Abstract
Ovarian cancer (OC) is the most lethal gynecological cancer globally. Its incidence and mortality consistently rise after menopause. While parity reduces the risk of OC, nulliparity during a woman’s fertile years increases it. Although the association between reproductive history and OC risk is well-established, the long-term impact of pregnancy on the postmenopausal human ovary has received little to no attention. Parity apparently delays the natural decline of the ovarian reserve, but this association also remains unexplored to date. Based on data from cellular, biochemical, and histological markers, as well as epidemiological studies, transcriptomic analyses, and gene knockout mouse models, we review compelling evidence suggesting a critical intraovarian interplay between the residual ovarian reserve and the ovarian surface epithelium (OSE) in the aged ovary. This interaction appears to be a key factor underlying the protective effect of parity on ovarian cancer (OC) risk. We propose that functional FSHR signaling in the remnant follicles of the aged multiparous ovary somehow counteracts the oxidative stress and subsequent chronic inflammation typically observed in the senescent ovary. This mechanism would minimize DNA damage, thereby lowering the probability of neoplastic transformation in the aged mammalian ovary. The precise mechanism by which pregnancy imprints such a long-term follicle–OSE crosstalk warrants further investigation.
1. Introduction
Ovarian cancer (OC) is the leading cause of gynecological cancer mortality worldwide [1]. While the precise etiology of OC remains incompletely understood, reproductive history is a well-established factor influencing OC risk [2,3]. Numerous epidemiological studies demonstrate that OC risk increases with uninterrupted ovulation (e.g., nulliparity) and decreases under physiological and pharmacological conditions that temporarily suppress ovulation, such as full-term pregnancies, oral contraceptive use, and breastfeeding [3,4,5]. Furthermore, the incidence and mortality of sporadic OC—that without a hereditary component—steadily rise after menopause, peaking during the post-menopausal period. This age-dependent decline in ovarian function is primarily attributed to a reduced follicular count, which correlates with diminished ovarian synthesis of estrogen and alpha-inhibin. The subsequent disruption of the hypothalamic–pituitary–ovarian (HPO) axis leads to persistently high circulating levels of the pituitary gonadotropins, follicle-stimulating hormone (FSH), and luteinizing hormone (LH). This hormonal shift induces systemic metabolic and inflammatory changes in women and promotes deleterious effects within the ovary, including oxidative stress, fibrosis, senescence, and chronic inflammation [6,7,8]. Crucially, how the OC risk-reducing effect of parity can overcome this multifaceted age-dependent ovarian decline from the fertile to the post-menopausal phase has not been elucidated to date. Moreover, there is a notable absence of data regarding the effects of past parity on molecular pathogenic markers in the human postmenopausal ovary. This review aims to synthesize existing literature to enhance our understanding of the factors promoting early ovarian carcinogenesis during the post-menopausal age, considering the ovary as a primary site of OC origin. The relevance of this topic is further underscored by the increasing trend in developed countries over recent decades for women to remain childless or postpone childbearing [9], which is projected to lead to rising rates of various health issues, including OC, during post-reproductive age.
2. Oxidative Stress as a Central Feature of Ovarian Aging
2.1. Reactive Oxygen Species in the Fertile-Age Ovary
Processes essential to the reproductive function of the ovary, including ovulation, follicle maturation, atresia, luteinization, and luteolysis, rely on transient but controlled pulses of reactive oxygen species (ROS) [10]. As an inherent aspect of aerobic metabolism, ROS are primarily generated as by-products of the mitochondrial electron transport chain (Figure 1A). The canonical ROS include hydrogen peroxide (H2O2), its precursor superoxide anion (•O2−), and the hydroxyl radical (•OH). The •OH is a product of the well-known Fenton reaction between H2O2 and Fe2+ ions. While basal ROS levels are essential for certain cellular functions, an excess of ROS leads to an unbalanced redox state known as oxidative stress (OS), which can inflict damage on cellular components. To counteract this threat, cells possess an antioxidant system comprising enzymes such as catalase (CAT), superoxide dismutase (SOD), and glutathione peroxidase (GPX). Additionally, ROS can be scavenged by the tripeptide glutathione (GSH), which is subsequently oxidized to glutathione disulfide (GSSG). The NADPH-dependent enzyme glutathione disulfide reductase (GSR) can reduce GSSG back to GSH. GSH can also be synthesized de novo from amino acid precursors by the enzymes GSH synthetase (GSS) and glutamate–cysteine ligase (GCL). GCL catalyzes the rate-limiting step of the de novo GSH synthesis. All mentioned ROS and their metabolizing enzymes have been identified in the mammalian ovary [11] (Figure 1A).
Ovulation is a ROS-dependent process that requires the cyclical rupture and subsequent repair of the ovarian surface epithelium (OSE) [12]. This “tear and repair” process can, as a trade-off, induce DNA damage in the OSE [13]. Furthermore, mature follicular fluid contains ROS [11], which can exacerbate oxidative damage to both the OSE and the fimbria upon ovulation. Similarly, follicle atresia, the degeneration of ovarian follicles, is frequently linked to OS across most of the recruited follicular cohort [14]. However, the dominant follicle, which is selected for ovulation, manages to preserve an adequate level of GSH to counteract oxidative damage, thereby allowing its progression towards ovulation [15,16]. Finally, the functional and structural regression of the corpus luteum (CL), a temporary intraovarian endocrine gland, is an apoptosis-mediated process determined by a delicate balance between the activity of antioxidant enzymes and the endogenous ROS generated from its steroidogenic activity [17].
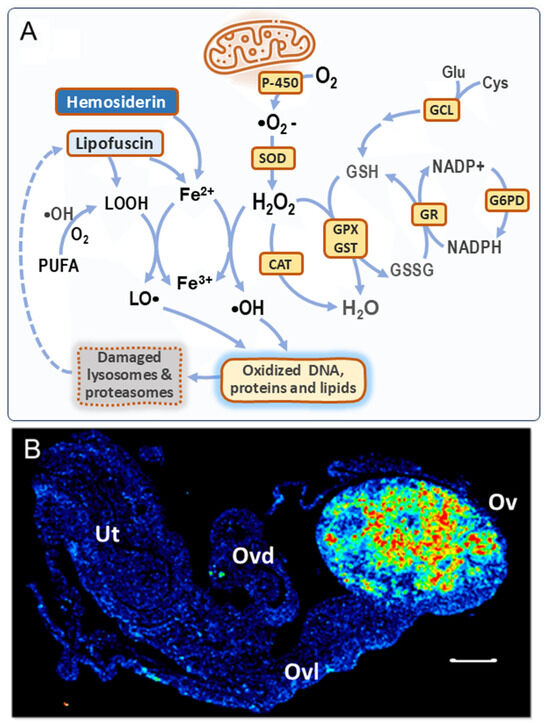
Figure 1.
Formation of ROS in the aged ovary. (A) Canonical ROS production by mitochondria, the antioxidant system, and additional ROS sources. P-450 = cytochrome oxidases; SOD = superoxide dismutase; CAT = catalase; GPX/GST = glutathione peroxidase/transferase; GR = glutathione reductase; G6PD = glucose-6-P-dehydrogenase; GCL = glutamate–cysteine ligase. In the aged ovary, both lipofuscin and hemosiderin can leak Fe2+, which reacts with H2O2 to produce •OH radicals (Fenton reaction). Lipid peroxides (LOOH) derived from lipofuscin and from polyunsaturated fatty acids (PUFAs) can also react with Fe2+ to form LO• radicals [18,19]. Both •OH and LO• radicals cause oxidative damage to biomolecules. Lipofuscin is formed as a consequence of dysfunctional proteasomes and lysosomes due to OS. Further details in the text. (B) Lipofuscin imaging in a 16 m.o. nulliparous mouse ovary, detected by autofluorescence; Ov = ovary, Ovd = oviduct, Ovl = ovarian ligament, and Ut = uterine horn; bar = 200 μm. Taken with permission from reference [20]. Lower lipofuscin levels were observed in young [21] and aged parous [20] mouse ovaries.
Figure 1.
Formation of ROS in the aged ovary. (A) Canonical ROS production by mitochondria, the antioxidant system, and additional ROS sources. P-450 = cytochrome oxidases; SOD = superoxide dismutase; CAT = catalase; GPX/GST = glutathione peroxidase/transferase; GR = glutathione reductase; G6PD = glucose-6-P-dehydrogenase; GCL = glutamate–cysteine ligase. In the aged ovary, both lipofuscin and hemosiderin can leak Fe2+, which reacts with H2O2 to produce •OH radicals (Fenton reaction). Lipid peroxides (LOOH) derived from lipofuscin and from polyunsaturated fatty acids (PUFAs) can also react with Fe2+ to form LO• radicals [18,19]. Both •OH and LO• radicals cause oxidative damage to biomolecules. Lipofuscin is formed as a consequence of dysfunctional proteasomes and lysosomes due to OS. Further details in the text. (B) Lipofuscin imaging in a 16 m.o. nulliparous mouse ovary, detected by autofluorescence; Ov = ovary, Ovd = oviduct, Ovl = ovarian ligament, and Ut = uterine horn; bar = 200 μm. Taken with permission from reference [20]. Lower lipofuscin levels were observed in young [21] and aged parous [20] mouse ovaries.
2.2. Oxidative Stress in the Aging Ovary Is Linked to Follicle Depletion
Biological aging is characterized as a progressive, time-dependent decline in cell and tissue function. OS is a recognized factor contributing to aging through the oxidative damage of cellular components across virtually all mammalian tissues [22]. In the ovary, this redox imbalance is coupled with the gradual exhaustion of follicles as age advances. This depletion ultimately impacts post-reproductive female physiology at a systemic level, thereby increasing the risk of chronic diseases, including reproductive cancers [23]. This age-dependent OS negatively affects several ovarian cell types, including oocytes, follicular cells (granulosa and theca), stromal cells, the OSE, and resident immune cells [21].
Mitochondrial dysfunction, alongside altered metabolism of glucose, GSH, and phospholipids, contributes to the primary redox imbalance observed in oocytes, follicles, and cumulus cells of aging human ovaries [6]. Similarly, the aging mouse ovary experiences follicle depletion linked to OS, characterized by low expression of both mitochondrial (e.g., peroxiredoxin 3 (Prdx3), thioredoxin 2 (Txn2)) and cytosolic (e.g., glutaredoxin (Glrx), glutathione S-transferase mu 2 (Gstm2)) antioxidant genes, as well as structural damage to ovarian molecular components, including proteins, lipids, and nucleic acids [24].
Lifestyle factors, such as tobacco smoking, a high-fat diet, obesity, and exposure to certain environmental pollutants, endocrine-disrupting agents, and cancer chemotherapy can negatively impact the quantity and quality of follicles, leading to concomitant OS and thus mimicking an early ovarian aging-like phenotype [25,26,27]. These conditions are associated with premature ovarian failure in women and can be modeled in mice through gene silencing or knockdown to identify candidate genes involved in follicle loss associated with redox imbalance. For instance, female mice null for the Gclm gene, which codes for the modifier subunit of GCL, exhibit low oocyte and ovarian GSH levels, resulting in OS and early depletion of ovarian follicles [28,29]. Similarly, female knockout mice for Nrf2 (nuclear factor, erythroid derived 2, like 2) display a low count of primordial follicles at middle age (10–12 months old) [30]. Nrf2 is a transcription factor that regulates the expression of antioxidant genes in many cell types and plays a relevant role during ovarian aging [31].
2.3. Oxidative DNA Damage and Disrupted DNA Repair in the Aged Ovary
An excess of ROS can chemically modify the genetic material at both the nucleotide bases and the sugar–phosphate backbone. Specifically, •OH and LO• radicals abstract hydrogen atoms from the bases and the sugar–phosphate backbone of DNA (Figure 1A). The most frequent types of oxidative DNA lesions include base modifications, base loss, single/double DNA strand breaks, protein–DNA adducts, and intra/interstrand DNA crosslinks. The guanine oxidation product, 8-hydroxy-2′-deoxyguanosine (8-OHdG), is widely employed as a marker of oxidative DNA damage [32].
Importantly, two synergistic aspects link cancer and aging concerning oxidative DNA damage: as aging increases OS, the capacity to sense and repair damaged DNA diminishes in aged cells. Specifically, both double-strand break (DSB) repair [33,34] and base-excision repair (BER)—which performs the removal of adducts such as alkylated and oxidized bases—are affected by age-dependent OS [35]. In the ovary, oxidative DNA damage is elevated in stromal ovarian cells of aged mice [20,24]. Furthermore, decreased expression of the DSB repair genes ATM, BRCA1, RAD51, ERCC2, and H2AX has been observed during follicle and oocyte aging in murine and human ovaries [36,37]. Consistent with these findings, a transcriptome study detected downregulation of over three dozen genes involved in the DNA damage response in aged (16 months old) mouse ovaries compared to young-adult (4 months old) mouse ovaries [38]. Seventeen of these were DSB repair genes, including Rad51, Rad54b, and Rad54l, which function via homologous recombination, as well as the Fanconi anemia complementation genes Fancd2 and Brca2, which colocalize in nuclear foci in response to DNA damage [39].
