
Redox Imbalance Is Associated with Neuronal Apoptosis in the Cortex of Neonates Gestated Under Chronic Hypoxia

 ,
,  , , and
, , and
Abstract

1. Introduction
2. Materials and Methods
2.1. Animals and Experimental Groups
2.2. Pre- and Postnatal Biometry Assessment
2.3. Protein Expression and Activity Assay in Total Brain
2.4. Immunolocalization of Proteins in the Cerebral Cortex
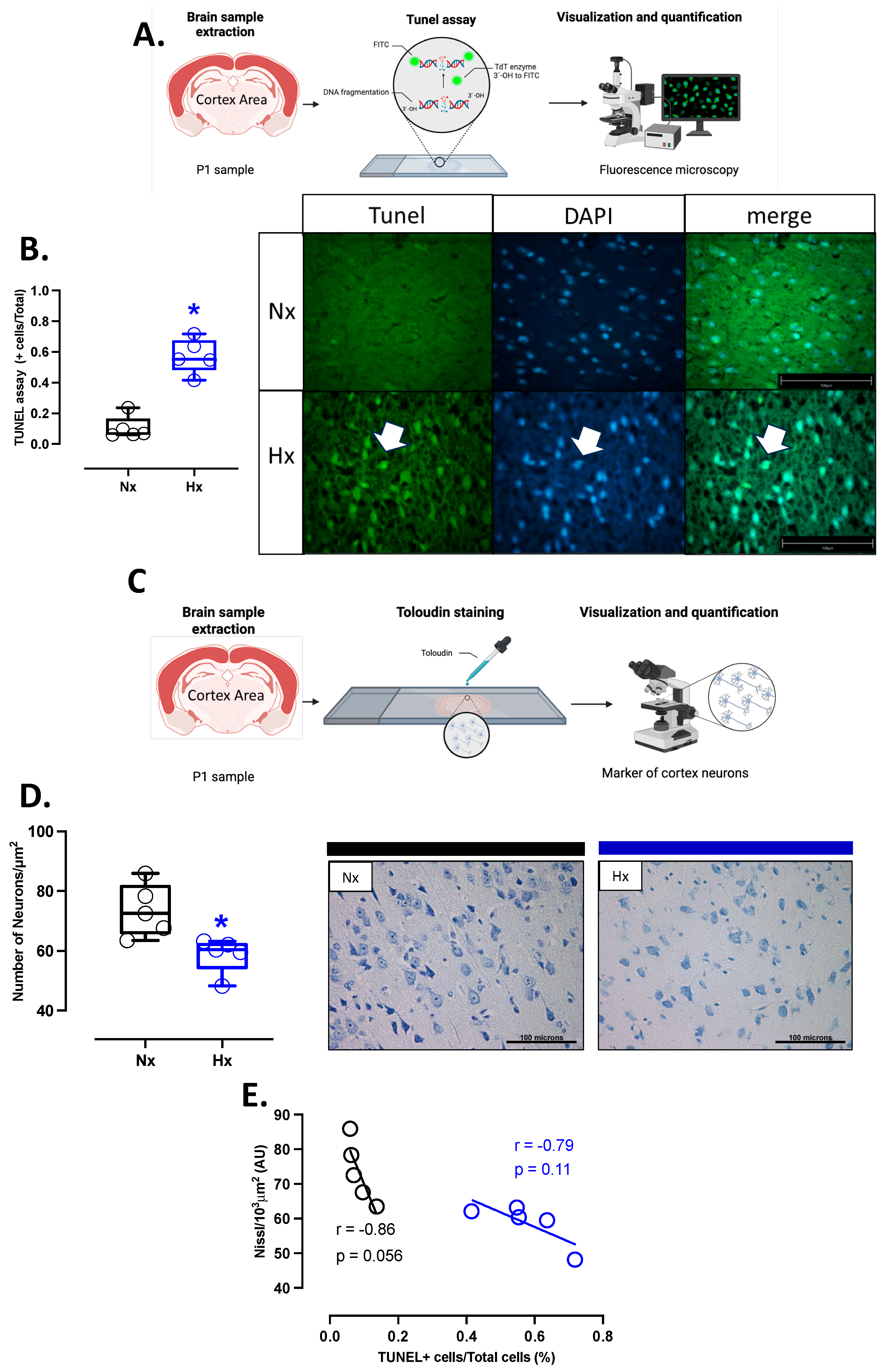
2.5. Apoptosis Detection in the Cerebral Cortex
2.6. Statistical Analyses
3. Results
3.1. Pre- and Postnatal Biometric Variables
3.2. Antioxidant Capacity of the Postnatal Brain
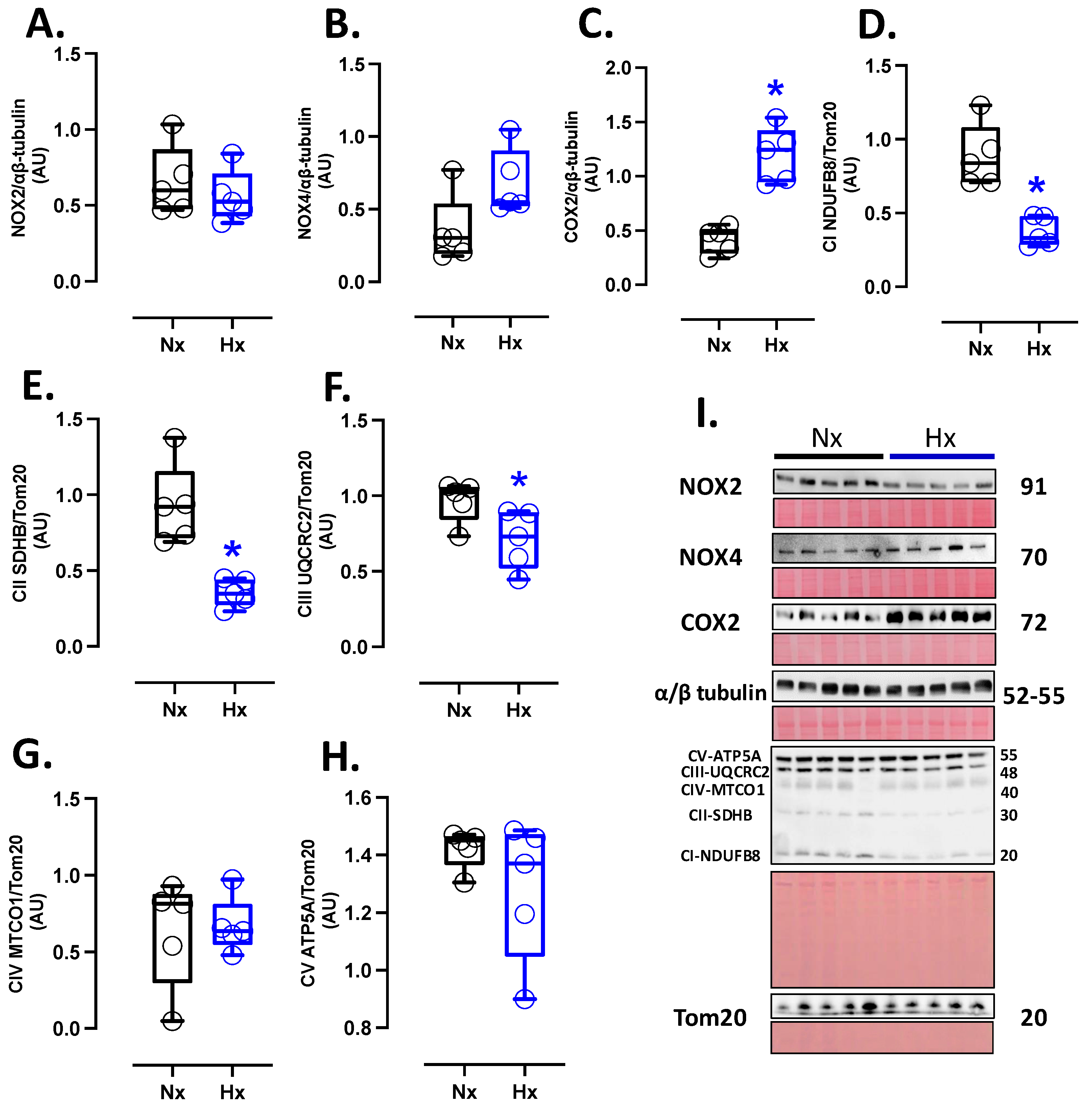
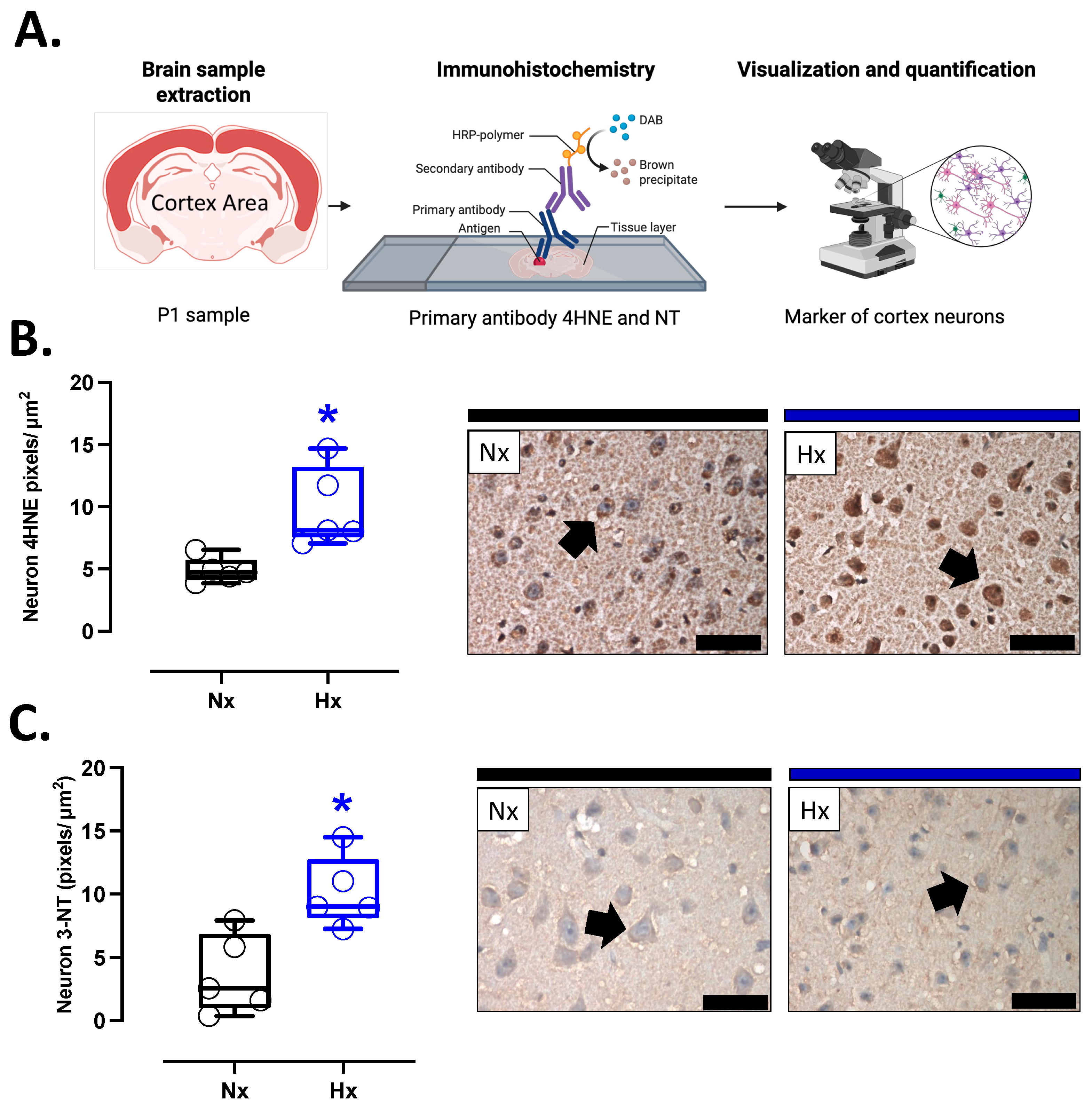
3.3. Pro-Oxidant Protein Levels and Oxidative Stress Marker in the Postnatal Brain
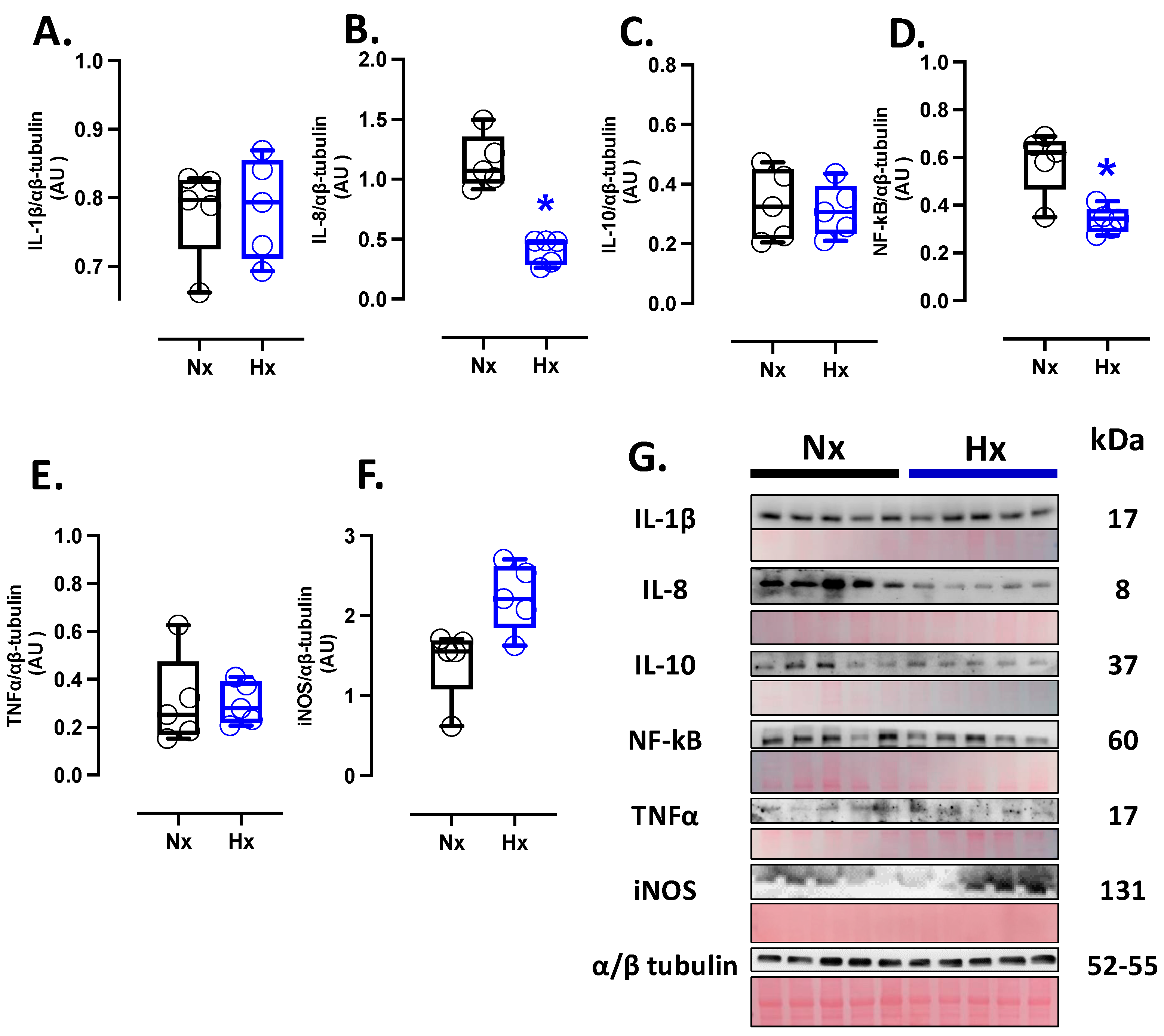
3.4. Inflammatory Protein Levels in the Postnatal Brain
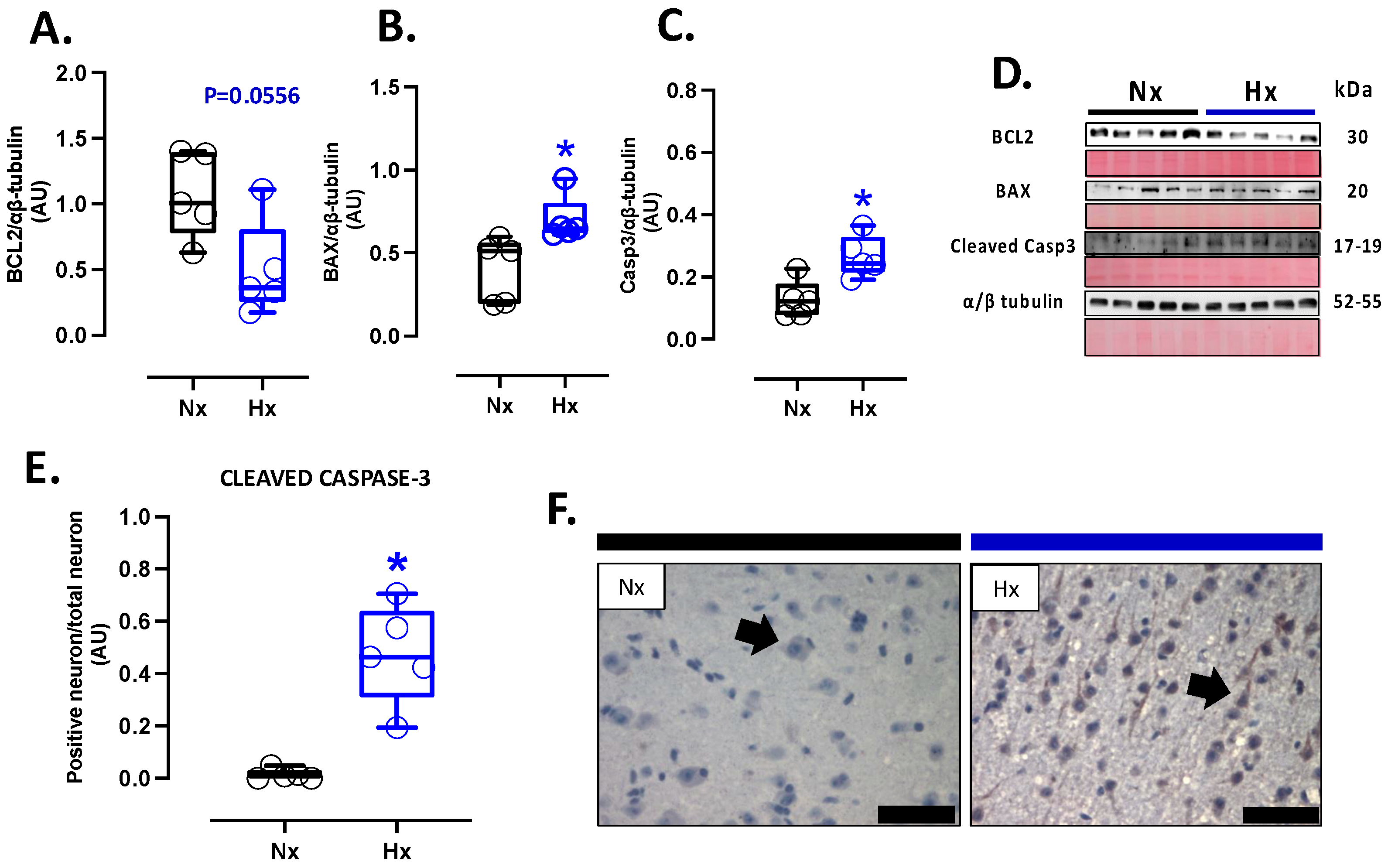
3.5. Apoptosis and Neuronal Density in Postnatal Brain Cortex
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| N | Normoxia |
| Hx | Hypoxia |
| GD | Gestational Days |
| FGR | Fetal Growth Restriction |
| ROS | Reactive Oxygen Species |
| NOX | NADPH Oxidase |
| SOD | Superoxide Dismutase |
| CAT | Catalase |
| GPX | Glutathione Peroxidase |
| H2O2 | Hydrogen Peroxide |
| GSH | Glutathione (reduced form) |
| GSSG | Glutathione (oxidized form) |
| 4-HNE | 4-Hydroxynonenal |
| NT | 3-Nitrotyrosine |
| TUNEL | TdT-mediated dUTP Nick-End Labeling |
| DAPI | 4′,6-Diamidino-2-phenylindole |
| COX2 | Cyclooxygenase-2 |
| IL-1β | Interleukin 1 beta |
| IL-8 | Interleukin 8 |
| IL-10 | Interleukin 10 |
| TNFα | Tumor Necrosis Factor Alpha |
| iNOS | Inducible Nitric Oxide Synthase |
| NF-κB | Nuclear Factor kappa-light-chain-enhancer of activated B cells |
| BAX | Bcl-2-associated X protein |
| Bcl-xL | B-cell lymphoma-extra large |
| Casp-3 | Caspase-3 |
| OXPHOS | Oxidative Phosphorylation |
| ATP | Adenosine Triphosphate |
| UA | Umbilical Artery |
| MCA | Middle Cerebral Artery |
| PI | Pulsatility Index |
| CPR | Cerebroplacental Ratio |
| HIE | Hypoxic–Ischemic Encephalopathy |
| APAF1 | Apoptotic Protease Activating Factor 1 |
| ELISA | Enzyme-Linked Immunosorbent Assay |
| SEM | Standard Error of the Mean |
| ARRIVE | Animal Research: Reporting of In Vivo Experiments |
