
Anti-TNFα and Anti-IL-1β Monoclonal Antibodies Preserve BV-2 Microglial Homeostasis Under Hypoxia by Mitigating Inflammatory Reactivity and ATF4/MAPK-Mediated Apoptosis


Abstract
1. Introduction
2. Materials and Methods
- 1.
- Chemicals and reagents
- 2.
- BV-2 cell line cultivation
- 3.
- Establishment of hypoxic model in vitro
- 4.
- Fluorescein–phalloidin staining
- 5.
- MTS assay
- 6.
- Cell treatment
- 7.
- Lactate dehydrogenase (LDH) assay
- 8.
- Intracellular ROS measurement
- 9.
- Mitochondrial membrane potential (MMP) assessment, JC-10
- 10.
- Immunofluorescence staining
- 11.
- TUNEL assay
- 12.
- Western blot analysis
- 13.
- Liquid chromatography–mass spectrometry (LC–MS/MS)-based proteomics analysis
- 14.
- Scratch wound migration assay
- 15.
- Statistical Analysis
3. Results
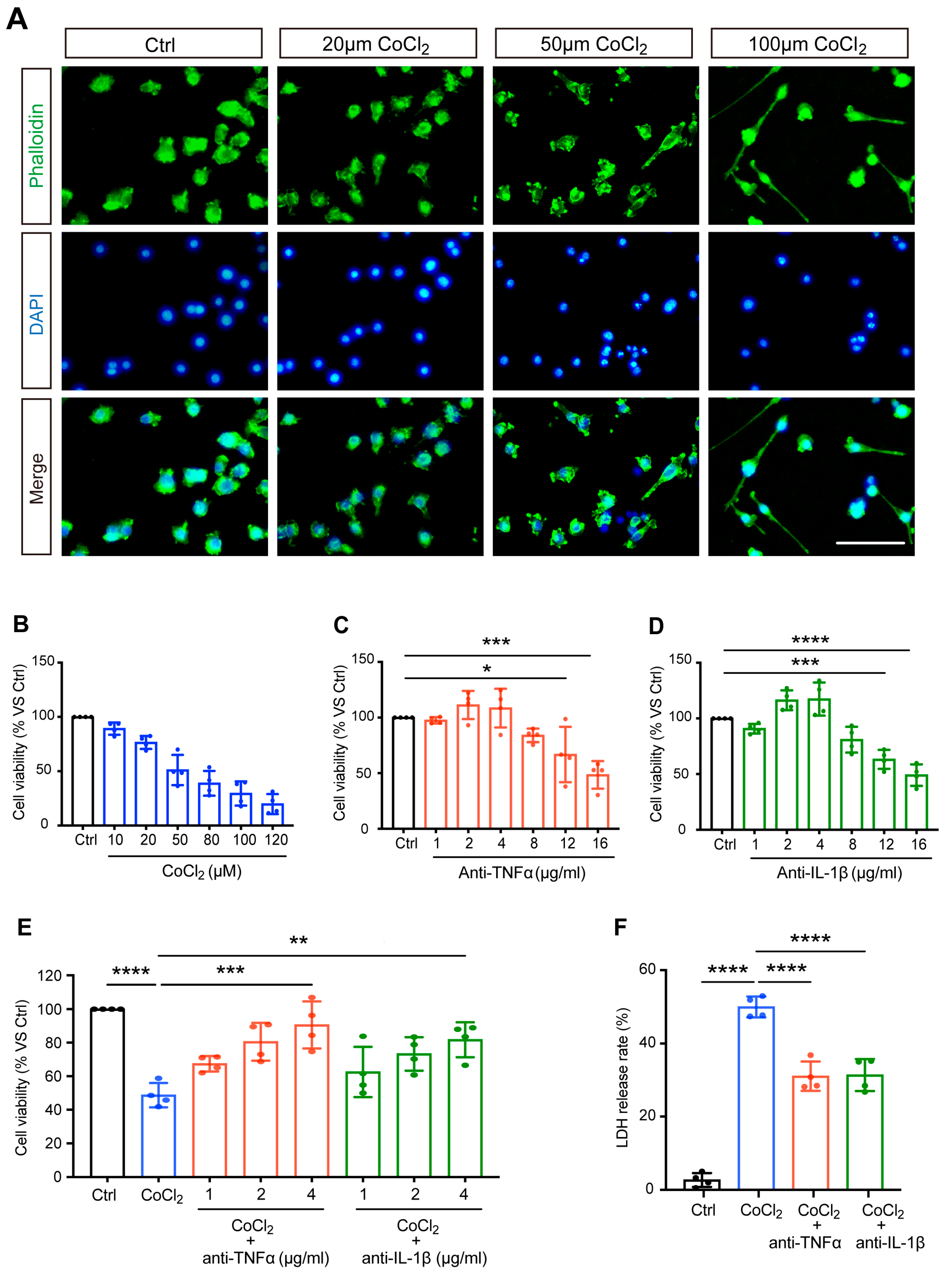
3.1. Anti-TNFα and Anti-IL-1β Increased Cell Viability and Reduced Cytotoxicity in CoCl2-Treated BV-2 Cells
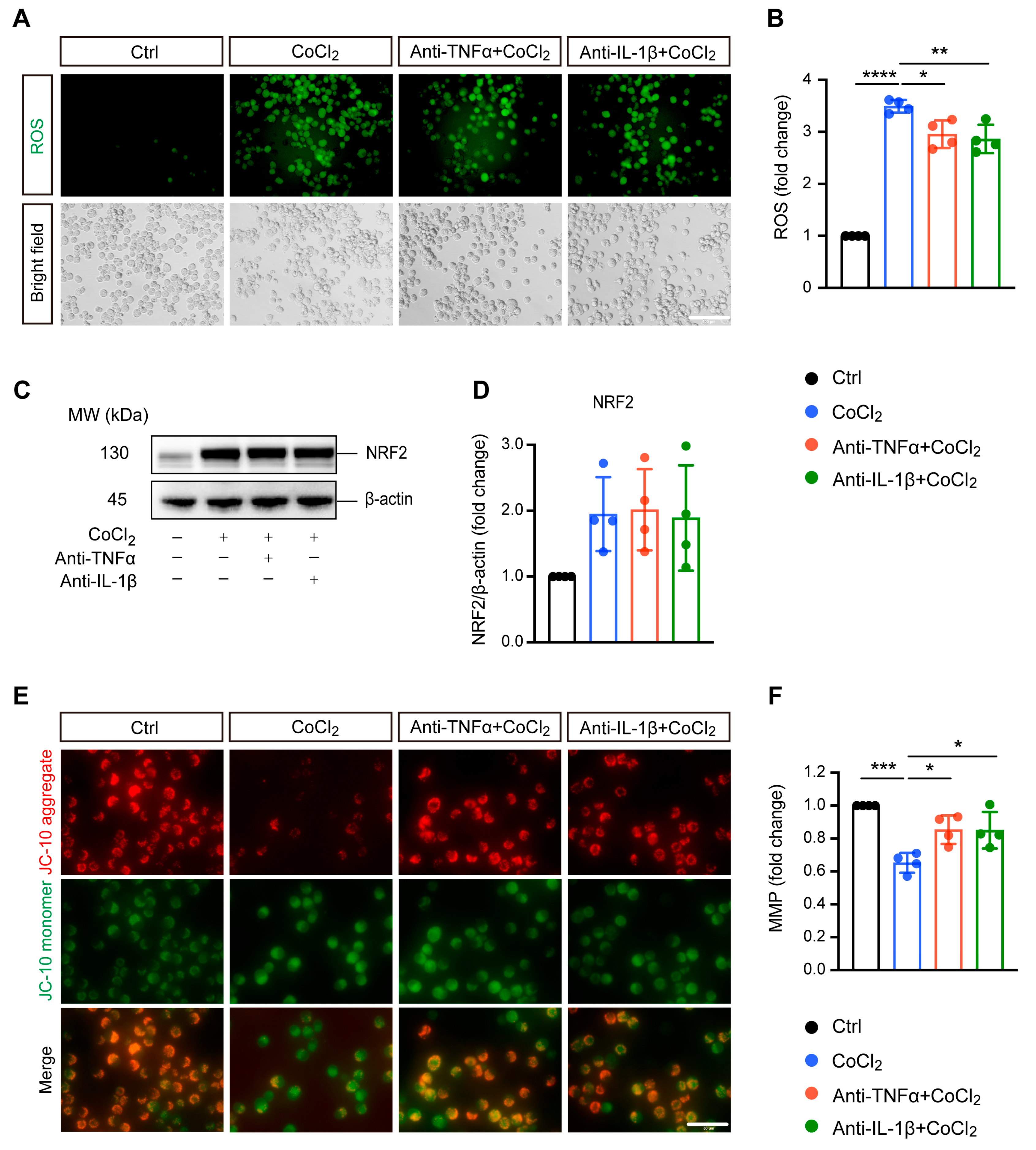
3.2. Anti-TNFα and Anti-IL-1β Treatment Alleviated CoCl2-Induced ROS Generation and Mitochondrial Impairment
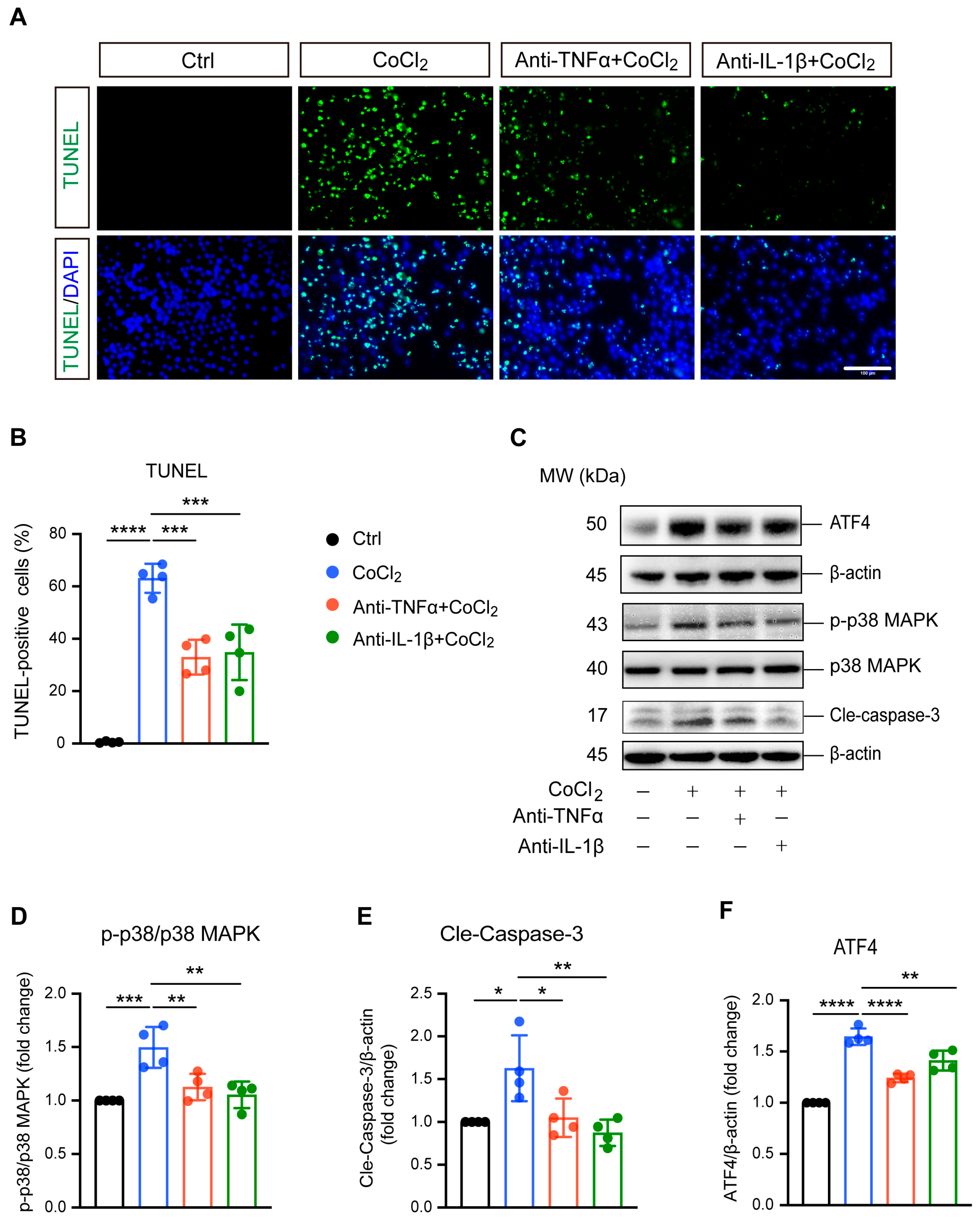
3.3. Anti-TNFα and Anti-IL-1β Reduced BV-2 Cell Apoptosis by Inhibiting ATF4 and p38 MAPK/Caspase-3 Pathways
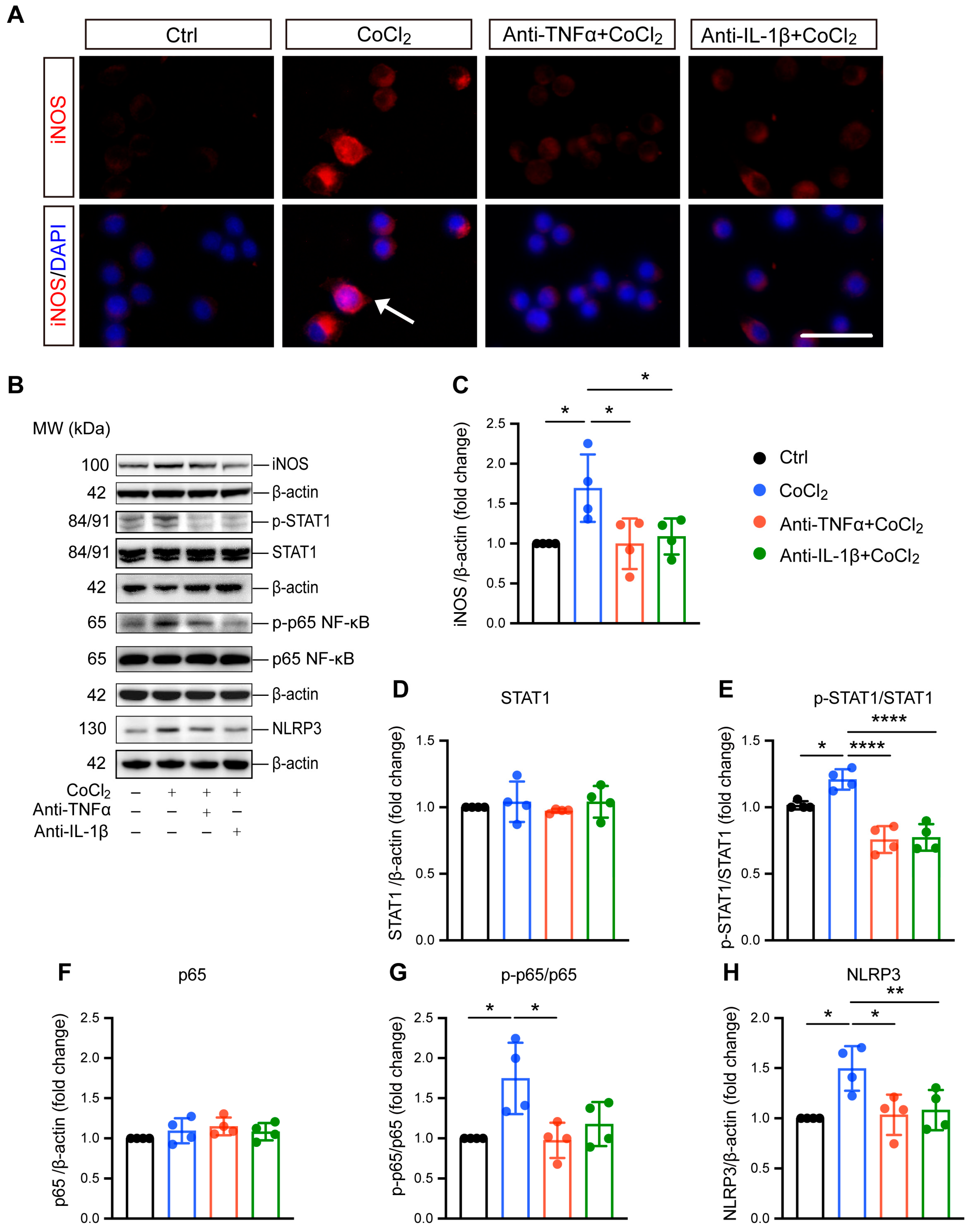
3.4. Anti-TNFα and Anti-IL-1β Suppress Hypoxia-Induced Microglial Reactivity by Inhibiting STAT1 and NF-κB/NLRP3 Pathways
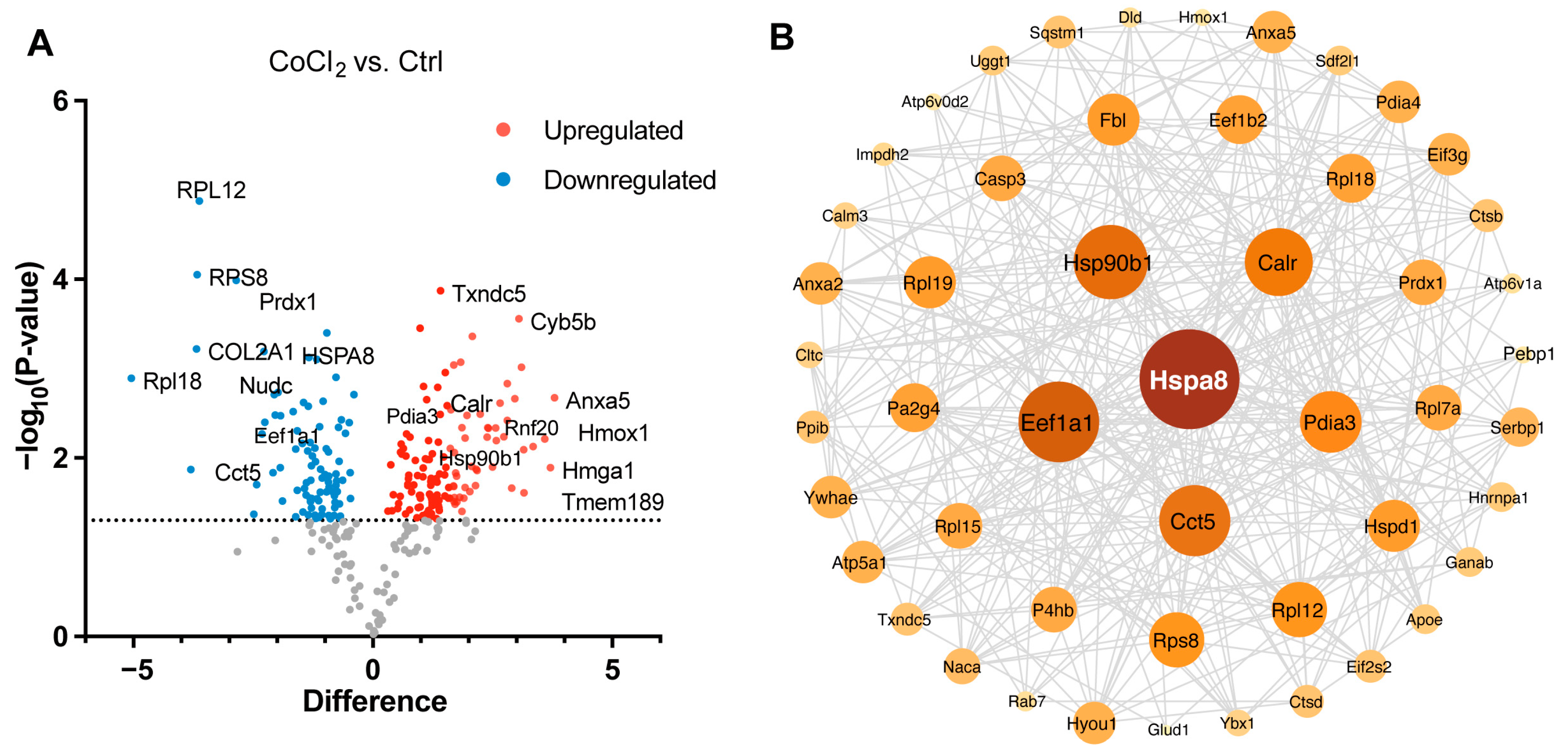
3.5. Hierarchical Clustering Analysis of Differentially Expressed Proteins in BV-2 Cells
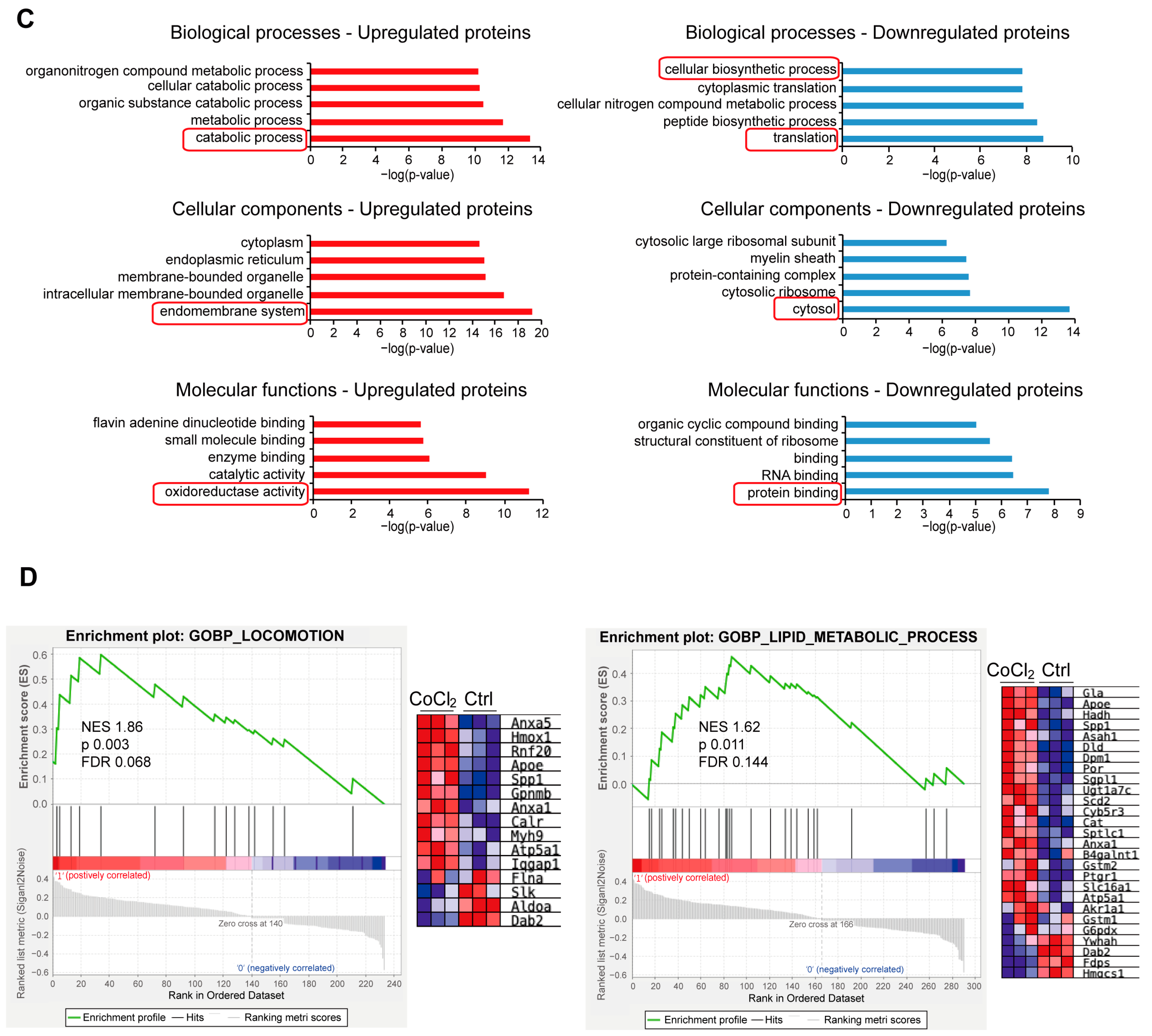
3.6. Gene Ontology Classification and Key Proteins Involved in Hypoxia-Damaged BV-2 Microglia
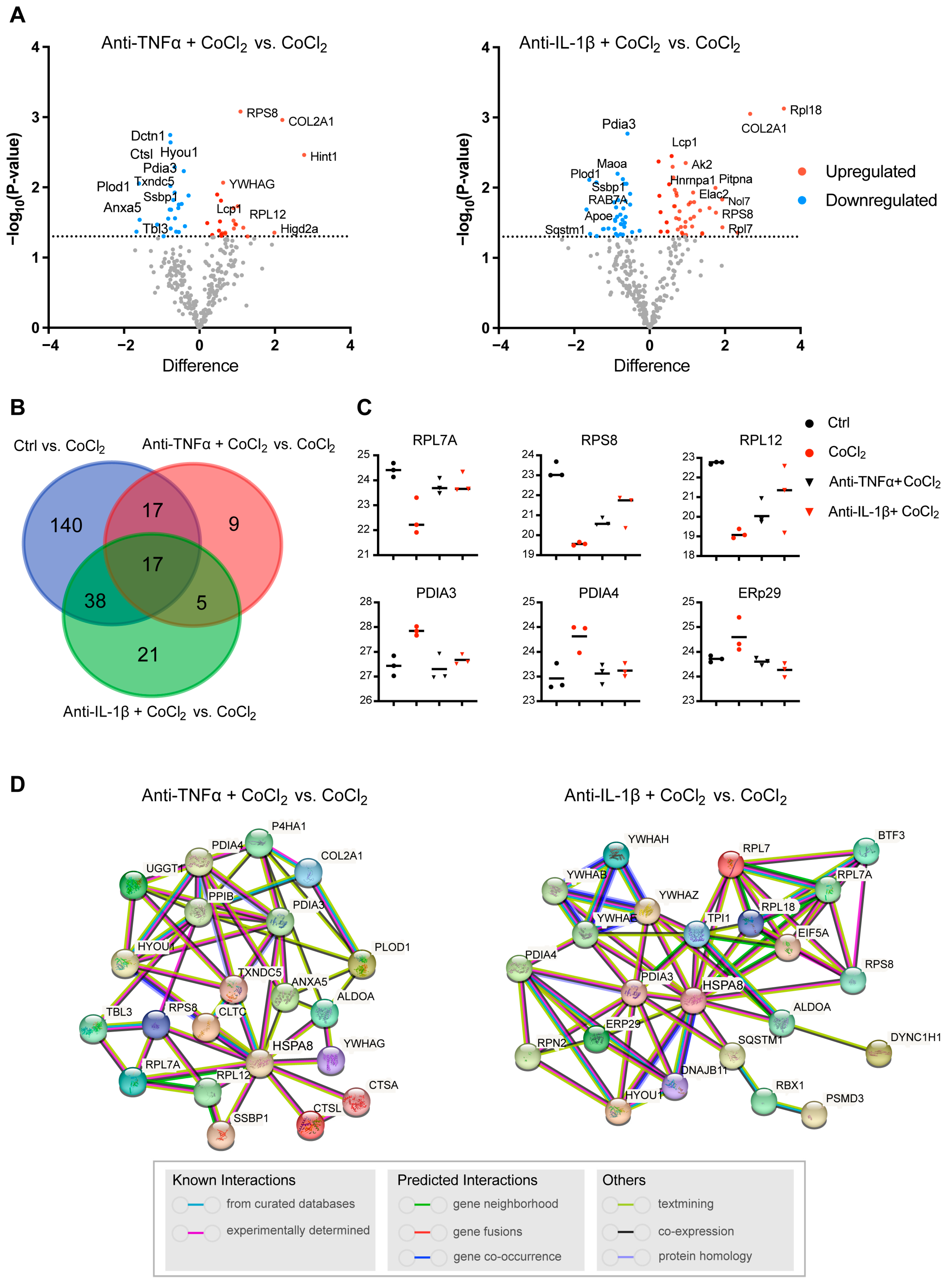
3.7. Network Analysis of Recovery Process Mediators
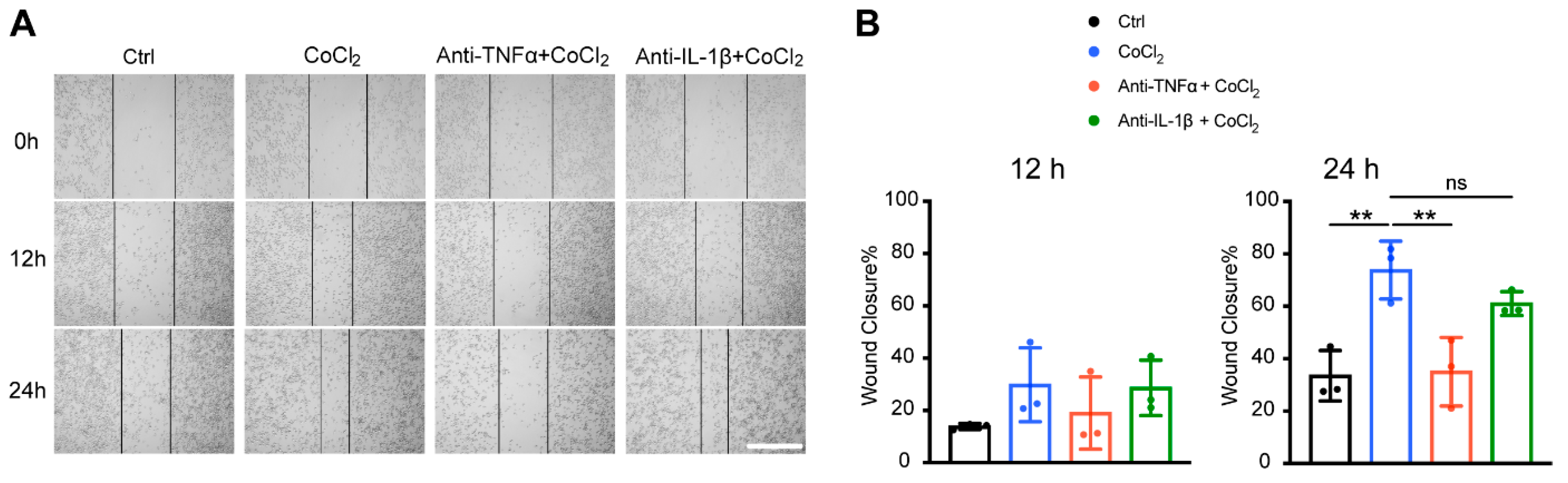
3.8. Anti-TNFα, but Not Anti-IL-1β, Inhibited Hypoxia-Promoted Microglia Migration
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| ANXA5 | Annexin A5 |
| APOE | Apolipoprotein E |
| ATF4 | Activating transcription factor 4 |
| CoCl2 | Cobalt chloride |
| Ctrl | Control |
| DEP | Differentially expressed protein |
| ER | Endoplasmic reticulum |
| HSPA8 | Heat shock protein family A (Hsp70) member 8 |
| HYOU1 | Hypoxia upregulated 1 |
| IL-1β | Interleukin-1 beta |
| IL-1R | Interleukin-1 receptor |
| iNOS | Inducible nitric oxide synthase |
| LPS | Lipopolysaccharide |
| MAPK | Mitogen-activated protein kinase |
| MMP | Mitochondrial membrane potential |
| MS | Mass spectrometry |
| NF-κB | Nuclear factor kappa B |
| NLRP3 | NLR family pyrin domain-containing 3 |
| NRF2 | Nuclear factor erythroid 2-related factor 2 |
| PDIA3 | Protein disulfide-isomerase A3 |
| PDIA4 | Protein disulfide-isomerase A4 |
| PLOD1 | Procollagen-lysine,2-oxoglutarate 5-dioxygenase 1 |
| PPI | Protein–protein interaction |
| PPIB | Peptidylprolyl isomerase B |
| ROS | Reactive oxygen species |
| RPL7A | Ribosomal protein L7a |
| RPL12 | Ribosomal protein L12 |
| RPL18 | Ribosomal protein L18 |
| RPS8 | Ribosomal protein S8 |
| STAT1 | Signal transducer and activator of transcription 1 |
| TNF-α | Tumor necrosis factor alpha |
| TNFR | Tumor necrosis factor receptor |
| TUNEL | Terminal deoxynucleotidyl transferase dUTP nick end labeling |
| TXNDC5 | Thioredoxin domain-containing protein 5 |
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Host | Working Dilution | Supplier | RRID |
|---|---|---|---|---|
| TNFα | Armenian hamster | 1:12.5 | Thermo Fisher Scientific, Waltham, MA, USA. | AB_468491 |