Interestingly, in the absence of pathogenic mutations directly affecting the homologous recombination repair function of the BRCA1/BRCA2-encoded proteins, the promoter methylation of these genes has been associated with tumorigenesis and poor prognosis in breast/ovarian cancer [40]. In fact, various types of epigenetic alterations have been recently proposed as one of the hallmarks of ovarian aging [41]. Despite the fact that genomic DNA methylation decreases with age, additional epigenetic factors (histone modifications, ncRNAs, or RNA methylation) and upstream transcriptional regulators might modulate this age-concerted downregulation of DNA repair genes. This is an emerging field of research since it links various age-related ovarian pathologies to the epigenetic changes occurring in the ovary that parallel the natural fertility decline in women.
2.4. Lipofuscin and Hemosiderin Accumulation Further Contribute to OS in the Aged Ovary
Oxidative DNA damage is one of the major drivers of the aged ovary to a senescent state [8]. Senescence is a cellular response to various stressors leading to irreversible cell cycle arrest, metabolic reprogramming, chromatin remodeling, resistance to cell death, and the production of inflammatory mediators and proteases. Senescence can play divergent roles as a tumor suppressor or tumor promoter [42]. In the early senescent phase, proliferation of cells containing damaged DNA is prevented by induction of p53 and the cyclin-dependent cell cycle kinase inhibitors p21 and p16, among other factors. As this proliferative blockade persists, senescent cells develop a secretory, pro-inflammatory phenotype that modifies the tissue microenvironment, thus promoting endogenous chronic inflammation through increased immune-cell recruitment [8]. Indeed, the transcriptome of the aged mouse ovary shows increased expression of genes involved in inflammatory/immune responses, cell adhesion, TNF synthesis, peptidase activity, wound healing, and immune cell markers [8,21,38]. The ovarian stroma is particularly prone to accumulating senescent cells [8]. In fact, ROS-induced senescent fibroblasts in the mouse ovarian stroma promote neoplastic transformation of c-myc immortalized OSE cells [18].
In addition to modifying DNA, OS can also inflict damage upon various organelles. In organs and tissues characterized by high rates of phagocytosis and autophagy, age-related dysfunction of lysosomes and proteasomes leads to the formation and accumulation of lipofuscin. This intracellular, oxidized, and cross-linked substance is considered a hallmark of senescence [43]. Lipofuscin has been detected in aged murine ovaries [20,21,44,45], specifically inside foamy, enlarged multinucleated macrophages (MNM) associated with stromal cells [44]. Cellular debris resulting from follicular atresia during uninterrupted fertile cycles may contribute to lipofuscin formation as the ovary ages. This phenomenon might explain why the ovary uniquely accumulates lipofuscin within the aged mouse reproductive tract, as depicted in Figure 1B [20].
Furthermore, lipofuscin possesses the ability to trap certain metals, predominantly ionic iron. Interestingly, iron also accumulates with advancing age in several organs in the form of hemosiderin. Hemosiderin is another intracellular, insoluble aggregate containing degraded ferritin plus lysosomal remnants, and it is also found within macrophages [44,46]. Ferritin is a multimeric, non-heme iron carrier and storage protein that predominantly binds ferric ion and is found increased in the serum of post-menopausal women [19]. In situations of iron overload, such as hemolysis after the rupture of blood microvessels (hemorrhage), macrophages engulf erythrocyte contents, mostly hemoglobin, leading to the deposition of hemosiderin [47,48]. Importantly, redox-active iron can be released during heme degradation [49] and from hemosiderin in acidic conditions, such as the lysosome; this also takes place in inflammation and hypoxia [50], both processes exacerbated in aging.
Iron accumulation can induce ferroptosis, an alternative type of cell death characterized by distinct mitochondrial shrinkage, increased ROS, depletion of GSH, and lipid peroxidation without DNA fragmentation or evident chromatin changes [51]. Decreased glutathione peroxidase 4 (GPX4) activity due to low GSH levels is a marker of ferroptosis, and this alteration has been recently described in patients with diminished OR and in human aged ovaries [52]. Relatedly, iron overload and hemosiderin aggregates are observed in the ovaries of an estrogen receptor 1 (Esr1)-deficient mouse model along with enlarged MNM, increased mast cells, and dysregulated expression of genes involved in iron transport, storage, and regulation [53]. In fact, the aged mouse ovary overexpresses the iron ion transporters NRAMP (Slc11a1) and ferroportin (Slc40a1) [38], the latter an exporter of ferrous iron to the plasma, which has been implicated in iron overload diseases [54]. As shown in Figure 1A, non-heme free ferrous ion (Fe2+) reacts with H2O2 to form •OH radicals. In turn, OH• reacts with polyunsaturated fatty acids (PUFAs) to form lipid peroxides (LOOH), which can also react with Fe2+ to form LO• radicals. Both •OH and LO• are able to induce oxidative damage to macromolecules and organelles. Therefore, as lipofuscin and hemosiderin apparently colocalize in the MNM of the aged mouse ovary [20], this might represent an additional source of redox-active iron that amplifies ROS levels in senescent cells [55], in addition to those produced by dysfunctional mitochondria [6]. As mentioned above, the OS perpetuated by such mechanisms ends up depleting GSH levels, thus resulting in iron-mediated damage, ferroptosis, and chronic inflammation [8].
3. Aging and Reproductive History Modulate Ovarian Cancer Risk
3.1. Ovarian Cancer Risk Increases at Menopause and Decreases by Prior Parity
Coincident with the age-dependent increase in OS and the diminished DNA repair capacity of the mammalian ovary, OC mortality and incidence steadily rise as women approach menopause—the hallmark of reproductive aging—which typically occurs at a mean age of 51 years [2]. As previously mentioned, the primary underlying cause of reproductive aging is the depletion of ovarian follicles, leading to decreased circulating levels of steroid hormones and a concomitant increase in the pituitary gonadotropins, FSH and LH, due to the disruption of the HPO axis [2].
Despite significant follicle depletion at menopause, the postmenopausal ovarian stroma retains a partial steroidogenic capacity [56,57,58]. Testosterone, androstenedione, and estradiol [59], as well as the receptors for estrogen (ER-alpha), androgen [60], and the enzymes 17-beta hydroxysteroid dehydrogenase type-1 (HSD17B1) and aromatase (CYP19A1), have been detected in various cell types of the postmenopausal ovary [61,62]. Consistent with increased circulating gonadotropin levels, their cognate receptors, FSHR and LHR, are expressed in human postmenopausal ovaries [63] and in aged female mice [21,38].
In addition to age, a woman’s reproductive history during her fertile life significantly influences her risk of developing OC later in the postmenopausal period. Specifically, periods that suppress ovulatory cycles, such as pregnancy, oral contraceptive use, and breastfeeding, are consistently linked to a significantly decreased OC risk [3,4,5]. Regarding parity, a meta-analysis of 32 studies reported a relative risk of 0.46 for women with three or more births compared to nulliparous women [5]. Notably, the reduction in OC risk conferred by pregnancy and oral contraception is directly proportional to the number of full-term births and years of use, respectively. Furthermore, the magnitude of this decreased risk is similar for both parity and oral contraceptive use [3,4,5].
This reduced OC risk is linked to the transient suppression of ovulatory cycles and supports the “incessant ovulation” theory as an etiological factor in ovarian carcinogenesis [2]. As discussed, the ROS-dependent, repetitive tear-repair damage to the OSE, coupled with the exposure of the OSE and fimbria to ROS-containing follicular fluid during each ovulatory event, is believed to increase the likelihood of DNA damage [12,13] that may remain unrepaired as aging progresses [20,36,37].
Conversely, limited attention has been paid to the long-term effects of pregnancy, oral contraceptive hormones, and breastfeeding in preventing ovarian carcinogenesis beyond simply pausing ovulation. High progesterone levels during pregnancy have been proposed to preclude the mitogenic action of low-to-moderate estrogen levels, thereby reducing the chances of malignant transformation [64]. However, precisely how this ovarian exposure to progesterone during fertile life confers a long-term reduced OC risk at menopause remains largely unknown.
Parity also reduces the risks of endometrial and breast cancers. A recent follow-up population study demonstrated a linear reduction in the incidence of ovarian, endometrial, and breast cancers for each additional childbirth across the entire fertile age range studied (<20 to 45 years old). This reduction occurred at similar rates for all three cancers, suggesting a possible common underlying biology [65]. However, while the link between parity and breast cancer risk has been explored in postmenopausal breast tissue, even identifying specific genes and pathways [66,67,68], analogous studies associating parity with OC through gene expression analyses of postmenopausal ovarian tissue are exceedingly scarce. Thus, the evidence reviewed here aims to address this knowledge gap by exploring how parity modulates long-term gene expression in the postmenopausal ovary.
3.2. Parity Delays the Natural Decline of the Ovarian Reserve
Age-dependent follicle depletion is accurately expressed as the decrease in the ovarian reserve (OR), a term defining the total number of primordial (non-growing) follicles that contain the entire pool of oocytes potentially fertilizable during the reproductive life of females. In mammals, the OR is typically established in utero around birth and declines steadily during early adult life, accelerating with advanced reproductive aging [69]. In women, this decline accelerates during their mid-to-late 30s. Over the first decade of life, all primordial follicles remain quiescent due to inhibitory mechanisms within both the oocytes and the pre-granulosa cells [70]. At menarche, a cohort of primordial follicles, containing oocytes in meiotic arrest, are activated upon the release of these inhibitory signals [71]. Once activated, typically only a single follicle develops and matures towards ovulation, while the remaining ones undergo degradation through atresia. From menarche to menopause, this process repeats periodically during fertile life, leading to progressive follicle depletion. The initial OR consists of approximately half a million to one million primordial follicles, yet only around 500 are ovulated during a woman’s fertile lifespan, and nearly 1000 primordial follicles remain in the human ovary at menopause [72].
As described previously, certain conditions and exposures have been associated with an accelerated rate of OR decline, mirroring the effects of aging [25,26,27]. Experimental evidence suggests that accelerated OR depletion results from the recurrent activation of dormant follicles, which leads to increased ovarian OS, early ovarian aging, and an augmented risk of postmenopausal pathologies [70,71]. Conversely, other interventions and agents that delay the OR depletion can attenuate ovarian aging and thereby promote healthy reproductive aging in females. These include caloric restriction; certain hormones, such as melatonin, ghrelin, and anti-Müllerian hormone (AMH); and energy-metabolism regulating compounds, such as metformin, rapamycin, and nicotinamide mononucleotide [27,73,74].
Emerging epidemiological evidence indicates that parity may be another factor delaying OR decline. A large cross-sectional analysis, encompassing 10 cohorts with over 3800 women aged 21–57 years, found significantly higher circulating levels of AMH—an established OR marker—in parous versus nulliparous women, particularly in those aged 40 and above [75]. Another cross-sectional study, which aimed to analyze both reproductive and lifestyle AMH determinants in 2320 women with a mean age of 37.3 (±9.2) years, found a positive association between higher parity and higher age-specific AMH levels [76]. Lastly, a smaller study involving 186 women aged 20–35 years observed higher plasma levels of AMH in multiparous compared to nulliparous women. Ovarian volume and antral follicle number—additional OR markers—were also elevated in the parous condition [77].
