


Oxidative Stress and Inflammation: Drivers of Tumorigenesis and Therapeutic Opportunities

 and
and
Abstract
1. Introduction
2. The Complex and Dual Relationships Between Oxidative Stress and Cancer
2.1. The Generation of ROS
2.2. The Mechanism of ROS Based on Oxidative Stress Promoting Tumor Development
2.3. Mechanisms of ROS Based on Oxidative Stress Impeding Tumor Development
2.4. Spatiotemporal Regulation of ROS Under Oxidative Stress
3. Inflammation in Cancer
3.1. The Link Between Inflammation and Cancer
3.2. Role of Key Inflammatory Cells in Tumorigenesis and Progression
3.3. Role of Key Inflammatory Cytokines in Tumorigenesis and Progression
4. Crosstalk Between Oxidative Stress and Inflammation in Cancer
5. Diagnostic and Therapeutic Implications
5.1. Biomarkers

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Biomarkers | Description of Correlation | Significance in the Occurrence and Development of Tumors | Significance in Clinical Diagnosis and Treatment | References |
|---|---|---|---|---|
| CRP | Inflammation | High levels of CRP are associated with an increased risk of cancers. | An indicator to evaluate a chronic inflammatory state, it is helpful for cancer risk assessment. | [288,289,290,291,292] |
| IL-6 | Inflammation | Promoting the growth and metastasis of tumor cells and inhibiting the surveillance of tumor cells by the immune system. | It can be used to monitor disease progression and treatment efficacy in patients with cancer. | [293,294,295,296] |
| COX-2 | Inflammation | It induces inflammation by promoting prostaglandin synthesis, which in turn supports tumor cell proliferation, invasion, and angiogenesis. | It can be used as an important reference for tumor diagnosis, prognosis, and treatment target selection. | [297,298,299,300] |
| MDA | Oxidative stress | Elevated concentrations indicate excessive oxidative stress in the body, which may impair normal cellular structure and function, thus creating the conditions for tumorigenesis. | The detection of MDA can help to understand the individual oxidative stress status and indirectly indicate the risk of cancer. | [301,302,303] |
| 8-OHdG | Oxidative stress | High levels of 8-OHdG mean the DNA has been subjected to more oxidative attack, increasing the probability of genetic mutations. | It is helpful to evaluate the degree of oxidative damage in individuals and has potential value for predicting cancer susceptibility. | [303,304,305] |
| 4-HNE | Oxidative stress | It combines with a variety of biological macromolecules, such as proteins and DNA, leading to cell dysfunction and participating in the occurrence and development of tumors. | The determination of 4-HNE can reflect the lipid peroxidation status in vivo, which is helpful for evaluating the stage of tumor development and its prognosis. | [306,307,308] |
| SOD | Antioxidant defense system | It can catalyze the disproportionation of superoxide anion free radicals into hydrogen peroxide and oxygen, reduce the damage caused by oxidative stress to cells, and inhibit the formation and development of tumors to a certain extent. | It can be used as an important index to evaluate the antioxidant capacity of the body, and it has guiding significance for tumor prevention and treatment. | [309,310] |
| GPX | Antioxidant defense system | By reducing peroxides to water or alcohol, cells are protected from oxidative damage and the likelihood of tumorigenesis is reduced. | The level of GPX activity is closely related to the risk and severity of cancer, so it can be used as one of the bases for early warning and intervention measures of cancer. | [310,311,312,313] |
5.2. Therapeutic Strategies Targeting Oxidative Stress–Inflammation Axis
5.2.1. Oxidative-Stress-Based Cancer Intervention Strategies
5.2.2. Inflammation-Based Cancer Intervention Strategies
| Name | Mechanism of Action; Effects on Inflammation | Effect on Tumors | References |
|---|---|---|---|
| NSAIDs | Inhibit COX-2 activity, reduce prostaglandin biosynthesis | Reduce the incidence of cancers; inhibit tumor cell survival, angiogenesis, and immune evasion; improve clinical outcomes | [17,358] |
| CCR2 inhibitors (PF-04136309, BMS-687681) | Cause remodeling of immune cells within the tumor | Enhance anti-tumor immunity and overcome resistance | [138] |
| Bisphosphonates | Cytotoxic effects on macrophages | Reduce breast and prostate cancer patients’ recurrence rates and improve overall survival | [424] |
| Trabectedin | Cytotoxicity toward monocyte-derived macrophages; inhibits pro-inflammatory mediator secretion | Inhibit advanced soft tissue sarcoma and ovarian cancer | [425] |
| CP-870,893 | Reprograms TAMs into M1-like effector cells | Facilitate the depletion of tumor stroma; inhibit tumor growth | [426] |
| Anti-IL-1β mAbs | A monoclonal antibody against IL-1β | Combined treatment enhances the efficacy of chemotherapy drugs | [431,432] |
| Siltuximab | A chimeric monoclonal antibody targeting IL-6 | Combined therapy enhances the efficacy of chemotherapy drugs and improves their safety | [433] |
| Infliximab | A monoclonal antibody against TNF-α | Reverse oxaliplatin resistance in CRC | [434] |
| Interferon-α | Regulate the differentiation and infiltration of immune cells | Improve DFS and OS in patients | [435] |
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Conflicts of Interest
Abbreviations
References
- Arafeh, R.; Shibue, T.; Dempster, J.M.; Hahn, W.C.; Vazquez, F. The present and future of the Cancer Dependency Map. Nat. Rev. Cancer 2025, 25, 59–73. [Google Scholar] [CrossRef] [PubMed]
- Ju, S.; Singh, M.K.; Han, S.; Ranbhise, J.; Ha, J.; Choe, W.; Yoon, K.S.; Yeo, S.G.; Kim, S.S.; Kang, I. Oxidative Stress and Cancer Therapy: Controlling Cancer Cells Using Reactive Oxygen Species. Int. J. Mol. Sci. 2024, 25, 12387. [Google Scholar] [CrossRef]
- Xie, Y.; Liu, F.; Wu, Y.; Zhu, Y.; Jiang, Y.; Wu, Q.; Dong, Z.; Liu, K. Inflammation in cancer: Therapeutic opportunities from new insights. Mol. Cancer 2025, 24, 51. [Google Scholar] [CrossRef] [PubMed]
- Dong, R.; Wang, J.; Guan, R.; Sun, J.; Jin, P.; Shen, J. Role of Oxidative Stress in the Occurrence, Development, and Treatment of Breast Cancer. Antioxidants 2025, 14, 104. [Google Scholar] [CrossRef] [PubMed]
- Fan, C.; Yang, X.; Yan, L.; Shi, Z. Oxidative stress is two-sided in the treatment of acute myeloid leukemia. Cancer Med. 2024, 13, e6806. [Google Scholar] [CrossRef]
- Kuo, C.L.; Ponneri Babuharisankar, A.; Lin, Y.C.; Lien, H.W.; Lo, Y.K.; Chou, H.Y.; Tangeda, V.; Cheng, L.C.; Cheng, A.N.; Lee, A.Y. Mitochondrial oxidative stress in the tumor microenvironment and cancer immunoescape: Foe or friend? J. Biomed. Sci. 2022, 29, 74. [Google Scholar] [CrossRef]
- Hayes, J.D.; Dinkova-Kostova, A.T.; Tew, K.D. Oxidative Stress in Cancer. Cancer Cell 2020, 38, 167–197. [Google Scholar] [CrossRef] [PubMed]
- Shahgoli, V.K.; Noorolyai, S.; Ahmadpour Youshanlui, M.; Saeidi, H.; Nasiri, H.; Mansoori, B.; Holmskov, U.; Baradaran, B. Inflammatory bowel disease, colitis, and cancer: Unmasking the chronic inflammation link. Int. J. Color. Dis. 2024, 39, 173. [Google Scholar] [CrossRef]
- Kapoor, G.; Prakash, S.; Jaiswal, V.; Singh, A.K. Chronic Inflammation and Cancer: Key Pathways and Targeted Therapies. Cancer Investig. 2025, 43, 1–23. [Google Scholar] [CrossRef]
- Nishida, A.; Andoh, A. The Role of Inflammation in Cancer: Mechanisms of Tumor Initiation, Progression, and Metastasis. Cells 2025, 14, 488. [Google Scholar] [CrossRef]
- Chen, A.; Huang, H.; Fang, S.; Hang, Q. ROS: A “booster” for chronic inflammation and tumor metastasis. Biochim. Biophys. Acta Rev. Cancer 2024, 1879, 189175. [Google Scholar] [CrossRef] [PubMed]
- Fiorilla, I.; Martinotti, S.; Todesco, A.M.; Bonsignore, G.; Cavaletto, M.; Patrone, M.; Ranzato, E.; Audrito, V. Chronic Inflammation, Oxidative Stress and Metabolic Plasticity: Three Players Driving the Pro-Tumorigenic Microenvironment in Malignant Mesothelioma. Cells 2023, 12, 2048. [Google Scholar] [CrossRef] [PubMed]
- Arora, S.; Bhardwaj, A.; Singh, S.; Srivastava, S.K.; McClellan, S.; Nirodi, C.S.; Piazza, G.A.; Grizzle, W.E.; Owen, L.B.; Singh, A.P. An undesired effect of chemotherapy: Gemcitabine promotes pancreatic cancer cell invasiveness through reactive oxygen species-dependent, nuclear factor kappaB- and hypoxia-inducible factor 1alpha-mediated up-regulation of CXCR4. J. Biol. Chem. 2013, 288, 21197–21207. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; Zhang, H. The Role of Proinflammatory Pathways in the Pathogenesis of Colitis-Associated Colorectal Cancer. Mediat. Inflamm. 2017, 2017, 5126048. [Google Scholar] [CrossRef] [PubMed]
- An, X.; Yu, W.; Liu, J.; Tang, D.; Yang, L.; Chen, X. Oxidative cell death in cancer: Mechanisms and therapeutic opportunities. Cell Death Dis. 2024, 15, 556. [Google Scholar] [CrossRef]
- Azmanova, M.; Pitto-Barry, A. Oxidative Stress in Cancer Therapy: Friend or Enemy? ChemBioChem 2022, 23, e202100641. [Google Scholar] [CrossRef]
- Laila, U.E.; Zhao, Z.L.; Liu, H.; Xu, Z.X. Aspirin in Cancer Therapy: Pharmacology and Nanotechnology Advances. Int. J. Nanomed. 2025, 20, 2327–2365. [Google Scholar] [CrossRef]
- Shah, M.; Parmar, R.; Patel, K.; Nagani, A. Indole-based COX-2 inhibitors: A decade of advances in inflammation, cancer, and Alzheimer’s therapy. Bioorg Chem. 2024, 153, 107931. [Google Scholar] [CrossRef]
- Lee, J.; Han, Y.; Wang, W.; Jo, H.; Kim, H.; Kim, S.; Yang, K.M.; Kim, S.J.; Dhanasekaran, D.N.; Song, Y.S. Phytochemicals in Cancer Immune Checkpoint Inhibitor Therapy. Biomolecules 2021, 11, 1107. [Google Scholar] [CrossRef]
- Liang, J.; Hansch, G.M.; Hubner, K.; Samstag, Y. Sulforaphane as anticancer agent: A double-edged sword? Tricky balance between effects on tumor cells and immune cells. Adv. Biol. Regul. 2019, 71, 79–87. [Google Scholar] [CrossRef]
- Baird, L.; Kensler, T.W.; Yamamoto, M. Novel NRF2-activated cancer treatments utilizing synthetic lethality. IUBMB Life 2022, 74, 1209–1231. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Hu, H.; Tan, S.; Dong, Q.; Fan, X.; Wang, Y.; Zhang, H.; He, J. The role of neutrophil extracellular traps in cancer progression, metastasis and therapy. Exp. Hematol. Oncol. 2022, 11, 99. [Google Scholar] [CrossRef] [PubMed]
- Valko, M.; Rhodes, C.J.; Moncol, J.; Izakovic, M.; Mazur, M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem. Biol. Interact. 2006, 160, 1–40. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, X.; Cueto, R.; Effi, C.; Zhang, Y.; Tan, H.; Qin, X.; Ji, Y.; Yang, X.; Wang, H. Biochemical basis and metabolic interplay of redox regulation. Redox Biol. 2019, 26, 101284. [Google Scholar] [CrossRef]
- Forman, H.J.; Zhang, H. Targeting oxidative stress in disease: Promise and limitations of antioxidant therapy. Nat. Rev. Drug Discov. 2021, 20, 689–709. [Google Scholar] [CrossRef]
- Glorieux, C.; Liu, S.; Trachootham, D.; Huang, P. Targeting ROS in cancer: Rationale and strategies. Nat. Rev. Drug Discov. 2024, 23, 583–606. [Google Scholar] [CrossRef]
- O’Malley, J.; Kumar, R.; Inigo, J.; Yadava, N.; Chandra, D. Mitochondrial Stress Response and Cancer. Trends Cancer 2020, 6, 688–701. [Google Scholar] [CrossRef]
- Chio, I.I.C.; Tuveson, D.A. ROS in Cancer: The Burning Question. Trends Mol. Med. 2017, 23, 411–429. [Google Scholar] [CrossRef]
- Gorrini, C.; Harris, I.S.; Mak, T.W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 2013, 12, 931–947. [Google Scholar] [CrossRef]
- Li, K.; Deng, Z.; Lei, C.; Ding, X.; Li, J.; Wang, C. The Role of Oxidative Stress in Tumorigenesis and Progression. Cells 2024, 13, 441. [Google Scholar] [CrossRef]
- Nakamura, H.; Takada, K. Reactive oxygen species in cancer: Current findings and future directions. Cancer Sci. 2021, 112, 3945–3952. [Google Scholar] [CrossRef] [PubMed]
- Tay, E.X.Y.; Chia, K.; Ong, D.S.T. Epigenetic plasticity and redox regulation of neural stem cell state and fate. Free Radic. Biol. Med. 2021, 170, 116–130. [Google Scholar] [CrossRef] [PubMed]
- Cheung, E.C.; Vousden, K.H. The role of ROS in tumour development and progression. Nat. Rev. Cancer 2022, 22, 280–297. [Google Scholar] [CrossRef] [PubMed]
- Sarmiento-Salinas, F.L.; Perez-Gonzalez, A.; Acosta-Casique, A.; Ix-Ballote, A.; Diaz, A.; Trevino, S.; Rosas-Murrieta, N.H.; Millan-Perez-Pena, L.; Maycotte, P. Reactive oxygen species: Role in carcinogenesis, cancer cell signaling and tumor progression. Life Sci. 2021, 284, 119942. [Google Scholar] [CrossRef] [PubMed]
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Chandel, N.S. Mitochondrial complex III: An essential component of universal oxygen sensing machinery? Respir. Physiol. Neurobiol. 2010, 174, 175–181. [Google Scholar] [CrossRef]
- Han, D.; Williams, E.; Cadenas, E. Mitochondrial respiratory chain-dependent generation of superoxide anion and its release into the intermembrane space. Biochem. J. 2001, 353, 411–416. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Kim, M.J.; Ha, S.J.; So, B.R.; Kim, C.K.; Kim, K.M.; Jung, S.K. NADPH Oxidase and Epidermal Growth Factor Receptor Are Promising Targets of Phytochemicals for Ultraviolet-Induced Skin Carcinogenesis. Antioxidants 2021, 10, 1909. [Google Scholar] [CrossRef]
- Wang, L.; Kuang, Z.; Zhang, D.; Gao, Y.; Ying, M.; Wang, T. Reactive oxygen species in immune cells: A new antitumor target. Biomed. Pharmacother. 2021, 133, 110978. [Google Scholar] [CrossRef]
- Liang, S.; Ma, H.Y.; Zhong, Z.; Dhar, D.; Liu, X.; Xu, J.; Koyama, Y.; Nishio, T.; Karin, D.; Karin, G.; et al. NADPH Oxidase 1 in Liver Macrophages Promotes Inflammation and Tumor Development in Mice. Gastroenterology 2019, 156, 1156–1172.e6. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, F.; Hamanaka, R.; Wheaton, W.W.; Weinberg, S.; Joseph, J.; Lopez, M.; Kalyanaraman, B.; Mutlu, G.M.; Budinger, G.R.; Chandel, N.S. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc. Natl. Acad. Sci. USA 2010, 107, 8788–8793. [Google Scholar] [CrossRef]
- Maya-Mendoza, A.; Ostrakova, J.; Kosar, M.; Hall, A.; Duskova, P.; Mistrik, M.; Merchut-Maya, J.M.; Hodny, Z.; Bartkova, J.; Christensen, C.; et al. Myc and Ras oncogenes engage different energy metabolism programs and evoke distinct patterns of oxidative and DNA replication stress. Mol. Oncol. 2015, 9, 601–616. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, K.; Takenaga, K.; Akimoto, M.; Koshikawa, N.; Yamaguchi, A.; Imanishi, H.; Nakada, K.; Honma, Y.; Hayashi, J. ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science 2008, 320, 661–664. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Garg, M.; Braunstein, G.; Koeffler, H.P. LAMC2 as a therapeutic target for cancers. Expert Opin. Ther. Targets 2014, 18, 979–982. [Google Scholar] [CrossRef]