| IL-1β | Armenian hamster | 1:12.5 | Thermo Fisher Scientific, Waltham, MA, USA. | AB_468396 |
| NLRP3 | Rabbit | 1:1000 | Thermo Fisher Scientific, Waltham, MA, USA. | AB_2224377 |
| β-actin | Rabbit | 1:2000 | Thermo Fisher Scientific, Waltham, MA, USA. | AB_10855480 |
| NF-κB p65 | Mouse | 1:1000 | Cell Signaling Technology, Danvers, MA, USA. | AB_10828935 |
| phospho-NF-κB | Rabbit | 1:1000 | Cell Signaling Technology, Danvers, MA, USA. | AB_331284 |
| STAT1 | Rabbit | 1:1000 | Cell Signaling Technology, Danvers, MA, USA. | AB_2198300 |
| phospho-STAT1 | Rabbit | 1:1000 | Cell Signaling Technology, Danvers, MA, USA. | AB_10950970 |
| iNOS | Rabbit | 1:1000 | Proteintech, Rosemont, IL, USA. | AB_2782960 |
| p38 MAPK | Rabbit | 1:1000 | Abclonal, Woburn, MA, USA. | AB_2863345 |
| phospho-p38 MAPK | Rabbit | 1:1000 | Abclonal, Woburn, MA, USA. | AB_2864024 |
| ATF4 | Rabbit | 1:1000 | GeneTex, Irvine, CA, USA. | AB_1240487 |
| NRF2 | Rabbit | 1:1000 | Proteintech, Rosemont, IL, USA. | AB_2782956 |
| anti-rabbit (HRP) secondary antibody | Goat | 1:8000 | Abcam, Cambridge, UK. | AB_955447 |
| iNOS | Mouse | 1:200 | Santa Cruz, Dallas, TX, USA. | AB_627810 |
| anti-mouse secondary antibody (Alexa Fluor® 488) | Goat | 1:400 | Thermo Fisher Scientific, Waltham, MA, USA. | AB_2895153 |
References
- Gnanasambandam, B.; Prince, J.; Limaye, S.; Moran, E.; Lee, B.; Huynh, J.; Irudayaraj, J.; Tsipursky, M. Addressing retinal hypoxia: Pathophysiology, therapeutic innovations, and future prospects. Ther. Adv. Ophthalmol. 2024, 16, 25158414241280187. [Google Scholar] [CrossRef] [PubMed]
- Tham, Y.C.; Li, X.; Wong, T.Y.; Quigley, H.A.; Aung, T.; Cheng, C.Y. Global prevalence of glaucoma and projections of glaucoma burden through 2040: A systematic review and meta-analysis. Ophthalmology 2014, 121, 2081–2090. [Google Scholar] [CrossRef] [PubMed]
- Flammer, J.; Orgul, S.; Costa, V.P.; Orzalesi, N.; Krieglstein, G.K.; Serra, L.M.; Renard, J.P.; Stefansson, E. The impact of ocular blood flow in glaucoma. Prog. Retin. Eye Res. 2002, 21, 359–393. [Google Scholar] [CrossRef] [PubMed]
- Donkor, N.; Gardner, J.J.; Bradshaw, J.L.; Cunningham, R.L.; Inman, D.M. Ocular Inflammation and Oxidative Stress as a Result of Chronic Intermittent Hypoxia: A Rat Model of Sleep Apnea. Antioxidants 2024, 13, 878. [Google Scholar] [CrossRef]
- Karlstetter, M.; Scholz, R.; Rutar, M.; Wong, W.T.; Provis, J.M.; Langmann, T. Retinal microglia: Just bystander or target for therapy? Prog. Retin. Eye Res. 2015, 45, 30–57. [Google Scholar] [CrossRef]
- Langmann, T. Microglia activation in retinal degeneration. J. Leukoc. Biol. 2007, 81, 1345–1351. [Google Scholar] [CrossRef]
- Kaur, C.; Sivakumar, V.; Zou, Z.; Ling, E.A. Microglia-derived proinflammatory cytokines tumor necrosis factor-alpha and interleukin-1beta induce Purkinje neuronal apoptosis via their receptors in hypoxic neonatal rat brain. Brain Struct. Funct. 2014, 219, 151–170. [Google Scholar] [CrossRef]
- Pearse, D.D.; Pereira, F.C.; Stolyarova, A.; Barakat, D.J.; Bunge, M.B. Inhibition of tumour necrosis factor-alpha by antisense targeting produces immunophenotypical and morphological changes in injury-activated microglia and macrophages. Eur. J. Neurosci. 2004, 20, 3387–3396. [Google Scholar] [CrossRef]
- Natoli, R.; Fernando, N.; Madigan, M.; Chu-Tan, J.A.; Valter, K.; Provis, J.; Rutar, M. Microglia-derived IL-1beta promotes chemokine expression by Muller cells and RPE in focal retinal degeneration. Mol. Neurodegener. 2017, 12, 31. [Google Scholar] [CrossRef]