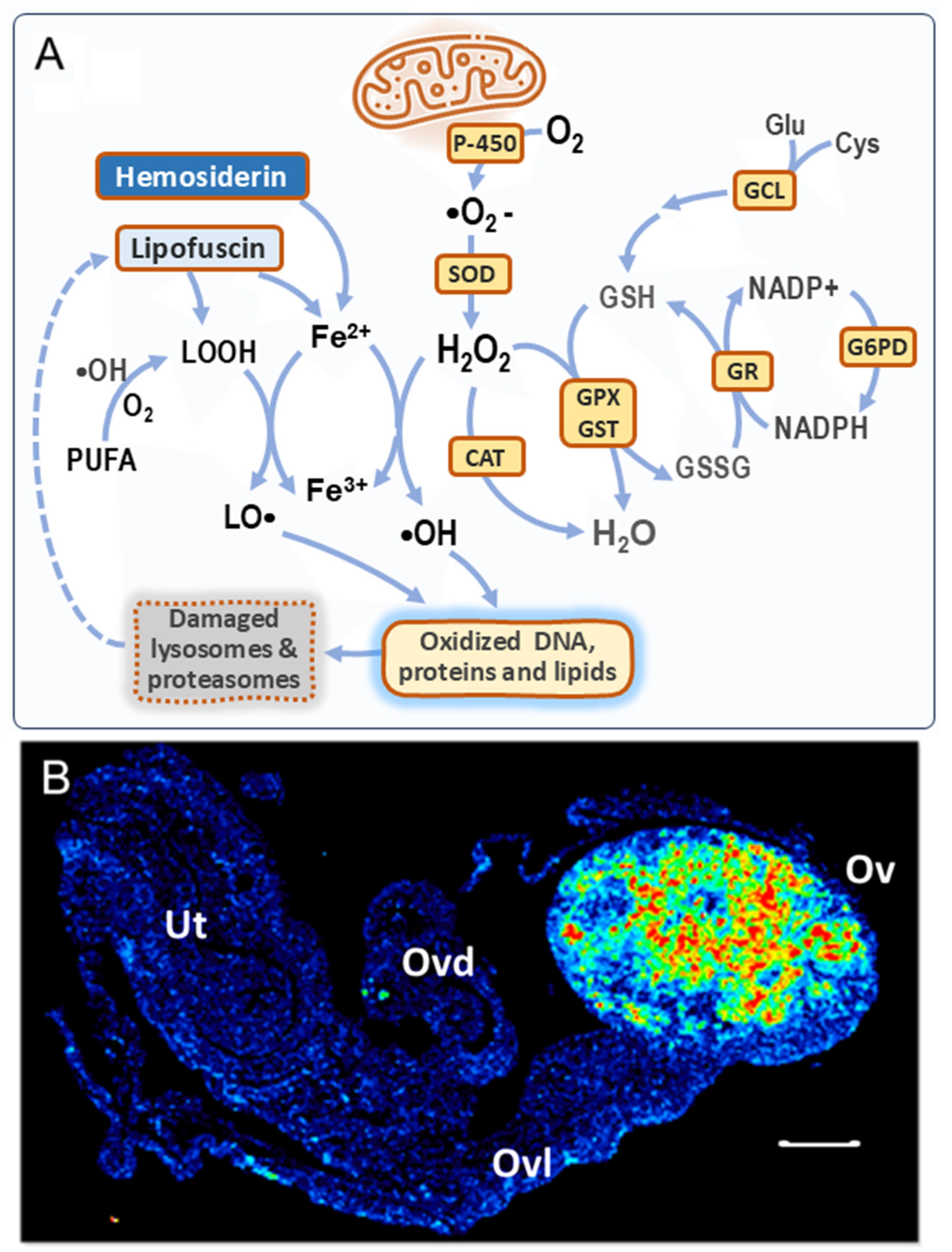
Consistent with the above epidemiological data, a higher residual follicle count was detected in multiparous compared to nulliparous mouse ovaries, both 21 months old (equivalent to post-menopause) [38]. Moreover, microarray analysis showed higher expression of 32 genes involved in follicle and oocyte homeostasis in the ovaries of aged multiparous mice compared to the aged nulliparous condition. Using a tool designed to explore single-cell RNA-seq data of a recent study on mouse ovarian aging [21], this 32-gene set was confirmed to be highly expressed in two primary types of granulosa cells (A, B) and oocytes and at lower levels in stroma B cells (Figure 2). Diverse granulosa cell types are constituents of follicles at different stages of development, including preantral, antral, mitotic, and atretic [21]. Twenty-one out of the thirty-two genes were expressed by granulosa cells A. In turn, from these 21, 11 showed predominant expression in mitotic, 3 in atretic, and 3 in antral granulosa cells [21]. The genes Fshr, Inhba, Foxo1, Nr5a2, Gdnf, and Efna5 are involved in hormone response, while the oocyte-expressed genes Nlrp5, Nlrp14, Padi6, Bmp15, Zp3, and Oas1d are the target genes of transcription factors involved in the maintenance and survival of primordial follicles [78], reflecting a follicle–oocyte interdependence. Given that parity seems to delay the natural OR decline, it sounds conceivable to attribute the reducing OC risk effect to a higher residual OR persisting in the mammalian parous ovary at post-menopausal age. The functionality of this residual OR in the senescent, post-reproductive ovary has not been characterized so far.
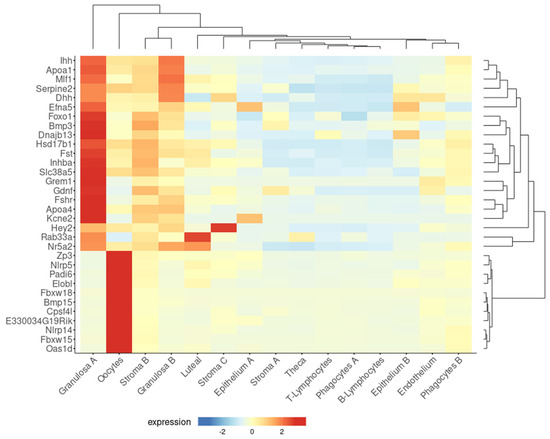
Figure 2.
A subset of genes upregulated in the aged multiparous ovary is predominantly expressed by granulosa cells and oocytes. The Shiny tool (https://omrf.shinyapps.io/ovarianagingscatlas/, last accessed on 24 January 2025) contains single-cell RNA-seq data for the 15 distinct cell types identified in the middle-aged mouse ovary [21] and was employed to predict which specific cell types predominantly express each of the 32 follicle/oocyte-related genes that were upregulated in the aged multiparous relative to the aged nulliparous mouse ovary [38]. Gene symbols were entered into the “bubbleplot-heatmap” tool and grouped by cluster names. Plot type settings were as follows: heatmap, scale gene expression, cluster genes, and cluster samples to visualize dendrograms in both axes.
To date, there is little information on how pregnancy might slow the natural OR decline. As with the presumed effects of transient ovulatory suppression [3,4,5] or high progesterone levels in decreasing OC risk [64], these pregnancy factors could somehow promote a long-term intraovarian homeostasis of follicles and oocytes. Clues may arise from the processes of CL formation, maintenance, and regression during pregnancy. The CL is considered a transient intraovarian “gland” originating from the remaining theca and mural granulosa cells of the mature Graafian follicle that differentiate into progesterone-producing luteal cells [79]. Limited amounts of progesterone are produced by granulosa cells during follicular development, but excess progesterone can inhibit follicular development at the primordial–primary transition [80]. During pregnancy, high progesterone levels derived from the CL could diffuse inside the ovary to inhibit the maturation of primordial follicles. Compared to the non-parous condition, this may result in a higher AMH level, i.e., a higher OR if measured postpartum, since serum AMH tends to decrease during gestation [81]. Upon delivery, the progesterone drop would allow resuming follicle development. A long-term epigenetic effect of progesterone on the OR cannot be ruled out, but this has not been experimentally addressed so far.
As described above, the association of the OR size with age and reproductive history comes from cross-sectional studies. To date, no longitudinal study has addressed the OR–parity association. Instead, some metabolic parameters and the exposure to certain environmental agents have been analyzed with this approach. A few examples include the association of AMH with serum lipid levels as a risk factor of cardiovascular disease, which was studied during 12 years of follow-up in a cohort of 1015 Iranian women [82]. In a recent study, the association of air pollutant exposure with longitudinal AMH levels was studied for over 20 years in 2574 women from the Netherlands [83]. Another study addressed the association of serum AMH levels with markers of bone density and turnover during ∼14 years of follow-up [84]. Future longitudinal studies in the above range of follow-up periods are expected to confirm the OR–parity association.
4. Follicle–OSE Interactions and FSHR Signaling
4.1. A Paracrine Crosstalk Between Follicles and the Ovarian Surface Epithelium
As discussed previously, the decline in the reproductive function of the ovary, specifically the depletion of germ cells and follicles, is intrinsically linked to an increased risk of ovarian cancer (OC). An additional characteristic of the aging process in the mammalian ovary is the proliferation of epithelial structures, such as clefts and inclusion cysts, which form from invaginations of the ovarian surface epithelium (OSE) into the ovarian stroma [85,86]. Compared to the OSE—which is actually a mixed or uncommitted epithelium—these cysts are enriched in epithelial markers, suggesting that a mesenchymal-to-epithelial transition likely led to their formation [87]. Importantly, clefts and inclusion cysts have been implicated in metaplasia and neoplastic transformation, thus being considered precursor lesions of OC [88].
Similar to the effects of aging, the ovaries of mice with genetically induced loss of germ cells and follicles typically develop OSE hyperplasia that invades the stroma, leading to the formation of tubular adenomas [89]. For instance, in Rev7 mutant mice, the complete loss of oocytes and follicles was concomitant with increased gonadotropin levels, ovarian DNA damage, and the development of tubulostromal adenomas [90]. Another illustrative case is the white spotting variant (Wv) mouse, which carries a spontaneous c-kit mutation resulting in early follicular loss. In fact, Wv ovaries exhibited an inverse relationship between the levels of alpha-inhibin (a granulosa cell marker) and cytokeratin-8 (an epithelial cell marker). Interestingly, alpha-inhibin was detected in stromal remnants of degenerated follicles, and even in a TP53 knockout background, a minimal number of ovarian follicles were able to suppress OSE hyperplasia in this model [91]. Further studies have suggested that the depletion of germ cells and follicles creates an ovarian environment conducive to the oncogenic transformation of OSE cells [92,93]. This inverse relationship between follicle content and OSE proliferation supports the existence of a growth-inhibiting paracrine factor produced by follicular granulosa cells [93]. This factor presumably diminishes or becomes absent upon follicular depletion due to age (or other aforementioned factors), thereby promoting epithelial proliferation.
A similar inverse association between the epithelial and follicle compartments was observed in the ovaries of the Fshr knockout (FORKO) mouse. This model exhibits a decreased OR and an early menopause-like phenotype, accompanied by OSE thickening, cytokeratin-positive stromal cysts, high expression of tight-junction proteins, and serous papillary cystadenomas [94,95]. Interestingly, a gene signature resembling that of the FORKO model was detected in a recent transcriptome analysis of the aged nulliparous mouse ovary [96]. In this reproductive condition, Fshr was downregulated with a concomitant overexpression of epithelial genes, such as the cytokeratins Krt7, Krt8, Krt18, and Krt23, and the cell junction genes Cldn3, Cldn11, Cldn15, F11r, and Ezr [96]. Moreover, when displayed in a cell-type context (Figure 3), a total of 47 genes upregulated in the aged nulliparous mouse ovary showed predominant expression in the epithelium-A and epithelium-B cell types [21], representing 42% (47/112) of all the genes upregulated in the nulliparous, high OC risk condition [96]. The cytokeratin and cell junction protein-coding genes, in addition to another gene subset including Rec8, Aldh1a2, Espn, Gng13, Cfi, Stard5, and Crygb, were also found coordinately dysregulated in the pre-neoplastic phase of the spontaneous transformation of cultured mouse OSE cells in a previous transcriptome study [97]. In summary, the inverse association between the sizes of the follicular and the OSE-derived epithelial components observed in the FORKO mouse ovary [94,95] also appears to occur in ovaries with divergent parity histories [38,96], thereby providing insight into the mechanistic bases of the protective effect of pregnancy on OC risk.
Figure 3.
Genes upregulated in the aged nulliparous mouse ovary are mostly of epithelial origin. As described in legend to Figure 2, the Shiny tool was used to track the expression of the 112 genes up-regulated in the aged nulliparous mouse ovary [96] across the 15 cell types identified in the middle-aged mouse ovary [21]. A total of 47 out of the 112 genes showed predominant expression in “epithelium A” and “epithelium B” cell types. Immune cells were omitted from the heatmap. Display parameters were those described in Figure 2.
Similar to the Fshr knockout mice, additional gene expression signatures from the mouse knockouts of Pten, Smad3, and Cdh1 were also significantly enriched in the transcriptomic profile of the aged nulliparous mouse ovary [96]. These genes are also linked to follicle homeostasis and have recognized roles as tumor suppressors. The downregulation of Pten in oocytes provokes a massive activation of follicles, resulting in OR depletion [70], whereas the transcription factor Smad3, a downstream member of TGF-beta signaling, has been found to inhibit mouse OSE proliferation [98].
4.2. FSHR Expression in the Aged Ovary: Does It Hold the Gonadotropin Theory of Ovarian Cancer?
Expressed by the granulosa cells, the Fshr gene codes for the FSH receptor, a G-protein-coupled receptor that plays a pivotal role in female reproductive function. In response to FSH, FSHR promotes follicular development and mediates estrogen synthesis in the ovaries during fertile age [99]. FSHR displays four alternative splicing isoforms, where FSHR1 acts through the canonical Gαs/cAMP/PKA pathway, and FSHR3 exhibits growth factor activity via MAPK–ERK signaling [100]. Consistent with age-dependent follicle depletion, Fshr expression decreases with age in human and mouse ovaries [21,38] but can still be detected in granulosa cells and other ovarian cell types, such as the OSE [63]. According to a single-cell RNA-seq analysis, Fshr expression in the middle-aged mouse ovary is predominantly follicular [21], i.e., highest in granulosa A cells, significantly lower in granulosa B and stroma B cells, and negligible in the epithelial cell types (Figure 2). The role of this low but detectable Fshr expression in the post-reproductive ovary remains largely unknown.