| CICUA | Institutional Committee for the Care and Use of Animals |
| NIH | National Institutes of Health |
References
- Gluckman, P.D.; Cutfield, W.; Hofman, P.; Hanson, M.A. The Fetal, Neonatal, and Infant Environments-the Long-Term Consequences for Disease Risk. Early Hum. Dev. 2005, 81, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Fowden, A.L.; Giussani, D.A.; Forhead, A.J. Intrauterine Programming of Physiological Systems: Causes and Consequences. Physiology 2006, 21, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Fajersztajn, L.; Veras, M.M. Hypoxia: From Placental Development to Fetal Programming. Birth Defects Res. 2017, 109, 1377–1385. [Google Scholar] [CrossRef]
- Sun, C.; Groom, K.M.; Oyston, C.; Chamley, L.W.; Clark, A.R.; James, J.L. The Placenta in Fetal Growth Restriction: What Is Going Wrong? Placenta 2020, 96, 10–18. [Google Scholar] [CrossRef]
- Giussani, D.A. The Fetal Brain Sparing Response to Hypoxia: Physiological Mechanisms. J. Physiol. 2016, 594, 1215. [Google Scholar] [CrossRef]
- Demirci-Çekiç, S.; Özkan, G.; Avan, A.N.; Uzunboy, S.; Çapanoğlu, E.; Apak, R. Biomarkers of Oxidative Stress and Antioxidant Defense. J. Pharm. Biomed. Anal. 2022, 209, 114477. [Google Scholar] [CrossRef]
- Chen, R.; Lai, U.H.; Zhu, L.; Singh, A.; Ahmed, M.; Forsyth, N.R. Reactive Oxygen Species Formation in the Brain at Different Oxygen Levels: The Role of Hypoxia Inducible Factors. Front. Cell Dev. Biol. 2018, 6, 132. [Google Scholar] [CrossRef] [PubMed]
- McGarry, T.; Biniecka, M.; Veale, D.J.; Fearon, U. Hypoxia, Oxidative Stress and Inflammation. Free Radic. Biol. Med. 2018, 125, 15–24. [Google Scholar] [CrossRef]
- Lou, Z.; Wang, A.P.; Duan, X.M.; Hu, G.H.; Song, G.L.; Zuo, M.L.; Yang, Z.B. Upregulation of NOX2 and NOX4 Mediated by TGF-β Signaling Pathway Exacerbates Cerebral Ischemia/Reperfusion Oxidative Stress Injury. Cell Physiol. Biochem. 2018, 46, 2103–2113. [Google Scholar] [CrossRef]
- Wang, J.; Liu, Y.; Shen, H.; Li, H.; Wang, Z.; Chen, G. Nox2 and Nox4 Participate in ROS-Induced Neuronal Apoptosis and Brain Injury During Ischemia-Reperfusion in Rats. Acta Neurochir. Suppl. 2020, 127, 47–54. [Google Scholar] [CrossRef]