- Sivakumar, V.; Foulds, W.S.; Luu, C.D.; Ling, E.A.; Kaur, C. Retinal ganglion cell death is induced by microglia derived pro-inflammatory cytokines in the hypoxic neonatal retina. J. Pathol. 2011, 224, 245–260. [Google Scholar] [CrossRef]
- Au, N.P.B.; Ma, C.H.E. Neuroinflammation, Microglia and Implications for Retinal Ganglion Cell Survival and Axon Regeneration in Traumatic Optic Neuropathy. Front. Immunol. 2022, 13, 860070. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Feng, S.; Qin, H.; Yan, L.; Zheng, C.; Yao, K. Microglia: The breakthrough to treat neovascularization and repair blood-retinal barrier in retinopathy. Front. Mol. Neurosci. 2023, 16, 1100254. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Zhao, G.L.; Xu, M.X.; Zhou, H.; Li, F.; Miao, Y.; Lei, B.; Yang, X.L.; Wang, Z. Interplay between Muller cells and microglia aggravates retinal inflammatory response in experimental glaucoma. J. Neuroinflammation 2021, 18, 303. [Google Scholar] [CrossRef]
- Zigon-Branc, S.; Barlic, A.; Knezevic, M.; Jeras, M.; Vunjak-Novakovic, G. Testing the potency of anti-TNF-α and anti-IL-1β drugs using spheroid cultures of human osteoarthritic chondrocytes and donor-matched chondrogenically differentiated mesenchymal stem cells. Biotechnol. Prog. 2018, 34, 1045–1058. [Google Scholar] [CrossRef]
- Li, P.; Zheng, Y.; Chen, X. Drugs for Autoimmune Inflammatory Diseases: From Small Molecule Compounds to Anti-TNF Biologics. Front. Pharmacol. 2017, 8, 460. [Google Scholar] [CrossRef]
- Peter, I.; Dubinsky, M.; Bressman, S.; Park, A.; Lu, C.; Chen, N.; Wang, A. Anti-Tumor Necrosis Factor Therapy and Incidence of Parkinson Disease Among Patients With Inflammatory Bowel Disease. JAMA Neurol. 2018, 75, 939–946. [Google Scholar] [CrossRef]
- Chen, X.; Hovanesian, V.; Naqvi, S.; Lim, Y.P.; Tucker, R.; Donahue, J.E.; Stopa, E.G.; Stonestreet, B.S. Systemic infusions of anti-interleukin-1beta neutralizing antibodies reduce short-term brain injury after cerebral ischemia in the ovine fetus. Brain Behav. Immun. 2018, 67, 24–35. [Google Scholar] [CrossRef]
- Blasi, E.; Barluzzi, R.; Bocchini, V.; Mazzolla, R.; Bistoni, F. Immortalization of murine microglial cells by a v-raf/v-myc carrying retrovirus. J. Neuroimmunol. 1990, 27, 229–237. [Google Scholar] [CrossRef]
- Perumal, N.; Strassburger, L.; Herzog, D.P.; Muller, M.B.; Pfeiffer, N.; Grus, F.H.; Manicam, C. Bioenergetic shift and actin cytoskeleton remodelling as acute vascular adaptive mechanisms to angiotensin II in murine retina and ophthalmic artery. Redox Biol. 2020, 34, 101597. [Google Scholar] [CrossRef]
- Di Mattia, M.; Mauro, A.; Delle Monache, S.; Pulcini, F.; Russo, V.; Berardinelli, P.; Citeroni, M.R.; Turriani, M.; Peserico, A.; Barboni, B. Hypoxia-Mimetic CoCl(2) Agent Enhances Pro-Angiogenic Activities in Ovine Amniotic Epithelial Cells-Derived Conditioned Medium. Cells 2022, 11, 461. [Google Scholar] [CrossRef]
- Moncao, C.C.D.; Scrideli, C.A.; Andrade, A.F.; Viapiano, M.S.; Carlotti, C.G.; Moreno, D.A.; Baroni, M.; Tone, L.G.; Teixeira, S.A. Indisulam Reduces Viability and Regulates Apoptotic Gene Expression in Pediatric High-Grade Glioma Cells. Biomedicines 2022, 11, 68. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Yoshinari, T. A multicenter, open-label, long-term study of three-year infliximab administration in Japanese patients with ankylosing spondylitis. Mod. Rheumatol. 2017, 27, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Chen, Y.; Pan, X.; Li, P.; Ren, Z.; Wang, X.; Chen, Y.; Shen, S.; Wang, T.; Lin, A. IL-1beta mediates Candida tropicalis-induced immunosuppressive function of MDSCs to foster colorectal cancer. Cell Commun. Signal 2024, 22, 408. [Google Scholar] [CrossRef]