As described, follicular Fshr expression restricts OSE hyperplasia, an observation linking parity history to a higher ovarian reserve (OR) and reduced OC risk. It therefore seems plausible to consider a protective role of Fshr expression in residual follicles of the aged parous ovary resulting from former pregnancies [38]. However, this idea might appear to contradict the “gonadotropin theory” of OC, which is based on the well-known increase of FSH and LH in circulation at menopause and the growth-promoting, migratory, and invasive effects of these gonadotropins on OC cells [101]. Such cancer-promoting actions are mediated by FSH signaling through the FSHR3 variant, which is predominantly expressed by the fertile-age OSE and in OC cells [100].
Nevertheless, both earlier and recent evidence challenge the gonadotropin theory. Firstly, FSH is subject to age-dependent post-translational modifications in the FSHβ subunit that modulate its action. In younger, ovulating women, the partially/hypo-glycosylated FSH21/18 form predominates, while the hyper-glycosylated FSH24 form increases with aging and is predominant in peri- and post-menopause [102]. The binding of FSH24 to the FSHR delays the dissociation of FSHR oligomers into monomers [103] and reduces the proliferative response of follicles [104]. Besides this age-altered action of FSH on granulosa cells, a study with OSE cells from healthy donors reported a growth inhibitory effect of FSH in postmenopausal OSE samples [105]. Additionally, gonadotropins, including FSH, have been used as ovulation-inducing drugs in fertility treatments, but their possible effects on OC risk are controversial. Earlier studies claimed an increased OC risk, but a recent meta-analysis indicated no increased risk of invasive OC regardless of fertility drug use. A specific increased risk of borderline OC was found only in nulliparous women [106]. Another literature review concluded that mostly observational studies have suggested a link between fertility drugs and various cancers, but a significant association is lacking due to limitations in sample sizes, adequate follow-up, and uncontrolled confounding factors in those studies [107].
Secondly, epidemiological analyses linking endogenous FSH levels and OC risk have provided conflicting results. While some reports suggest that high FSH levels are significantly associated with increased OC risk in pre- and post-menopausal women [108], other studies fail to confirm a direct correlation between FSH levels and OC risk. Among the latter, a nested case-control study of 88 patients with primary invasive epithelial OC compared to 168 matched, cancer-free control women showed that FSH levels were not significantly different between women who were subsequently diagnosed with OC and control women. Moreover, when FSH levels were ranked in tertiles (low, medium, high), women in the highest tertile did not have a higher OC risk relative to the lowest tertile [109]. Another nested case-control study in 67 epithelial OC patients showed that high prediagnostic FSH levels were associated with lower OC risk. In this case, FSH levels were ranked in tertiles (low, medium, or high) according to the distribution of FSH levels among control women [110]. A recent analysis in 370 premenopausal women indicated that higher FSH was directly associated with an increased risk of type-II OC but inversely associated with type-I OC [111].
Part of the reason for conflicting data regarding the role of the FSH/FSHR axis on OC risk can be context-dependent. While hormones such as FSH can regulate repair, differentiation, and stress responses to maintain the homeostasis of normal ovarian tissue, in the context of OC and other endocrine-related malignancies, FSH can play a role as a tumor promoter by influencing OC onset, progression, and metastasis. As mentioned above, this OC promoter action of FSH can be mediated by the alternative FSHR3 variant [100]. In addition, FSHR has been proposed as a therapeutic target [112], but, again, this contradicts a recent report indicating that Fshr expression seems to act as a good prognostic factor in OC [113]. Moreover, high FSHR expression in tumor cells and tumor vasculature was associated with a longer progression-free survival and overall survival. Conversely, low FSHR protein expression was associated with shorter survival parameters in high-grade OC, while blockage of FSHR expression increased the invasive ability of OC cell lines OVCAR-3 cells and COV362 [113]. The latter findings might have therapeutic applications. Small molecules such as Org41841, a type of thienopyr(im)idine that increases human FSHR expression in the plasma membrane [114], could be tested as a therapeutic co-adjuvant in OC patients. Such approaches would be worth attempting in the future.
4.3. A Proposed Role for FSHR Signaling in Protection Against OC in the Aged Parous Ovary
Fifteen years ago, Huhtaniemi conducted a critical analysis of the gonadotropin effect on carcinogenesis. His primary conclusion was that the “gonadotropin theory” was predominantly based on in vitro studies and that “the clinical and experimental evidence in vivo was fragmentary, weak, and controversial” [115]. Much of the data reviewed here from various sources confirm that a convincing and definitive link between FSH and increased ovarian cancer (OC) risk is still lacking. In contrast, a significant body of novel evidence discussed herein suggests that FSHR expression in the aged ovary is dependent on a higher residual ovarian reserve (OR) resulting from past pregnancies compared to the aged nulliparous condition. Importantly, this higher FSHR expression in parous women could account for their observed lower OC risk.
Then how could these remnant follicles contribute to protecting the aged ovary? Two possible, non-exclusive scenarios can be proposed. The first was introduced in Section 4.1 above: the granulosa cells (and oocytes) from residual follicles could secrete a paracrine factor that diffuses inside the ovarian stroma to suppress the OSE proliferation of normal, pre-neoplastic, or inclusion cysts [93]. Inhibin-alpha and TGF-beta have been proposed as candidates for this role [91,92,93], and a recent work shows that AMH secreted by ovarian follicles inhibits the proliferation of mouse OSE cells [116]. These proteins belong to the TGF-beta/BMP superfamily, a class of growth factors implicated in the formation, assembly, and activation of primordial follicles [117]. In coherence with this idea, the TGF-beta/BMP family members Inhba, Bmp3, Fst, and Grem1 were among the overexpressed genes in the aged multiparous mouse ovary [38]. Inhba codes for the inhibin subunit beta A, a monomer of the dimeric protein complexes activin-A, activin-AB, and inhibin-A. As their names suggest, the systemic action of these dimers is to either activate or inhibit pituitary FSH secretion, respectively. During aging and menopause, circulating inhibin-A (inhibin-alpha) levels decrease along with estrogens [118], while activin-A increases parallel to the FSH rise [119]. In human and mouse ovaries of fertile age, activins promote the assembly of primordial follicles, thereby influencing the size of the primordial follicle pool [120]. Thus, the concomitant expression of the TGF-beta/BMP antagonists, follistatin (Fst) and gremlin-1 (Grem1), would be part of a mechanism to counteract the growth-promoting effect of activins, a role proposed in human postmenopausal ovaries [58]. Besides the mentioned TGF-beta/BMP members, the genes Ihh, Hey2, Efna5, Serpine2, and Zp3 were also overexpressed in the aged multiparous mouse ovary compared to its nulliparous counterpart [38]. According to the Gene Ontology (GO) database, these genes are also involved in growth regulation.
The ovarian FSH/FSHR axis offers a second potential explanation for the role of remnant follicles in reducing ovarian cancer (OC) risk. We hypothesize that FSHR signaling plays a protective role against OS in the parous ovary in a manner similar to how it promotes the survival of growing follicles during folliculogenesis. In this process, most of the recruited follicles undergo atresia due to OS, with the exception of the dominant follicle, which maintains a sufficient GSH level to reach the antral state. This antioxidant protection is dependent on FSH/FSHR signaling in granulosa cells [11,15,16]. Importantly, as androgens predominate in the aged ovary [56,58,59] and prevent ovulation, such an active FSH/FSHR axis in the parous condition would maintain granulosa cells of residual follicles in a quiescent state with an adequate GSH level. In fact, the postmenopausal ovary has been regarded as an androgen-producing and gonadotropin-driven organ [121], a notion implying functional FSH/FSHR signaling and supported by the observation that androgens like dehydroepiandrosterone enhance follicular viability and responsiveness in an animal model of menopause [122]. Thus, a key aspect of this hypothetical scenario is how GSH levels are sustained by the remnant follicles of the aged parous ovary to counteract age-dependent OS. In this regard, an interesting question would be to evaluate if the menopause-predominant hyperglycosylated FSH, which has a reduced proliferative effect [104], is still capable of maintaining a sufficient GSH level. High expression and activity of GCL in follicular granulosa cells determines increased synthesis of GSH [123]; therefore, GCL activity could serve as a readout of FSH functionality and GSH levels.
Further clues supporting the link between parity history and GSH levels come from a recent study suggesting elevated expression of the glutathione transferases Gstm6 and Gsta3 and glutathione peroxidase Gpx3 in the aged nulliparous mouse ovary [96]. This represents a high-risk OC condition with reduced Fshr expression, analogous to the FORKO model. GSH acts as a substrate for GPX and GST enzymes, which detoxify H2O2 and xenobiotic compounds (Figure 1A). Assuming a positive correlation between transcript and GPX/GST enzyme levels, excessive H2O2 and lipid hydroperoxides (LOOH) would lead to GSH depletion in the aged nulliparous condition. Consequently, low GSH levels would result in a reduced GSH/GSSG ratio, a typical marker of OS and an additional readout of FSHR functionality. Other redox-related genes overexpressed in the aged nulliparous ovary were alcohol dehydrogenase (Adh7) and the aldehyde dehydrogenases Aldh1a2 and Aldh1a7 [96], which detoxify reactive aldehydes derived from lipid peroxides [124] (i.e., LOOH), as also depicted in Figure 1A. Finally, a behavioral model of ovariectomized (menopause-like) Fshr knockout mice (Fshr−/−) resulted in severe OS with a low GSH/GSSG ratio in the whole brain of these animals [125]. Although a different organ and disease context, this finding resembles and supports the hypothesized mechanism that we propose to occur in the parous ovary.
5. Concluding Remarks and Perspectives
This review compiles information from diverse sources regarding reproductive history as a major OC risk factor and the biological processes underlying this effect. Multiple lines of evidence are presented in the context of age-related ovarian decline and the potential protective action of parity. The text emphasizes the complex interactions among OS, aging, and reproductive history in modulating OC risk. Though full-term pregnancies and nulliparity significantly influence the risk, the precise causes of OC remain unclear. The transition from fertile age to menopause involves significant hormonal changes, notably in the gonadotropins FSH and LH. These shifts impact ovarian function and are associated with systemic metabolic and inflammatory changes that contribute to both ovarian aging and increased cancer susceptibility.
OS is a key factor in normal ovarian function, with ROS playing a central role in processes such as ovulation, follicle maturation, and luteal regression. While basal levels of ROS are necessary for certain cellular functions, their overproduction leads to OS and subsequent DNA damage, thereby increasing the risk of OC. In the aging ovary, OS is directly related to the OR decline, the follicle depletion, and the increase of both reproductive aging and cancer susceptibility. Environmental factors, such as smoking, obesity, and exposure to toxicants, can further exacerbate OS, promoting ovarian aging and, in some cases, leading to premature ovarian failure. Aged ovarian cells exhibit impaired DNA repair mechanisms, increasing susceptibility to mutations and genomic instability, which are hallmarks of carcinogenesis. Senescence in the aged ovary indicates a cellular state that could either inhibit or promote tumorigenesis, with the precise role of senescence in cancer initiation remaining context-dependent.
As previously mentioned, cumulative evidence suggests that pregnancy, oral contraceptive use, and breastfeeding—conditions that temporarily suppress ovulation—are significantly associated with a reduced risk of OC later in menopause. Parity, specifically, reduces OC risk proportionally to the number of full-term pregnancies. This protective effect is likely attributable to the hormonal changes during pregnancy, particularly elevated progesterone levels, and their influence on the reproductive tract, especially the ovary. A less explored effect is the influence of parity on delaying the natural decay of the OR, meaning the preservation of a greater number of primordial follicles. Indeed, women who have given birth tend to have higher AMH levels, a well-established OR marker, particularly in women over 40 years of age. The missing, but key, link between ovarian progesterone exposure and the long-term improved follicle homeostasis is largely unknown and requires further research. In animal models, parity is linked to a higher number of residual follicles in the aging ovary, with gene expression profiles suggesting preserved follicle and oocyte homeostasis (Figure 2). These residual follicles may confer protection against OC by preventing OSE proliferation via paracrine signaling. On the other hand, the aged nulliparous ovary displays increased epithelial structures (Figure 3 and Figure 4) and multiple signs of cell stress beyond those induced by aging. Between these divergent risk conditions, we propose that FSHR expression in the aged parous ovary might play a critical role in maintaining follicle viability and regulating OS, thereby reducing OC risk. A scheme compiling all the above-described evidence and proposed hypotheses is shown in Figure 4.
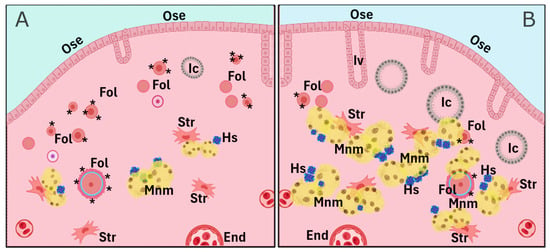
Figure 4.