- Bishayee, A.; Sethi, G. Bioactive natural products in cancer prevention and therapy: Progress and promise. Semin. Cancer Biol. 2016, 40–41, 1–3. [Google Scholar] [CrossRef]
- Moore, J.M.; Correa, R.; Rosenberg, S.M.; Hastings, P.J. Persistent damaged bases in DNA allow mutagenic break repair in Escherichia coli. PLoS Genet. 2017, 13, e1006733. [Google Scholar] [CrossRef]
- Russo, M.; Crisafulli, G.; Sogari, A.; Reilly, N.M.; Arena, S.; Lamba, S.; Bartolini, A.; Amodio, V.; Magri, A.; Novara, L.; et al. Adaptive mutability of colorectal cancers in response to targeted therapies. Science 2019, 366, 1473–1480. [Google Scholar] [CrossRef]
- Messina, S.; De Simone, G.; Ascenzi, P. Cysteine-based regulation of redox-sensitive Ras small GTPases. Redox Biol. 2019, 26, 101282. [Google Scholar] [CrossRef] [PubMed]
- Liou, G.Y.; Doppler, H.; DelGiorno, K.E.; Zhang, L.; Leitges, M.; Crawford, H.C.; Murphy, M.P.; Storz, P. Mutant KRas-Induced Mitochondrial Oxidative Stress in Acinar Cells Upregulates EGFR Signaling to Drive Formation of Pancreatic Precancerous Lesions. Cell Rep. 2016, 14, 2325–2336. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Sawalha, A.H. The Role of Oxidative Stress in Epigenetic Changes Underlying Autoimmunity. Antioxid Redox Signal 2022, 36, 423–440. [Google Scholar] [CrossRef]
- Zhang, R.; Kang, K.A.; Kim, K.C.; Na, S.Y.; Chang, W.Y.; Kim, G.Y.; Kim, H.S.; Hyun, J.W. Oxidative stress causes epigenetic alteration of CDX1 expression in colorectal cancer cells. Gene 2013, 524, 214–219. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Ni, X. ROS-mediated DNA methylation pattern alterations in carcinogenesis. Curr. Drug Targets 2015, 16, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Ye, Z.; Zou, Z.; Xiao, G.; Luo, G.; Yang, H. Clinicopathological significance of RUNX3 gene hypermethylation in hepatocellular carcinoma. Tumour Biol. 2014, 35, 10333–10340. [Google Scholar] [CrossRef]
- Moloney, J.N.; Cotter, T.G. ROS signalling in the biology of cancer. Semin. Cell Dev. Biol. 2018, 80, 50–64. [Google Scholar] [CrossRef]
- Du, W.; Jiang, P.; Mancuso, A.; Stonestrom, A.; Brewer, M.D.; Minn, A.J.; Mak, T.W.; Wu, M.; Yang, X. TAp73 enhances the pentose phosphate pathway and supports cell proliferation. Nat. Cell Biol. 2013, 15, 991–1000. [Google Scholar] [CrossRef]
- Edderkaoui, M.; Hong, P.; Vaquero, E.C.; Lee, J.K.; Fischer, L.; Friess, H.; Buchler, M.W.; Lerch, M.M.; Pandol, S.J.; Gukovskaya, A.S. Extracellular matrix stimulates reactive oxygen species production and increases pancreatic cancer cell survival through 5-lipoxygenase and NADPH oxidase. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 289, G1137–G1147. [Google Scholar] [CrossRef]
- Gao, P.; Zhang, H.; Dinavahi, R.; Li, F.; Xiang, Y.; Raman, V.; Bhujwalla, Z.M.; Felsher, D.W.; Cheng, L.; Pevsner, J.; et al. HIF-dependent antitumorigenic effect of antioxidants in vivo. Cancer Cell 2007, 12, 230–238. [Google Scholar] [CrossRef]
- Trachootham, D.; Lu, W.; Ogasawara, M.A.; Nilsa, R.D.; Huang, P. Redox regulation of cell survival. Antioxid. Redox Signal. 2008, 10, 1343–1374. [Google Scholar] [CrossRef] [PubMed]
- Storz, P. Reactive oxygen species in tumor progression. Front. Biosci. 2005, 10, 1881–1896. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Shen, Z.; Yang, Q.; Sui, F.; Pu, J.; Ma, J.; Ma, S.; Yao, D.; Ji, M.; Hou, P. Vitamin C kills thyroid cancer cells through ROS-dependent inhibition of MAPK/ERK and PI3K/AKT pathways via distinct mechanisms. Theranostics 2019, 9, 4461–4473. [Google Scholar] [CrossRef] [PubMed]
- Pani, G.; Galeotti, T.; Chiarugi, P. Metastasis: Cancer cell’s escape from oxidative stress. Cancer Metastasis Rev. 2010, 29, 351–378. [Google Scholar] [CrossRef]
- Zuo, J.; Zhao, M.; Liu, B.; Han, X.; Li, Y.; Wang, W.; Zhang, Q.; Lv, P.; Xing, L.; Shen, H.; et al. TNF-alpha-mediated upregulation of SOD-2 contributes to cell proliferation and cisplatin resistance in esophageal squamous cell carcinoma. Oncol. Rep. 2019, 42, 1497–1506. [Google Scholar]
- Satooka, H.; Hara-Chikuma, M. Aquaporin-3 Controls Breast Cancer Cell Migration by Regulating Hydrogen Peroxide Transport and Its Downstream Cell Signaling. Mol. Cell Biol. 2016, 36, 1206–1218. [Google Scholar] [CrossRef]
- Piskounova, E.; Agathocleous, M.; Murphy, M.M.; Hu, Z.; Huddlestun, S.E.; Zhao, Z.; Leitch, A.M.; Johnson, T.M.; DeBerardinis, R.J.; Morrison, S.J. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature 2015, 527, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.A.; Huang, R.Y.; Jackson, R.A.; Thiery, J.P. Emt: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.M.; Muthuramalingam, K.; Cho, M. Redox Regulation of NOX Isoforms on FAK(Y397)/SRC(Y416) Phosphorylation Driven Epithelial-to-Mesenchymal Transition in Malignant Cervical Epithelial Cells. Cells 2020, 9, 1555. [Google Scholar] [CrossRef]
- Nikitovic, D.; Corsini, E.; Kouretas, D.; Tsatsakis, A.; Tzanakakis, G. ROS-major mediators of extracellular matrix remodeling during tumor progression. Food Chem. Toxicol. 2013, 61, 178–186. [Google Scholar] [CrossRef]
- Lee, K.H.; Kim, S.W.; Kim, J.R. Reactive oxygen species regulate urokinase plasminogen activator expression and cell invasion via mitogen-activated protein kinase pathways after treatment with hepatocyte growth factor in stomach cancer cells. J. Exp. Clin. Cancer Res. 2009, 28, 73. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Han, Q.; Yang, Z.; Ni, Y.; Agbana, Y.L.; Bai, H.; Yi, Z.; Yi, X.; Kuang, Y.; Zhu, Y. G6PD facilitates clear cell renal cell carcinoma invasion by enhancing MMP2 expression through ROS-MAPK axis pathway. Int. J. Oncol. 2020, 57, 197–212. [Google Scholar] [CrossRef] [PubMed]
- Xi, C.; Hu, Y.; Buckhaults, P.; Moskophidis, D.; Mivechi, N.F. Heat shock factor Hsf1 cooperates with ErbB2 (Her2/Neu) protein to promote mammary tumorigenesis and metastasis. J. Biol. Chem. 2012, 287, 35646–35657. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Cao, L.; Han, L.; Xu, Q.; Ma, Q. Superoxide dismutase promotes the epithelial-mesenchymal transition of pancreatic cancer cells via activation of the H2O2/ERK/NF-kappaB axis. Int. J. Oncol. 2015, 46, 2613–2620. [Google Scholar] [CrossRef] [PubMed]
- Cheung, E.C.; DeNicola, G.M.; Nixon, C.; Blyth, K.; Labuschagne, C.F.; Tuveson, D.A.; Vousden, K.H. Dynamic ROS Control by TIGAR Regulates the Initiation and Progression of Pancreatic Cancer. Cancer Cell 2020, 37, 168–182 e164. [Google Scholar] [CrossRef]
- Wu, B.; Cheng, Y.; Li, L.; Du, Z.; Liu, Q.; Tan, X.; Li, X.; Zhao, G.; Li, E. Role of the sulfur-containing amino acid-ROS axis in cancer chemotherapeutic drug resistance. Drug Resist. Updat. 2025, 81, 101238. [Google Scholar] [CrossRef]
- Zhou, N.; Chen, J.; Ling, Z.; Zhang, C.; Zhou, Y.; Wang, D.; Zhou, L.; Wang, Z.; Sun, N.; Wang, X.; et al. Aryl hydrocarbon receptor sulfenylation promotes glycogenolysis and rescues cancer chemoresistance. J. Clin. Investig. 2023, 133, e170753. [Google Scholar] [CrossRef] [PubMed]
- Cui, Q.; Wang, J.Q.; Assaraf, Y.G.; Ren, L.; Gupta, P.; Wei, L.; Ashby, C.R., Jr.; Yang, D.H.; Chen, Z.S. Modulating ROS to overcome multidrug resistance in cancer. Drug Resist. Updat. 2018, 41, 1–25. [Google Scholar] [CrossRef]
- Dong, S.; Liang, S.; Cheng, Z.; Zhang, X.; Luo, L.; Li, L.; Zhang, W.; Li, S.; Xu, Q.; Zhong, M.; et al. ROS/PI3K/Akt and Wnt/beta-catenin signalings activate HIF-1alpha-induced metabolic reprogramming to impart 5-fluorouracil resistance in colorectal cancer. J. Exp. Clin. Cancer Res. 2022, 41, 15. [Google Scholar] [CrossRef]
- Dayem, A.A.; Choi, H.Y.; Kim, J.H.; Cho, S.G. Role of oxidative stress in stem, cancer, and cancer stem cells. Cancers 2010, 2, 859–884. [Google Scholar] [CrossRef]
- Jiang, J.; Wang, K.; Chen, Y.; Chen, H.; Nice, E.C.; Huang, C. Redox regulation in tumor cell epithelial-mesenchymal transition: Molecular basis and therapeutic strategy. Signal Transduct. Target. Ther. 2017, 2, 17036. [Google Scholar] [CrossRef] [PubMed]
- Shanmugasundaram, K.; Nayak, B.K.; Friedrichs, W.E.; Kaushik, D.; Rodriguez, R.; Block, K. NOX4 functions as a mitochondrial energetic sensor coupling cancer metabolic reprogramming to drug resistance. Nat. Commun. 2017, 8, 997. [Google Scholar] [CrossRef]
- Zhang, J.; Ali, M.Y.; Chong, H.B.; Tien, P.C.; Woods, J.; Noble, C.; Vornbaumen, T.; Ordulu, Z.; Possemato, A.P.; Harry, S.; et al. Oxidation of retromer complex controls mitochondrial translation. Nature 2025, 641, 1048–1058. [Google Scholar] [CrossRef]
- Liu, C.; Yuan, Y.; Zhan, Y.; Zou, M.; Wu, L.; Zhang, C.; Chen, B.; Zeng, H.; Yang, R.; Hu, T.; et al. Role of the USP family in autophagy regulation and cancer progression. Apoptosis 2025, 30, 1133–1151. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, Y.; Luo, Y.; Liu, Y.; Yi, Y.; Li, J.; Pan, Y.; Li, W.; You, W.; Hu, Q.; et al. USP5-Beclin 1 axis overrides p53-dependent senescence and drives Kras-induced tumorigenicity. Nat. Commun. 2022, 13, 7799. [Google Scholar] [CrossRef]
- Ullah, H.; Di Minno, A.; Santarcangelo, C.; Khan, H.; Daglia, M. Improvement of Oxidative Stress and Mitochondrial Dysfunction by beta-Caryophyllene: A Focus on the Nervous System. Antioxidants 2021, 10, 546. [Google Scholar] [CrossRef] [PubMed]
- AlBasher, G.; AlKahtane, A.A.; Alarifi, S.; Ali, D.; Alessia, M.S.; Almeer, R.S.; Abdel-Daim, M.M.; Al-Sultan, N.K.; Al-Qahtani, A.A.; Ali, H.; et al. Methotrexate-induced apoptosis in human ovarian adenocarcinoma SKOV-3 cells via ROS-mediated bax/bcl-2-cyt-c release cascading. Onco Targets Ther. 2019, 12, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Cui, L.; Ye, J.; Yang, G.; Lu, G.; Fang, X.; Zeng, Z.; Zhou, J. Dioscin facilitates ROS-induced apoptosis via the p38-MAPK/HSP27-mediated pathways in lung squamous cell carcinoma. Int. J. Biol. Sci. 2020, 16, 2883–2894. [Google Scholar] [CrossRef] [PubMed]
- Pessoa, J. Cytochrome c in cancer therapy and prognosis. Biosci. Rep. 2022, 42, BSR20222171. [Google Scholar] [CrossRef]
- Liu, X.; Hussain, R.; Mehmood, K.; Tang, Z.; Zhang, H.; Li, Y. Mitochondrial-Endoplasmic Reticulum Communication-Mediated Oxidative Stress and Autophagy. Biomed. Res. Int. 2022, 2022, 6459585. [Google Scholar] [CrossRef]
- Kim, S.H.; Kim, K.Y.; Yu, S.N.; Park, S.G.; Yu, H.S.; Seo, Y.K.; Ahn, S.C. Monensin Induces PC-3 Prostate Cancer Cell Apoptosis via ROS Production and Ca2+ Homeostasis Disruption. Anticancer. Res. 2016, 36, 5835–5843. [Google Scholar] [CrossRef] [PubMed]
- Rai, A.; Patwardhan, R.S.; Jayakumar, S.; Pachpatil, P.; Das, D.; Panigrahi, G.C.; Gota, V.; Patwardhan, S.; Sandur, S.K. Clobetasol propionate, a Nrf-2 inhibitor, sensitizes human lung cancer cells to radiation-induced killing via mitochondrial ROS-dependent ferroptosis. Acta Pharmacol. Sin. 2024, 45, 1506–1519. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Wang, Y.; Li, X.; Zhou, J.; Yang, W.; Wang, X.; Jiao, S.; Zuo, W.; You, Z.; Ying, W.; et al. O-GlcNAcylation regulates the stability of transferrin receptor (TFRC) to control the ferroptosis in hepatocellular carcinoma cells. Redox Biol. 2024, 73, 103182. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Tavana, O.; Chu, B.; Erber, L.; Chen, Y.; Baer, R.; Gu, W. NRF2 Is a Major Target of ARF in p53-Independent Tumor Suppression. Mol. Cell 2017, 68, 224–232.e4. [Google Scholar] [CrossRef]
- Zheng, W.; Hu, J.; Lv, Y.; Bai, B.; Shan, L.; Chen, K.; Dai, S.; Zhu, H. Pyrvinium pamoate inhibits cell proliferation through ROS-mediated AKT-dependent signaling pathway in colorectal cancer. Med. Oncol. 2021, 38, 21. [Google Scholar] [CrossRef]
- Toyoshima, H.; Hunter, T. p27, a novel inhibitor of G1 cyclin-Cdk protein kinase activity, is related to p21. Cell 1994, 78, 67–74. [Google Scholar] [CrossRef]
- Coats, S.; Flanagan, W.M.; Nourse, J.; Roberts, J.M. Requirement of p27Kip1 for restriction point control of the fibroblast cell cycle. Science 1996, 272, 877–880. [Google Scholar] [CrossRef]
- Nevins, J.R. Toward an understanding of the functional complexity of the E2F and retinoblastoma families. Cell Growth Differ. 1998, 9, 585–593. [Google Scholar] [PubMed]
- Brown, K.; Xie, S.; Qiu, X.; Mohrin, M.; Shin, J.; Liu, Y.; Zhang, D.; Scadden, D.T.; Chen, D. SIRT3 reverses aging-associated degeneration. Cell Rep. 2013, 3, 319–327. [Google Scholar] [CrossRef]
- Kaul, S.C.; Aida, S.; Yaguchi, T.; Kaur, K.; Wadhwa, R. Activation of wild type p53 function by its mortalin-binding, cytoplasmically localizing carboxyl terminus peptides. J. Biol. Chem. 2005, 280, 39373–39379. [Google Scholar] [CrossRef]
- Trinh, D.L.N.; Elwi, A.N.; Kim, S.W. Direct interaction between p53 and Tid1 proteins affects p53 mitochondrial localization and apoptosis. Oncotarget 2010, 1, 396–404. [Google Scholar] [CrossRef]
- Arena, G.; Cisse, M.Y.; Pyrdziak, S.; Chatre, L.; Riscal, R.; Fuentes, M.; Arnold, J.J.; Kastner, M.; Gayte, L.; Bertrand-Gaday, C.; et al. Mitochondrial MDM2 Regulates Respiratory Complex I Activity Independently of p53. Mol. Cell 2018, 69, 594–609.e8. [Google Scholar] [CrossRef]
- Fasano, C.; Disciglio, V.; Bertora, S.; Lepore Signorile, M.; Simone, C. FOXO3a from the Nucleus to the Mitochondria: A Round Trip in Cellular Stress Response. Cells 2019, 8, 1110. [Google Scholar] [CrossRef]
- Hu, C.; Ni, Z.; Li, B.S.; Yong, X.; Yang, X.; Zhang, J.W.; Zhang, D.; Qin, Y.; Jie, M.M.; Dong, H.; et al. hTERT promotes the invasion of gastric cancer cells by enhancing FOXO3a ubiquitination and subsequent ITGB1 upregulation. Gut 2017, 66, 31–42. [Google Scholar] [CrossRef]
- Li, X.; Gao, J.; Wu, C.; Wang, C.; Zhang, R.; He, J.; Xia, Z.J.; Joshi, N.; Karp, J.M.; Kuai, R. Precise modulation and use of reactive oxygen species for immunotherapy. Sci. Adv. 2024, 10, eadl0479. [Google Scholar] [CrossRef]
- Hui, C.; Marquez, C.; Lau, B.; Das, M.; Myall, N.J.; Roy, M.; Wakelee, H.A.; Neal, J.W.; Kovalchuk, N.; Chin, A.; et al. Patient Selection and Outcomes for Hypofractionated Accelerated Radiation and Concurrent Chemotherapy for Non-Small-Cell Lung Cancer. Clin. Lung Cancer 2024, 25, e92–e100.e4. [Google Scholar] [CrossRef]
- Zhu, S.; Zhang, T.; Zheng, L.; Liu, H.; Song, W.; Liu, D.; Li, Z.; Pan, C.X. Combination strategies to maximize the benefits of cancer immunotherapy. J. Hematol. Oncol. 2021, 14, 156. [Google Scholar] [CrossRef]