- Wu, Y.; Chen, M.; Jiang, J. Mitochondrial dysfunction in neurodegenerative diseases and drug targets via apoptotic signaling. Mitochondrion 2019, 49, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Sun, L.; Chen, X.; Zhang, D. Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural Regen. Res. 2013, 8, 2003–2014. [Google Scholar] [CrossRef]
- Lange, P.S.; Chavez, J.C.; Pinto, J.T.; Coppola, G.; Sun, C.W.; Townes, T.M.; Geschwind, D.H.; Ratan, R.R. ATF4 is an oxidative stress-inducible, prodeath transcription factor in neurons in vitro and in vivo. J. Exp. Med. 2008, 205, 1227–1242. [Google Scholar] [CrossRef]
- Wendimu, M.Y.; Hooks, S.B. Microglia Phenotypes in Aging and Neurodegenerative Diseases. Cells 2022, 11, 2091. [Google Scholar] [CrossRef]
- Zhao, Y.; Ma, C.; Chen, C.; Li, S.; Wang, Y.; Yang, T.; Stetler, R.A.; Bennett, M.V.L.; Dixon, C.E.; Chen, J.; et al. STAT1 contributes to microglial/macrophage inflammation and neurological dysfunction in a mouse model of traumatic brain injury. J. Neurosci. 2022, 42, 7466–7481. [Google Scholar] [CrossRef]
- Slusarczyk, J.; Trojan, E.; Glombik, K.; Piotrowska, A.; Budziszewska, B.; Kubera, M.; Popiolek-Barczyk, K.; Lason, W.; Mika, J.; Basta-Kaim, A. Targeting the NLRP3 Inflammasome-Related Pathways via Tianeptine Treatment-Suppressed Microglia Polarization to the M1 Phenotype in Lipopolysaccharide-Stimulated Cultures. Int. J. Mol. Sci. 2018, 19, 1965. [Google Scholar] [CrossRef]
- Kitazawa, M.; Cheng, D.; Tsukamoto, M.R.; Koike, M.A.; Wes, P.D.; Vasilevko, V.; Cribbs, D.H.; LaFerla, F.M. Blocking IL-1 signaling rescues cognition, attenuates tau pathology, and restores neuronal beta-catenin pathway function in an Alzheimer’s disease model. J. Immunol. 2011, 187, 6539–6549. [Google Scholar] [CrossRef]
- Diaz-Canestro, C.; Reiner, M.F.; Bonetti, N.R.; Liberale, L.; Merlini, M.; Wust, P.; Amstalden, H.; Briand-Schumacher, S.; Semerano, A.; Giacalone, G.; et al. AP-1 (Activated Protein-1) Transcription Factor JunD Regulates Ischemia/Reperfusion Brain Damage via IL-1beta (Interleukin-1beta). Stroke 2019, 50, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.Q.; Fang, Z.; Chen, X.L.; Yang, S.; Zhou, Y.F.; Mao, L.; Xia, Y.P.; Jin, H.J.; Li, Y.N.; You, M.F.; et al. Microglia-derived TNF-alpha mediates endothelial necroptosis aggravating blood brain-barrier disruption after ischemic stroke. Cell Death Dis. 2019, 10, 487. [Google Scholar] [CrossRef] [PubMed]
- Muccini, A.M.; Tran, N.T.; Hale, N.; McKenzie, M.; Snow, R.J.; Walker, D.W.; Ellery, S.J. The Effects of In Utero Fetal Hypoxia and Creatine Treatment on Mitochondrial Function in the Late Gestation Fetal Sheep Brain. Oxid. Med. Cell Longev. 2022, 2022, 3255296. [Google Scholar] [CrossRef] [PubMed]
- Plascencia-Villa, G.; Perry, G. Exploring Molecular Targets for Mitochondrial Therapies in Neurodegenerative Diseases. Int. J. Mol. Sci. 2023, 24, 12486. [Google Scholar] [CrossRef]
- Ying, Z.; Ye, N.; Ma, Q.; Chen, F.; Li, N.; Zhen, X. Targeted to neuronal organelles for CNS drug development. Adv. Drug Deliv. Rev. 2023, 200, 115025. [Google Scholar] [CrossRef]