Comparative scheme of parity-related cell and tissue markers depicting the long-term effect of parity on the aged ovary. The parous condition (A) contains a higher number of remnant follicles (Fol) and lower counts of lipofuscin-containing multinucleated macrophages (Mnm), hemosiderin (Hs) aggregates, epithelial invaginations (Iv), and inclusion cysts (Ic), while the nulliparous condition (B) shows a lower follicle count and increased numbers of Iv, Ic, Mnm, and Hs. Further cell types and structures include the ovarian surface epithelium (Ose), stromal cells (Str), and endothelial cells (End). Asterisks in follicles depict the FSH/FSHR axis. Figure was created using a generic template from BioRender (www.biorender.com, last accessed 3 February 2025).
In summary, further research is needed to better understand the molecular mechanisms underlying the effects of parity, hormonal changes, inflammation, and FSHR signaling in aging ovaries. Such knowledge could contribute to designing preventive strategies and therapeutic interventions for OC. Given the decreasing fertility trends coupled with increased life expectancy in developed countries, this research becomes particularly relevant as increasing rates of postmenopausal health issues, including OC, would be expected.
Author Contributions
Conceptualization, U.U.; formal analysis, U.U. and E.A.C.; investigation, U.U., A.M. and E.A.C.; writing and original draft preparation, U.U.; review and editing, U.U., A.M. and E.A.C.; project administration and funding acquisition, E.A.C. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by ANID, FONDAP 152220002 (CECAN).
Data Availability Statement
No new data was created. All data presented is available from their respective sources, including links and references.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| AMH | Anti-Mullerian hormone |
| BER | Base excision repair |
| CAT | Catalase |
| CL | Corpus luteum |
| DSB | Double-strand break |
| FORKO | FSHR knockout |
| FSH | Follicle-stimulating hormone |
| FSHR | Follicle-stimulating hormone receptor |
| GLC | Glutamate–cysteine ligase educed glutathione |
| GPX | Glutathione peroxidase |
| GSH | Reduced glutathione |
| GSR | Glutathione disulphide reductase |
| GSS | Glutathione synthetase |
| GSSG | Glutathione disulphide |
| GST | Glutathione transferase |
| HPO | Hypothalamic pituitary ovarian |
| LH | Luteinizing hormone |
| LHR | Luteinizing hormone receptor |
| LOOH | Lipid peroxide |
| MAPK-ERK | Mitogen-activated protein kinase–extracellular signal-regulated kinase |
| MNM | Multinucleated macrophage |
| OC | Ovarian cancer |
| OR | Ovarian reserve |
| OS | Oxidative stress |
| OSE | Ovarian surface epithelium |
| PUFA | Polyunsaturated fatty acid |
| PFS | Progression-free survival |
| ROS | Reactive oxygen species |
| SOD | Superoxide dismutase |
| TGF-beta/BMP | Transforming growth factor beta/bone morphogenetic protein |
| Wv | White spotting variant |
References
- Webb, P.M.; Jordan, S.J. Global epidemiology of epithelial ovarian cancer. Nat. Rev. Clin. Oncol. 2024, 21, 389–400. [Google Scholar] [CrossRef] [PubMed]
- Hunn, J.; Rodriguez, G.C. Ovarian cancer: Etiology, risk factors, and epidemiology. Clin. Obstet. Gynecol. 2012, 55, 3–23. [Google Scholar] [CrossRef]
- Toufakis, V.; Katuwal, S.; Pukkala, E.; Tapanainen, J.S. Impact of parity on the incidence of ovarian cancer subtypes: A population-based case-control study. Acta Oncol. 2021, 60, 850–855. [Google Scholar] [CrossRef] [PubMed]
- Havrilesky, L.J.; Moorman, P.G.; Lowery, W.J.; Gierisch, J.M.; Coeytaux, R.R.; Urrutia, R.P.; Dinan, M.; McBroom, A.J.; Hasselblad, V.; Sanders, G.D.; et al. Oral contraceptive pills as primary prevention for ovarian cancer: A systematic review and meta-analysis. Obstet. Gynecol. 2013, 122, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.K.; Ma, S.H.; Choi, J.Y.; Hwang, Y.; Ahn, C.; Kim, B.G.; Kim, Y.M.; Kim, J.W.; Kang, S.; Kim, J.; et al. The Effect of Breastfeeding Duration and Parity on the Risk of Epithelial Ovarian Cancer: A Systematic Review and Meta-analysis. J. Prev. Med. Public Health 2016, 49, 349–366. [Google Scholar] [CrossRef]
- Smits, M.A.J.; Schomakers, B.V.; van Weeghel, M.; Wever, E.J.M.; Wüst, R.C.I.; Dijk, F.; Janssens, G.E.; Goddijn, M.; Mastenbroek, S.; Houtkooper, R.H.; et al. Human ovarian aging is characterized by oxidative damage and mitochondrial dysfunction. Hum. Reprod. 2023, 38, 2208–2220. [Google Scholar] [CrossRef]
- Yan, F.; Zhao, Q.; Li, Y.; Zheng, Z.; Kong, X.; Shu, C.; Liu, Y.; Shi, Y. The role of oxidative stress in ovarian aging: A review. J. Ovarian Res. 2022, 15, 100. [Google Scholar] [CrossRef]
- Hense, J.D.; Isola, J.V.V.; Garcia, D.N.; Magalhães, L.S.; Masternak, M.M.; Stout, M.B.; Schneider, A. The role of cellular senescence in ovarian aging. NPJ Aging 2024, 10, 35. [Google Scholar] [CrossRef]
- Beaujouan, É.; Reimondos, A.; Gray, E.; Evans, A.; Sobotka, T. Declining realisation of reproductive intentions with age. Hum. Reprod. 2019, 34, 1906–1914. [Google Scholar] [CrossRef]
- Das, A.; Roychoudhury, S. Reactive Oxygen Species in the Reproductive System: Sources and Physiological Roles. Adv. Exp. Med. Biol. 2022, 1358, 9–40. [Google Scholar]
- Wang, S.; He, G.; Chen, M.; Zuo, T.; Xu, W.; Liu, X. The Role of Antioxidant Enzymes in the Ovaries. Oxid. Med. Cell. Longev. 2017, 2017, 4371714. [Google Scholar] [CrossRef] [PubMed]
- Shkolnik, K.; Tadmor, A.; Ben-Dor, S.; Nevo, N.; Galiani, D.; Dekel, N. Reactive oxygen species are indispensable in ovulation. Proc. Natl. Acad. Sci. USA 2011, 108, 1462–1467. [Google Scholar] [CrossRef]
- Murdoch, W.J.; Martinchick, J.F. Oxidative damage to DNA of ovarian surface epithelial cells affected by ovulation: Carcinogenic implication and chemoprevention. Exp. Biol. Med. 2004, 229, 546–552. [Google Scholar] [CrossRef]
- Matsuda, F.; Inoue, N.; Manabe, N.; Ohkura, S. Follicular growth and atresia in mammalian ovaries: Regulation by survival and death of granulosa cells. J. Reprod. Dev. 2012, 58, 44–50. [Google Scholar] [CrossRef]
- Shen, M.; Jiang, Y.; Guan, Z.; Cao, Y.; Li, L.; Liu, H.; Sun, S.C. Protective mechanism of FSH against oxidative damage in mouse ovarian granulosa cells by repressing autophagy. Autophagy 2017, 13, 1364–1385. [Google Scholar] [CrossRef]
- Tsai-Turton, M.; Luderer, U. Opposing effects of glutathione depletion and follicle-stimulating hormone on reactive oxygen species and apoptosis in cultured preovulatory rat follicles. Endocrinology 2006, 147, 1224–1236. [Google Scholar] [CrossRef] [PubMed]
- Al-Gubory, K.H.; Garrel, C.; Faure, P.; Sugino, N. Roles of antioxidant enzymes in corpus luteum rescue from reactive oxygen species-induced oxidative stress. Reprod. Biomed. Online 2012, 25, 551–560. [Google Scholar] [CrossRef] [PubMed]
- Lawrenson, K.; Grun, B.; Benjamin, E.; Jacobs, I.J.; Dafou, D.; Gayther, S.A. Senescent fibroblasts promote neoplastic transformation of partially transformed ovarian epithelial cells in a three-dimensional model of early stage ovarian cancer. Neoplasia 2010, 12, 317–325. [Google Scholar] [CrossRef]
- Jian, J.; Pelle, E.; Huang, X. Iron and menopause: Does increased iron affect the health of postmenopausal women? Antioxid. Redox Signal. 2009, 11, 2939–2943. [Google Scholar] [CrossRef]
- Urzúa, U.; Chacon, C.; Espinoza, R.; Martínez, S.; Hernandez, N. Parity-Dependent Hemosiderin and Lipofuscin Accumulation in the Reproductively Aged Mouse Ovary. Anal. Cell. Pathol. 2018, 2018, 1289103. [Google Scholar] [CrossRef]
- Isola, J.V.V.; Ocañas, S.R.; Hubbart, C.R.; Ko, S.; Mondal, S.A.; Hense, J.D.; Carter, H.N.C.; Schneider, A.; Kovats, S.; Alberola-Ila, J.; et al. A single-cell atlas of the aging mouse ovary. Nat. Aging 2024, 4, 145–162. [Google Scholar] [CrossRef]
- Finkel, T.; Holbrook, N.J. Oxidants, oxidative stress and the biology of ageing. Nature 2000, 408, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Calaf, G.M.; Urzua, U.; Termini, L.; Aguayo, F. Oxidative stress in female cancers. Oncotarget 2018, 9, 23824–23842. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.; Luderer, U. Oxidative damage increases and antioxidant gene expression decreases with aging in the mouse ovary. Biol. Reprod. 2011, 84, 775–782. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Hernández, V.; Montaño, L.M.; Caldelas, I.; Marmolejo-Valencia, A. A High-Fat and High-Carbohydrate Diet Promotes Reminiscent Hallmarks of an Aging Ovary in the Rabbit Model. Biomedicines 2022, 10, 3068. [Google Scholar] [CrossRef]