- Gaur, P.; Prasad, S.; Kumar, B.; Sharma, S.K.; Vats, P. High-Altitude Hypoxia Induced Reactive Oxygen Species Generation, Signaling, and Mitigation Approaches. Int. J. Biometeorol. 2021, 65, 601–615. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.Q.; Zhang, L. Hypoxia and Mitochondrial Dysfunction in Pregnancy Complications. Antioxidants 2021, 10, 405. [Google Scholar] [CrossRef] [PubMed]
- Herrera, E.A.; Krause, B.; Ebensperger, G.; Reyes, R.V.; Casanello, P.; Parra-Cordero, M.; Llanos, A.J. The Placental Pursuit for an Adequate Oxidant Balance between the Mother and the Fetus. Front. Pharmacol. 2014, 5, 149. [Google Scholar] [CrossRef]
- Ahamad, T.; Ansari, W.A.; Negi, D.S.; Khan, M.F. Non-Enzymatic Natural Reactive Oxygen Scavengers (ROS): A Review on Structures and Mode of Action. Era’s J. Med. Res. 2019, 6, 103–112. [Google Scholar] [CrossRef]
- Ozsurekci, Y.; Aykac, K. Oxidative Stress Related Diseases in Newborns. Oxid. Med. Cell Longev. 2016, 2016, 2768365. [Google Scholar] [CrossRef]
- Panfoli, I.; Candiano, G.; Malova, M.; De Angelis, L.; Cardiello, V.; Buonocore, G.; Ramenghi, L.A. Oxidative Stress as a Primary Risk Factor for Brain Damage in Preterm Newborns. Front. Pediatr. 2018, 6, 369. [Google Scholar] [CrossRef]
- Baud, O.; Greene, A.E.; Li, J.; Wang, H.; Volpe, J.J.; Rosenberg, P.A. Glutathione Peroxidase-Catalase Cooperativity Is Required for Resistance to Hydrogen Peroxide by Mature Rat Oligodendrocytes. J. Neurosci. 2004, 24, 1531–1540. [Google Scholar] [CrossRef]
- Hulbert, A.J.; Pamplona, R.; Buffenstein, R.; Buttemer, W.A. Life and Death: Metabolic Rate, Membrane Composition, and Life Span of Animals. Physiol. Rev. 2007, 87, 1175–1213. [Google Scholar] [CrossRef]
- Hassan, W.; Noreen, H.; Rehman, S.; Kamal, M.A.; da Rocha, J.B.T. Association of Oxidative Stress with Neurological Disorders. Curr. Neuropharmacol. 2022, 20, 1046–1072. [Google Scholar] [CrossRef]
- Astorga, C.R.; González-Candia, A.; Candia, A.A.; Figueroa, E.G.; Cañas, D.; Ebensperger, G.; Reyes, R.V.; Llanos, A.J.; Herrera, E.A. Melatonin Decreases Pulmonary Vascular Remodeling and Oxygen Sensitivity in Pulmonary Hypertensive Newborn Lambs. Front. Physiol. 2018, 9, 348028. [Google Scholar] [CrossRef]