- Bhootada, Y.; Kotla, P.; Zolotukhin, S.; Gorbatyuk, O.; Bebok, Z.; Athar, M.; Gorbatyuk, M. Limited ATF4 Expression in Degenerating Retinas with Ongoing ER Stress Promotes Photoreceptor Survival in a Mouse Model of Autosomal Dominant Retinitis Pigmentosa. PLoS ONE 2016, 11, e0154779. [Google Scholar] [CrossRef]
- Gully, J.C.; Sergeyev, V.G.; Bhootada, Y.; Mendez-Gomez, H.; Meyers, C.A.; Zolotukhin, S.; Gorbatyuk, M.S.; Gorbatyuk, O.S. Up-regulation of activating transcription factor 4 induces severe loss of dopamine nigral neurons in a rat model of Parkinson’s disease. Neurosci. Lett. 2016, 627, 36–41. [Google Scholar] [CrossRef]
- Aime, P.; Karuppagounder, S.S.; Rao, A.; Chen, Y.; Burke, R.E.; Ratan, R.R.; Greene, L.A. The drug adaptaquin blocks ATF4/CHOP-dependent pro-death Trib3 induction and protects in cellular and mouse models of Parkinson’s disease. Neurobiol. Dis. 2020, 136, 104725. [Google Scholar] [CrossRef]
- Tezel, G.; Chauhan, B.C.; LeBlanc, R.P.; Wax, M.B. Immunohistochemical assessment of the glial mitogen-activated protein kinase activation in glaucoma. Invest. Ophthalmol. Vis. Sci. 2003, 44, 3025–3033. [Google Scholar] [CrossRef]
- Meng, J.; Wang, D.M.; Luo, L.L. CTRP3 acts as a novel regulator in depressive-like behavior associated inflammation and apoptosis by meditating p38 and JNK MAPK signaling. Biomed. Pharmacother. 2019, 120, 109489. [Google Scholar] [CrossRef]
- Grab, J.; Rybniker, J. The Expanding Role of p38 Mitogen-Activated Protein Kinase in Programmed Host Cell Death. Microbiol. Insights 2019, 12, 1178636119864594. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Deng, B.; Yan, P.; Wu, H.; Li, C.; Zhu, H.; Du, J.; Hou, L. Neuroprotective effect of ketamine against TNF-alpha-induced necroptosis in hippocampal neurons. J. Cell Mol. Med. 2021, 25, 3449–3459. [Google Scholar] [CrossRef] [PubMed]
- Savard, A.; Brochu, M.E.; Chevin, M.; Guiraut, C.; Grbic, D.; Sebire, G. Neuronal self-injury mediated by IL-1beta and MMP-9 in a cerebral palsy model of severe neonatal encephalopathy induced by immune activation plus hypoxia-ischemia. J. Neuroinflammation 2015, 12, 111. [Google Scholar] [CrossRef] [PubMed]
- Rossi, S.; Furlan, R.; De Chiara, V.; Muzio, L.; Musella, A.; Motta, C.; Studer, V.; Cavasinni, F.; Bernardi, G.; Martino, G.; et al. Cannabinoid CB1 receptors regulate neuronal TNF-alpha effects in experimental autoimmune encephalomyelitis. Brain Behav. Immun. 2011, 25, 1242–1248. [Google Scholar] [CrossRef]
- Mandolesi, G.; Musella, A.; Gentile, A.; Grasselli, G.; Haji, N.; Sepman, H.; Fresegna, D.; Bullitta, S.; De Vito, F.; Musumeci, G.; et al. Interleukin-1beta alters glutamate transmission at purkinje cell synapses in a mouse model of multiple sclerosis. J. Neurosci. 2013, 33, 12105–12121. [Google Scholar] [CrossRef]
- Tremblay, M.E.; Stevens, B.; Sierra, A.; Wake, H.; Bessis, A.; Nimmerjahn, A. The role of microglia in the healthy brain. J. Neurosci. 2011, 31, 16064–16069. [Google Scholar] [CrossRef]
- Vidal-Itriago, A.; Radford, R.A.W.; Aramideh, J.A.; Maurel, C.; Scherer, N.M.; Don, E.K.; Lee, A.; Chung, R.S.; Graeber, M.B.; Morsch, M. Microglia morphophysiological diversity and its implications for the CNS. Front. Immunol. 2022, 13, 997786. [Google Scholar] [CrossRef]
- Paolicelli, R.C.; Sierra, A.; Stevens, B.; Tremblay, M.E.; Aguzzi, A.; Ajami, B.; Amit, I.; Audinat, E.; Bechmann, I.; Bennett, M.; et al. Microglia states and nomenclature: A field at its crossroads. Neuron 2022, 110, 3458–3483. [Google Scholar] [CrossRef]
- Butturini, E.; Boriero, D.; Carcereri de Prati, A.; Mariotto, S. STAT1 drives M1 microglia activation and neuroinflammation under hypoxia. Arch. Biochem. Biophys. 2019, 669, 22–30. [Google Scholar] [CrossRef]