- Zhou, J.; Lin, L.; Liu, L.; Wang, J.; Xia, G.; Wang, C. The transcriptome reveals the molecular regulatory network of primordial follicle depletion in obese mice. Fertil. Steril. 2023, 120, 899–910. [Google Scholar] [CrossRef]
- Markowska, A.; Antoszczak, M.; Mrkowska, J.; Huczyński, A. Gynotoxic Effects of Chemotherapy and Potential Protective Mechanisms. Cancers 2024, 16, 2288. [Google Scholar] [CrossRef]
- Nakamura, B.N.; Fielder, T.J.; Hoang, Y.D.; Lim, J.; McConnachie, L.A.; Kavanagh, T.J.; Luderer, U. Lack of maternal glutamate cysteine ligase modifier subunit (Gclm) decreases oocyte glutathione concentrations and disrupts preimplantation development in mice. Endocrinology 2011, 152, 2806–2815. [Google Scholar] [CrossRef]
- Lim, J.; Nakamura, B.N.; Mohar, I.; Kavanagh, T.J.; Luderer, U. Glutamate cysteine ligase modifier subunit (Gclm) null mice have increased ovarian oxidative stress and accelerated age-related ovarian failure. Endocrinology 2015, 156, en20151206. [Google Scholar] [CrossRef]
- Lim, J.; Ortiz, L.; Nakamura, B.N.; Hoang, Y.D.; Banuelos, J.; Flores, V.N.; Chan, J.Y.; Luderer, U. Effects of deletion of the transcription factor Nrf2 and benzo [a]pyrene treatment on ovarian follicles and ovarian surface epithelial cells in mice. Reprod. Toxicol. 2015, 58, 24–32. [Google Scholar] [CrossRef]
- Gao, X.; Wang, B.; Huang, Y.; Wu, M.; Li, Y.; Li, Y.; Zhu, X.; Wu, M. Role of the Nrf2 Signaling Pathway in Ovarian Aging: Potential Mechanism and Protective Strategies. Int. J. Mol. Sci. 2023, 24, 13327. [Google Scholar] [CrossRef]
- Berquist, B.R.; Wilson, D.M., 3rd. Pathways for repairing and tolerating the spectrum of oxidative DNA lesions. Cancer Lett. 2012, 327, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhang, W.; Chen, Y.; Guo, W.; Zhang, J.; Tang, H.; Xu, Z.; Zhang, H.; Tao, Y.; Wang, F.; et al. Impaired DNA double-strand break repair contributes to the age-associated rise of genomic instability in humans. Cell Death Differ. 2016, 23, 1765–1777. [Google Scholar] [CrossRef] [PubMed]
- Sedelnikova, O.A.; Horikawa, I.; Redon, C.; Nakamura, A.; Zimonjic, D.B.; Popescu, N.C.; Bonner, W.M. Delayed kinetics of DNA double-strand break processing in normal and pathological aging. Aging Cell 2008, 7, 89–100. [Google Scholar] [CrossRef]
- Leandro, G.S.; Sykora, P.; Bohr, V.A. The impact of base excision DNA repair in age-related neurodegenerative diseases. Mutat. Res. 2015, 776, 31–39. [Google Scholar] [CrossRef]
- Titus, S.; Li, F.; Stobezki, R.; Akula, K.; Unsal, E.; Jeong, K.; Dickler, M.; Robson, M.; Moy, F.; Goswami, S.; et al. Impairment of BRCA1-related DNA double-strand break repair leads to ovarian aging in mice and humans. Sci. Transl. Med. 2013, 5, 172ra21. [Google Scholar] [CrossRef] [PubMed]
- Govindaraj, V.; Keralapura Basavaraju, R.; Rao, A.J. Changes in the expression of DNA double strand break repair genes in primordial follicles from immature and aged rats. Reprod. Biomed. Online 2015, 30, 303–310. [Google Scholar] [CrossRef]
- Urzúa, U.; Chacón, C.; Norambuena, M.; Lizama, L.; Sarmiento, S.; Asaki, E.; Powell, J.I.; Ampuero, S. The Ovarian Transcriptome of Reproductively Aged Multiparous Mice: Candidate Genes for Ovarian Cancer Protection. Biomolecules 2020, 10, 113. [Google Scholar] [CrossRef]
- Li, X.; Heyer, W.D. Homologous recombination in DNA repair and DNA damage tolerance. Cell Res. 2008, 18, 99–113. [Google Scholar] [CrossRef]
- Panagopoulou, M.; Panou, T.; Gkountakos, A.; Tarapatzi, G.; Karaglani, M.; Tsamardinos, I.; Chatzaki, E. BRCA1 & BRCA2 methylation as a prognostic and predictive biomarker in cancer: Implementation in liquid biopsy in the era of precision medicine. Clin. Epigenetics 2024, 16, 178. [Google Scholar]
- Wu, C.; Chen, D.; Stout, M.B.; Wu, M.; Wang, S. Hallmarks of ovarian aging. Trends Endocrinol. Metab. 2025, 36, 418–439. [Google Scholar] [CrossRef] [PubMed]
- McHugh, D.; Gil, J. Senescence and aging: Causes, consequences, and therapeutic avenues. J. Cell Biol. 2018, 217, 65–77. [Google Scholar] [CrossRef]
- Jung, T.; Bader, N.; Grune, T. Lipofuscin: Formation, distribution, and metabolic consequences. Ann. N. Y. Acad. Sci. 2007, 1119, 97–111. [Google Scholar] [CrossRef]
- Asano, Y. Age-related accumulation of non-heme ferric and ferrous iron in mouse ovarian stroma visualized by sensitive non-heme iron histochemistry. J. Histochem. Cytochem. 2012, 60, 229–242. [Google Scholar] [CrossRef] [PubMed]
- Watson, J.M.; Marion, S.L.; Rice, P.F.; Utzinger, U.; Brewer, M.A.; Hoyer, P.B.; Barton, J.K. Two-photon excited fluorescence imaging of endogenous contrast in a mouse model of ovarian cancer. Lasers Surg. Med. 2013, 45, 155–166. [Google Scholar] [CrossRef]
- Koorts, A.M.; Viljoen, M. Ferritin and ferritin isoforms I: Structure-function relationships, synthesis, degradation and secretion. Arch. Physiol. Biochem. 2007, 113, 30–54. [Google Scholar] [CrossRef] [PubMed]
- Theil, E.C. Ferritin iron minerals are chelator targets, antioxidants, and coated, dietary iron. Ann. N. Y. Acad. Sci. 2010, 1202, 197–204. [Google Scholar] [CrossRef]
- Humar, R.; Schaer, D.J.; Vallelian, F. Erythrophagocytes in hemolytic anemia, wound healing, and cancer. Trends Mol. Med. 2022, 28, 906–915. [Google Scholar] [CrossRef]
- Chifman, J.; Laubenbacher, R.; Torti, S.V. A systems biology approach to iron metabolism. Adv. Exp. Med. Biol. 2014, 844, 201–225. [Google Scholar]
- Ozaki, M.; Kawabata, T.; Awai, M. Iron release from haemosiderin and production of iron-catalysed hydroxyl radicals in vitro. Biochem. J. 1988, 250, 589–595. [Google Scholar] [CrossRef]
- Li, J.; Cao, F.; Yin, H.L.; Huang, Z.J.; Lin, Z.T.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, present and future. Cell Death Dis. 2020, 11, 88. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Wang, H.; Fang, J.; Jiang, R.; Kong, Y.; Zhang, T.; Yang, G.; Jin, H.; Shi, S.; Song, N.; et al. Ovarian aging-associated downregulation of GPX4 expression regulates ovarian follicular development by affecting granulosa cell functions and oocyte quality. FASEB J. 2025, 39, e70469. [Google Scholar] [CrossRef]
- Schröder, S.K.; Krizanac, M.; Kim, P.; Kessel, J.C.; Weiskirchen, R. Ovaries of estrogen receptor 1-deficient mice show iron overload and signs of aging. Front. Endocrinol. 2024, 15, 1325386. [Google Scholar] [CrossRef] [PubMed]
- Pelucchi, S.; Mariani, R.; Salvioni, A.; Bonfadini, S.; Riva, A.; Bertola, F.; Trombini, P.; Piperno, A. Novel mutations of the ferroportin gene (SLC40A1): Analysis of 56 consecutive patients with unexplained iron overload. Clin. Genet. 2008, 73, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Höhn, A.; Jung, T.; Grimm, S.; Grune, T. Lipofuscin-bound iron is a major intracellular source of oxidants: Role in senescent cells. Free Radic. Biol. Med. 2010, 48, 1100–1108. [Google Scholar] [CrossRef]
- Havelock, J.C.; Rainey, W.E.; Bradshaw, K.D.; Carr, B.R. The post-menopausal ovary displays a unique pattern of steroidogenic enzyme expression. Hum. Reprod. 2006, 21, 309–317. [Google Scholar] [CrossRef]
- Longcope, C. Endocrine function of the postmenopausal ovary. J. Soc. Gynecol. Investig. 2001, 8 (Suppl. S1), S67–S68. [Google Scholar] [CrossRef]
- Jabara, S.; Christenson, L.K.; Wang, C.Y.; McAllister, J.M.; Javitt, N.B.; Dunaif, A.; Strauss, J.F., 3rd. Stromal cells of the human postmenopausal ovary display a distinctive biochemical and molecular phenotype. J. Clin. Endocrinol. Metab. 2003, 88, 484–492. [Google Scholar] [CrossRef]
- Brodowski, J.; Brodowska, A.; Laszczynska, M.; Chlubek, D.; Starczewski, A. Hormone concentrations in the homogenates of ovarian tissue and blood serum in postmenopausal women not using hormone therapy. Gynecol. Endocrinol. 2012, 28, 396–399. [Google Scholar] [CrossRef]