- Candia, A.A.; Jiménez, T.; Navarrete, Á.; Beñaldo, F.; Silva, P.; García-Herrera, C.; Sferruzzi-Perri, A.N.; Krause, B.J.; González-Candia, A.; Herrera, E.A. Developmental Ultrasound Characteristics in Guinea Pigs: Similarities with Human Pregnancy. Vet. Sci. 2023, 10, 144. [Google Scholar] [CrossRef]
- Torres, F.; González-Candia, A.; Montt, C.; Ebensperger, G.; Chubretovic, M.; Serõn-Ferré, M.; Reyes, R.V.; Llanos, A.J.; Herrera, E.A. Melatonin Reduces Oxidative Stress and Improves Vascular Function in Pulmonary Hypertensive Newborn Sheep. J. Pineal Res. 2015, 58, 362–373. [Google Scholar] [CrossRef]
- Gonzalez-Candia, A.; Veliz, M.; Carrasco-Pozo, C.; Castillo, R.L.; Cárdenas, J.C.; Ebensperger, G.; Reyes, R.V.; Llanos, A.J.; Herrera, E.A. Antenatal Melatonin Modulates an Enhanced Antioxidant/pro-Oxidant Ratio in Pulmonary Hypertensive Newborn Sheep. Redox Biol. 2019, 22, 101128. [Google Scholar] [CrossRef]
- Yang, X.; Zhang, J.; Cheng, Y.; Cui, M.; Jiang, Z.; Fan, C.; Chen, J.; Qi, L.; Liu, H.; Bao, D. Tenofovir Disoproxil Fumarate Mediates Neuronal Injury by Inducing Neurotoxicity. Eur. J. Clin. Microbiol. Infect. Dis. 2023, 42, 1195–1205. [Google Scholar] [CrossRef]
- Lau, C.; Rogers, J.M.; Desai, M.; Ross, M.G. Fetal Programming of Adult Disease: Implications for Prenatal Care. Obstet. Gynecol. 2011, 117, 978–985. [Google Scholar] [CrossRef] [PubMed]
- Coe, C.L.; Lubach, G.R. Fetal programming: Prenatal origins of health and illness. Curr. Dir. Psychol. Sci. 2008, 17, 36–41. [Google Scholar] [CrossRef]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.D.; Mazur, M.; Telser, J. Free Radicals and Antioxidants in Normal Physiological Functions and Human Disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef] [PubMed]
- Frank, L.; Groseclose, E.E. Preparation for Birth into an O2-Rich Environment: The Antioxidant Enzymes in the Developing Rabbit Lung. Pediatr. Res. 1984, 18, 240–244. [Google Scholar] [CrossRef]
- Davis, J.M.; Auten, R.L. Maturation of the Antioxidant System and the Effects on Preterm Birth. Semin. Fetal Neonatal Med. 2010, 15, 191–195. [Google Scholar] [CrossRef]
- Morrison, J.L.; Botting, K.J.; Darby, J.R.T.; David, A.L.; Dyson, R.M.; Gatford, K.L.; Gray, C.; Herrera, E.A.; Hirst, J.J.; Kim, B.; et al. Guinea pig models for translation of the developmental origins of health and disease hypothesis into the clinic. J. Physiol. 2018, 596, 5535–5569. [Google Scholar] [CrossRef]