- Feng, X.; Li, M.; Lin, Z.; Lu, Y.; Zhuang, Y.; Lei, J.; Liu, X.; Zhao, H. Tetramethylpyrazine promotes axonal remodeling and modulates microglial polarization via JAK2-STAT1/3 and GSK3-NFkappaB pathways in ischemic stroke. Neurochem. Int. 2023, 170, 105607. [Google Scholar] [CrossRef]
- Boriero, D.; Carcereri de Prati, A.; Antonini, L.; Ragno, R.; Sohji, K.; Mariotto, S.; Butturini, E. The anti-STAT1 polyphenol myricetin inhibits M1 microglia activation and counteracts neuronal death. FEBS J. 2021, 288, 2347–2359. [Google Scholar] [CrossRef] [PubMed]
- Guo, K.; Mou, X.; Huang, J.; Xiong, N.; Li, H. Trans-caryophyllene suppresses hypoxia-induced neuroinflammatory responses by inhibiting NF-kappaB activation in microglia. J. Mol. Neurosci. 2014, 54, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Sul, O.J.; Ra, S.W. Quercetin Prevents LPS-Induced Oxidative Stress and Inflammation by Modulating NOX2/ROS/NF-kB in Lung Epithelial Cells. Molecules 2021, 26, 6949. [Google Scholar] [CrossRef]
- Companys-Alemany, J.; Turcu, A.L.; Vazquez, S.; Pallas, M.; Grinan-Ferre, C. Glial cell reactivity and oxidative stress prevention in Alzheimer’s disease mice model by an optimized NMDA receptor antagonist. Sci. Rep. 2022, 12, 17908. [Google Scholar] [CrossRef]
- Verstrepen, L.; Bekaert, T.; Chau, T.L.; Tavernier, J.; Chariot, A.; Beyaert, R. TLR-4, IL-1R and TNF-R signaling to NF-kappaB: Variations on a common theme. Cell Mol. Life Sci. 2008, 65, 2964–2978. [Google Scholar] [CrossRef]
- Kitanaka, T.; Nakano, R.; Kitanaka, N.; Kimura, T.; Okabayashi, K.; Narita, T.; Sugiya, H. JNK activation is essential for activation of MEK/ERK signaling in IL-1beta-induced COX-2 expression in synovial fibroblasts. Sci. Rep. 2017, 7, 39914. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-kappaB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef]
- Verma, N.; Chaudhury, I.; Kumar, D.; Das, R.H. Silencing of TNF-alpha receptors coordinately suppresses TNF-alpha expression through NF-kappaB activation blockade in THP-1 macrophage. FEBS Lett. 2009, 583, 2968–2974. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, Y.; Sun, R.; Wu, X.; Chu, X.; Zhou, S.; Hu, X.; Gao, L.; Kong, Q. The treatment value of IL-1beta monoclonal antibody under the targeting location of alpha-methyl-L-tryptophan and superparamagnetic iron oxide nanoparticles in an acute temporal lobe epilepsy model. J. Transl. Med. 2018, 16, 337. [Google Scholar] [CrossRef]
- Hong, Z.Y.; Shi, X.R.; Zhu, K.; Wu, T.T.; Zhu, Y.Z. SCM-198 inhibits microglial overactivation and attenuates Abeta(1-40)-induced cognitive impairments in rats via JNK and NF-small ka, CyrillicB pathways. J. Neuroinflammation 2014, 11, 147. [Google Scholar] [CrossRef]
- Yang, Y.; Hao, T.; Yao, X.; Che, Y.; Liu, Y.; Fang, M.; Wang, Y.; Zhou, D.; Chai, H.; Li, N.; et al. Crebanine ameliorates ischemia-reperfusion brain damage by inhibiting oxidative stress and neuroinflammation mediated by NADPH oxidase 2 in microglia. Phytomedicine 2023, 120, 155044. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, W.W.; Skalicka-Wozniak, K.; Budzynska, B.; El Sayed, N.S. NLRP3 inflammasome inhibition and M1-to-M2 microglial polarization shifting via scoparone-inhibited TLR4 axis in ovariectomy/D-galactose Alzheimer’s disease rat model. Int. Immunopharmacol. 2023, 119, 110239. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Deng, Y.; Gan, X.; Li, Y.; Huang, W.; Lu, L.; Wei, L.; Su, L.; Luo, J.; Zou, B.; et al. NLRP12 collaborates with NLRP3 and NLRC4 to promote pyroptosis inducing ganglion cell death of acute glaucoma. Mol. Neurodegener. 2020, 15, 26. [Google Scholar] [CrossRef] [PubMed]