- Mendez, C.; Morales-Vasquez, F.; Perez-Montiel, D.; Gomora, M.J.; Espinola-Zetina, C.; Hernandez-Martinez, A.; Lopez-Basave, H.; Pedernera, E. Estrogen and androgen receptor expression in surface epithelium and inclusion cyst in the ovary of premenopausal and postmenopausal women. J. Ovarian Res. 2013, 6, 85. [Google Scholar] [CrossRef]
- Inkster, S.E.; Brodie, A.M. Expression of aromatase cytochrome P-450 in premenopausal and postmenopausal human ovaries: An immunocytochemical study. J. Clin. Endocrinol. Metab. 1991, 73, 717–726. [Google Scholar] [CrossRef]
- Brodowska, A.; Brodowski, J.; Laszczyńska, M.; Słuczanowska-Głąbowska, S.; Rumianowski, B.; Rotter, I.; Starczewski, A.; Ratajczak, M.Z. Immunoexpression of aromatase cytochrome P450 and 17β-hydroxysteroid dehydrogenase in women’s ovaries after menopause. J. Ovarian Res. 2014, 7, 52. [Google Scholar] [CrossRef] [PubMed]
- Brodowska, A.; Laszczyńska, M.; Brodowski, J.; Masiuk, M.; Starczewski, A. Analysis of pituitary gonadotropin concentration in blood serum and immunolocalization and immunoexpression of follicle stimulating hormone and luteinising hormone receptors in ovaries of postmenopausal women. Histol. Histopathol. 2012, 27, 241–248. [Google Scholar] [PubMed]
- Schindler, A.E. Benefits and risks of ovarian function and reproduction for cancer development and prevention. Gynecol. Endocrinol. 2011, 27, 1043–1047. [Google Scholar] [CrossRef] [PubMed]
- Lund, E.; Busund, L.R.; Holden, L. Curvilinear incidence models for parity in the entire fertility range for cancers of the breast, ovary, and endometrium: A follow-up of the Norwegian 1960 Census. Int. J. Cancer 2025, 156, 2118–2126. [Google Scholar] [CrossRef]
- Russo, J.; Santucci-Pereira, J.; de Cicco, R.L.; Sheriff, F.; Russo, P.A.; Peri, S.; Slifker, M.; Ross, E.; Mello, M.L.S.; Vidal, B.C.; et al. Pregnancy-induced chromatin remodeling in the breast of postmenopausal women. Int. J. Cancer 2012, 131, 1059–1070. [Google Scholar] [CrossRef]
- Peri, S.; de Cicco, R.L.; Santucci-Pereira, J.; Slifker, M.; Ross, E.A.; Russo, I.H.; Russo, P.A.; Arslan, A.A.; Belitskaya-Lévy, I.; Zeleniuch-Jacquotte, A.; et al. Defining the genomic signature of the parous breast. BMC Med. Genom. 2012, 5, 46. [Google Scholar] [CrossRef]
- Dearth, R.K.; Delgado, D.A.; Hiney, J.K.; Pathiraja, T.; Oesterreich, S.; Medina, D.; Dees, W.L.; Lee, A.V. Parity-induced decrease in systemic growth hormone alters mammary gland signaling: A potential role in pregnancy protection from breast cancer. Cancer Prev. Res. 2010, 3, 312–321. [Google Scholar] [CrossRef]
- Findlay, J.K.; Hutt, K.J.; Hickey, M.; Anderson, R.A. How Is the Number of Primordial Follicles in the Ovarian Reserve Established? Biol. Reprod. 2015, 93, 111. [Google Scholar] [CrossRef]
- Zhao, Y.; Feng, H.; Zhang, Y.; Zhang, J.V.; Wang, X.; Liu, D.; Wang, T.; Li, R.H.W.; Ng, E.H.Y.; Yeung, W.S.B.; et al. Current Understandings of Core Pathways for the Activation of Mammalian Primordial Follicles. Cells 2021, 10, 1491. [Google Scholar] [CrossRef]
- Adhikari, D.; Liu, K. Molecular mechanisms underlying the activation of mammalian primordial follicles. Endocr. Rev. 2009, 30, 438–464. [Google Scholar] [CrossRef] [PubMed]
- Wallace, W.H.; Kelsey, T.W. Human ovarian reserve from conception to the menopause. PLoS ONE 2010, 5, e8772. [Google Scholar] [CrossRef]
- Veiga, G.B.; Zanini, B.M.; Garcia, D.N.; Hense, J.D.; Barreto, M.M.; Isola, J.V.V.; Mondadori, R.G.; Masternak, M.M.; Stout, M.B.; Schneider, A. Effects of calorie, protein, and branched chain amino acid restriction on ovarian aging in mice. Reprod. Biol. 2024, 24, 100856. [Google Scholar] [CrossRef]
- Schneider, A.; Saccon, T.D.; Garcia, D.N.; Zanini, B.M.; Isola, J.V.V.; Hense, J.D.; Alvarado-Rincón, J.A.; Cavalcante, M.B.; Mason, J.B.; Stout, M.B.; et al. The Interconnections Between Somatic and Ovarian Aging in Murine Models. J. Gerontol. A Biol. Sci. Med. Sci. 2021, 76, 1579–1586. [Google Scholar] [CrossRef]
- Clendenen, T.V.; Ge, W.; Koenig, K.L.; Afanasyeva, Y.; Agnoli, C.; Bertone-Johnson, E.; Brinton, L.A.; Darvishian, F.; Dorgan, J.F.; Eliassen, A.H.; et al. Breast Cancer Risk Factors and Circulating Anti-Müllerian Hormone Concentration in Healthy Premenopausal Women. J. Clin. Endocrinol. Metab. 2021, 106, e4542–e4553. [Google Scholar] [CrossRef]
- Dólleman, M.; Verschuren, W.M.; Eijkemans, M.J.; Dollé, M.E.; Jansen, E.H.; Broekmans, F.J.; van der Schouw, Y.T. Reproductive and lifestyle determinants of anti-Müllerian hormone in a large population-based study. J. Clin. Endocrinol. Metab. 2013, 98, 2106–2115. [Google Scholar] [CrossRef] [PubMed]
- Moini, A.; Hedayatshodeh, M.; Hosseini, R.; Rastad, H. Association between parity and ovarian reserve in reproductive age women. Eur. J. Obstet. Gynecol. Reprod. Biol. 2016, 207, 184–187. [Google Scholar] [CrossRef]
- Lim, E.J.; Choi, Y. Transcription factors in the maintenance and survival of primordial follicles. Clin. Exp. Reprod. Med. 2012, 39, 127–131. [Google Scholar] [CrossRef] [PubMed]
- Stocco, C.; Telleria, C.; Gibori, G. The molecular control of corpus luteum formation, function, and regression. Endocr. Rev. 2007, 28, 117–149. [Google Scholar] [CrossRef]
- Zhu, Q.; Li, Y.; Ma, J.; Ma, H.; Liang, X. Potential factors result in diminished ovarian reserve: A comprehensive review. J. Ovarian Res. 2023, 16, 208. [Google Scholar] [CrossRef]
- McCredie, S.; Ledger, W.; Venetis, C.A. Anti-Müllerian hormone kinetics in pregnancy and post-partum: A systematic review. Reprod. Biomed. Online 2017, 34, 522–533. [Google Scholar] [CrossRef] [PubMed]
- Tehrani, F.R.; Erfani, H.; Cheraghi, L.; Tohidi, M.; Azizi, F. Lipid profiles and ovarian reserve status: A longitudinal study. Hum. Reprod. 2014, 29, 2522–2529. [Google Scholar] [CrossRef] [PubMed]
- Zou, R.; Durkin, A.; de Kat, A.; van der Schouw, Y.T.; Hoek, G.; Verschuren, W.M.M.; Vermeulen, R.; Lenters, V. Air pollution and anti-Müllerian hormone: The Doetinchem Cohort Study. Environ. Int. 2025, 201, 109565. [Google Scholar] [CrossRef]
- Wang, S.; Yu, E.W.; Hivert, M.F.; Rifas-Shiman, S.L.; Shifren, J.L.; Kazemi, M.; Oken, E.; Chavarro, J.E. Associations of AMH in mid-reproductive years with bone mineral density and turnover markers in mid-life. J. Clin. Endocrinol. Metab. 2024, 110, dgae694. [Google Scholar] [CrossRef] [PubMed]
- Tan, O.L.; Hurst, P.R.; Fleming, J.S. Location of inclusion cysts in mouse ovaries in relation to age, pregnancy, and total ovulation number: Implications for ovarian cancer? J. Pathol. 2005, 205, 483–490. [Google Scholar] [CrossRef]
- Cai, K.Q.; Klein-Szanto, A.; Karthik, D.; Edelson, M.; Daly, M.B.; Ozols, R.F.; Lynch, H.T.; Godwin, A.K.; Xu, X.X. Age-dependent morphological alterations of human ovaries from populations with and without BRCA mutations. Gynecol. Oncol. 2006, 103, 719–728. [Google Scholar] [CrossRef]
- Okamoto, S.; Okamoto, A.; Nikaido, T.; Saito, M.; Takao, M.; Yanaihara, N.; Takakura, S.; Ochiai, K.; Tanaka, T. Mesenchymal to epithelial transition in the human ovarian surface epithelium focusing on inclusion cysts. Oncol. Rep. 2009, 21, 1209–1214. [Google Scholar] [CrossRef]
- Banet, N.; Kurman, R.J. Two types of ovarian cortical inclusion cysts: Proposed origin and possible role in ovarian serous carcinogenesis. Int. J. Gynecol. Pathol. 2015, 34, 3–8. [Google Scholar] [CrossRef]
- Vanderhyden, B.C. Loss of ovarian function and the risk of ovarian cancer. Cell Tissue Res. 2005, 322, 117–124. [Google Scholar] [CrossRef]
- Abbasi, A.; Khalaj, M.; Akiyama, K.; Mukai, Y.; Matsumoto, H.; Acosta, T.J.; Said, N.; Yoshida, M.; Kunieda, T. Lack of Rev7 function results in development of tubulostromal adenomas in mouse ovary. Mol. Cell. Endocrinol. 2015, 412, 19–25. [Google Scholar] [CrossRef]
- Cai, K.Q.; Wang, Y.; Smith, E.R.; Smedberg, J.L.; Yang, D.H.; Yang, W.L.; Xu, X.X. Global deletion of Trp53 reverts ovarian tumor phenotype of the germ cell-deficient white spotting variant (Wv) mice. Neoplasia 2015, 17, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.R.; Xu, X.X. Ovarian ageing, follicle depletion, and cancer: A hypothesis for the aetiology of epithelial ovarian cancer involving follicle depletion. Lancet Oncol. 2008, 9, 1108–1111. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Cai, K.Q.; Smith, E.R.; Yeasky, T.M.; Moore, R.; Ganjei-Azar, P.; Klein-Szanto, A.J.; Godwin, A.K.; Hamilton, T.C.; Xu, X.X. Follicle Depletion Provides a Permissive Environment for Ovarian Carcinogenesis. Mol. Cell. Biol. 2016, 36, 2418–2430. [Google Scholar] [CrossRef]