- Uchoa, M.F.; de Souza, L.F.; dos Santos, D.B.; Peres, T.V.; Mello, D.F.; Leal, R.B.; Farina, M.; Dafre, A.L. Modulation of Brain Glutathione Reductase and Peroxiredoxin 2 by α-Tocopheryl Phosphate. Cell Mol. Neurobiol. 2016, 36, 1015–1022. [Google Scholar] [CrossRef] [PubMed]
- Terraneo, L.; Paroni, R.; Bianciardi, P.; Giallongo, T.; Carelli, S.; Gorio, A.; Samaja, M. Brain Adaptation to Hypoxia and Hyperoxia in Mice. Redox Biol. 2016, 11, 12. [Google Scholar] [CrossRef] [PubMed]
- Bedard, K.; Krause, K.H. The NOX Family of ROS-Generating NADPH Oxidases: Physiology and Pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Candia, A.; Herrera, E.A. High Altitude Pregnancies and Vascular Dysfunction: Observations From Latin American Studies. Front. Physiol. 2021, 12, 786038. [Google Scholar] [CrossRef]
- Fathali, N.; Ostrowski, R.P.; Lekic, T.; Jadhav, V.; Tong, W.; Tang, J.; Zhang, J.H. Cyclooxygenase-2 Inhibition Provides Lasting Protection against Neonatal Hypoxic-Ischemic Brain Injury. Crit. Care Med. 2010, 38, 572. [Google Scholar] [CrossRef]
- Odegaard, A.O.; Jacobs, D.R.; Sanchez, O.A.; Goff, D.C.; Reiner, A.P.; Gross, M.D. Oxidative Stress, Inflammation, Endothelial Dysfunction and Incidence of Type 2 Diabetes. Cardiovasc. Diabetol. 2016, 15, 51. [Google Scholar] [CrossRef]
- Rodríguez, M.; Valez, V.; Cimarra, C.; Blasina, F.; Radi, R. Hypoxic-Ischemic Encephalopathy and Mitochondrial Dysfunction: Facts, Unknowns, and Challenges. Antioxid. Redox Signal 2020, 33, 247–262. [Google Scholar] [CrossRef]
- Mallard, C.; Loeliger, M.; Copolov, D.; Rees, S. Reduced Number of Neurons in the Hippocampus and the Cerebellum in the Postnatal Guinea-Pig Following Intrauterine Growth-Restriction. Neuroscience 2000, 100, 327–333. [Google Scholar] [CrossRef]
- Guo, R.; Hou, W.; Dong, Y.; Yu, Z.; Stites, J.; Weiner, C.P. Brain Injury Caused by Chronic Fetal Hypoxemia Is Mediated by Inflammatory Cascade Activation. Reprod. Sci. 2010, 17, 540–548. [Google Scholar] [CrossRef]
- Wixey, J.A.; Chand, K.K.; Colditz, P.B.; Bjorkman, S.T. Review: Neuroinflammation in Intrauterine Growth Restriction. Placenta 2017, 54, 117–124. [Google Scholar] [CrossRef]
