
Renin–Angiotensin System Autoantibody Network in Parkinson’s Disease Patients

 , , and
, , and
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Description of the Study Population
2.2. Sample Collection and Storage
2.3. Clinical Follow-Up and Data Collection
2.4. Quantification of Autoantibodies
2.5. Quantification of Cytokines and 27-Hydroxycholesterol
2.6. Statistical Analysis
3. Results
3.1. Patient Characteristics
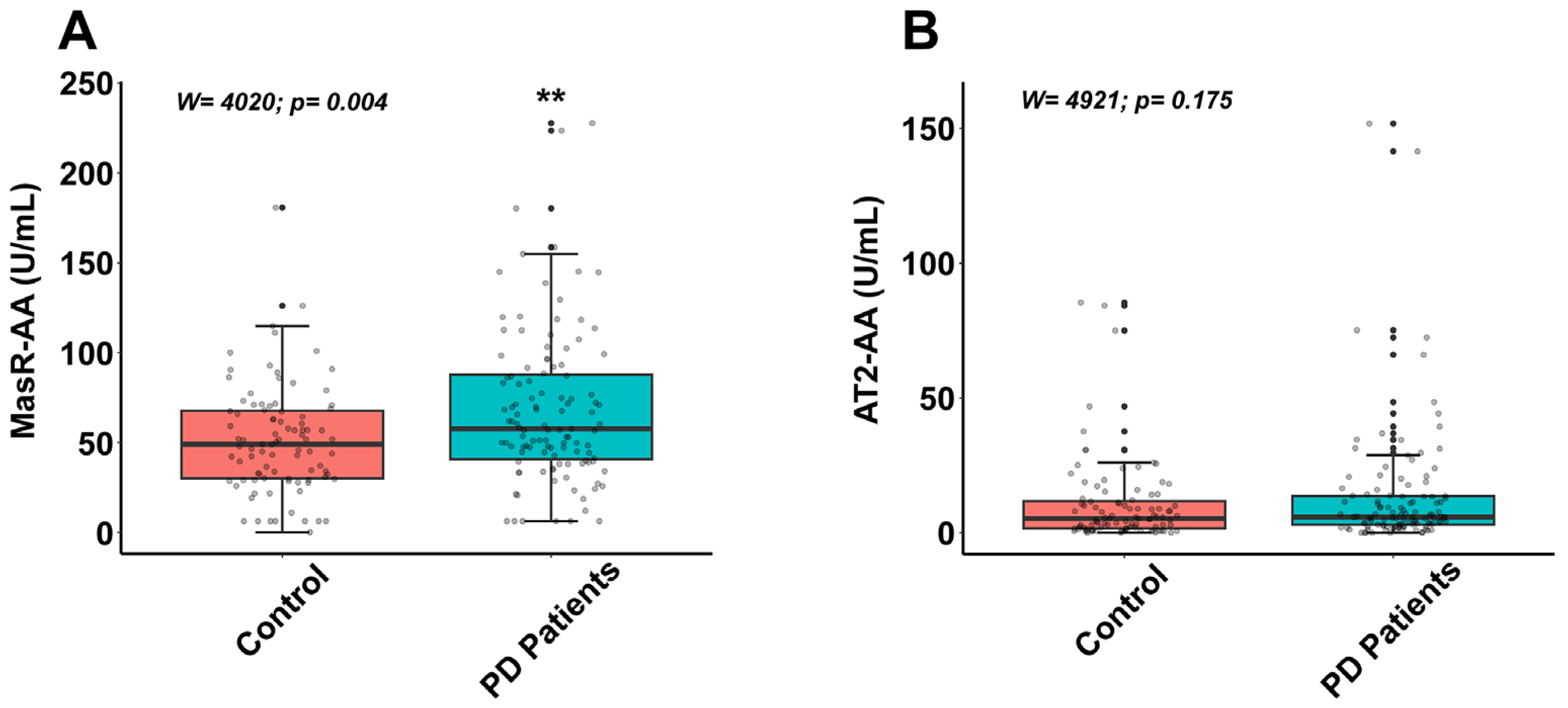