- Fu, Q.; Zhang, S.; Shen, S.; Gu, Z.; Chen, J.; Song, D.; Sun, P.; Wang, C.; Guo, Z.; Xiao, Y.; et al. Radiotherapy-triggered reduction of platinum-based chemotherapeutic prodrugs in tumours. Nat. Biomed. Eng. 2024, 8, 1425–1435. [Google Scholar] [CrossRef]
- Dai, Q.; Wang, L.; Ren, E.; Chen, H.; Gao, X.; Cheng, H.; An, Y.; Chu, C.; Liu, G. Ruthenium-Based Metal-Organic Nanoradiosensitizers Enhance Radiotherapy by Combining ROS Generation and CO Gas Release. Angew. Chem. Int. Ed. Engl. 2022, 61, e202211674. [Google Scholar] [CrossRef]
- Guo, S.; Burcus, N.I.; Scott, M.; Jing, Y.; Semenov, I. The role of reactive oxygen species in the immunity induced by nano-pulse stimulation. Sci. Rep. 2021, 11, 23745. [Google Scholar] [CrossRef]
- Wang, Z.; Ge, S.; Liao, T.; Yuan, M.; Qian, W.; Chen, Q.; Liang, W.; Cheng, X.; Zhou, Q.; Ju, Z.; et al. Integrative single-cell metabolomics and phenotypic profiling reveals metabolic heterogeneity of cellular oxidation and senescence. Nat. Commun. 2025, 16, 2740. [Google Scholar] [CrossRef]
- Xu, Q.; Wang, M.; Zhang, F.; Chen, G.; Shu, Z.; Li, L.; Zhang, F.; Wang, Y.; Wang, Y.; Duan, X.; et al. A Bimetallic Electro-Sensitizer Improves ROS Therapy by Relieving Autophagy-Induced ROS Tolerance and Immune Suppression. Small 2024, 20, e2402312. [Google Scholar] [CrossRef]
- Zhu, Y.; Yu, J.; Gu, J.; Xue, C.; Zhang, L.; Chen, J.; Shen, L. Relaxed 3D genome conformation facilitates the pluripotent to totipotent-like state transition in embryonic stem cells. Nucleic Acids Res. 2021, 49, 12167–12177. [Google Scholar] [CrossRef]
- Xu, S.; Liu, Y.; Yang, S.; Fei, W.; Qin, J.; Lu, W.; Xu, J. FXN targeting induces cell death in ovarian cancer stem-like cells through PRDX3-Mediated oxidative stress. iScience 2024, 27, 110506. [Google Scholar] [CrossRef]
- Chen, C.Y.; Ye, Y.Z.; Huang, Y.H.; Tzeng, Y.M.; Gurbanov, R.; Wang, W.L.; Chang, W.W. Ovatodiolide inhibits endometrial cancer stemness via reactive oxygen species-mediated DNA damage and cell cycle arrest. Chem. Biol. Interact. 2024, 403, 111244. [Google Scholar] [CrossRef]
- Liu, S.; Qiu, Y.; Xiang, R.; Huang, P. Characterization of H2O2-Induced Alterations in Global Transcription of mRNA and lncRNA. Antioxidants 2022, 11, 495. [Google Scholar] [CrossRef]
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting cancer cells by ROS-mediated mechanisms: A radical therapeutic approach? Nat. Rev. Drug Discov. 2009, 8, 579–591. [Google Scholar] [CrossRef]
- Xue, D.; Zhou, X.; Qiu, J. Emerging role of NRF2 in ROS-mediated tumor chemoresistance. Biomed. Pharmacother. 2020, 131, 110676. [Google Scholar] [CrossRef]
- Cabrera-Serrano, A.J.; Sanchez-Maldonado, J.M.; Gonzalez-Olmedo, C.; Carretero-Fernandez, M.; Diaz-Beltran, L.; Gutierrez-Bautista, J.F.; Garcia-Verdejo, F.J.; Galvez-Montosa, F.; Lopez-Lopez, J.A.; Garcia-Martin, P.; et al. Crosstalk Between Autophagy and Oxidative Stress in Hematological Malignancies: Mechanisms, Implications, and Therapeutic Potential. Antioxidants 2025, 14, 264. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Qi, H.; Liu, Y.; Duan, C.; Liu, X.; Xia, T.; Chen, D.; Piao, H.L.; Liu, H.X. The double-edged roles of ROS in cancer prevention and therapy. Theranostics 2021, 11, 4839–4857. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Li, A.; Wang, Y.; Zhang, Y. Intratumoral microbiota: Roles in cancer initiation, development and therapeutic efficacy. Signal Transduct. Target. Ther. 2023, 8, 35. [Google Scholar] [CrossRef]
- Han, R.; Zhao, M.; Wang, Z.; Liu, H.; Zhu, S.; Huang, L.; Wang, Y.; Wang, L.; Hong, Y.; Sha, Y.; et al. Super-efficient in Vivo Two-Photon Photodynamic Therapy with a Gold Nanocluster as a Type I Photosensitizer. ACS Nano 2020, 14, 9532–9544. [Google Scholar] [CrossRef]
- Ni, M.; Zhou, J.; Zhu, Z.; Xu, Q.; Yin, Z.; Wang, Y.; Zheng, Z.; Zhao, H. Shikonin and cisplatin synergistically overcome cisplatin resistance of ovarian cancer by inducing ferroptosis via upregulation of HMOX1 to promote Fe2+ accumulation. Phytomedicine 2023, 112, 154701. [Google Scholar] [CrossRef]
- Zhao, Z.; Wu, Y.; Liang, X.; Liu, J.; Luo, Y.; Zhang, Y.; Li, T.; Liu, C.; Luo, X.; Chen, J.; et al. Sonodynamic Therapy of NRP2 Monoclonal Antibody-Guided MOFs@COF Targeted Disruption of Mitochondrial and Endoplasmic Reticulum Homeostasis to Induce Autophagy-Dependent Ferroptosis. Adv. Sci. 2023, 10, e2303872. [Google Scholar] [CrossRef]
- Yuan, J.; Khan, S.U.; Yan, J.; Lu, J.; Yang, C.; Tong, Q. Baicalin enhances the efficacy of 5-Fluorouracil in gastric cancer by promoting ROS-mediated ferroptosis. Biomed. Pharmacother. 2023, 164, 114986. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, X.; Cheng, F.; Cao, W.; Geng, Y.; Chen, Z.; Wei, W.; Zhang, L. Xanthatin induce DDP-resistance lung cancer cells apoptosis through regulation of GLUT1 mediated ROS accumulation. Drug Dev. Res. 2023, 84, 1266–1278. [Google Scholar] [CrossRef]
- Dilenko, H.; Barton Tomankova, K.; Valkova, L.; Hosikova, B.; Kolarikova, M.; Malina, L.; Bajgar, R.; Kolarova, H. Graphene-Based Photodynamic Therapy and Overcoming Cancer Resistance Mechanisms: A Comprehensive Review. Int. J. Nanomed. 2024, 19, 5637–5680. [Google Scholar] [CrossRef]
- Zhou, Y.; Chen, Y.; Xuan, C.; Li, X.; Tan, Y.; Yang, M.; Cao, M.; Chen, C.; Huang, X.; Hu, R. DPP9 regulates NQO1 and ROS to promote resistance to chemotherapy in liver cancer cells. Redox Biol. 2024, 75, 103292. [Google Scholar] [CrossRef]
- Kopecka, J.; Trouillas, P.; Gasparovic, A.C.; Gazzano, E.; Assaraf, Y.G.; Riganti, C. Phospholipids and cholesterol: Inducers of cancer multidrug resistance and therapeutic targets. Drug Resist. Updat. 2020, 49, 100670. [Google Scholar] [CrossRef]
- Jin, P.; Jiang, J.; Zhou, L.; Huang, Z.; Nice, E.C.; Huang, C.; Fu, L. Mitochondrial adaptation in cancer drug resistance: Prevalence, mechanisms, and management. J. Hematol. Oncol. 2022, 15, 97. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Chang, L.; Tang, L.; Jiang, H.; Xu, X.; Zhang, X.; Chen, J.; Dong, L.; Xu, Q.; Cao, R.; et al. Long noncoding RNA GDIL acts as a scaffold for CHAC1 and XRN2 to promote platinum resistance of colorectal cancer through inhibition of glutathione degradation. Cell Death Dis. 2025, 16, 62. [Google Scholar] [CrossRef] [PubMed]
- Ge, W.; Zhao, K.; Wang, X.; Li, H.; Yu, M.; He, M.; Xue, X.; Zhu, Y.; Zhang, C.; Cheng, Y.; et al. iASPP Is an Antioxidative Factor and Drives Cancer Growth and Drug Resistance by Competing with Nrf2 for Keap1 Binding. Cancer Cell 2017, 32, 561–573.e6. [Google Scholar] [CrossRef] [PubMed]
- Oren, Y.; Tsabar, M.; Cuoco, M.S.; Amir-Zilberstein, L.; Cabanos, H.F.; Hutter, J.C.; Hu, B.; Thakore, P.I.; Tabaka, M.; Fulco, C.P.; et al. Cycling cancer persister cells arise from lineages with distinct programs. Nature 2021, 596, 576–582. [Google Scholar] [CrossRef]
- Hu, A.; Pu, Y.; Xu, N.; Yang, H.; Hu, X.; Sun, R.; Jin, R.; Nie, Y. Hierarchically decorated magnetic nanoparticles amplify the oxidative stress and promote the chemodynamic/magnetic hyperthermia/immune therapy. Acta Biomater. 2024, 173, 457–469. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.; Ai, X.; Yang, K.; Yang, Z.; Fei, F.; Liao, X.; Qiu, Z.; Gimple, R.C.; Yuan, H.; Huang, H.; et al. Targeting Microglial Metabolic Rewiring Synergizes with Immune-Checkpoint Blockade Therapy for Glioblastoma. Cancer Discov. 2023, 13, 974–1001. [Google Scholar] [CrossRef]
- Duan, G.; Huang, C.; Zhao, J.; Zhang, Y.; Zhao, W.; Dai, H. Investigating subtypes of lung adenocarcinoma by oxidative stress and immunotherapy related genes. Sci. Rep. 2023, 13, 20930. [Google Scholar] [CrossRef]
- Yang, T.; Wang, G.; Zhang, M.; Hu, X.; Li, Q.; Yun, F.; Xing, Y.; Song, X.; Zhang, H.; Hu, G.; et al. Triggering endogenous Z-RNA sensing for anti-tumor therapy through ZBP1-dependent necroptosis. Cell Rep. 2023, 42, 113377. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Chen, S.; He, X.; Yuan, Y.; Wei, X. Targeting inflammation as cancer therapy. J. Hematol. Oncol. 2024, 17, 13. [Google Scholar] [CrossRef]
- Nihira, N.T.; Kudo, R.; Ohta, T. Inflammation and tumor immune escape in response to DNA damage. Semin. Cancer Biol. 2025, 110, 36–45. [Google Scholar] [CrossRef]
- Krugliak Cleveland, N.; Torres, J.; Rubin, D.T. What Does Disease Progression Look Like in Ulcerative Colitis, and How Might It Be Prevented? Gastroenterology 2022, 162, 1396–1408. [Google Scholar] [CrossRef]
- Shah, S.C.; Itzkowitz, S.H. Colorectal Cancer in Inflammatory Bowel Disease: Mechanisms and Management. Gastroenterology 2022, 162, 715–730.e3. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.C.; Chiang, T.H.; Chou, C.K.; Tu, Y.K.; Liao, W.C.; Wu, M.S.; Graham, D.Y. Association Between Helicobacter pylori Eradication and Gastric Cancer Incidence: A Systematic Review and Meta-analysis. Gastroenterology 2016, 150, 1113–1124.e5. [Google Scholar] [CrossRef] [PubMed]
- Tian, T.; Song, C.; Jiang, L.; Dai, J.; Lin, Y.; Xu, X.; Yu, C.; Ge, Z.; Ding, Y.; Wen, Y.; et al. Hepatitis B virus infection and the risk of cancer among the Chinese population. Int. J. Cancer 2020, 147, 3075–3084. [Google Scholar] [CrossRef] [PubMed]
- Kawanishi, S.; Ohnishi, S.; Ma, N.; Hiraku, Y.; Murata, M. Crosstalk between DNA Damage and Inflammation in the Multiple Steps of Carcinogenesis. Int. J. Mol. Sci. 2017, 18, 1808. [Google Scholar] [CrossRef] [PubMed]
- Suresh, V.; Dash, P.; Suklabaidya, S.; Murmu, K.C.; Sasmal, P.K.; Jogdand, G.M.; Parida, D.; Sethi, M.; Das, B.; Mohapatra, D.; et al. MIF confers survival advantage to pancreatic CAFs by suppressing interferon pathway-induced p53-dependent apoptosis. FASEB J. 2022, 36, e22449. [Google Scholar] [CrossRef] [PubMed]
- Propper, D.J.; Balkwill, F.R. Harnessing cytokines and chemokines for cancer therapy. Nat. Rev. Clin. Oncol. 2022, 19, 237–253. [Google Scholar] [CrossRef]
- Li, L.; Yu, R.; Cai, T.; Chen, Z.; Lan, M.; Zou, T.; Wang, B.; Wang, Q.; Zhao, Y.; Cai, Y. Effects of immune cells and cytokines on inflammation and immunosuppression in the tumor microenvironment. Int. Immunopharmacol. 2020, 88, 106939. [Google Scholar] [CrossRef]
- Wu, M.; Ma, M.; Tan, Z.; Zheng, H.; Liu, X. Neutrophil: A New Player in Metastatic Cancers. Front. Immunol. 2020, 11, 565165. [Google Scholar] [CrossRef]
- Albini, A.; Bruno, A.; Noonan, D.M.; Mortara, L. Contribution to Tumor Angiogenesis From Innate Immune Cells Within the Tumor Microenvironment: Implications for Immunotherapy. Front. Immunol. 2018, 9, 527. [Google Scholar] [CrossRef]
- Singhal, S.; Bhojnagarwala, P.S.; O’Brien, S.; Moon, E.K.; Garfall, A.L.; Rao, A.S.; Quatromoni, J.G.; Stephen, T.L.; Litzky, L.; Deshpande, C.; et al. Origin and Role of a Subset of Tumor-Associated Neutrophils with Antigen-Presenting Cell Features in Early-Stage Human Lung Cancer. Cancer Cell 2016, 30, 120–135. [Google Scholar] [CrossRef]
- Mishalian, I.; Bayuh, R.; Eruslanov, E.; Michaeli, J.; Levy, L.; Zolotarov, L.; Singhal, S.; Albelda, S.M.; Granot, Z.; Fridlender, Z.G. Neutrophils recruit regulatory T-cells into tumors via secretion of CCL17—A new mechanism of impaired antitumor immunity. Int. J. Cancer 2014, 135, 1178–1186. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.L.; Zhou, Z.J.; Hu, Z.Q.; Huang, X.W.; Wang, Z.; Chen, E.B.; Fan, J.; Cao, Y.; Dai, Z.; Zhou, J. Tumor-Associated Neutrophils Recruit Macrophages and T-Regulatory Cells to Promote Progression of Hepatocellular Carcinoma and Resistance to Sorafenib. Gastroenterology 2016, 150, 1646–1658.e17. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.; Ko, S.Y.; Mohamed, M.S.; Kenny, H.A.; Lengyel, E.; Naora, H. Neutrophils facilitate ovarian cancer premetastatic niche formation in the omentum. J. Exp. Med. 2019, 216, 176–194. [Google Scholar] [CrossRef] [PubMed]
- Demkow, U. Neutrophil Extracellular Traps (NETs) in Cancer Invasion, Evasion and Metastasis. Cancers 2021, 13, 4495. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, F.; Chen, L.; Fang, C.; Li, S.; Yuan, S.; Qian, X.; Yin, Y.; Yu, B.; Fu, B.; et al. Neutrophil Extracellular Traps (NETs) Promote Non-Small Cell Lung Cancer Metastasis by Suppressing lncRNA MIR503HG to Activate the NF-kappaB/NLRP3 Inflammasome Pathway. Front. Immunol. 2022, 13, 867516. [Google Scholar]
- Deng, J.; Kang, Y.; Cheng, C.C.; Li, X.; Dai, B.; Katz, M.H.; Men, T.; Kim, M.P.; Koay, E.A.; Huang, H.; et al. DDR1-induced neutrophil extracellular traps drive pancreatic cancer metastasis. JCI Insight. 2021, 6, e146133. [Google Scholar] [CrossRef]
- Khan, U.; Chowdhury, S.; Billah, M.M.; Islam, K.M.D.; Thorlacius, H.; Rahman, M. Neutrophil Extracellular Traps in Colorectal Cancer Progression and Metastasis. Int. J. Mol. Sci. 2021, 22, 7260. [Google Scholar] [CrossRef]
- Xiao, Y.; Cong, M.; Li, J.; He, D.; Wu, Q.; Tian, P.; Wang, Y.; Yang, S.; Liang, C.; Liang, Y.; et al. Cathepsin C promotes breast cancer lung metastasis by modulating neutrophil infiltration and neutrophil extracellular trap formation. Cancer Cell 2021, 39, 423–437.e7. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Wang, Z.; Li, L.; Zhang, Z.; Jin, X.; Wu, P.; Sun, S.; Pan, J.; Su, K.; Jia, F.; et al. Aged neutrophils form mitochondria-dependent vital NETs to promote breast cancer lung metastasis. J. Immunother. Cancer 2021, 9, e002875. [Google Scholar] [CrossRef]
- Arelaki, S.; Arampatzioglou, A.; Kambas, K.; Papagoras, C.; Miltiades, P.; Angelidou, I.; Mitsios, A.; Kotsianidis, I.; Skendros, P.; Sivridis, E.; et al. Gradient Infiltration of Neutrophil Extracellular Traps in Colon Cancer and Evidence for Their Involvement in Tumour Growth. PLoS ONE 2016, 11, e0154484. [Google Scholar] [CrossRef]
- Millrud, C.R.; Kagedal, A.; Kumlien Georen, S.; Winqvist, O.; Uddman, R.; Razavi, R.; Munck-Wikland, E.; Cardell, L.O. NET-producing CD16high CD62Ldim neutrophils migrate to tumor sites and predict improved survival in patients with HNSCC. Int. J. Cancer 2017, 140, 2557–2567. [Google Scholar] [CrossRef] [PubMed]
- Schedel, F.; Mayer-Hain, S.; Pappelbaum, K.I.; Metze, D.; Stock, M.; Goerge, T.; Loser, K.; Sunderkotter, C.; Luger, T.A.; Weishaupt, C. Evidence and impact of neutrophil extracellular traps in malignant melanoma. Pigment. Cell Melanoma Res. 2020, 33, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Mackaness, G.B. Cellular resistance to infection. J. Exp. Med. 1962, 116, 381–406. [Google Scholar] [CrossRef] [PubMed]