- Wan, L.; Luo, K.; Chen, P. Mechanisms Underlying Neurologic Injury in Intrauterine Growth Restriction. J. Child. Neurol. 2021, 36, 776–784. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Liu, J.; Cheung, P.Y.; Chen, C. Long-Term Cognitive Impairment and Myelination Deficiency in a Rat Model of Perinatal Hypoxic-Ischemic Brain Injury. Brain Res. 2009, 1301, 100–109. [Google Scholar] [CrossRef]
- Fan, L.W.; Lin, S.; Pang, Y.; Rhodes, P.G.; Cai, Z. Minocycline Attenuates Hypoxia-Ischemia-Induced Neurological Dysfunction and Brain Injury in the Juvenile Rat. Eur. J. Neurosci. 2006, 24, 341–350. [Google Scholar] [CrossRef]
- Girard, S.; Kadhim, H.; Roy, M.; Lavoie, K.; Brochu, M.E.; Larouche, A.; Sébire, G. Role of Perinatal Inflammation in Cerebral Palsy. Pediatr. Neurol. 2009, 40, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Kossmann, T.; Stahel, P.F.; Lenzlinger, P.M.; Redl, H.; Dubs, R.W.; Trentz, O.; Schlag, G.; Morganti-Kossmann, M.C. Interleukin-8 Released into the Cerebrospinal Fluid after Brain Injury Is Associated with Blood-Brain Barrier Dysfunction and Nerve Growth Factor Production. J. Cereb. Blood Flow. Metab. 1997, 17, 280–289. [Google Scholar] [CrossRef]
- Aly, H.; Khashaba, M.T.; El-Ayouty, M.; El-Sayed, O.; Hasanein, B.M. IL-1beta, IL-6 and TNF-Alpha and Outcomes of Neonatal Hypoxic Ischemic Encephalopathy. Brain Dev. 2006, 28, 178–182. [Google Scholar] [CrossRef] [PubMed]
- Sadowska, G.B.; Chen, X.; Zhang, J.; Lim, Y.P.; Cummings, E.E.; Makeyev, O.; Besio, W.G.; Gaitanis, J.; Padbury, J.F.; Banks, W.A.; et al. Interleukin-1β Transfer across the Blood–Brain Barrier in the Ovine Fetus. J. Cereb. Blood Flow Metab. 2015, 35, 1388. [Google Scholar] [CrossRef]
- Giannopoulou, I.; Pagida, M.A.; Briana, D.D.; Panayotacopoulou, M.T. Perinatal Hypoxia as a Risk Factor for Psychopathology Later in Life: The Role of Dopamine and Neurotrophins. Hormones 2018, 17, 25–32. [Google Scholar] [CrossRef]
- Malamitsi-Puchner, A.; Nikolaou, K.E.; Economou, E.; Boutsikou, M.; Boutsikou, T.; Kyriakakou, M.; Puchner, K.P.; Hassiakos, D. Intrauterine Growth Restriction and Circulating Neurotrophin Levels at Term. Early Hum. Dev. 2007, 83, 465–469. [Google Scholar] [CrossRef]
- Chung, Y.; So, K.; Kim, E.; Kim, S.; Jeon, Y. Immunoreactivity of Neurogenic Factor in the Guinea Pig Brain after Prenatal Hypoxia. Ann. Anat. 2015, 200, 66–72. [Google Scholar] [CrossRef]