- Stein, K.C.; Morales-Polanco, F.; van der Lienden, J.; Rainbolt, T.K.; Frydman, J. Ageing exacerbates ribosome pausing to disrupt cotranslational proteostasis. Nature 2022, 601, 637–642. [Google Scholar] [CrossRef]
- Charif, S.E.; Vassallu, M.F.; Salvanal, L.; Igaz, L.M. Protein synthesis modulation as a therapeutic approach for amyotrophic lateral sclerosis and frontotemporal dementia. Neural Regen. Res. 2022, 17, 1423–1430. [Google Scholar] [CrossRef]
- Ding, Q.; Markesbery, W.R.; Chen, Q.; Li, F.; Keller, J.N. Ribosome dysfunction is an early event in Alzheimer’s disease. J. Neurosci. 2005, 25, 9171–9175. [Google Scholar] [CrossRef]
- Wang, C.Y.; Xie, J.W.; Wang, T.; Xu, Y.; Cai, J.H.; Wang, X.; Zhao, B.L.; An, L.; Wang, Z.Y. Hypoxia-triggered m-calpain activation evokes endoplasmic reticulum stress and neuropathogenesis in a transgenic mouse model of Alzheimer’s disease. CNS Neurosci. Ther. 2013, 19, 820–833. [Google Scholar] [CrossRef]
- Yadav, S.K.; Jauhari, A.; Singh, N.; Pandey, A.; Sarkar, S.; Pandey, S.; Garg, R.K.; Parmar, D.; Yadav, S. Transcriptomics and Proteomics Approach for the Identification of Altered Blood microRNAs and Plasma Proteins in Parkinson’s Disease. Cell Mol. Neurobiol. 2023, 43, 3527–3553. [Google Scholar] [CrossRef]
- Ma, M.; Cheng, Y.; Hou, X.; Li, Z.; Wang, M.; Ma, B.; Cheng, Q.; Ding, Z.; Feng, H. Serum biomarkers in patients with drug-resistant epilepsy: A proteomics-based analysis. Front. Neurol. 2024, 15, 1383023. [Google Scholar] [CrossRef]
- Wolzak, K.; Vermunt, L.; Campo, M.D.; Jorge-Oliva, M.; van Ziel, A.M.; Li, K.W.; Smit, A.B.; Chen-Ploktkin, A.; Irwin, D.J.; Lemstra, A.W.; et al. Protein disulfide isomerases as CSF biomarkers for the neuronal response to tau pathology. Alzheimers Dement. 2023, 19, 3563–3574. [Google Scholar] [CrossRef]
- Muraoka, S.; DeLeo, A.M.; Sethi, M.K.; Yukawa-Takamatsu, K.; Yang, Z.; Ko, J.; Hogan, J.D.; Ruan, Z.; You, Y.; Wang, Y.; et al. Proteomic and biological profiling of extracellular vesicles from Alzheimer’s disease human brain tissues. Alzheimers Dement. 2020, 16, 896–907. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Li, J.; Weng, L. Identification and Validation of Aging-Related Genes in Alzheimer’s Disease. Front. Neurosci. 2022, 16, 905722. [Google Scholar] [CrossRef]
- Krasemann, S.; Madore, C.; Cialic, R.; Baufeld, C.; Calcagno, N.; El Fatimy, R.; Beckers, L.; O’Loughlin, E.; Xu, Y.; Fanek, Z.; et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 2017, 47, 566–581.e569. [Google Scholar] [CrossRef]
- Urquhart, K.R.; Zhao, Y.; Baker, J.A.; Lu, Y.; Yan, L.; Cook, M.N.; Jones, B.C.; Hamre, K.M.; Lu, L. A novel heat shock protein alpha 8 (Hspa8) molecular network mediating responses to stress- and ethanol-related behaviors. Neurogenetics 2016, 17, 91–105. [Google Scholar] [CrossRef]
- Wu, E.; He, W.; Wu, C.; Chen, Z.; Zhou, S.; Wu, X.; Hu, Z.; Jia, K.; Pan, J.; Wang, L.; et al. HSPA8 acts as an amyloidase to suppress necroptosis by inhibiting and reversing functional amyloid formation. Cell Res. 2023, 33, 851–866. [Google Scholar] [CrossRef]
- Zhao, N.; Hao, X.N.; Huang, J.M.; Song, Z.M.; Tao, Y. Crosstalk Between Microglia and Muller Glia in the Age-Related Macular Degeneration: Role and Therapeutic Value of Neuroinflammation. Aging Dis. 2023, 15, 1132–1154. [Google Scholar] [CrossRef]