- Eum, H.H.; Kwon, M.; Ryu, D.; Jo, A.; Chung, W.; Kim, N.; Hong, Y.; Son, D.S.; Kim, S.T.; Lee, J.; et al. Tumor-promoting macrophages prevail in malignant ascites of advanced gastric cancer. Exp. Mol. Med. 2020, 52, 1976–1988. [Google Scholar] [CrossRef]
- Bernsmeier, C.; van der Merwe, S.; Perianin, A. Innate immune cells in cirrhosis. J. Hepatol. 2020, 73, 186–201. [Google Scholar] [CrossRef] [PubMed]
- Bruns, H.; Buttner, M.; Fabri, M.; Mougiakakos, D.; Bittenbring, J.T.; Hoffmann, M.H.; Beier, F.; Pasemann, S.; Jitschin, R.; Hofmann, A.D.; et al. Vitamin D-dependent induction of cathelicidin in human macrophages results in cytotoxicity against high-grade B cell lymphoma. Sci. Transl. Med. 2015, 7, 282ra247. [Google Scholar] [CrossRef] [PubMed]
- Larionova, I.; Kazakova, E.; Gerashchenko, T.; Kzhyshkowska, J. New Angiogenic Regulators Produced by TAMs: Perspective for Targeting Tumor Angiogenesis. Cancers 2021, 13, 3253. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, D.B.; Severn, C.E.; Twomey, C.; Greenhough, A.; Cash, J.; Toye, A.M.; Mellor, H.; Martin, P. Live imaging of wound angiogenesis reveals macrophage orchestrated vessel sprouting and regression. EMBO J. 2018, 37, e97786. [Google Scholar] [CrossRef]
- Ramirez-Pedraza, M.; Fernandez, M. Interplay Between Macrophages and Angiogenesis: A Double-Edged Sword in Liver Disease. Front. Immunol. 2019, 10, 2882. [Google Scholar] [CrossRef]
- Wynn, T.A.; Chawla, A.; Pollard, J.W. Macrophage biology in development, homeostasis and disease. Nature 2013, 496, 445–455. [Google Scholar] [CrossRef]
- Palucka, K.; Banchereau, J. Cancer immunotherapy via dendritic cells. Nat. Rev. Cancer 2012, 12, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Aras, S.; Zaidi, M.R. TAMeless traitors: Macrophages in cancer progression and metastasis. Br. J. Cancer 2017, 117, 1583–1591. [Google Scholar] [CrossRef] [PubMed]
- Chemnitz, J.M.; Parry, R.V.; Nichols, K.E.; June, C.H.; Riley, J.L. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J. Immunol. 2004, 173, 945–954. [Google Scholar] [CrossRef] [PubMed]
- Li, B.H.; Garstka, M.A.; Li, Z.F. Chemokines and their receptors promoting the recruitment of myeloid-derived suppressor cells into the tumor. Mol. Immunol. 2020, 117, 201–215. [Google Scholar] [CrossRef]
- Kumar, V.; Patel, S.; Tcyganov, E.; Gabrilovich, D.I. The Nature of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Trends Immunol. 2016, 37, 208–220. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ding, Y.; Guo, N.; Wang, S. MDSCs: Key Criminals of Tumor Pre-metastatic Niche Formation. Front. Immunol. 2019, 10, 172. [Google Scholar] [CrossRef] [PubMed]
- Bruno, A.; Mortara, L.; Baci, D.; Noonan, D.M.; Albini, A. Myeloid Derived Suppressor Cells Interactions With Natural Killer Cells and Pro-angiogenic Activities: Roles in Tumor Progression. Front. Immunol. 2019, 10, 771. [Google Scholar] [CrossRef]
- Wen, Y.; Zhu, Y.; Zhang, C.; Yang, X.; Gao, Y.; Li, M.; Yang, H.; Liu, T.; Tang, H. Chronic inflammation, cancer development and immunotherapy. Front. Pharmacol. 2022, 13, 1040163. [Google Scholar] [CrossRef]
- Saleh, R.; Elkord, E. FoxP3+ T regulatory cells in cancer: Prognostic biomarkers and therapeutic targets. Cancer Lett. 2020, 490, 174–185. [Google Scholar] [CrossRef]
- Chen, M.L.; Pittet, M.J.; Gorelik, L.; Flavell, R.A.; Weissleder, R.; von Boehmer, H.; Khazaie, K. Regulatory T cells suppress tumor-specific CD8 T cell cytotoxicity through TGF-beta signals in vivo. Proc. Natl. Acad. Sci. USA 2005, 102, 419–424. [Google Scholar] [CrossRef]
- Khalaf, K.; Hana, D.; Chou, J.T.; Singh, C.; Mackiewicz, A.; Kaczmarek, M. Aspects of the Tumor Microenvironment Involved in Immune Resistance and Drug Resistance. Front. Immunol. 2021, 12, 656364. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Yu, Y.; Wang, X.; Zhang, T. Tumor-Associated Macrophages in Tumor Immunity. Front. Immunol. 2020, 11, 583084. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.N.; Lou, D.F.; Li, D.Y.; Jiang, W.; Dong, J.Y.; Gao, W.; Chen, H.C. Elevated levels of IL-17A and IL-35 in plasma and bronchoalveolar lavage fluid are associated with checkpoint inhibitor pneumonitis in patients with non-small cell lung cancer. Oncol. Lett. 2020, 20, 611–622. [Google Scholar] [CrossRef]
- Timperi, E.; Pacella, I.; Schinzari, V.; Focaccetti, C.; Sacco, L.; Farelli, F.; Caronna, R.; Del Bene, G.; Longo, F.; Ciardi, A.; et al. Regulatory T cells with multiple suppressive and potentially pro-tumor activities accumulate in human colorectal cancer. Oncoimmunology 2016, 5, e1175800. [Google Scholar] [CrossRef] [PubMed]
- Shaopeng, Z.; Yang, Z.; Yuan, F.; Chen, H.; Zhengjun, Q. Regulation of regulatory T cells and tumor-associated macrophages in gastric cancer tumor microenvironment. Cancer Med. 2024, 13, e6959. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, S.; Mikami, N.; Wing, J.B.; Tanaka, A.; Ichiyama, K.; Ohkura, N. Regulatory T Cells and Human Disease. Annu. Rev. Immunol. 2020, 38, 541–566. [Google Scholar] [CrossRef] [PubMed]
- Yue, T.; Li, J.; Zhu, J.; Zuo, S.; Wang, X.; Liu, Y.; Liu, J.; Liu, X.; Wang, P.; Chen, S. Hydrogen Sulfide Creates a Favorable Immune Microenvironment for Colon Cancer. Cancer Res. 2023, 83, 595–612. [Google Scholar] [CrossRef]
- Li, F.; Guo, Z.; Lizee, G.; Yu, H.; Wang, H.; Si, T. Clinical prognostic value of CD4+CD25+FOXP3+regulatory T cells in peripheral blood of Barcelona Clinic Liver Cancer (BCLC) stage B hepatocellular carcinoma patients. Clin. Chem. Lab. Med. 2014, 52, 1357–1365. [Google Scholar] [CrossRef]
- O’Carroll, S.J.; Kho, D.T.; Wiltshire, R.; Nelson, V.; Rotimi, O.; Johnson, R.; Angel, C.E.; Graham, E.S. Pro-inflammatory TNFalpha and IL-1beta differentially regulate the inflammatory phenotype of brain microvascular endothelial cells. J. Neuroinflammation 2015, 12, 131. [Google Scholar] [CrossRef]
- Hillyer, P.; Mordelet, E.; Flynn, G.; Male, D. Chemokines, chemokine receptors and adhesion molecules on different human endothelia: Discriminating the tissue-specific functions that affect leucocyte migration. Clin. Exp. Immunol. 2003, 134, 431–441. [Google Scholar] [CrossRef] [PubMed]
- Weis, S.; Cui, J.; Barnes, L.; Cheresh, D. Endothelial barrier disruption by VEGF-mediated Src activity potentiates tumor cell extravasation and metastasis. J. Cell Biol. 2004, 167, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Burdick, M.M.; Henson, K.A.; Delgadillo, L.F.; Choi, Y.E.; Goetz, D.J.; Tees, D.F.; Benencia, F. Expression of E-selectin ligands on circulating tumor cells: Cross-regulation with cancer stem cell regulatory pathways? Front. Oncol. 2012, 2, 103. [Google Scholar] [CrossRef] [PubMed]
- Hauselmann, I.; Roblek, M.; Protsyuk, D.; Huck, V.; Knopfova, L.; Grassle, S.; Bauer, A.T.; Schneider, S.W.; Borsig, L. Monocyte Induction of E-Selectin-Mediated Endothelial Activation Releases VE-Cadherin Junctions to Promote Tumor Cell Extravasation in the Metastasis Cascade. Cancer Res. 2016, 76, 5302–5312. [Google Scholar] [CrossRef] [PubMed]
- Hiratsuka, S.; Goel, S.; Kamoun, W.S.; Maru, Y.; Fukumura, D.; Duda, D.G.; Jain, R.K. Endothelial focal adhesion kinase mediates cancer cell homing to discrete regions of the lungs via E-selectin up-regulation. Proc. Natl. Acad. Sci. USA 2011, 108, 3725–3730. [Google Scholar] [CrossRef]
- Kang, S.A.; Blache, C.A.; Bajana, S.; Hasan, N.; Kamal, M.; Morita, Y.; Gupta, V.; Tsolmon, B.; Suh, K.S.; Gorenstein, D.G.; et al. The effect of soluble E-selectin on tumor progression and metastasis. BMC Cancer 2016, 16, 331. [Google Scholar]
- Shea, D.J.; Li, Y.W.; Stebe, K.J.; Konstantopoulos, K. E-selectin-mediated rolling facilitates pancreatic cancer cell adhesion to hyaluronic acid. FASEB J. 2017, 31, 5078–5086. [Google Scholar] [CrossRef]
- Tichet, M.; Prod’Homme, V.; Fenouille, N.; Ambrosetti, D.; Mallavialle, A.; Cerezo, M.; Ohanna, M.; Audebert, S.; Rocchi, S.; Giacchero, D.; et al. Tumour-derived SPARC drives vascular permeability and extravasation through endothelial VCAM1 signalling to promote metastasis. Nat. Commun. 2015, 6, 6993. [Google Scholar] [CrossRef]
- Zamarron, B.F.; Chen, W. Dual roles of immune cells and their factors in cancer development and progression. Int. J. Biol. Sci. 2011, 7, 651–658. [Google Scholar] [CrossRef]
- Amin, M.N.; Siddiqui, S.A.; Ibrahim, M.; Hakim, M.L.; Ahammed, M.S.; Kabir, A.; Sultana, F. Inflammatory cytokines in the pathogenesis of cardiovascular disease and cancer. SAGE Open Med. 2020, 8, 2050312120965752. [Google Scholar] [CrossRef]
- Fahey, E.; Doyle, S.L. IL-1 Family Cytokine Regulation of Vascular Permeability and Angiogenesis. Front. Immunol. 2019, 10, 1426. [Google Scholar] [CrossRef]
- Malik, A.; Kanneganti, T.D. Function and regulation of IL-1alpha in inflammatory diseases and cancer. Immunol. Rev. 2018, 281, 124–137. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.; Mei, Y.; Lei, L.; Binte Hanafi, Z.; Jin, Z.; Liu, Y.; Song, Y.; Zhang, Y.; Hu, B.; Liu, C.; et al. Immune suppressive function of IL-1alpha release in the tumor microenvironment regulated by calpain 1. Oncoimmunology 2022, 11, 2088467. [Google Scholar] [CrossRef]
- Carmi, Y.; Dotan, S.; Rider, P.; Kaplanov, I.; White, M.R.; Baron, R.; Abutbul, S.; Huszar, M.; Dinarello, C.A.; Apte, R.N.; et al. The role of IL-1beta in the early tumor cell-induced angiogenic response. J. Immunol. 2013, 190, 3500–3509. [Google Scholar] [CrossRef]
- Voronov, E.; Carmi, Y.; Apte, R.N. The role IL-1 in tumor-mediated angiogenesis. Front. Physiol. 2014, 5, 114. [Google Scholar] [CrossRef]
- Haabeth, O.A.; Lorvik, K.B.; Yagita, H.; Bogen, B.; Corthay, A. Interleukin-1 is required for cancer eradication mediated by tumor-specific Th1 cells. Oncoimmunology 2016, 5, e1039763. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Shapiro, B.; Vucic, E.A.; Vogt, S.; Bar-Sagi, D. Tumor Cell-Derived IL1beta Promotes Desmoplasia and Immune Suppression in Pancreatic Cancer. Cancer Res. 2020, 80, 1088–1101. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Narazaki, M.; Kishimoto, T. IL-6 in inflammation, immunity, and disease. Cold Spring Harb. Perspect. Biol. 2014, 6, a016295. [Google Scholar] [CrossRef]
- Yao, X.; Huang, J.; Zhong, H.; Shen, N.; Faggioni, R.; Fung, M.; Yao, Y. Targeting interleukin-6 in inflammatory autoimmune diseases and cancers. Pharmacol. Ther. 2014, 141, 125–139. [Google Scholar] [CrossRef]
- Qin, B.; Zhou, Z.; He, J.; Yan, C.; Ding, S. IL-6 Inhibits Starvation-induced Autophagy via the STAT3/Bcl-2 Signaling Pathway. Sci. Rep. 2015, 5, 15701. [Google Scholar] [CrossRef] [PubMed]
- Shi, R.; Chen, M.; Litifu, B. Serum interleukin-6 and survivin levels predict clinical response to etanercept treatment in patients with established rheumatoid arthritis. Mod. Rheumatol. 2018, 28, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Montero, P.; Londono-Vallejo, A.; Vernot, J.P. Senescence-associated IL-6 and IL-8 cytokines induce a self- and cross-reinforced senescence/inflammatory milieu strengthening tumorigenic capabilities in the MCF-7 breast cancer cell line. Cell Commun. Signal 2017, 15, 17. [Google Scholar] [CrossRef] [PubMed]
- Sapochnik, M.; Haedo, M.R.; Fuertes, M.; Ajler, P.; Carrizo, G.; Cervio, A.; Sevlever, G.; Stalla, G.K.; Arzt, E. Autocrine IL-6 mediates pituitary tumor senescence. Oncotarget 2017, 8, 4690–4702. [Google Scholar] [CrossRef] [PubMed]
- Ray, K.; Ujvari, B.; Ramana, V.; Donald, J. Cross-talk between EGFR and IL-6 drives oncogenic signaling and offers therapeutic opportunities in cancer. Cytokine Growth Factor. Rev. 2018, 41, 18–27. [Google Scholar] [CrossRef]
- Bharti, R.; Dey, G.; Das, A.K.; Mandal, M. Differential expression of IL-6/IL-6R and MAO-A regulates invasion/angiogenesis in breast cancer. Br. J. Cancer 2018, 118, 1442–1452. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Hu, J.; Wu, X.; Liang, Z. PMA treated THP-1-derived-IL-6 promotes EMT of SW48 through STAT3/ERK-dependent activation of Wnt/beta-catenin signaling pathway. Biomed. Pharmacother. 2018, 108, 618–624. [Google Scholar] [CrossRef]
- Liu, W.; Wang, H.; Bai, F.; Ding, L.; Huang, Y.; Lu, C.; Chen, S.; Li, C.; Yue, X.; Liang, X.; et al. IL-6 promotes metastasis of non-small-cell lung cancer by up-regulating TIM-4 via NF-kappaB. Cell Prolif. 2020, 53, e12776. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wong, K.; Ouyang, W.; Rutz, S. Targeting IL-10 Family Cytokines for the Treatment of Human Diseases. Cold Spring Harb. Perspect. Biol. 2019, 11, a028548. [Google Scholar] [CrossRef]
- Sawant, D.V.; Yano, H.; Chikina, M.; Zhang, Q.; Liao, M.; Liu, C.; Callahan, D.J.; Sun, Z.; Sun, T.; Tabib, T.; et al. Adaptive plasticity of IL-10+ and IL-35+ Treg cells cooperatively promotes tumor T cell exhaustion. Nat. Immunol. 2019, 20, 724–735. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, W.; O’Garra, A. IL-10 Family Cytokines IL-10 and IL-22: From Basic Science to Clinical Translation. Immunity 2019, 50, 871–891. [Google Scholar] [CrossRef]
- Jang, D.I.; Lee, A.H.; Shin, H.Y.; Song, H.R.; Park, J.H.; Kang, T.B.; Lee, S.R.; Yang, S.H. The Role of Tumor Necrosis Factor Alpha (TNF-alpha) in Autoimmune Disease and Current TNF-alpha Inhibitors in Therapeutics. Int. J. Mol. Sci. 2021, 22, 2719. [Google Scholar] [CrossRef]
- Mercogliano, M.F.; De Martino, M.; Venturutti, L.; Rivas, M.A.; Proietti, C.J.; Inurrigarro, G.; Frahm, I.; Allemand, D.H.; Deza, E.G.; Ares, S.; et al. TNFalpha-Induced Mucin 4 Expression Elicits Trastuzumab Resistance in HER2-Positive Breast Cancer. Clin. Cancer Res. 2017, 23, 636–648. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, F.; Rochotte, J.; Colacios, C.; Montfort, A.; Tilkin-Mariame, A.F.; Touriol, C.; Rochaix, P.; Lajoie-Mazenc, I.; Andrieu-Abadie, N.; Levade, T.; et al. Blocking Tumor Necrosis Factor alpha Enhances CD8 T-cell-Dependent Immunity in Experimental Melanoma. Cancer Res. 2015, 75, 2619–2628. [Google Scholar] [CrossRef] [PubMed]
- Salomon, B.L.; Leclerc, M.; Tosello, J.; Ronin, E.; Piaggio, E.; Cohen, J.L. Tumor Necrosis Factor alpha and Regulatory T Cells in Oncoimmunology. Front. Immunol. 2018, 9, 444. [Google Scholar] [CrossRef] [PubMed]
- Landskron, G.; De la Fuente, M.; Thuwajit, P.; Thuwajit, C.; Hermoso, M.A. Chronic inflammation and cytokines in the tumor microenvironment. J. Immunol. Res. 2014, 2014, 149185. [Google Scholar] [CrossRef] [PubMed]
- Torrey, H.; Butterworth, J.; Mera, T.; Okubo, Y.; Wang, L.; Baum, D.; Defusco, A.; Plager, S.; Warden, S.; Huang, D.; et al. Targeting TNFR2 with antagonistic antibodies inhibits proliferation of ovarian cancer cells and tumor-associated Tregs. Sci. Signal 2017, 10, eaaf8608. [Google Scholar] [CrossRef]
