMP/PKA in the regulation of insulin secretion. Diabetes 2012, 61, 2842–2850. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Lu, Y.; Huang, X.; He, Q.; Man, Y.; Zhou, Y.; Wang, S.; Li, J. Suppression of NADPH oxidase 2 substantially restores glucose-induced dysfunction of pancreatic NIT-1 cells. FEBS J. 2010, 277, 5061–5071. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Zhang, X.; Huang, X.; Lu, Y.; Tang, W.; Man, Y.; Wang, S.; Xi, J.; Li, J. NADPH oxidase 2-derived reactive oxygen species mediate FFAs-induced dysfunction and apoptosis of beta-cells via JNK, p38 MAPK and p53 pathways. PLoS ONE 2010, 5, e15726. [Google Scholar] [CrossRef]
- Flannick, J.; Thorleifsson, G.; Beer, N.L.; Jacobs, S.B.; Grarup, N.; Burtt, N.P.; Mahajan, A.; Fuchsberger, C.; Atzmon, G.; Benediktsson, R.; et al. Loss-of-function mutations in SLC30A8 protect against type 2 diabetes. Nat. Genet. 2014, 46, 357–363. [Google Scholar] [CrossRef]
- Li, L.; Bai, S.; Sheline, C.T. hZnT8 (Slc30a8) Transgenic Mice That Overexpress the R325W Polymorph Have Reduced Islet Zn2+ and Proinsulin Levels, Increased Glucose Tolerance After a High-Fat Diet, and Altered Levels of Pancreatic Zinc Binding Proteins. Diabetes 2017, 66, 551–559. [Google Scholar]
- Sergi, D.; Naumovski, N.; Heilbronn, L.K.; Abeywardena, M.; O’Callaghan, N.; Lionetti, L.; Luscombe-Marsh, N. Mitochondrial (Dys)function and Insulin Resistance: From Pathophysiological Molecular Mechanisms to the Impact of Diet. Front. Physiol. 2019, 10, 532. [Google Scholar] [CrossRef]
- Pacher, P.; Szabo, C. Role of poly(ADP-ribose) polymerase-1 activation in the pathogenesis of diabetic complications: Endothelial dysfunction, as a common underlying theme. Antioxid. Redox Signal. 2005, 7, 1568–1580. [Google Scholar] [CrossRef]
- Manna, P.T.; Munsey, T.S.; Abuarab, N.; Li, F.; Asipu, A.; Howell, G.; Sedo, A.; Yang, W.; Naylor, J.; Beech, D.J.; et al. TRPM2 mediated intracellular Zn2+ release triggers pancreatic beta cell death. Biochem. J. 2015, 466, 537–546. [Google Scholar] [CrossRef]
- Xiang, F.L.; Lu, X.; Strutt, B.; Hill, D.J.; Feng, Q. NOX2 deficiency protects against streptozotocin-induced beta-cell destruction and development of diabetes in mice. Diabetes 2010, 59, 2603–2611. [Google Scholar] [CrossRef] [PubMed]
- Priel, T.; Aricha-Tamir, B.; Sekler, I. Clioquinol attenuates zinc-dependent beta-cell death and the onset of insulitis and hyperglycemia associated with experimental type I diabetes in mice. Eur. J. Pharmacol. 2007, 565, 232–239. [Google Scholar] [CrossRef] [PubMed]
- Charron, M.J.; Bonner-Weir, S. Implicating PARP and NAD+ depletion in type I diabetes. Nat. Med. 1999, 5, 269–270. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shitaw, E.E.; AlAhmad, M.; Sivaprasadarao, A. Inter-Organelle Crosstalk in Oxidative Distress: A Unified TRPM2-NOX2 Mediated Vicious Cycle Involving Ca2+, Zn2+, and ROS Amplification. Antioxidants 2025, 14, 776. https://doi.org/10.3390/antiox14070776
Shitaw EE, AlAhmad M, Sivaprasadarao A. Inter-Organelle Crosstalk in Oxidative Distress: A Unified TRPM2-NOX2 Mediated Vicious Cycle Involving Ca2+, Zn2+, and ROS Amplification. Antioxidants. 2025; 14(7):776. https://doi.org/10.3390/antiox14070776
Chicago/Turabian StyleShitaw, Esra Elhashmi, Maali AlAhmad, and Asipu Sivaprasadarao. 2025. "Inter-Organelle Crosstalk in Oxidative Distress: A Unified TRPM2-NOX2 Mediated Vicious Cycle Involving Ca2+, Zn2+, and ROS Amplification" Antioxidants 14, no. 7: 776. https://doi.org/10.3390/antiox14070776
APA StyleShitaw, E. E., AlAhmad, M., & Sivaprasadarao, A. (2025). Inter-Organelle Crosstalk in Oxidative Distress: A Unified TRPM2-NOX2 Mediated Vicious Cycle Involving Ca2+, Zn2+, and ROS Amplification. Antioxidants, 14(7), 776. https://doi.org/10.3390/antiox14070776