- Tolcos, M.; Mcgregor, H.; Walker, D.; Rees, S. Chronic Prenatal Exposure to Carbon Monoxide Results in a Reduction in Tyrosine Hydroxylase-Immunoreactivity and an Increase in Choline Acetyltransferase-Immunoreactivity in the Fetal Medulla: Implications for Sudden Infant Death Syndrome. J. Neuropathol. Exp. Neurol. 2000, 59, 218–228. [Google Scholar] [CrossRef] [PubMed]
- Kannan, K.; Jain, S.K. Oxidative Stress and Apoptosis. Pathophysiology 2000, 7, 153–163. [Google Scholar] [CrossRef]
- Popa-Wagner, A.; Mitran, S.; Sivanesan, S.; Chang, E.; Buga, A.M. ROS and Brain Diseases: The Good, the Bad, and the Ugly. Oxid. Med. Cell Longev. 2013, 2013, 963520. [Google Scholar] [CrossRef] [PubMed]
- Ultanir, S.; Shetty, A.S.; Constantin Badea, T.; Sun, M.; Wang, B.; Zeng, H.; Liu, J. Effects of Prenatal Hypoxia on Nervous System Development and Related Diseases. Front. Neurosci. 2021, 15, 755554. [Google Scholar] [CrossRef]
- Gu, M.; Mei, X.L.; Zhao, Y.N. Sepsis and Cerebral Dysfunction: BBB Damage, Neuroinflammation, Oxidative Stress, Apoptosis and Autophagy as Key Mediators and the Potential Therapeutic Approaches. Neurotox. Res. 2021, 39, 489–503. [Google Scholar] [CrossRef] [PubMed]
- Silva, F.F.V.e.; Padín-Iruegas, M.E.; Caponio, V.C.A.; Lorenzo-Pouso, A.I.; Saavedra-Nieves, P.; Chamorro-Petronacci, C.M.; Suaréz-Peñaranda, J.; Pérez-Sayáns, M. Caspase 3 and Cleaved Caspase 3 Expression in Tumorogenesis and Its Correlations with Prognosis in Head and Neck Cancer: A Systematic Review and Meta-Analysis. Int. J. Mol. Sci. 2022, 23, 11937. [Google Scholar] [CrossRef]
- Delivoria-Papadopoulos, M.; Mishra, O.P. Mechanisms of Perinatal Cerebral Injury in Fetus and Newborn. Ann. N. Y. Acad. Sci. 2000, 900, 159–168. [Google Scholar] [CrossRef]
- Blomgren, K.; Hagberg, H. Free Radicals, Mitochondria, and Hypoxia-Ischemia in the Developing Brain. Free Radic. Biol. Med. 2006, 40, 388–397. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Prenatal Biometry and Doppler Across Pregnancy | ||||||
| Nx | Hx | Nx | Hx | Nx | Hx | |
| 30–35 GD | 40–45 GD | 60–65 GD | ||||
| Biparietal Diameter (mm) | 8.82 ± 0.26 | 7.13 ± 0.47 * | 13.66 ± 0.32 | 11.48 ± 0.60 * | 18.89 ± 0.22 | 17.65 ± 0.41 |