- Batlle, E.; Massague, J. Transforming Growth Factor-beta Signaling in Immunity and Cancer. Immunity 2019, 50, 924–940. [Google Scholar] [CrossRef]
- Crane, J.L.; Cao, X. Bone marrow mesenchymal stem cells and TGF-beta signaling in bone remodeling. J. Clin. Investig. 2014, 124, 466–472. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Hisamatsu, T.; Haemmerle, M.; Cho, M.S.; Pradeep, S.; Rupaimoole, R.; Rodriguez-Aguayo, C.; Lopez-Berestein, G.; Wong, S.T.C.; Sood, A.K.; et al. Role of Platelet-Derived Tgfbeta1 in the Progression of Ovarian Cancer. Clin. Cancer Res. 2017, 23, 5611–5621. [Google Scholar] [CrossRef]
- Melzer, C.; Hass, R.; von der Ohe, J.; Lehnert, H.; Ungefroren, H. The role of TGF-beta and its crosstalk with RAC1/RAC1b signaling in breast and pancreas carcinoma. Cell Commun. Signal 2017, 15, 19. [Google Scholar] [CrossRef]
- Melzer, C.; von der Ohe, J.; Otterbein, H.; Ungefroren, H.; Hass, R. Changes in uPA, PAI-1, and TGF-beta Production during Breast Cancer Cell Interaction with Human Mesenchymal Stroma/Stem-Like Cells (MSC). Int. J. Mol. Sci. 2019, 20, 2630. [Google Scholar] [CrossRef]
- Esquivel-Velazquez, M.; Ostoa-Saloma, P.; Palacios-Arreola, M.I.; Nava-Castro, K.E.; Castro, J.I.; Morales-Montor, J. The role of cytokines in breast cancer development and progression. J. Interferon Cytokine Res. 2015, 35, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Baker, D.; Ten Dijke, P. TGF-beta-Mediated Epithelial-Mesenchymal Transition and Cancer Metastasis. Int. J. Mol. Sci. 2019, 20, 2767. [Google Scholar] [CrossRef] [PubMed]
- Tauriello, D.V.F.; Sancho, E.; Batlle, E. Overcoming TGFbeta-mediated immune evasion in cancer. Nat. Rev. Cancer 2022, 22, 25–44. [Google Scholar] [CrossRef] [PubMed]
- Laine, A.; Labiad, O.; Hernandez-Vargas, H.; This, S.; Sanlaville, A.; Leon, S.; Dalle, S.; Sheppard, D.; Travis, M.A.; Paidassi, H.; et al. Regulatory T cells promote cancer immune-escape through integrin alphavbeta8-mediated TGF-beta activation. Nat. Commun. 2021, 12, 6228. [Google Scholar] [CrossRef] [PubMed]
- Gato-Canas, M.; Zuazo, M.; Arasanz, H.; Ibanez-Vea, M.; Lorenzo, L.; Fernandez-Hinojal, G.; Vera, R.; Smerdou, C.; Martisova, E.; Arozarena, I.; et al. PDL1 Signals through Conserved Sequence Motifs to Overcome Interferon-Mediated Cytotoxicity. Cell Rep. 2017, 20, 1818–1829. [Google Scholar] [CrossRef] [PubMed]
- Gong, W.; Donnelly, C.R.; Heath, B.R.; Bellile, E.; Donnelly, L.A.; Taner, H.F.; Broses, L.; Brenner, J.C.; Chinn, S.B.; Ji, R.R.; et al. Cancer-specific type-I interferon receptor signaling promotes cancer stemness and effector CD8+ T-cell exhaustion. Oncoimmunology 2021, 10, 1997385. [Google Scholar] [CrossRef]
- Pidugu, V.K.; Wu, M.M.; Yen, A.H.; Pidugu, H.B.; Chang, K.W.; Liu, C.J.; Lee, T.C. IFIT1 and IFIT3 promote oral squamous cell carcinoma metastasis and contribute to the anti-tumor effect of gefitinib via enhancing p-EGFR recycling. Oncogene 2019, 38, 3232–3247. [Google Scholar] [CrossRef]
- Chen, J.; Cao, Y.; Markelc, B.; Kaeppler, J.; Vermeer, J.A.; Muschel, R.J. Type I IFN protects cancer cells from CD8+ T cell-mediated cytotoxicity after radiation. J. Clin. Investig. 2019, 129, 4224–4238. [Google Scholar] [CrossRef]
- Cunningham, C.R.; Champhekar, A.; Tullius, M.V.; Dillon, B.J.; Zhen, A.; de la Fuente, J.R.; Herskovitz, J.; Elsaesser, H.; Snell, L.M.; Wilson, E.B.; et al. Type I and Type II Interferon Coordinately Regulate Suppressive Dendritic Cell Fate and Function during Viral Persistence. PLoS Pathog. 2016, 12, e1005356. [Google Scholar] [CrossRef]
- Lee, M.S.; Kim, B.; Oh, G.T.; Kim, Y.J. OASL1 inhibits translation of the type I interferon-regulating transcription factor IRF7. Nat. Immunol. 2013, 14, 346–355. [Google Scholar] [CrossRef]
- Boukhaled, G.M.; Harding, S.; Brooks, D.G. Opposing Roles of Type I Interferons in Cancer Immunity. Annu. Rev. Pathol. 2021, 16, 167–198. [Google Scholar] [CrossRef] [PubMed]
- Jomova, K.; Alomar, S.Y.; Valko, R.; Liska, J.; Nepovimova, E.; Kuca, K.; Valko, M. Flavonoids and their role in oxidative stress, inflammation, and human diseases. Chem. Biol. Interact. 2025, 413, 111489. [Google Scholar] [CrossRef] [PubMed]
- Reuter, S.; Gupta, S.C.; Chaturvedi, M.M.; Aggarwal, B.B. Oxidative stress, inflammation, and cancer: How are they linked? Free Radic. Biol. Med. 2010, 49, 1603–1616. [Google Scholar] [CrossRef] [PubMed]
- Hamarsheh, S.; Osswald, L.; Saller, B.S.; Unger, S.; De Feo, D.; Vinnakota, J.M.; Konantz, M.; Uhl, F.M.; Becker, H.; Lubbert, M.; et al. Oncogenic KrasG12D causes myeloproliferation via NLRP3 inflammasome activation. Nat. Commun. 2020, 11, 1659. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Tu, Y.; Long, Z.; Liu, J.; Kong, D.; Peng, J.; Wu, H.; Zheng, G.; Zhao, J.; Chen, Y.; et al. Reactive Oxygen Species Bridge the Gap between Chronic Inflammation and Tumor Development. Oxid. Med. Cell Longev. 2022, 2022, 2606928. [Google Scholar] [CrossRef]
- Yu, Y.; Liu, S.; Yang, L.; Song, P.; Liu, Z.; Liu, X.; Yan, X.; Dong, Q. Roles of reactive oxygen species in inflammation and cancer. MedComm (2020) 2024, 5, e519. [Google Scholar] [CrossRef] [PubMed]
- Liguori, I.; Russo, G.; Curcio, F.; Bulli, G.; Aran, L.; Della-Morte, D.; Gargiulo, G.; Testa, G.; Cacciatore, F.; Bonaduce, D.; et al. Oxidative stress, aging, and diseases. Clin. Interv. Aging 2018, 13, 757–772. [Google Scholar] [CrossRef]
- McGarry, T.; Biniecka, M.; Veale, D.J.; Fearon, U. Hypoxia, oxidative stress and inflammation. Free Radic. Biol. Med. 2018, 125, 15–24. [Google Scholar] [CrossRef]
- Qi, L.; Dong, Y.M.; Chao, H.; Zhao, P.; Ma, S.L.; Li, G. Glyphosate based-herbicide disrupts energy metabolism and activates inflammatory response through oxidative stress in mice liver. Chemosphere 2023, 315, 137751. [Google Scholar] [CrossRef]
- Sun, Q.; Gong, L.; Qi, R.; Qing, W.; Zou, M.; Ke, Q.; Zhang, L.; Tang, X.; Nie, Q.; Yang, Y.; et al. Oxidative stress-induced KLF4 activates inflammatory response through IL17RA and its downstream targets in retinal pigment epithelial cells. Free Radic. Biol. Med. 2020, 147, 271–281. [Google Scholar] [CrossRef]
- Hussain, S.P.; Harris, C.C. Inflammation and cancer: An ancient link with novel potentials. Int. J. Cancer 2007, 121, 2373–2380. [Google Scholar] [CrossRef] [PubMed]
- Perillo, B.; Di Donato, M.; Pezone, A.; Di Zazzo, E.; Giovannelli, P.; Galasso, G.; Castoria, G.; Migliaccio, A. ROS in cancer therapy: The bright side of the moon. Exp. Mol. Med. 2020, 52, 192–203. [Google Scholar] [CrossRef]
- Khan, A.Q.; Rashid, K.; AlAmodi, A.A.; Agha, M.V.; Akhtar, S.; Hakeem, I.; Raza, S.S.; Uddin, S. Reactive oxygen species (ROS) in cancer pathogenesis and therapy: An update on the role of ROS in anticancer action of benzophenanthridine alkaloids. Biomed. Pharmacother. 2021, 143, 112142. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.J.; Liu, Z.G. Crosstalk of reactive oxygen species and NF-kappaB signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Baud, V.; Karin, M. Is NF-kappaB a good target for cancer therapy? Hopes and pitfalls. Nat. Rev. Drug Discov. 2009, 8, 33–40. [Google Scholar] [CrossRef]
- Li, Y.; Zhao, B.; Peng, J.; Tang, H.; Wang, S.; Peng, S.; Ye, F.; Wang, J.; Ouyang, K.; Li, J.; et al. Inhibition of NF-kappaB signaling unveils novel strategies to overcome drug resistance in cancers. Drug Resist. Updat. 2024, 73, 101042. [Google Scholar] [CrossRef] [PubMed]
- Gambardella, J.; Ciccarelli, M.; Del Giudice, C.; Fiordelisi, A.; De Rosa, M.; Sala, M.; Pacelli, R.; Campiglia, P.; Trimarco, B.; Iaccarino, G.; et al. A Novel Small Peptide Inhibitor of NFkappaB, RH10, Blocks Oxidative Stress-Dependent Phenotypes in Cancer. Oxid. Med. Cell Longev. 2018, 2018, 5801807. [Google Scholar] [CrossRef] [PubMed]
- Gambardella, J.; Fiordelisi, A.; Santulli, G.; Ciccarelli, M.; Cerasuolo, F.A.; Sala, M.; Sommella, E.; Campiglia, P.; Illario, M.; Iaccarino, G.; et al. Exploiting GRK2 Inhibition as a Therapeutic Option in Experimental Cancer Treatment: Role of p53-Induced Mitochondrial Apoptosis. Cancers 2020, 12, 3530. [Google Scholar] [CrossRef]
- Soprano, M.; Sorriento, D.; Rusciano, M.R.; Maione, A.S.; Limite, G.; Forestieri, P.; D’Angelo, D.; D’Alessio, M.; Campiglia, P.; Formisano, P.; et al. Oxidative Stress Mediates the Antiproliferative Effects of Nelfinavir in Breast Cancer Cells. PLoS ONE 2016, 11, e0155970. [Google Scholar] [CrossRef]
- Sorriento, D.; Campanile, A.; Santulli, G.; Leggiero, E.; Pastore, L.; Trimarco, B.; Iaccarino, G. A new synthetic protein, TAT-RH, inhibits tumor growth through the regulation of NFkappaB activity. Mol. Cancer 2009, 8, 97. [Google Scholar] [CrossRef]
- Xu, P.; Xue, Y.N.; Ji, H.H.; Tan, C.; Guo, S. H2O2-induced oxidative stress disrupts mitochondrial functions and impairs migratory potential of human epidermal melanocytes. Exp. Dermatol. 2020, 29, 733–741. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Lin, Z.; Lu, D.; Li, H. Oxidative stress-mediated tetrabromobisphenol A disrupts mitochondrial function in HepG2 cells and activates ferroptosis signalling to induce apoptosis. J. Environ. Manag. 2025, 382, 125360. [Google Scholar] [CrossRef] [PubMed]
- Gu, F.; Wang, Z.; Ding, H.; Tao, X.; Zhang, J.; Dai, K.; Li, X.; Shen, H.; Li, H.; Chen, Z.; et al. Microglial mitochondrial DNA release contributes to neuroinflammation after intracerebral hemorrhage through activating AIM2 inflammasome. Exp. Neurol. 2024, 382, 114950. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Zhang, D.; Hu, D.; Zhou, X.; Zhou, Y. The role of mitochondria in NLRP3 inflammasome activation. Mol. Immunol. 2018, 103, 115–124. [Google Scholar] [CrossRef]
- Kim, J.; Kim, H.S.; Chung, J.H. Molecular mechanisms of mitochondrial DNA release and activation of the cGAS-STING pathway. Exp. Mol. Med. 2023, 55, 510–519. [Google Scholar] [CrossRef] [PubMed]
- Silver, J.; Hood, L. Structure and evolution of transplantation antigens: Partial amino-acid sequences of H-2K and H-2D alloantigens. Proc. Natl. Acad. Sci. USA 1976, 73, 599–603. [Google Scholar] [CrossRef]
- Bardelcikova, A.; Soltys, J.; Mojzis, J. Oxidative Stress, Inflammation and Colorectal Cancer: An Overview. Antioxidants 2023, 12, 901. [Google Scholar] [CrossRef]
- Hanahan, D.; Coussens, L.M. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef]
- Afify, S.M.; Hassan, G.; Seno, A.; Seno, M. Cancer-inducing niche: The force of chronic inflammation. Br. J. Cancer 2022, 127, 193–201. [Google Scholar] [CrossRef]
- Zheng, S.; Song, Q.; Zhang, P. Metabolic Modifications, Inflammation, and Cancer Immunotherapy. Front. Oncol. 2021, 11, 703681. [Google Scholar] [CrossRef]
- Colarusso, C.; Terlizzi, M.; Di Caprio, S.; Falanga, A.; D’Andria, E.; d’Emmanuele di Villa Bianca, R.; Sorrentino, R. Conventional Chemotherapy and Inflammation: What Is the Role of the Inflammasome in the Tumor Microenvironment? Biomedicines 2025, 13, 203. [Google Scholar] [CrossRef]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.P.; Hofseth, L.J.; Harris, C.C. Radical causes of cancer. Nat. Rev. Cancer 2003, 3, 276–285. [Google Scholar] [CrossRef]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef]
- Fujiwara, Y.; Karol, A.B.; Joshi, H.; Reford, E.; Izadmehr, S.; Doroshow, D.B.; Galsky, M.D. C-reactive protein (CRP) as a prognostic biomarker in patients with urothelial carcinoma: A systematic review and meta-analysis. Crit. Rev. Oncol. Hematol. 2024, 197, 104352. [Google Scholar] [CrossRef] [PubMed]
- Valavanidis, A.; Vlachogianni, T.; Fiotakis, C. 8-hydroxy-2′ -deoxyguanosine (8-OHdG): A critical biomarker of oxidative stress and carcinogenesis. J. Environ. Sci. Health C Environ. Carcinog. Ecotoxicol. Rev. 2009, 27, 120–139. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.C.; Yu, J. Gut microbiota in colorectal cancer development and therapy. Nat. Rev. Clin. Oncol. 2023, 20, 429–452. [Google Scholar] [CrossRef] [PubMed]
- Knupfer, H.; Preiss, R. Serum interleukin-6 levels in colorectal cancer patients--a summary of published results. Int. J. Color. Dis. 2010, 25, 135–140. [Google Scholar] [CrossRef]
- Traverso, N.; Ricciarelli, R.; Nitti, M.; Marengo, B.; Furfaro, A.L.; Pronzato, M.A.; Marinari, U.M.; Domenicotti, C. Role of glutathione in cancer progression and chemoresistance. Oxid. Med. Cell Longev. 2013, 2013, 972913. [Google Scholar] [CrossRef]
- Kostic, A.D.; Chun, E.; Robertson, L.; Glickman, J.N.; Gallini, C.A.; Michaud, M.; Clancy, T.E.; Chung, D.C.; Lochhead, P.; Hold, G.L.; et al. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host Microbe 2013, 14, 207–215. [Google Scholar] [CrossRef]
- Dizdaroglu, M.; Jaruga, P. Mechanisms of free radical-induced damage to DNA. Free Radic. Res. 2012, 46, 382–419. [Google Scholar] [CrossRef] [PubMed]
- Burns, M.B.; Lackey, L.; Carpenter, M.A.; Rathore, A.; Land, A.M.; Leonard, B.; Refsland, E.W.; Kotandeniya, D.; Tretyakova, N.; Nikas, J.B.; et al. APOBEC3B is an enzymatic source of mutation in breast cancer. Nature 2013, 494, 366–370. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.L.; Chiou, C.C.; Chang, P.Y.; Wu, J.T. Urinary 8-OHdG: A marker of oxidative stress to DNA and a risk factor for cancer, atherosclerosis and diabetics. Clin. Chim. Acta 2004, 339, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Yang, C.; Wang, S.; Xiang, Z.; Dou, R.; Lin, Z.; Zheng, J.; Xiong, B. SDC2 and TFPI2 Methylation in Stool Samples as an Integrated Biomarker for Early Detection of Colorectal Cancer. Cancer Manag. Res. 2021, 13, 3601–3617. [Google Scholar] [CrossRef]
- Kang, M.; Jeong, S.; Park, S.; Nam, S.; Chung, J.W.; Kim, K.O.; An, J.; Kim, J.H. Significance of 8-OHdG Expression as a Predictor of Survival in Colorectal Cancer. Cancers 2023, 15, 4613. [Google Scholar] [CrossRef]
- Ai, R.; Tao, Y.; Hao, Y.; Jiang, L.; Dan, H.; Ji, N.; Zeng, X.; Zhou, Y.; Chen, Q. Microenvironmental regulation of the progression of oral potentially malignant disorders towards malignancy. Oncotarget 2017, 8, 81617–81635. [Google Scholar] [CrossRef] [PubMed]
- Kessenbrock, K.; Wang, C.Y.; Werb, Z. Matrix metalloproteinases in stem cell regulation and cancer. Matrix Biol. 2015, 44–46, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Bjorkman, K.; Kaprio, T.; Beilmann-Lehtonen, I.; Stenman, U.H.; Bockelman, C.; Haglund, C. TATI, TAT-2, and CRP as Prognostic Factors in Colorectal Cancer. Oncology 2022, 100, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.R.; Li, X.; Han, C.; Chang, Y.; Fu, Y.; Feng, G.C.; Lei, Y.; Li, H.Y.; Tang, P.M.; Ji, S.R.; et al. C-Reactive Protein Induces Immunosuppression by Activating FcgammaR2B in Pulmonary Macrophages to Promote Lung Metastasis. Cancer Res. 2024, 84, 4184–4198. [Google Scholar] [CrossRef]