- Chen, X.; Aravindakshan, J.; Yang, Y.; Sairam, M.R. Early alterations in ovarian surface epithelial cells and induction of ovarian epithelial tumors triggered by loss of FSH receptor. Neoplasia 2007, 9, 521–531. [Google Scholar] [CrossRef] [PubMed]
- Aravindakshan, J.; Chen, X.; Sairam, M.R. Differential expression of claudin family proteins in mouse ovarian serous papillary epithelial adenoma in aging FSH receptor-deficient mutants. Neoplasia 2006, 8, 984–994. [Google Scholar] [CrossRef]
- Chacón, C.; Mounieres, C.; Ampuero, S.; Urzúa, U. Transcriptomic Analysis of the Aged Nulliparous Mouse Ovary Suggests a Stress State That Promotes Pro-Inflammatory Lipid Signaling and Epithelial Cell Enrichment. Int. J. Mol. Sci. 2023, 25, 513. [Google Scholar] [CrossRef]
- Urzúa, U.; Ampuero, S.; Roby, K.F.; Owens, G.A.; Munroe, D.J. Dysregulation of mitotic machinery genes precedes genome instability during spontaneous pre-malignant transformation of mouse ovarian surface epithelial cells. BMC Genom. 2016, 17 (Suppl. S8), 728. [Google Scholar] [CrossRef]
- Symonds, D.; Tomic, D.; Borgeest, C.; McGee, E.; Flaws, J.A. Smad3 regulates proliferation of the mouse ovarian surface epithelium. Anat. Rec. A Discov. Mol. Cell. Evol. Biol. 2003, 273, 681–686. [Google Scholar] [CrossRef]
- George, J.W.; Dille, E.A.; Heckert, L.L. Current concepts of follicle-stimulating hormone receptor gene regulation. Biol. Reprod. 2011, 84, 7–17. [Google Scholar] [CrossRef]
- Bhartiya, D.; Singh, J. FSH-FSHR3-stem cells in ovary surface epithelium: Basis for adult ovarian biology, failure, aging, and cancer. Reproduction 2015, 149, R35–R48. [Google Scholar] [CrossRef]
- Mertens-Walker, I.; Baxter, R.C.; Marsh, D.J. Gonadotropin signaling in epithelial ovarian cancer. Cancer Lett. 2012, 324, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Bousfield, G.R.; Butnev, V.Y.; Rueda-Santos, M.A.; Brown, A.; Hall, A.S.; Harvey, D.J. Macro- and Microheterogeneity in Pituitary and Urinary Follicle-Stimulating Hormone Glycosylation. J. Glycom. Lipidom. 2014, 4, 1000125. [Google Scholar]
- Agwuegbo, U.T.; Colley, E.; Albert, A.P.; Butnev, V.Y.; Bousfield, G.R.; Jonas, K.C. Differential FSH Glycosylation Modulates FSHR Oligomerization and Subsequent cAMP Signaling. Front. Endocrinol. 2021, 12, 765727. [Google Scholar] [CrossRef] [PubMed]
- Johnson, G.P.; Onabanjo, C.G.A.; Hardy, K.; Butnev, V.Y.; Bousfield, G.R.; Jonas, K.C. Follicle-Stimulating Hormone Glycosylation Variants Distinctly Modulate Pre-antral Follicle Growth and Survival. Endocrinology 2022, 163, bqac161. [Google Scholar] [CrossRef] [PubMed]
- Ivarsson, K.; Sundfeldt, K.; Brännström, M.; Hellberg, P.; Janson, P.O. Diverse effects of FSH and LH on proliferation of human ovarian surface epithelial cells. Hum. Reprod. 2001, 16, 18–23. [Google Scholar] [CrossRef]
- Yu, L.; Sun, J.; Wang, Q.; Yu, W.; Wang, A.; Zhu, S.; Xu, W.; Wang, X. Ovulation induction drug and ovarian cancer: An updated systematic review and meta-analysis. J. Ovarian Res. 2023, 16, 22. [Google Scholar] [CrossRef]
- Momenimovahed, Z.; Taheri, S.; Tiznobaik, A.; Salehiniya, H. Do the Fertility Drugs Increase the Risk of Cancer? A Review Study. Front. Endocrinol. 2019, 10, 313. [Google Scholar] [CrossRef]
- Parchwani, D.; Dholariya, S.J.; Takodara, S.; Singh, R.; Sharma, V.K.; Saxena, A.; Patel, D.D.; Radadiya, M. Analysis of Prediagnostic Circulating Levels of Gonadotropins and Androgens with Risk of Epithelial Ovarian Cancer. J. Lab. Physicians 2022, 14, 47–56. [Google Scholar] [CrossRef]
- Arslan, A.A.; Zeleniuch-Jacquotte, A.; Lundin, E.; Micheli, A.; Lukanova, A.; Afanasyeva, Y.; Lenner, P.; Krogh, V.; Muti, P.; Rinaldi, S.; et al. Serum follicle-stimulating hormone and risk of epithelial ovarian cancer in postmenopausal women. Cancer Epidemiol. Biomark. Prev. 2003, 12, 1531–1535. [Google Scholar]
- McSorley, M.A.; Alberg, A.J.; Allen, D.S.; Allen, N.E.; Brinton, L.A.; Dorgan, J.F.; Kaaks, R.; Rinaldi, S.; Helzlsouer, K.J. Prediagnostic circulating follicle stimulating hormone concentrations and ovarian cancer risk. Int. J. Cancer 2009, 125, 674–679. [Google Scholar] [CrossRef]
- Irvin, S.R.; Weiderpass, E.; Stanczyk, F.Z.; Brinton, L.A.; Trabert, B.; Langseth, H.; Wentzensen, N. Association of Anti-Mullerian Hormone, Follicle-Stimulating Hormone, and Inhibin B with Risk of Ovarian Cancer in the Janus Serum Bank. Cancer Epidemiol. Biomark. Prev. 2020, 29, 636–642. [Google Scholar] [CrossRef] [PubMed]
- Feng, F.; Liu, T.; Hou, X.; Lin, X.; Zhou, S.; Tian, Y.; Qi, X. Targeting the FSH/FSHR axis in ovarian cancer: Advanced treatment using nanotechnology and immunotherapy. Front. Endocrinol. 2024, 15, 1489767. [Google Scholar] [CrossRef]
- Cheung, J.; Lokman, N.A.; Abraham, R.D.; Macpherson, A.M.; Lee, E.; Grutzner, F.; Ghinea, N.; Oehler, M.K.; Ricciardelli, C. Reduced Gonadotrophin Receptor Expression Is Associated with a More Aggressive Ovarian Cancer Phenotype. Int. J. Mol. Sci. 2020, 22, 71. [Google Scholar] [CrossRef] [PubMed]
- Janovick, J.A.; Maya-Núñez, G.; Ulloa-Aguirre, A.; Huhtaniemi, I.T.; Dias, J.A.; Verbost, P.; Conn, P.M. Increased plasma membrane expression of human follicle-stimulating hormone receptor by a small molecule thienopyr(im)idine. Mol. Cell. Endocrinol. 2009, 298, 84–88. [Google Scholar] [CrossRef]
- Huhtaniemi, I. Are gonadotrophins tumorigenic—A critical review of clinical and experimental data. Mol. Cell. Endocrinol. 2010, 329, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.R.; Ye, D.; Luo, S.; Xu, I.R.L.; Xu, X.X. AMH regulates a mosaic population of AMHR2-positive cells in the ovarian surface epithelium. J. Biol. Chem. 2024, 300, 107897. [Google Scholar] [CrossRef]
- Pangas, S.A. Regulation of the ovarian reserve by members of the transforming growth factor beta family. Mol. Reprod. Dev. 2012, 79, 666–679. [Google Scholar] [CrossRef]
- Pellicer, A.; Marí, M.; de los Santos, M.J.; Simón, C.; Remohí, J.; Tarín, J.J. Effects of aging on the human ovary: The secretion of immunoreactive alpha-inhibin and progesterone. Fertil. Steril. 1994, 61, 663–668. [Google Scholar] [CrossRef]
- Reame, N.E.; Wyman, T.L.; Phillips, D.J.; de Kretser, D.M.; Padmanabhan, V. Net increase in stimulatory input resulting from a decrease in inhibin B and an increase in activin A may contribute in part to the rise in follicular phase follicle-stimulating hormone of aging cycling women. J. Clin. Endocrinol. Metab. 1998, 83, 3302–3307. [Google Scholar]
- Knight, P.G.; Satchell, L.; Glister, C. Intra-ovarian roles of activins and inhibins. Mol. Cell. Endocrinol. 2012, 359, 53–65. [Google Scholar] [CrossRef]
- Adashi, E.Y. The climacteric ovary as a functional gonadotropin-driven androgen-producing gland. Fertil. Steril. 1994, 62, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Hassa, H.; Aydin, Y.; Ozatik, O.; Erol, K.; Ozatik, Y. Effects of dehydroepiandrosterone (DHEA) on follicular dynamics in a diminished ovarian reserve in vivo model. Syst. Biol. Reprod. Med. 2015, 61, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Hoang, Y.D.; Nakamura, B.N.; Luderer, U. Follicle-stimulating hormone and estradiol interact to stimulate glutathione synthesis in rat ovarian follicles and granulosa cells. Biol. Reprod. 2009, 81, 636–646. [Google Scholar] [CrossRef] [PubMed]
- Hauck, A.K.; Bertler, D.A. Oxidative stress and lipotoxicity. J. Lipid Res. 2016, 57, 1976–1986. [Google Scholar] [CrossRef]
- Bi, W.K.; Shao, S.S.; Li, Z.W.; Ruan, Y.W.; Luan, S.S.; Dong, Z.H.; Wang, J.; Wu, S.S.; Guo, T.; Ma, S.Z.; et al. FSHR ablation induces depression-like behaviors. Acta Pharmacol. Sin. 2020, 41, 1033–1040. [Google Scholar] [CrossRef]
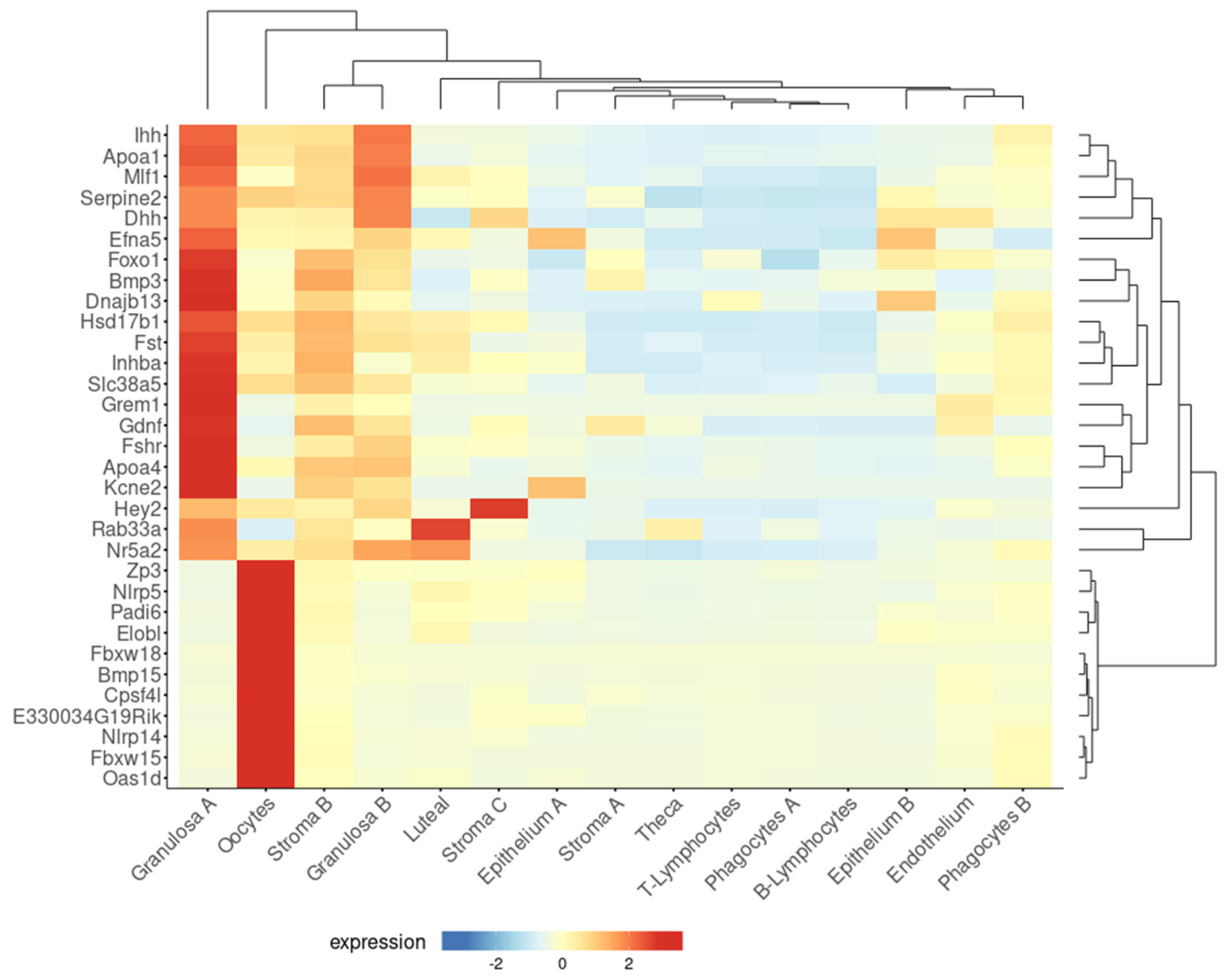
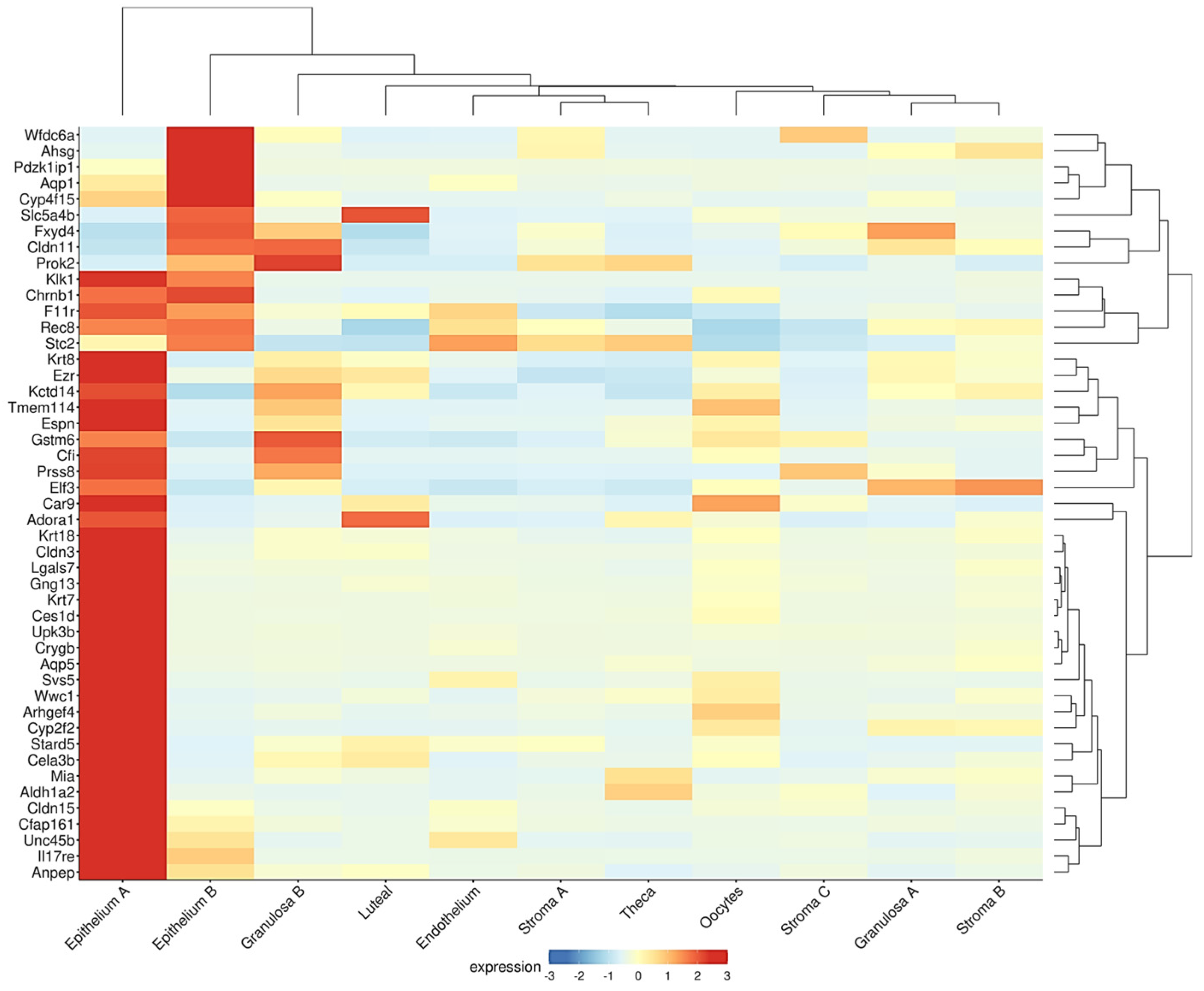
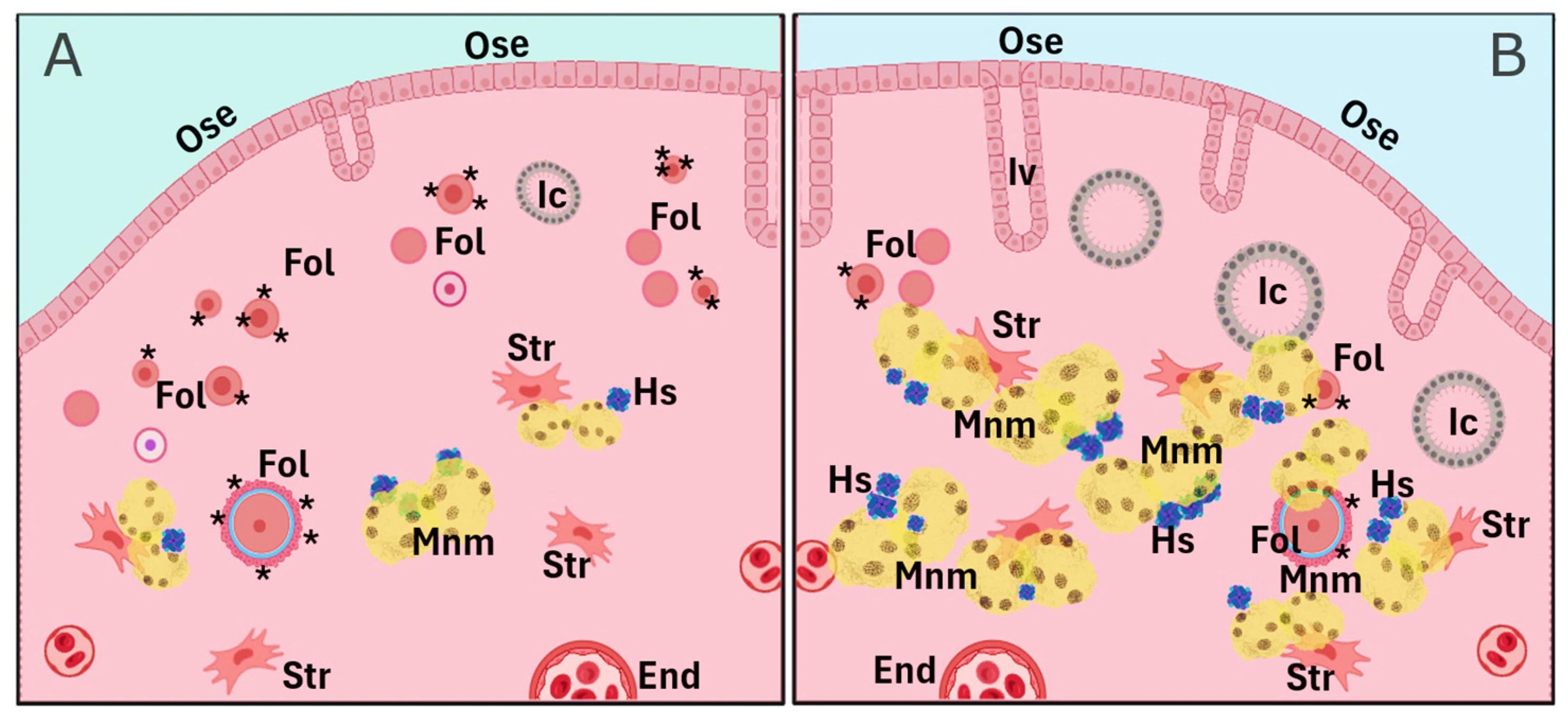
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).