- Wang, H.; Wang, X.; Shen, Y.; Wang, Y.; Yang, T.; Sun, J.; Liu, S. SENP1 modulates chronic intermittent hypoxia-induced inflammation of microglia and neuronal injury by inhibiting TOM1 pathway. Int. Immunopharmacol. 2023, 119, 110230. [Google Scholar] [CrossRef]
- Jeon, H.; Kim, J.H.; Kim, J.H.; Lee, W.H.; Lee, M.S.; Suk, K. Plasminogen activator inhibitor type 1 regulates microglial motility and phagocytic activity. J. Neuroinflammation 2012, 9, 149. [Google Scholar] [CrossRef]
- Torrente, Y.; El Fahime, E.; Caron, N.J.; Del Bo, R.; Belicchi, M.; Pisati, F.; Tremblay, J.P.; Bresolin, N. Tumor necrosis factor-alpha (TNF-alpha) stimulates chemotactic response in mouse myogenic cells. Cell Transplant. 2003, 12, 91–100. [Google Scholar] [CrossRef]
- Vera, M.J.; Guajardo, F.; Urra, F.A.; Tobar, N.; Martinez, J. TNF-Alpha Promotes an Inflammatory Mammary Microenvironment That Favors Macrophage and Epithelial Migration in a CCL2- and Mitochondrial-ROS-Dependent Manner. Antioxidants 2023, 12, 813. [Google Scholar] [CrossRef]
- Lee, M.J.; Kim, J.; Kim, M.Y.; Bae, Y.S.; Ryu, S.H.; Lee, T.G.; Kim, J.H. Proteomic analysis of tumor necrosis factor-alpha-induced secretome of human adipose tissue-derived mesenchymal stem cells. J. Proteome Res. 2010, 9, 1754–1762. [Google Scholar] [CrossRef] [PubMed]
- Kulbe, H.; Thompson, R.; Wilson, J.L.; Robinson, S.; Hagemann, T.; Fatah, R.; Gould, D.; Ayhan, A.; Balkwill, F. The inflammatory cytokine tumor necrosis factor-alpha generates an autocrine tumor-promoting network in epithelial ovarian cancer cells. Cancer Res. 2007, 67, 585–592. [Google Scholar] [CrossRef] [PubMed]
- Chuang, M.J.; Sun, K.H.; Tang, S.J.; Deng, M.W.; Wu, Y.H.; Sung, J.S.; Cha, T.L.; Sun, G.H. Tumor-derived tumor necrosis factor-alpha promotes progression and epithelial-mesenchymal transition in renal cell carcinoma cells. Cancer Sci. 2008, 99, 905–913. [Google Scholar] [CrossRef] [PubMed]
- Chou, C.C.; Wang, C.P.; Chen, J.H.; Lin, H.H. Anti-Atherosclerotic Effect of Hibiscus Leaf Polyphenols against Tumor Necrosis Factor-alpha-Induced Abnormal Vascular Smooth Muscle Cell Migration and Proliferation. Antioxidants 2019, 8, 620. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, L.; Guan, C.; Wang, S.; Pfeiffer, N.; Grus, F.H. Anti-TNFα and Anti-IL-1β Monoclonal Antibodies Preserve BV-2 Microglial Homeostasis Under Hypoxia by Mitigating Inflammatory Reactivity and ATF4/MAPK-Mediated Apoptosis. Antioxidants 2025, 14, 363. https://doi.org/10.3390/antiox14030363
Zhang L, Guan C, Wang S, Pfeiffer N, Grus FH. Anti-TNFα and Anti-IL-1β Monoclonal Antibodies Preserve BV-2 Microglial Homeostasis Under Hypoxia by Mitigating Inflammatory Reactivity and ATF4/MAPK-Mediated Apoptosis. Antioxidants. 2025; 14(3):363. https://doi.org/10.3390/antiox14030363
Chicago/Turabian StyleZhang, Linglin, Chaoqiang Guan, Sudena Wang, Norbert Pfeiffer, and Franz H. Grus. 2025. "Anti-TNFα and Anti-IL-1β Monoclonal Antibodies Preserve BV-2 Microglial Homeostasis Under Hypoxia by Mitigating Inflammatory Reactivity and ATF4/MAPK-Mediated Apoptosis" Antioxidants 14, no. 3: 363. https://doi.org/10.3390/antiox14030363
APA StyleZhang, L., Guan, C., Wang, S., Pfeiffer, N., & Grus, F. H. (2025). Anti-TNFα and Anti-IL-1β Monoclonal Antibodies Preserve BV-2 Microglial Homeostasis Under Hypoxia by Mitigating Inflammatory Reactivity and ATF4/MAPK-Mediated Apoptosis. Antioxidants, 14(3), 363. https://doi.org/10.3390/antiox14030363