- Jiang, J.; Peng, Z.; Wang, J.; Chen, M.; Wan, Y.; Huang, H.; Liu, Z.; Wang, J.; Hou, J. C-reactive protein impairs immune response of CD8+ T cells via FcgammaRIIb-p38MAPK-ROS axis in multiple myeloma. J. Immunother. Cancer 2023, 11, e007593. [Google Scholar] [CrossRef]
- Nurmi, A.M.; Mustonen, H.K.; Stenman, U.H.; Seppanen, H.E.; Haglund, C.H. Combining CRP and CA19-9 in a novel prognostic score in pancreatic ductal adenocarcinoma. Sci. Rep. 2021, 11, 781. [Google Scholar] [CrossRef] [PubMed]
- Plebani, M. Why C-reactive protein is one of the most requested tests in clinical laboratories? Clin. Chem. Lab. Med. 2023, 61, 1540–1545. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Lu, K.; Hou, Y.; You, Z.; Shu, C.; Wei, X.; Wu, T.; Shi, N.; Zhang, G.; Wu, J.; et al. YY1 complex in M2 macrophage promotes prostate cancer progression by upregulating IL-6. J. Immunother. Cancer 2023, 11, e006020. [Google Scholar] [CrossRef] [PubMed]
- Hirano, T. IL-6 in inflammation, autoimmunity and cancer. Int. Immunol. 2021, 33, 127–148. [Google Scholar] [CrossRef]
- Rupert, J.E.; Narasimhan, A.; Jengelley, D.H.A.; Jiang, Y.; Liu, J.; Au, E.; Silverman, L.M.; Sandusky, G.; Bonetto, A.; Cao, S.; et al. Tumor-derived IL-6 and trans-signaling among tumor, fat, and muscle mediate pancreatic cancer cachexia. J. Exp. Med. 2021, 218, e20190450. [Google Scholar] [CrossRef]
- Soler, M.F.; Abaurrea, A.; Azcoaga, P.; Araujo, A.M.; Caffarel, M.M. New perspectives in cancer immunotherapy: Targeting IL-6 cytokine family. J. Immunother. Cancer 2023, 11, e007530. [Google Scholar] [CrossRef]
- Jalilian, E.; Abolhasani-Zadeh, F.; Afgar, A.; Samoudi, A.; Zeinalynezhad, H.; Langroudi, L. Neutralizing tumor-related inflammation and reprogramming of cancer-associated fibroblasts by Curcumin in breast cancer therapy. Sci. Rep. 2023, 13, 20770. [Google Scholar] [CrossRef] [PubMed]
- Sohrab, S.S.; Raj, R.; Nagar, A.; Hawthorne, S.; Paiva-Santos, A.C.; Kamal, M.A.; El-Daly, M.M.; Azhar, E.I.; Sharma, A. Chronic Inflammation’s Transformation to Cancer: A Nanotherapeutic Paradigm. Molecules 2023, 28, 4413. [Google Scholar] [CrossRef]
- Stamatakis, K.; Torres-Gerica, P.; Jimenez-Segovia, A.; Ramos-Munoz, E.; Crespo-Toro, L.; Fuentes, P.; Toribio, M.L.; Callejas-Hernandez, F.; Carrato, A.; Garcia Bermejo, M.L.; et al. Cyclooxygenase 2 Effector Genes as Potential Inflammation-Related Biomarkers for Colorectal Cancer Circulating Tumor Cells Detection by Liquid Biopsy. Front. Pharmacol. 2021, 12, 806395. [Google Scholar] [CrossRef]
- Wang, D.; Cabalag, C.S.; Clemons, N.J.; DuBois, R.N. Cyclooxygenases and Prostaglandins in Tumor Immunology and Microenvironment of Gastrointestinal Cancer. Gastroenterology 2021, 161, 1813–1829. [Google Scholar] [CrossRef]
- Chen, M.; Wang, Q.; Wang, Y.; Xuan, Y.; Shen, M.; Hu, X.; Li, Y.; Guo, Y.; Wang, J.; Tan, F. Thiostrepton induces oxidative stress, mitochondrial dysfunction and ferroptosis in HaCaT cells. Cell Signal 2024, 121, 111285. [Google Scholar] [CrossRef] [PubMed]
- Galiniak, S.; Molon, M.; Biesiadecki, M.; Mokrzynska, A.; Balawender, K. Oxidative Stress Markers in Urine and Serum of Patients with Bladder Cancer. Antioxidants 2023, 12, 277. [Google Scholar] [CrossRef] [PubMed]
- Jelic, M.D.; Mandic, A.D.; Maricic, S.M.; Srdjenovic, B.U. Oxidative stress and its role in cancer. J. Cancer Res. Ther. 2021, 17, 22–28. [Google Scholar] [CrossRef]
- Brassea-Perez, E.; Hernandez-Camacho, C.J.; Labrada-Martagon, V.; Vazquez-Medina, J.P.; Gaxiola-Robles, R.; Zenteno-Savin, T. Oxidative stress induced by phthalates in mammals: State of the art and potential biomarkers. Environ. Res. 2022, 206, 112636. [Google Scholar] [CrossRef]
- Marino, M.; Rendine, M.; Venturi, S.; Porrini, M.; Gardana, C.; Klimis-Zacas, D.; Riso, P.; Del Bo, C. Red raspberry (Rubus idaeus) preserves intestinal barrier integrity and reduces oxidative stress in Caco-2 cells exposed to a proinflammatory stimulus. Food Funct. 2024, 15, 6943–6954. [Google Scholar] [CrossRef]
- Cherkas, A.; Zarkovic, N. 4-Hydroxynonenal in Redox Homeostasis of Gastrointestinal Mucosa: Implications for the Stomach in Health and Diseases. Antioxidants 2018, 7, 118. [Google Scholar] [CrossRef] [PubMed]
- Dalleau, S.; Baradat, M.; Gueraud, F.; Huc, L. Cell death and diseases related to oxidative stress: 4-hydroxynonenal (HNE) in the balance. Cell Death Differ. 2013, 20, 1615–1630. [Google Scholar] [CrossRef]
- Gall Troselj, K.; Tomljanovic, M.; Jaganjac, M.; Matijevic Glavan, T.; Cipak Gasparovic, A.; Milkovic, L.; Borovic Sunjic, S.; Buttari, B.; Profumo, E.; Saha, S.; et al. Oxidative Stress and Cancer Heterogeneity Orchestrate NRF2 Roles Relevant for Therapy Response. Molecules 2022, 27, 1468. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.X.; Wang, Y.B.; Wu, J.; Ding, G.S.; Wu, Y.; Wei, L.H.; Wang, F.; Zhang, Z.M. Clinical significance of serum oxidative stress and serum uric acid levels before surgery for hepatitis B-related liver cancer. World J. Gastrointest. Surg. 2023, 15, 1995–2002. [Google Scholar] [CrossRef]
- Jomova, K.; Alomar, S.Y.; Alwasel, S.H.; Nepovimova, E.; Kuca, K.; Valko, M. Several lines of antioxidant defense against oxidative stress: Antioxidant enzymes, nanomaterials with multiple enzyme-mimicking activities, and low-molecular-weight antioxidants. Arch. Toxicol. 2024, 98, 1323–1367. [Google Scholar] [CrossRef]
- Brzozowa-Zasada, M.; Ianaro, A.; Piecuch, A.; Michalski, M.; Matysiak, N.; Steplewska, K. Immunohistochemical Expression of Glutathione Peroxidase-2 (Gpx-2) and Its Clinical Relevance in Colon Adenocarcinoma Patients. Int. J. Mol. Sci. 2023, 24, 14650. [Google Scholar] [CrossRef] [PubMed]
- Guo, B.; Liao, W.; Wang, S. The clinical significance of glutathione peroxidase 2 in glioblastoma multiforme. Transl. Neurosci. 2021, 12, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Lawinski, M.; Zadka, K.; Ksepka, N.; Matin, M.; Wysocki, K.; Karkocha, D.; Gradowska, A.; Atanasov, A.G.; Slodkowski, M.; Wierzbicka, A.; et al. Does Resveratrol Impact Oxidative Stress Markers in Patients with Head and Neck Cancer Receiving Home Enteral Nutrition? Nutrients 2025, 17, 504. [Google Scholar] [CrossRef]
- de Oliveira, S.T.; Bessani, M.P.; Scandolara, T.B.; Silva, J.C.; Kawassaki, A.C.B.; Fagotti, P.A.F.; Maito, V.T.; de Souza, J.A.; Rech, D.; Panis, C. Systemic lipid peroxidation profile from patients with breast cancer changes according to the lymph nodal metastasis status. Oncoscience 2022, 9, 1–10. [Google Scholar] [CrossRef]
- Guo, L.; Wang, Y.; Yang, W.; Wang, C.; Guo, T.; Yang, J.; Shao, Z.; Cai, G.; Cai, S.; Zhang, L.; et al. Molecular Profiling Provides Clinical Insights Into Targeted and Immunotherapies as Well as Colorectal Cancer Prognosis. Gastroenterology 2023, 165, 414–428.e7. [Google Scholar] [CrossRef]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [PubMed]
- Martin-Perez, M.; Urdiroz-Urricelqui, U.; Bigas, C.; Benitah, S.A. The role of lipids in cancer progression and metastasis. Cell Metab. 2022, 34, 1675–1699. [Google Scholar] [CrossRef]
- Zhang, H.L.; Hu, B.X.; Li, Z.L.; Du, T.; Shan, J.L.; Ye, Z.P.; Peng, X.D.; Li, X.; Huang, Y.; Zhu, X.Y.; et al. PKCbetaII phosphorylates ACSL4 to amplify lipid peroxidation to induce ferroptosis. Nat. Cell Biol. 2022, 24, 88–98. [Google Scholar] [CrossRef]
- Zhong, H.; Yin, H. Role of lipid peroxidation derived 4-hydroxynonenal (4-HNE) in cancer: Focusing on mitochondria. Redox Biol. 2015, 4, 193–199. [Google Scholar] [CrossRef]
- Yang, M.; Cui, W.; Lv, X.; Xiong, G.; Sun, C.; Xuan, H.; Ma, W.; Cui, X.; Cheng, Y.; Han, L.; et al. S100P is a ferroptosis suppressor to facilitate hepatocellular carcinoma development by rewiring lipid metabolism. Nat. Commun. 2025, 16, 509. [Google Scholar] [CrossRef]
- Hu, D.; Li, Y.; Li, R.; Wang, M.; Zhou, K.; He, C.; Wei, Q.; Qian, Z. Recent advances in reactive oxygen species (ROS)-responsive drug delivery systems for photodynamic therapy of cancer. Acta Pharm. Sin. B 2024, 14, 5106–5131. [Google Scholar] [CrossRef] [PubMed]
- Gerber, T.S.; Witzel, H.R.; Weinmann, A.; Bartsch, F.; Schindeldecker, M.; Galle, P.R.; Lang, H.; Roth, W.; Ridder, D.A.; Straub, B.K. Reduced Lipid Peroxidation Predicts Unfavorable Prognosis in Hepatocellular Carcinoma, but Not Intrahepatic Cholangiocarcinoma. Biomedicines 2023, 11, 2471. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, Y.; Gong, J.; Liu, W.; Zhao, H.; Xue, W.; Ren, Z.; Bao, J.; Lin, Z. Development of a breast cancer invasion score to predict tumor aggressiveness and prognosis via PI3K/AKT/mTOR pathway analysis. Cell Death Discov. 2025, 11, 157. [Google Scholar] [CrossRef] [PubMed]
- Zhong, T.; Li, Y.; Jin, M.; Liu, J.; Wu, Z.; Zhu, F.; Zhao, L.; Fan, Y.; Xu, L.; Ji, J. Downregulation of 4-HNE and FOXO4 collaboratively promotes NSCLC cell migration and tumor growth. Cell Death Dis. 2024, 15, 546. [Google Scholar] [CrossRef]
- Panda, B.; Tripathy, A.; Patra, S.; Kullu, B.; Tabrez, S.; Jena, M. Imperative connotation of SODs in cancer: Emerging targets and multifactorial role of action. IUBMB Life 2024, 76, 592–613. [Google Scholar] [CrossRef]
- Vilchis-Landeros, M.M.; Vazquez-Meza, H.; Vazquez-Carrada, M.; Uribe-Ramirez, D.; Matuz-Mares, D. Antioxidant Enzymes and Their Potential Use in Breast Cancer Treatment. Int. J. Mol. Sci. 2024, 25, 5675. [Google Scholar] [CrossRef]
- Strycharz-Dudziak, M.; Kielczykowska, M.; Drop, B.; Swiatek, L.; Kliszczewska, E.; Musik, I.; Polz-Dacewicz, M. Total Antioxidant Status (TAS), Superoxide Dismutase (SOD), and Glutathione Peroxidase (GPx) in Oropharyngeal Cancer Associated with EBV Infection. Oxid. Med. Cell Longev. 2019, 2019, 5832410. [Google Scholar] [CrossRef]
- Yuzhalin, A.E.; Kutikhin, A.G. Inherited variations in the SOD and GPX gene families and cancer risk. Free Radic. Res. 2012, 46, 581–599. [Google Scholar] [CrossRef]
- Abd Al Moaty, M.N.; El Ashry, E.S.H.; Awad, L.F.; Mostafa, A.; Abu-Serie, M.M.; Teleb, M. Harnessing ROS-Induced Oxidative Stress for Halting Colorectal Cancer via Thiazolidinedione-Based SOD Inhibitors. ACS Omega 2022, 7, 21267–21279. [Google Scholar] [CrossRef]
- Sui, X.; Zhang, R.; Liu, S.; Duan, T.; Zhai, L.; Zhang, M.; Han, X.; Xiang, Y.; Huang, X.; Lin, H.; et al. RSL3 Drives Ferroptosis Through GPX4 Inactivation and ROS Production in Colorectal Cancer. Front. Pharmacol. 2018, 9, 1371. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.C.; Li, H.H.; Lin, J.H.; Chiang, M.C.; Hsu, T.W.; Li, A.F.; Yen, D.H.; Hsu, H.S.; Hung, S.C. Esophageal Cancer Stem-like Cells Resist Ferroptosis-Induced Cell Death by Active Hsp27-GPX4 Pathway. Biomolecules 2021, 12, 48. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Zheng, Y.; He, H.; Yang, L.; Yang, J.; Li, M.; Dai, W.; Huang, H. FBXO31 sensitizes cancer stem cells-like cells to cisplatin by promoting ferroptosis and facilitating proteasomal degradation of GPX4 in cholangiocarcinoma. Liver Int. 2022, 42, 2871–2888. [Google Scholar] [CrossRef] [PubMed]
- Cecerska-Heryc, E.; Surowska, O.; Heryc, R.; Serwin, N.; Napiontek-Balinska, S.; Dolegowska, B. Are antioxidant enzymes essential markers in the diagnosis and monitoring of cancer patients—A review. Clin. Biochem. 2021, 93, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Zhang, Y.; Duan, Y.; Zhang, X.; Yao, J.; Yang, D.; Wei, Z.; Zhu, Z.; Xu, J.; Hu, Z.; et al. Superoxide dismutase promotes gastric tumorigenesis mediated by Helicobacter pylori and enhances resistance to 5-fluorouracil in gastric cancer. iScience 2025, 28, 111553. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Yang, W.; Yang, L.; Wang, T.; Li, C.; Yu, J.; Zhang, P.; Yin, Y.; Li, R.; Tao, K. Nrf2 inhibition increases sensitivity to chemotherapy of colorectal cancer by promoting ferroptosis and pyroptosis. Sci. Rep. 2023, 13, 14359. [Google Scholar] [CrossRef]
- Iglesias-Matesanz, P.; Lacalle-Gonzalez, C.; Lopez-Blazquez, C.; Ochieng’ Otieno, M.; Garcia-Foncillas, J.; Martinez-Useros, J. Glutathione Peroxidases: An Emerging and Promising Therapeutic Target for Pancreatic Cancer Treatment. Antioxidants 2024, 13, 1405. [Google Scholar] [CrossRef]
- Wu, H.; Liao, X.; Huang, W.; Hu, H.; Lan, L.; Yang, Q.; An, Y. Examining the prognostic and clinicopathological significance of GPX4 in human cancers: A meta-analysis. Free Radic. Res. 2025, 59, 239–249. [Google Scholar] [CrossRef]
- Lin, C.H.; Yang, P.J.; Lin, S.H.; Yeh, K.T.; Tsao, T.C.; Chen, Y.E.; Lin, S.H.; Yang, S.F. Association between EGFR Gene Mutation and Antioxidant Gene Polymorphism of Non-Small-Cell Lung Cancer. Diagnostics 2020, 10, 692. [Google Scholar] [CrossRef]
- Chen, X.; Kang, R.; Kroemer, G.; Tang, D. Broadening horizons: The role of ferroptosis in cancer. Nat. Rev. Clin. Oncol. 2021, 18, 280–296. [Google Scholar] [CrossRef]
- Monson, S.; Chen, P.J.; Gangi, A.; Waters, K.; Billet, S.; Hendifar, A.; Lu, S.; Zell, J.A.; Gong, J. Chemopreventive strategies for sporadic colorectal cancer: A narrative review. Transl. Gastroenterol. Hepatol. 2025, 10, 11. [Google Scholar] [CrossRef]
- Wang, Q.; Bin, C.; Xue, Q.; Gao, Q.; Huang, A.; Wang, K.; Tang, N. GSTZ1 sensitizes hepatocellular carcinoma cells to sorafenib-induced ferroptosis via inhibition of NRF2/GPX4 axis. Cell Death Dis. 2021, 12, 426. [Google Scholar] [CrossRef] [PubMed]
- Iborra, M.; Moret, I.; Rausell, F.; Bastida, G.; Aguas, M.; Cerrillo, E.; Nos, P.; Beltran, B. Role of oxidative stress and antioxidant enzymes in Crohn’s disease. Biochem. Soc. Trans. 2011, 39, 1102–1106. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.; Gupta, N.; Khan, R.; Kumar, R.; Sharma, M.; Kumar, L.; Sharma, A. Interrelationship between angiogenesis, inflammation and oxidative stress in Indian patients with multiple myeloma. Clin. Transl. Oncol. 2016, 18, 132–137. [Google Scholar] [CrossRef] [PubMed]
- Kinugasa, H.; Whelan, K.A.; Tanaka, K.; Natsuizaka, M.; Long, A.; Guo, A.; Chang, S.; Kagawa, S.; Srinivasan, S.; Guha, M.; et al. Mitochondrial SOD2 regulates epithelial-mesenchymal transition and cell populations defined by differential CD44 expression. Oncogene 2015, 34, 5229–5239. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, J.D.; Debeljak, M.; Riel, S.; Millena, A.C.; Eshleman, J.R.; Paller, C.J.; Odero-Marah, V. Val16A SOD2 Polymorphism Promotes Epithelial-Mesenchymal Transition Antagonized by Muscadine Grape Skin Extract in Prostate Cancer Cells. Antioxidants 2021, 10, 213. [Google Scholar] [CrossRef]