| Abdominal circumference (mm) | 3.45 ± 0.15 | 2.64 ± 0.19 | 6.02 ± 0.43 | 4.39 ± 0.26 * | 8.78 ± 0.35 | 7.21 ± 0.30 * |
| Cranial Circumference (mm) | 3.16 ± 0.09 | 2.80 ± 0.09 * | 4.52 ± 0.10 | 3.92 ± 0.07 | 5.79 ± 0.09 | 5.31 ± 0.09 |
| Cerebral medial artery (PI) | NA | NA | NA | NA | 1.09 ± 0.06 | 1.55 ± 0.28 |
| Umbilical arteria (PI) | NA | NA | NA | NA | 1.07 ± 0.06 | 0.97 ± 0.07 |
| Cerebro-placental ratio (AU) | NA | NA | NA | NA | 1.04 ± 0.10 | 1.63 ± 0.24 * |
| Postnatal Biometry (at Birth) | ||||||
| Nx | Hx | |||||
| Birth weight (g) | 114.20 ± 14.14 | 80.48 ± 4.51 * | ||||
| Biparietal Diameter/weight (AU) | 0.26 ± 0.02 | 0.20 ± 0.01 * | ||||
| Cerebral weight (g) | 2.05 ± 0.04 | 1.93 ± 0.10 | ||||
| Cerebral/liver weight (AU) | 20.0 ± 2.3 | 20.6 ± 6.8 | ||||
| Cerebral/ birth weight (AU) | 56.3 ± 16 | 41.6 ± 7.1 | ||||
| % Brain water content | 82.81 ± 0.22 | 84.35 ± 0.54 * | ||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Figueroa, E.G.; Castillo, R.L.; Paz, A.A.; Monsalves-Alvarez, M.; Salas-Pérez, F.; Calle, X.; Jiménez, T.A.; Herrera, E.A.; Gonzaléz-Candia, A. Redox Imbalance Is Associated with Neuronal Apoptosis in the Cortex of Neonates Gestated Under Chronic Hypoxia. Antioxidants 2025, 14, 736. https://doi.org/10.3390/antiox14060736
Figueroa EG, Castillo RL, Paz AA, Monsalves-Alvarez M, Salas-Pérez F, Calle X, Jiménez TA, Herrera EA, Gonzaléz-Candia A. Redox Imbalance Is Associated with Neuronal Apoptosis in the Cortex of Neonates Gestated Under Chronic Hypoxia. Antioxidants. 2025; 14(6):736. https://doi.org/10.3390/antiox14060736
Chicago/Turabian StyleFigueroa, Esteban G., Rodrigo L. Castillo, Adolfo A. Paz, Matías Monsalves-Alvarez, Francisca Salas-Pérez, Ximena Calle, Tamara A. Jiménez, Emilio A. Herrera, and Alejandro Gonzaléz-Candia. 2025. "Redox Imbalance Is Associated with Neuronal Apoptosis in the Cortex of Neonates Gestated Under Chronic Hypoxia" Antioxidants 14, no. 6: 736. https://doi.org/10.3390/antiox14060736
APA StyleFigueroa, E. G., Castillo, R. L., Paz, A. A., Monsalves-Alvarez, M., Salas-Pérez, F., Calle, X., Jiménez, T. A., Herrera, E. A., & Gonzaléz-Candia, A. (2025). Redox Imbalance Is Associated with Neuronal Apoptosis in the Cortex of Neonates Gestated Under Chronic Hypoxia. Antioxidants, 14(6), 736. https://doi.org/10.3390/antiox14060736