- Kim, E.S.; Kim, S.Y.; Moon, A. C-Reactive Protein Signaling Pathways in Tumor Progression. Biomol. Ther. 2023, 31, 473–483. [Google Scholar] [CrossRef]
- Sawada, N.; Iwasaki, M.; Yamaji, T.; Goto, A.; Shimazu, T.; Inoue, M.; Tanno, K.; Sakata, K.; Yamagishi, K.; Iso, H.; et al. The Japan Public Health Center-based Prospective Study for the Next Generation (JPHC-NEXT): Study Design and Participants. J. Epidemiol. 2020, 30, 46–54. [Google Scholar] [CrossRef]
- Hart, P.C.; Rajab, I.M.; Alebraheem, M.; Potempa, L.A. C-Reactive Protein and Cancer-Diagnostic and Therapeutic Insights. Front. Immunol. 2020, 11, 595835. [Google Scholar] [CrossRef]
- Kostner, A.H.; Kersten, C.; Lowenmark, T.; Ydsten, K.A.; Peltonen, R.; Isoniemi, H.; Haglund, C.; Gunnarsson, U.; Isaksson, B. The prognostic role of systemic inflammation in patients undergoing resection of colorectal liver metastases: C-reactive protein (CRP) is a strong negative prognostic biomarker. J. Surg. Oncol. 2016, 114, 895–899. [Google Scholar] [CrossRef]
- Yang, M.; Lin, S.Q.; Liu, X.Y.; Tang, M.; Hu, C.L.; Wang, Z.W.; Zhang, Q.; Zhang, X.; Song, M.M.; Ruan, G.T.; et al. Association between C-reactive protein-albumin-lymphocyte (CALLY) index and overall survival in patients with colorectal cancer: From the investigation on nutrition status and clinical outcome of common cancers study. Front. Immunol. 2023, 14, 1131496. [Google Scholar] [CrossRef]
- Chen, D.; Ma, Y.; Li, J.; Wen, L.; Liu, L.; Su, J.; Wu, J.; Wang, P.; Zhang, G.; Huang, C.; et al. Prognostic and clinicopathological significance of C-reactive protein-albumin-lymphocyte(CALLY) in patients with digestive system neoplasms: A systematic review and meta-analysis. World J. Surg. Oncol. 2025, 23, 114. [Google Scholar] [CrossRef] [PubMed]
- Tajdari, M.; Peyrovinasab, A.; Bayanati, M.; Ismail Mahboubi Rabbani, M.; Abdolghaffari, A.H.; Zarghi, A. Dual COX-2/TNF-alpha Inhibitors as Promising Anti-inflammatory and Cancer Chemopreventive Agents: A Review. Iran. J. Pharm. Res. 2024, 23, e151312. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, J.; Sun, W.; Cao, J.; Ma, Z. COX-2 in lung cancer: Mechanisms, development, and targeted therapies. Chronic Dis. Transl. Med. 2024, 10, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Wang, A.; Yin, B.; Wu, D.; Han, S.; Zhang, W.; Liu, J.; Sun, K. SND1 promotes the proliferation of osteosarcoma cells by upregulating COX-2/PGE2 expression via activation of NF-kappaB. Oncol. Rep. 2019, 41, 579–589. [Google Scholar]
- Abe, A.; Fukui, H.; Fujii, S.; Fujita, M.; Mukawa, K.; Ichikawa, K.; Tomita, S.; Ono, Y.; Imai, Y.; Imura, J.; et al. Involvement of cyclooxygenase-2 and vascular endothelial growth factor in vascularization and lymph node metastasis of colorectal cancers with submucosal invasion. J. Gastroenterol. Hepatol. 2007, 22, 1071–1077. [Google Scholar] [CrossRef] [PubMed]
- Kosumi, K.; Hamada, T.; Zhang, S.; Liu, L.; da Silva, A.; Koh, H.; Twombly, T.S.; Mima, K.; Morikawa, T.; Song, M.; et al. Prognostic association of PTGS2 (COX-2) over-expression according to BRAF mutation status in colorectal cancer: Results from two prospective cohorts and CALGB 89803 (Alliance) trial. Eur. J. Cancer 2019, 111, 82–93. [Google Scholar] [CrossRef]
- Mahboubi-Rabbani, M.; Abdolghaffari, A.H.; Ghesmati, M.; Amini, A.; Zarghi, A. Selective COX-2 inhibitors as anticancer agents: A patent review (2018–2023). Expert Opin. Ther. Pat. 2024, 34, 733–757. [Google Scholar] [CrossRef] [PubMed]
- Burn, J.; Sheth, H.; Elliott, F.; Reed, L.; Macrae, F.; Mecklin, J.P.; Moslein, G.; McRonald, F.E.; Bertario, L.; Evans, D.G.; et al. Cancer prevention with aspirin in hereditary colorectal cancer (Lynch syndrome), 10-year follow-up and registry-based 20-year data in the CAPP2 study: A double-blind, randomised, placebo-controlled trial. Lancet 2020, 395, 1855–1863. [Google Scholar] [CrossRef]
- Jiang, W.; Yan, Y.; Chen, M.; Luo, G.; Hao, J.; Pan, J.; Hu, S.; Guo, P.; Li, W.; Wang, R.; et al. Aspirin enhances the sensitivity of colon cancer cells to cisplatin by abrogating the binding of NF-kappaB to the COX-2 promoter. Aging 2020, 12, 611–627. [Google Scholar] [CrossRef]
- Yang, Y.; Ma, L.; Xu, Y.; Liu, Y.; Li, W.; Cai, J.; Zhang, Y. Enalapril overcomes chemoresistance and potentiates antitumor efficacy of 5-FU in colorectal cancer by suppressing proliferation, angiogenesis, and NF-kappaB/STAT3-regulated proteins. Cell Death Dis. 2020, 11, 477. [Google Scholar] [CrossRef]
- Ramisetti, S.V.; Patra, T.; Munirathnam, V.; Sainath, J.V.; Veeraiyan, D.; Namani, A. NRF2 Signaling Pathway in Chemo/Radio/Immuno-Therapy Resistance of Lung Cancer: Looking Beyond the Tip of the Iceberg. Arch. Bronconeumol. 2024, 60 (Suppl. S2), S59–S66. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Ma, Y.; Li, D.; Wei, J.; Chen, K.; Zhang, E.; Liu, G.; Chu, X.; Liu, X.; Liu, W.; et al. Cancer associated fibroblasts and metabolic reprogramming: Unraveling the intricate crosstalk in tumor evolution. J. Hematol. Oncol. 2024, 17, 80. [Google Scholar] [CrossRef] [PubMed]
- Galasso, L.; Termite, F.; Mignini, I.; Esposto, G.; Borriello, R.; Vitale, F.; Nicoletti, A.; Paratore, M.; Ainora, M.E.; Gasbarrini, A.; et al. Unraveling the Role of Fusobacterium nucleatum in Colorectal Cancer: Molecular Mechanisms and Pathogenic Insights. Cancers 2025, 17, 368. [Google Scholar] [CrossRef]
- Hong, B.Y.; Chhaya, A.; Robles, A.; Cervantes, J.; Tiwari, S. The role of Fusobacterium nucleatum in the pathogenesis of colon cancer. J. Investig. Med. 2024, 72, 819–827. [Google Scholar] [CrossRef]
- Dadgar-Zankbar, L.; Elahi, Z.; Shariati, A.; Khaledi, A.; Razavi, S.; Khoshbayan, A. Exploring the role of Fusobacterium nucleatum in colorectal cancer: Implications for tumor proliferation and chemoresistance. Cell Commun. Signal 2024, 22, 547. [Google Scholar] [CrossRef]
- Ma, Y.; Chen, H.; Li, H.; Zheng, M.; Zuo, X.; Wang, W.; Wang, S.; Lu, Y.; Wang, J.; Li, Y.; et al. Intratumor microbiome-derived butyrate promotes lung cancer metastasis. Cell Rep. Med. 2024, 5, 101488. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, Y.; Lin, Y.; Cao, C.; Chen, D.; Huang, X.; Li, C.; Xu, H.; Lai, H.; Chen, H.; et al. Roles of the gut microbiota in hepatocellular carcinoma: From the gut dysbiosis to the intratumoral microbiota. Cell Death Discov. 2025, 11, 140. [Google Scholar] [CrossRef]
- Zitvogel, L.; Daillere, R.; Roberti, M.P.; Routy, B.; Kroemer, G. Anticancer effects of the microbiome and its products. Nat. Rev. Microbiol. 2017, 15, 465–478. [Google Scholar] [CrossRef]
- Zhang, H.; Li, Y.; Huang, J.; Shen, L.; Xiong, Y. Precise targeting of lipid metabolism in the era of immuno-oncology and the latest advances in nano-based drug delivery systems for cancer therapy. Acta Pharm. Sin. B 2024, 14, 4717–4737. [Google Scholar] [CrossRef]
- Hecht, F.; Zocchi, M.; Alimohammadi, F.; Harris, I.S. Regulation of antioxidants in cancer. Mol. Cell 2024, 84, 23–33. [Google Scholar] [CrossRef]
- Woldeselassie, M.; Tamene, A. Therapeutic controversies over use of antioxidant supplements during cancer treatment: A scoping review. Front. Nutr. 2024, 11, 1480780. [Google Scholar] [CrossRef] [PubMed]
- Gomaa, M.B.M.; Abdelhameed, K.M.A.; Sobhy, S.E.; Konper, H.M.A.; Hassanein, Z.A.E.; Saleh, A.A.; Jamal, M.T.; Hafez, E.E. Antioxidant activity, antibacterial behavior, and anticancer impact of Egyptian propolis. Open Vet. J. 2025, 15, 126–138. [Google Scholar] [CrossRef] [PubMed]
- Kaur, K.; Al-Khazaleh, A.K.; Bhuyan, D.J.; Li, F.; Li, C.G. A Review of Recent Curcumin Analogues and Their Antioxidant, Anti-Inflammatory, and Anticancer Activities. Antioxidants 2024, 13, 1092. [Google Scholar] [CrossRef]
- Blot, W.J.; Li, J.Y.; Taylor, P.R.; Guo, W.; Dawsey, S.; Wang, G.Q.; Yang, C.S.; Zheng, S.F.; Gail, M.; Li, G.Y.; et al. Nutrition intervention trials in Linxian, China: Supplementation with specific vitamin/mineral combinations, cancer incidence, and disease-specific mortality in the general population. J. Natl. Cancer Inst. 1993, 85, 1483–1492. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Y.L.; Dawsey, S.M.; Kamangar, F.; Fan, J.H.; Abnet, C.C.; Sun, X.D.; Johnson, L.L.; Gail, M.H.; Dong, Z.W.; Yu, B.; et al. Total and cancer mortality after supplementation with vitamins and minerals: Follow-up of the Linxian General Population Nutrition Intervention Trial. J. Natl. Cancer Inst. 2009, 101, 507–518. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Shu, X.O.; Li, H.; Yang, G.; Cai, H.; Ji, B.T.; Gao, J.; Gao, Y.T.; Zheng, W.; Xiang, Y.B. Vitamin intake and liver cancer risk: A report from two cohort studies in China. J. Natl. Cancer Inst. 2012, 104, 1173–1181. [Google Scholar] [CrossRef]
- Alpha-Tocopherol, B.C.C.P.S.G. The effect of vitamin E and beta carotene on the incidence of lung cancer and other cancers in male smokers. N. Engl. J. Med. 1994, 330, 1029–1035. [Google Scholar]
- Bates, C.A.; Vincent, M.J.; Buerger, A.N.; Santamaria, A.B.; Maier, A.; Jack, M. Investigating the relationship between beta-carotene intake from diet and supplements, smoking, and lung cancer risk. Food Chem. Toxicol. 2024, 194, 115104. [Google Scholar] [CrossRef]
- Krannich, F.; Mucke, R.; Buntzel, J.; Schomburg, L.; Micke, O.; Hubner, J.; Dorfler, J. A systematic review of Selenium as a complementary treatment in cancer patients. Complement. Ther. Med. 2024, 86, 103095. [Google Scholar] [CrossRef]
- Klein, E.A.; Thompson, I.M., Jr.; Tangen, C.M.; Crowley, J.J.; Lucia, M.S.; Goodman, P.J.; Minasian, L.M.; Ford, L.G.; Parnes, H.L.; Gaziano, J.M.; et al. Vitamin E and the risk of prostate cancer: The Selenium and Vitamin E Cancer Prevention Trial (SELECT). JAMA 2011, 306, 1549–1556. [Google Scholar] [CrossRef]
- Luo, M.; Li, L. Association Between Vitamin Intake and Colorectal Cancer: Evidence from NHANES Data. J. Gastrointest. Cancer 2024, 55, 1581–1587. [Google Scholar] [CrossRef] [PubMed]
- Seraphin, G.; Rieger, S.; Hewison, M.; Capobianco, E.; Lisse, T.S. The impact of vitamin D on cancer: A mini review. J. Steroid Biochem. Mol. Biol. 2023, 231, 106308. [Google Scholar] [CrossRef] [PubMed]
- Al-Qahtani, S.D.; Bin-Melaih, H.H.; Atiya, E.M.; Fahmy, U.A.; Binmahfouz, L.S.; Neamatallah, T.; Al-Abbasi, F.A.; Abdel-Naim, A.B. Self-Nanoemulsifying Drug Delivery System of 2-Methoxyestradiol Exhibits Enhanced Anti-Proliferative and Pro-Apoptotic Activities in MCF-7 Breast Cancer Cells. Life 2022, 12, 1369. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Sun, L.; Zhang, Y.; Wang, Y.; Zheng, J. Imbalanced GSH/ROS and sequential cell death. J. Biochem. Mol. Toxicol. 2022, 36, e22942. [Google Scholar] [CrossRef]
- Zhao, X.Y.; Zhang, R.R.; Ye, Q.; Qiu, F.; Xu, H.Y.; Wei, F.G.; Zhang, H. 4-Hydroxyphenyl Retinamide Preferentially Targets FLT3 Mutated Acute Myeloid Leukemia via ROS Induction and NF-kappaB Inhibition. Curr. Med. Sci. 2020, 40, 810–816. [Google Scholar] [CrossRef] [PubMed]
- Townsend, D.M.; He, L.; Hutchens, S.; Garrett, T.E.; Pazoles, C.J.; Tew, K.D. NOV-002, a glutathione disulfide mimetic, as a modulator of cellular redox balance. Cancer Res. 2008, 68, 2870–2877. [Google Scholar] [CrossRef] [PubMed]
- Kumbul, Y.C.; Naziroglu, M. Paclitaxel Promotes Oxidative Stress-Mediated Human Laryngeal Squamous Tumor Cell Death through the Stimulation of Calcium and Zinc Signaling Pathways: No Synergic Action of Melatonin. Biol. Trace Elem. Res. 2022, 200, 2084–2098. [Google Scholar] [CrossRef]
- Mosca, L.; Ilari, A.; Fazi, F.; Assaraf, Y.G.; Colotti, G. Taxanes in cancer treatment: Activity, chemoresistance and its overcoming. Drug Resist. Updat. 2021, 54, 100742. [Google Scholar] [CrossRef]
- Ozalp, G.R.; Ustuner, B.; Avci, G.; Bari, O.; Yilmaz, M.M.; Denk, B.; Aktar, A. Vincristine-associated total antioxidant and oxidant status of ovaries and in vitro nuclear oocyte maturation in dogs with canine transmissible venereal tumor. Anim. Reprod. Sci. 2023, 253, 107260. [Google Scholar] [CrossRef]
- Mendez, N.; Alarcon, P.; Millan, C.; Burgos, R.A.; Morera, F.J.; Ojeda, J. Vincristine, carboplatin and cisplatin increase oxidative burst induced by PAF in canine neutrophils. Vet. Immunol. Immunopathol. 2020, 221, 110011. [Google Scholar] [CrossRef]
- Varricchi, G.; Ameri, P.; Cadeddu, C.; Ghigo, A.; Madonna, R.; Marone, G.; Mercurio, V.; Monte, I.; Novo, G.; Parrella, P.; et al. Antineoplastic Drug-Induced Cardiotoxicity: A Redox Perspective. Front. Physiol. 2018, 9, 167. [Google Scholar] [CrossRef] [PubMed]
- Simunek, T.; Sterba, M.; Popelova, O.; Adamcova, M.; Hrdina, R.; Gersl, V. Anthracycline-induced cardiotoxicity: Overview of studies examining the roles of oxidative stress and free cellular iron. Pharmacol. Rep. 2009, 61, 154–171. [Google Scholar] [CrossRef] [PubMed]
- Hwang, I.T.; Chung, Y.M.; Kim, J.J.; Chung, J.S.; Kim, B.S.; Kim, H.J.; Kim, J.S.; Yoo, Y.D. Drug resistance to 5-FU linked to reactive oxygen species modulator 1. Biochem. Biophys. Res. Commun. 2007, 359, 304–310. [Google Scholar] [CrossRef] [PubMed]
- Chun, K.S.; Joo, S.H. Modulation of Reactive Oxygen Species to Overcome 5-Fluorouracil Resistance. Biomol. Ther. 2022, 30, 479–489. [Google Scholar] [CrossRef] [PubMed]
- Zhong, T.; Yu, J.; Pan, Y.; Zhang, N.; Qi, Y.; Huang, Y. Recent Advances of Platinum-Based Anticancer Complexes in Combinational Multimodal Therapy. Adv. Healthc. Mater. 2023, 12, e2300253. [Google Scholar] [CrossRef]
- Ahmad, S.; Isab, A.A.; Al-Arfaj, A.R. Antitumor potential of platinum(II) complexes of selenium donor ligands. Metallomics 2023, 15, mfad020. [Google Scholar] [CrossRef]
- Montero, A.J.; Diaz-Montero, C.M.; Deutsch, Y.E.; Hurley, J.; Koniaris, L.G.; Rumboldt, T.; Yasir, S.; Jorda, M.; Garret-Mayer, E.; Avisar, E.; et al. Phase 2 study of neoadjuvant treatment with NOV-002 in combination with doxorubicin and cyclophosphamide followed by docetaxel in patients with HER-2 negative clinical stage II-IIIc breast cancer. Breast Cancer Res. Treat. 2012, 132, 215–223. [Google Scholar] [CrossRef]
- Teyssonneau, D.; Dariane, C.; Barret, E.; Beauval, J.B.; Brureau, L.; Fiard, G.; Fromont, G.; Crehange, G.; Gauthe, M.; Ruffion, A.; et al. PARP inhibitors in prostate cancers, is it time for combinations? Ther. Adv. Med. Oncol. 2024, 16, 17588359241242959. [Google Scholar] [CrossRef]
- Kummar, S.; Chen, A.; Parchment, R.E.; Kinders, R.J.; Ji, J.; Tomaszewski, J.E.; Doroshow, J.H. Advances in using PARP inhibitors to treat cancer. BMC Med. 2012, 10, 25. [Google Scholar] [CrossRef]
- Morganti, S.; Marra, A.; De Angelis, C.; Toss, A.; Licata, L.; Giugliano, F.; Taurelli Salimbeni, B.; Berton Giachetti, P.P.M.; Esposito, A.; Giordano, A.; et al. PARP Inhibitors for Breast Cancer Treatment: A Review. JAMA Oncol. 2024, 10, 658–670. [Google Scholar] [CrossRef]
- Evers, B.; Drost, R.; Schut, E.; de Bruin, M.; van der Burg, E.; Derksen, P.W.; Holstege, H.; Liu, X.; van Drunen, E.; Beverloo, H.B.; et al. Selective inhibition of BRCA2-deficient mammary tumor cell growth by AZD2281 and cisplatin. Clin. Cancer Res. 2008, 14, 3916–3925. [Google Scholar] [CrossRef] [PubMed]
- Rottenberg, S.; Jaspers, J.E.; Kersbergen, A.; van der Burg, E.; Nygren, A.O.; Zander, S.A.; Derksen, P.W.; de Bruin, M.; Zevenhoven, J.; Lau, A.; et al. High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc. Natl. Acad. Sci. USA 2008, 105, 17079–17084. [Google Scholar] [CrossRef] [PubMed]
- Michels, J.; Vitale, I.; Galluzzi, L.; Adam, J.; Olaussen, K.A.; Kepp, O.; Senovilla, L.; Talhaoui, I.; Guegan, J.; Enot, D.P.; et al. Cisplatin resistance associated with PARP hyperactivation. Cancer Res. 2013, 73, 2271–2280. [Google Scholar] [CrossRef] [PubMed]
- Guo, T.; Xu, J. Cancer-associated fibroblasts: A versatile mediator in tumor progression, metastasis, and targeted therapy. Cancer Metastasis Rev. 2024, 43, 1095–1116. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Wang, Z.; Gu, M.; Wei, P.; Wang, X.; Li, W. Cancer-associated fibroblasts: Heterogeneity and their role in the tumor immune response. Clin. Exp. Med. 2024, 24, 126. [Google Scholar] [CrossRef] [PubMed]
- Qin, Q.; Yu, R.; Eriksson, J.E.; Tsai, H.I.; Zhu, H. Cancer-associated fibroblasts in pancreatic ductal adenocarcinoma therapy: Challenges and opportunities. Cancer Lett. 2024, 591, 216859. [Google Scholar] [CrossRef]
- Zheng, L.; Cai, W.; Ke, Y.; Hu, X.; Yang, C.; Zhang, R.; Wu, H.; Liu, D.; Yu, H.; Wu, C. Cancer-associated fibroblasts: A pivotal regulator of tumor microenvironment in the context of radiotherapy. Cell Commun. Signal 2025, 23, 147. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.H.; Pauklin, S. ROS and TGFbeta: From pancreatic tumour growth to metastasis. J. Exp. Clin. Cancer Res. 2021, 40, 152. [Google Scholar] [CrossRef]
- Hou, Y.; Wang, H.; Wu, J.; Guo, H.; Chen, X. Dissecting the pleiotropic roles of reactive oxygen species (ROS) in lung cancer: From carcinogenesis toward therapy. Med. Res. Rev. 2024, 44, 1566–1595. [Google Scholar] [CrossRef]
- Ren, H.; Wang, M.; Ma, X.; An, L.; Guo, Y.; Ma, H. METTL3 in cancer-associated fibroblasts-derived exosomes promotes the proliferation and metastasis and suppresses ferroptosis in colorectal cancer by eliciting ACSL3 m6A modification. Biol. Direct 2024, 19, 68. [Google Scholar] [CrossRef]
- Jankowski, J.A.Z.; de Caestecker, J.; Love, S.B.; Reilly, G.; Watson, P.; Sanders, S.; Ang, Y.; Morris, D.; Bhandari, P.; Brooks, C.; et al. Esomeprazole and aspirin in Barrett’s oesophagus (AspECT): A randomised factorial trial. Lancet 2018, 392, 400–408. [Google Scholar] [CrossRef] [PubMed]
- Mostafa, T.M.; Alm El-Din, M.A.; Rashdan, A.R. Celecoxib as an adjuvant to chemotherapy for patients with metastatic colorectal cancer: A randomized controlled clinical study. Saudi Med. J. 2022, 43, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Qian, X.; Yang, H.; Ye, Z.; Gao, B.; Qian, Z.; Ding, Y.; Mao, Z.; Du, Y.; Wang, W. Celecoxib Augments Paclitaxel-Induced Immunogenic Cell Death in Triple-Negative Breast Cancer. ACS Nano 2024, 18, 15864–15877. [Google Scholar] [CrossRef]
- Guo, Q.; Li, Q.; Wang, J.; Liu, M.; Wang, Y.; Chen, Z.; Ye, Y.; Guan, Q.; Zhou, Y. A comprehensive evaluation of clinical efficacy and safety of celecoxib in combination with chemotherapy in metastatic or postoperative recurrent gastric cancer patients: A preliminary, three-center, clinical trial study. Medicine 2019, 98, e16234. [Google Scholar] [CrossRef] [PubMed]
- Nowak, J.A.; Twombly, T.; Ma, C.; Shi, Q.; Haruki, K.; Fujiyoshi, K.; Vayrynen, J.; Zhao, M.; Knight, J.; Mane, S.; et al. Improved Survival With Adjuvant Cyclooxygenase 2 Inhibition in PIK3CA-Activated Stage III Colon Cancer: CALGB/SWOG 80702 (Alliance). J. Clin. Oncol. 2024, 42, 2853–2859. [Google Scholar] [CrossRef] [PubMed]
- Meyerhardt, J.A.; Shi, Q.; Fuchs, C.S.; Meyer, J.; Niedzwiecki, D.; Zemla, T.; Kumthekar, P.; Guthrie, K.A.; Couture, F.; Kuebler, P.; et al. Effect of Celecoxib vs Placebo Added to Standard Adjuvant Therapy on Disease-Free Survival Among Patients With Stage III Colon Cancer: The CALGB/SWOG 80702 (Alliance) Randomized Clinical Trial. JAMA 2021, 325, 1277–1286. [Google Scholar] [CrossRef]
- Hamy, A.S.; Tury, S.; Wang, X.; Gao, J.; Pierga, J.Y.; Giacchetti, S.; Brain, E.; Pistilli, B.; Marty, M.; Espie, M.; et al. Celecoxib With Neoadjuvant Chemotherapy for Breast Cancer Might Worsen Outcomes Differentially by COX-2 Expression and ER Status: Exploratory Analysis of the REMAGUS02 Trial. J. Clin. Oncol. 2019, 37, 624–635. [Google Scholar] [CrossRef] [PubMed]
- Chu, X.; Tian, Y.; Lv, C. Decoding the spatiotemporal heterogeneity of tumor-associated macrophages. Mol. Cancer 2024, 23, 150. [Google Scholar] [CrossRef]
- Marelli, G.; Sica, A.; Vannucci, L.; Allavena, P. Inflammation as target in cancer therapy. Curr. Opin. Pharmacol. 2017, 35, 57–65. [Google Scholar] [CrossRef]
- Pozzi, S.; Satchi-Fainaro, R. The role of CCL2/CCR2 axis in cancer and inflammation: The next frontier in nanomedicine. Adv. Drug Deliv. Rev. 2024, 209, 115318. [Google Scholar] [CrossRef]
- Loberg, R.D.; Ying, C.; Craig, M.; Day, L.L.; Sargent, E.; Neeley, C.; Wojno, K.; Snyder, L.A.; Yan, L.; Pienta, K.J. Targeting CCL2 with systemic delivery of neutralizing antibodies induces prostate cancer tumor regression in vivo. Cancer Res. 2007, 67, 9417–9424. [Google Scholar] [CrossRef] [PubMed]
- Moisan, F.; Francisco, E.B.; Brozovic, A.; Duran, G.E.; Wang, Y.C.; Chaturvedi, S.; Seetharam, S.; Snyder, L.A.; Doshi, P.; Sikic, B.I. Enhancement of paclitaxel and carboplatin therapies by CCL2 blockade in ovarian cancers. Mol. Oncol. 2014, 8, 1231–1239. [Google Scholar] [CrossRef] [PubMed]
- Nywening, T.M.; Wang-Gillam, A.; Sanford, D.E.; Belt, B.A.; Panni, R.Z.; Cusworth, B.M.; Toriola, A.T.; Nieman, R.K.; Worley, L.A.; Yano, M.; et al. Targeting tumour-associated macrophages with CCR2 inhibition in combination with FOLFIRINOX in patients with borderline resectable and locally advanced pancreatic cancer: A single-centre, open-label, dose-finding, non-randomised, phase 1b trial. Lancet Oncol. 2016, 17, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Veeraballi, S.; Bandaru, S.S.; Kiwan, C.; Chan, K.H.; Shaaban, H.S. A multifaceted role of bisphosphonates from palliative care to anti-cancer therapy in solid tumors. J. Oncol. Pharm. Pr. 2025, 31, 107–118. [Google Scholar] [CrossRef]
- Boccia, S.M.; Sassu, C.M.; Ergasti, R.; Vertechy, L.; Apostol, A.I.; Palluzzi, E.; Fagotti, A.; Scambia, G.; Marchetti, C. Focus on Trabectedin in Ovarian Cancer: What Do We Still Need to Know? Drug Des. Devel Ther. 2024, 18, 2021–2032. [Google Scholar] [CrossRef] [PubMed]
- Beatty, G.L.; Chiorean, E.G.; Fishman, M.P.; Saboury, B.; Teitelbaum, U.R.; Sun, W.; Huhn, R.D.; Song, W.; Li, D.; Sharp, L.L.; et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science 2011, 331, 1612–1616. [Google Scholar] [CrossRef] [PubMed]
- Beatty, G.L.; Torigian, D.A.; Chiorean, E.G.; Saboury, B.; Brothers, A.; Alavi, A.; Troxel, A.B.; Sun, W.; Teitelbaum, U.R.; Vonderheide, R.H.; et al. A phase I study of an agonist CD40 monoclonal antibody (CP-870,893) in combination with gemcitabine in patients with advanced pancreatic ductal adenocarcinoma. Clin. Cancer Res. 2013, 19, 6286–6295. [Google Scholar] [CrossRef]
- Soto-Pantoja, D.R.; Terabe, M.; Ghosh, A.; Ridnour, L.A.; DeGraff, W.G.; Wink, D.A.; Berzofsky, J.A.; Roberts, D.D. CD47 in the tumor microenvironment limits cooperation between antitumor T-cell immunity and radiotherapy. Cancer Res. 2014, 74, 6771–6783. [Google Scholar] [CrossRef]
- Weiskopf, K.; Ring, A.M.; Ho, C.C.; Volkmer, J.P.; Levin, A.M.; Volkmer, A.K.; Ozkan, E.; Fernhoff, N.B.; van de Rijn, M.; Weissman, I.L.; et al. Engineered SIRPalpha variants as immunotherapeutic adjuvants to anticancer antibodies. Science 2013, 341, 88–91. [Google Scholar] [CrossRef]
- Kureshi, C.T.; Dougan, S.K. Cytokines in cancer. Cancer Cell 2025, 43, 15–35. [Google Scholar] [CrossRef]
- Kaplanov, I.; Carmi, Y.; Kornetsky, R.; Shemesh, A.; Shurin, G.V.; Shurin, M.R.; Dinarello, C.A.; Voronov, E.; Apte, R.N. Blocking IL-1beta reverses the immunosuppression in mouse breast cancer and synergizes with anti-PD-1 for tumor abrogation. Proc. Natl. Acad. Sci. USA 2019, 116, 1361–1369. [Google Scholar] [CrossRef] [PubMed]
- Aggen, D.H.; Ager, C.R.; Obradovic, A.Z.; Chowdhury, N.; Ghasemzadeh, A.; Mao, W.; Chaimowitz, M.G.; Lopez-Bujanda, Z.A.; Spina, C.S.; Hawley, J.E.; et al. Blocking IL1 Beta Promotes Tumor Regression and Remodeling of the Myeloid Compartment in a Renal Cell Carcinoma Model: Multidimensional Analyses. Clin. Cancer Res. 2021, 27, 608–621. [Google Scholar] [CrossRef] [PubMed]
- Fizazi, K.; De Bono, J.S.; Flechon, A.; Heidenreich, A.; Voog, E.; Davis, N.B.; Qi, M.; Bandekar, R.; Vermeulen, J.T.; Cornfeld, M.; et al. Randomised phase II study of siltuximab (CNTO 328), an anti-IL-6 monoclonal antibody, in combination with mitoxantrone/prednisone versus mitoxantrone/prednisone alone in metastatic castration-resistant prostate cancer. Eur. J. Cancer 2012, 48, 85–93. [Google Scholar] [CrossRef]
- Huang, D.; Xue, J.; Li, S.; Yang, D. Oxaliplatin and infliximab synergize to induce regression of colon cancer. Oncol. Lett. 2018, 15, 1517–1522. [Google Scholar] [CrossRef] [PubMed]
- Eigentler, T.K.; Gutzmer, R.; Hauschild, A.; Heinzerling, L.; Schadendorf, D.; Nashan, D.; Holzle, E.; Kiecker, F.; Becker, J.; Sunderkotter, C.; et al. Adjuvant treatment with pegylated interferon alpha-2a versus low-dose interferon alpha-2a in patients with high-risk melanoma: A randomized phase III DeCOG trial. Ann. Oncol. 2016, 27, 1625–1632. [Google Scholar] [CrossRef] [PubMed]
- Bottomley, A.; Coens, C.; Suciu, S.; Santinami, M.; Kruit, W.; Testori, A.; Marsden, J.; Punt, C.; Sales, F.; Gore, M.; et al. Adjuvant therapy with pegylated interferon alfa-2b versus observation in resected stage III melanoma: A phase III randomized controlled trial of health-related quality of life and symptoms by the European Organisation for Research and Treatment of Cancer Melanoma Group. J. Clin. Oncol. 2009, 27, 2916–2923. [Google Scholar]
- Sabit, H.; Arneth, B.; Pawlik, T.M.; Abdel-Ghany, S.; Ghazy, A.; Abdelazeem, R.M.; Alqosaibi, A.; Al-Dhuayan, I.S.; Almulhim, J.; Alrabiah, N.A.; et al. Leveraging Single-Cell Multi-Omics to Decode Tumor Microenvironment Diversity and Therapeutic Resistance. Pharmaceuticals 2025, 18, 75. [Google Scholar] [CrossRef]
- Peng, X.; Fang, J.; Lou, C.; Yang, L.; Shan, S.; Wang, Z.; Chen, Y.; Li, H.; Li, X. Engineered nanoparticles for precise targeted drug delivery and enhanced therapeutic efficacy in cancer immunotherapy. Acta Pharm. Sin. B 2024, 14, 3432–3456. [Google Scholar] [CrossRef]
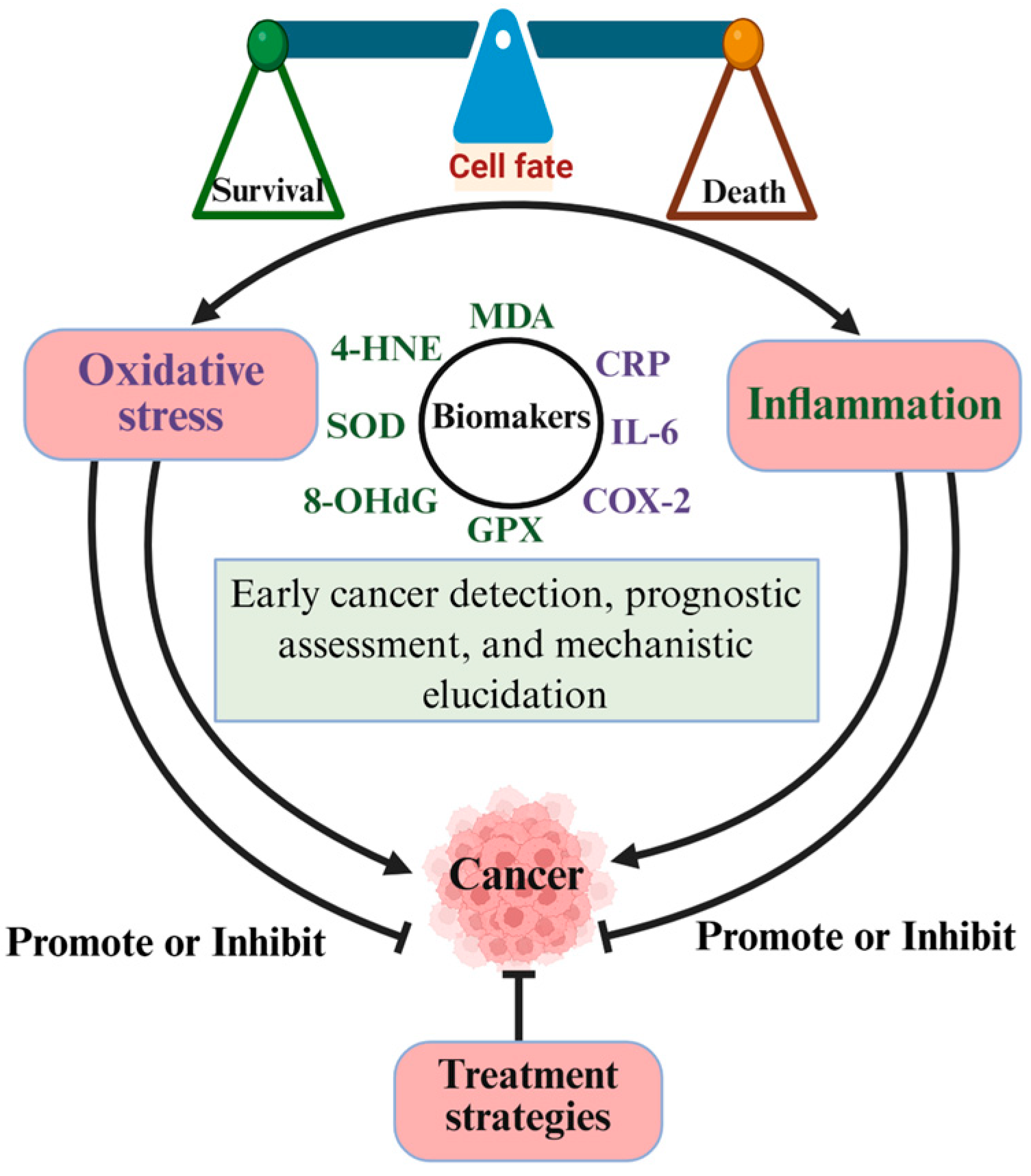
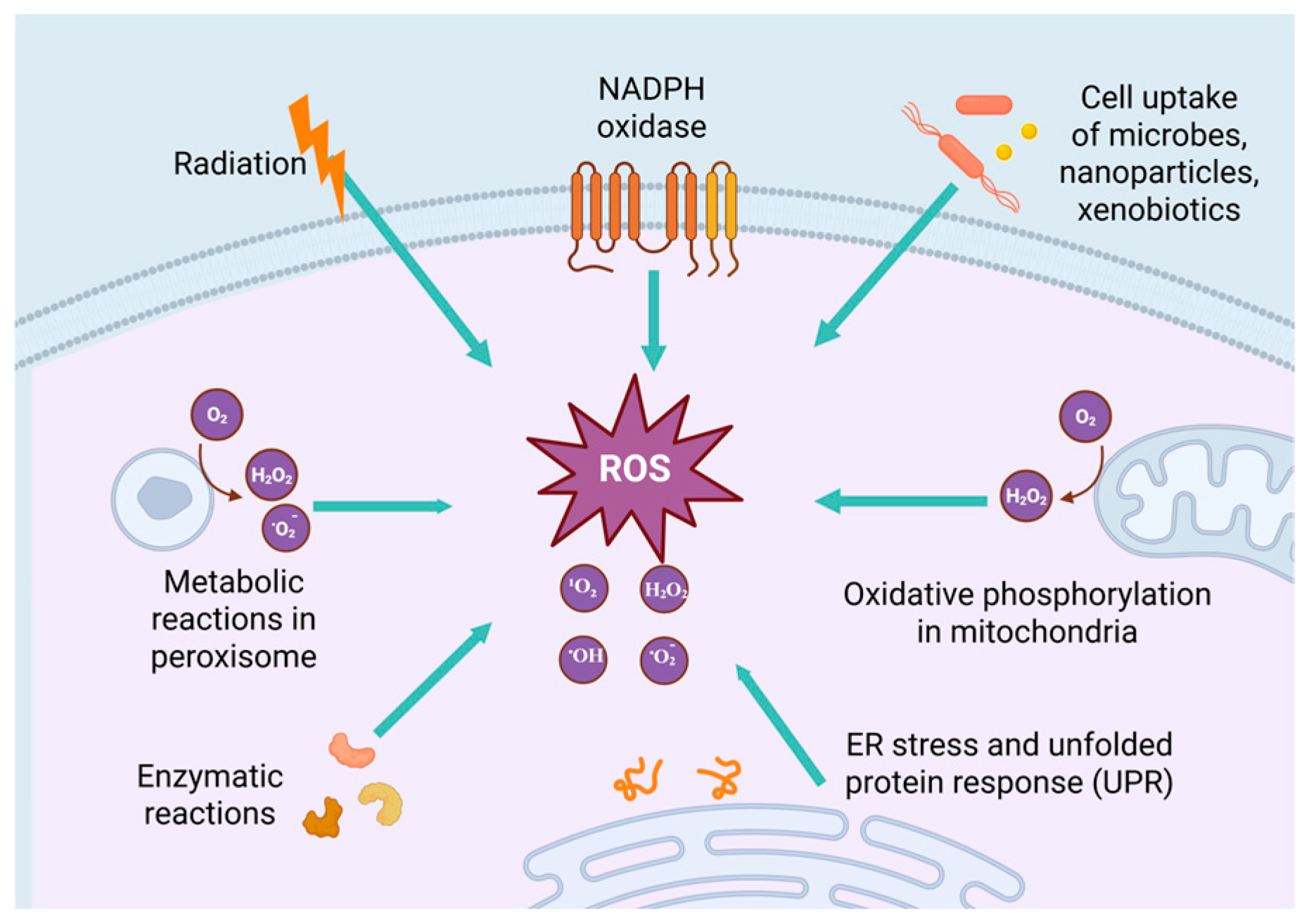
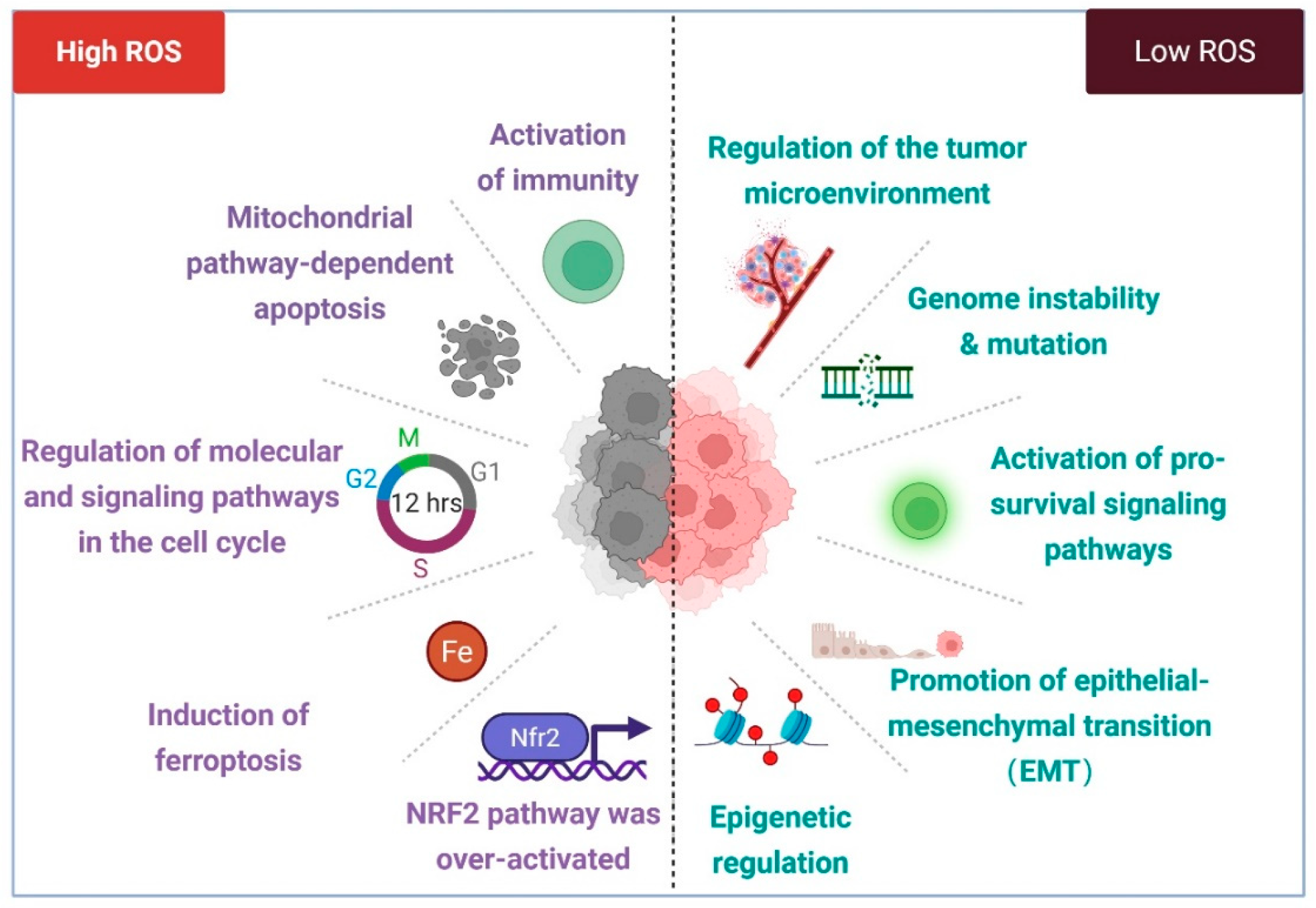
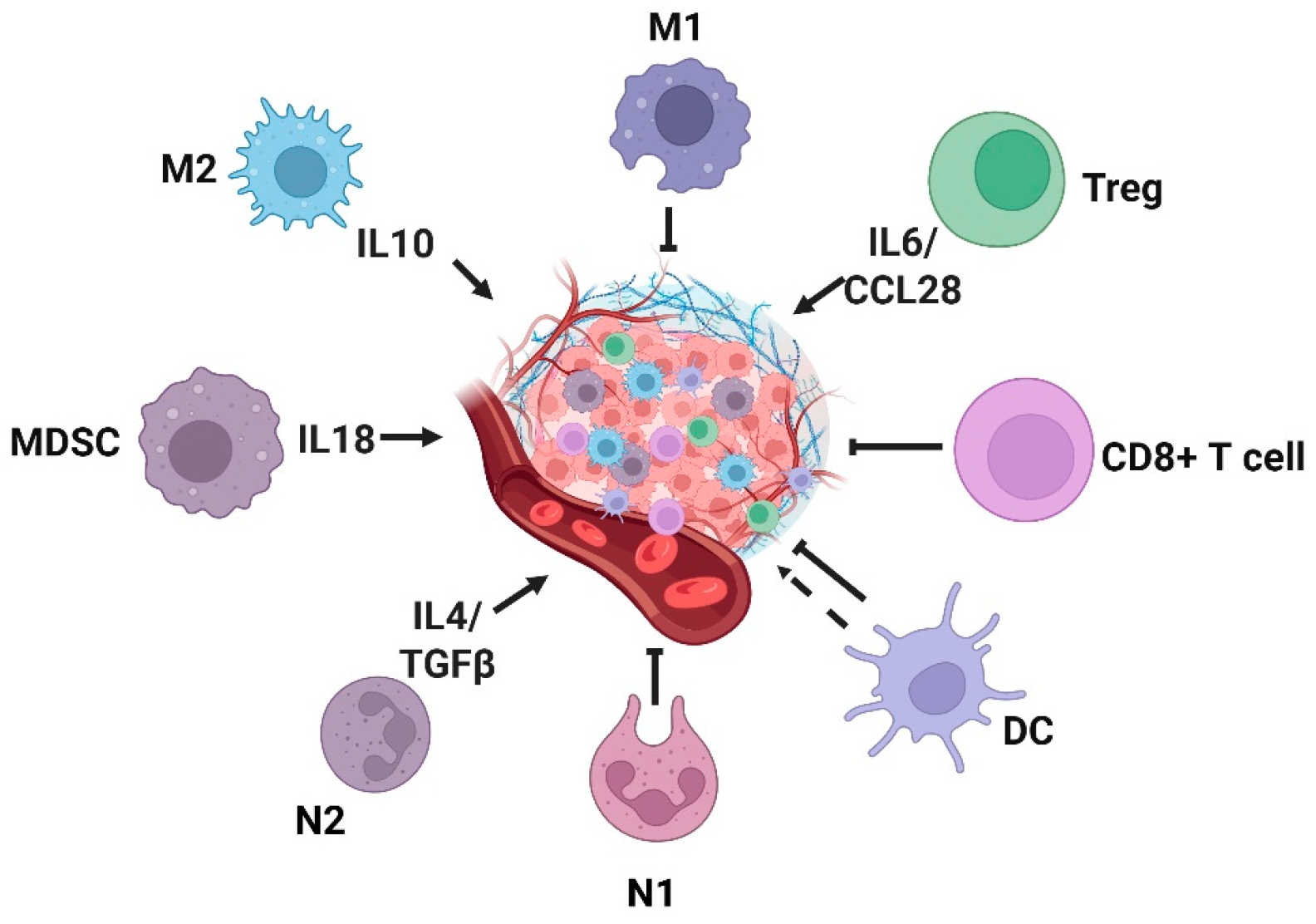
| Name | Mechanism of Action; Effects on Oxidative Stress | Effect on Tumors | References |
|---|---|---|---|
| β-carotene | Antioxidant effect | Reduce all-cause mortality in esophageal and gastric cancer patients | [374,375] |
| Vitamins A/D/E | Antioxidant effect | Reduce incidence of cancers and improve patient outcomes | [376,381,382] |
| Selenium | Antioxidant effect | Mitigate prostate cancer progression | [379] |
| Nutrient components (genistein, sulforaphane, curcumin, tannins, flavonoids, etc.) | Antioxidant effect | Inhibit the survival of breast, liver, and pancreatic cancers | [29,372,373] |
| As2O3 | Reacts with cysteine residues, inhibits mitochondrial respiratory function, induces ROS overproduction | Induce apoptosis across multiple cancer types, including leukemia, multiple myeloma, and breast cancer | [383,384,385] |
| NOV-002 | Glutathione disulphide mimetic; alters intracellular GSSG/GSH ratio | Inhibit tumor cell invasion, proliferation, and survival | [386] |
| Taxanes (paclitaxel, docetaxel) | Downregulation of antioxidant glutathione peroxidase and glutathione | Induce tumor cell apoptosis; inhibit angiogenesis, cell proliferation, and drug resistance | [387,388] |
| Vinca alkaloids (vincristine, vinblastine) | Induce an imbalance in the cellular oxidation/antioxidant state | Inhibit angiogenesis and tumor growth | [389] |
| Antimetabolites (fluorouracil, fludarabine) | Induce the generation of free radicals | Induce cancer cell apoptosis and inhibit the progression of CRC | [383,390,391] |
| Anthracyclines (doxorubicin, epirubicin, daunorubicin) | DNA Integration and Topoisomerase II inhibition; induce excessive ROS and RNS | Have a wide range of anti-tumor effects | [384,392] |
| Platinum-based anticancer agents (cisplatin, carboplatin, oxaliplatin) | Disrupt cellular antioxidant systems | Induce DNA damage, cycle arrest, and apoptosis in tumor cells | [393] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, M.; Xiao, Y.; Miao, J.; Zhang, X.; Liu, M.; Zhu, L.; Liu, H.; Shen, X.; Wang, J.; Xie, B.; et al. Oxidative Stress and Inflammation: Drivers of Tumorigenesis and Therapeutic Opportunities. Antioxidants 2025, 14, 735. https://doi.org/10.3390/antiox14060735
Wang M, Xiao Y, Miao J, Zhang X, Liu M, Zhu L, Liu H, Shen X, Wang J, Xie B, et al. Oxidative Stress and Inflammation: Drivers of Tumorigenesis and Therapeutic Opportunities. Antioxidants. 2025; 14(6):735. https://doi.org/10.3390/antiox14060735
Chicago/Turabian StyleWang, Meimei, Yaping Xiao, Jie Miao, Xin Zhang, Meng Liu, Longchao Zhu, Hongxin Liu, Xiaoyan Shen, Jihui Wang, Biao Xie, and et al. 2025. "Oxidative Stress and Inflammation: Drivers of Tumorigenesis and Therapeutic Opportunities" Antioxidants 14, no. 6: 735. https://doi.org/10.3390/antiox14060735
APA StyleWang, M., Xiao, Y., Miao, J., Zhang, X., Liu, M., Zhu, L., Liu, H., Shen, X., Wang, J., Xie, B., & Wang, D. (2025). Oxidative Stress and Inflammation: Drivers of Tumorigenesis and Therapeutic Opportunities. Antioxidants, 14(6), 735. https://doi.org/10.3390/antiox14060735