3.2. Autoantibodies for Components of Renin–Angiotensin System in Controls and Parkinson’s Disease Patients
3.3. Correlation Networks of RAS Autoantibodies in Controls and Parkinson’s Disease Patients
3.4. Sex-Dependent Correlation Networks of RAS Autoantibodies in Controls and Parkinson’s Disease Patients
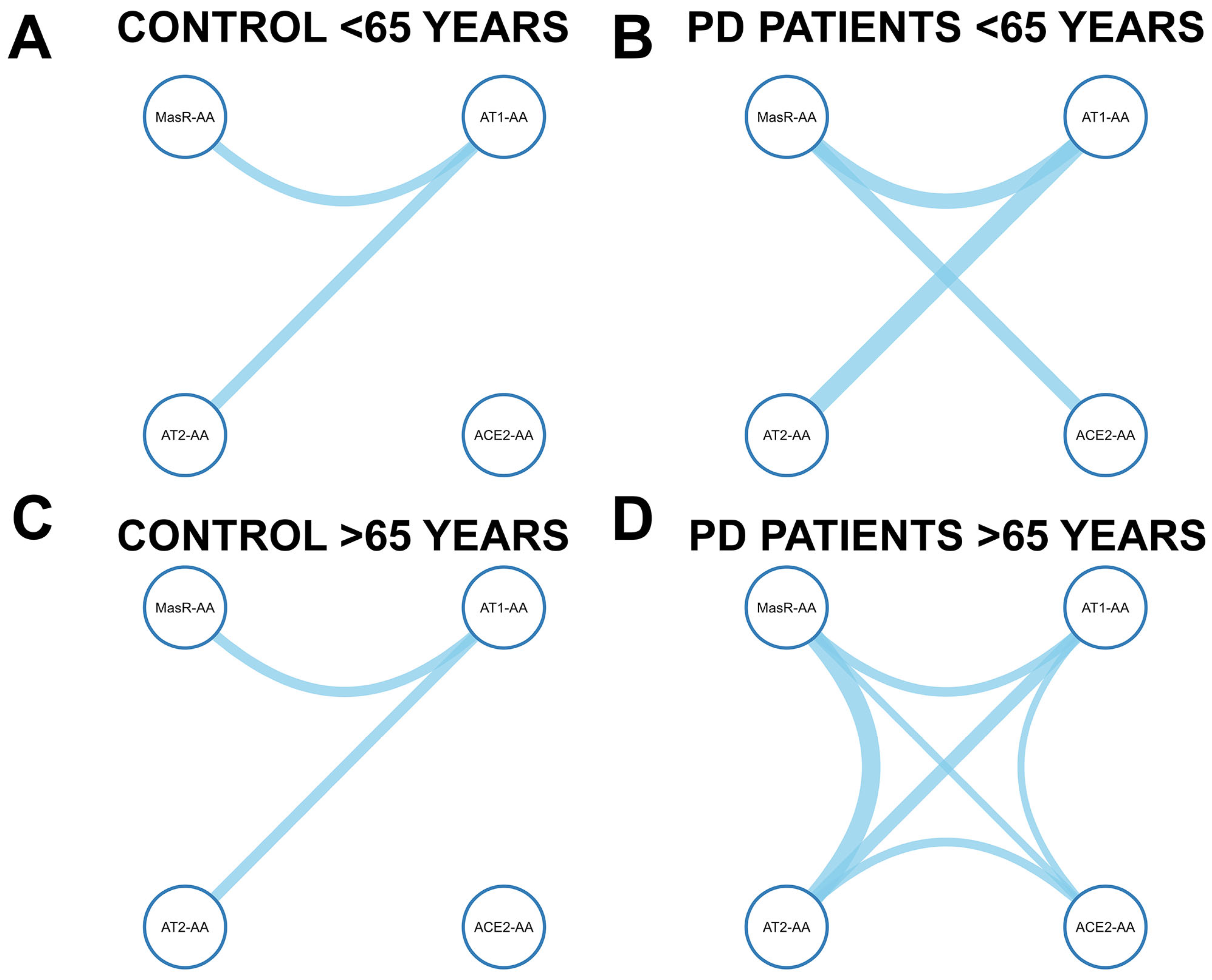
3.5. Age-Dependent Correlation Networks of RAS Autoantibodies in Controls and Parkinson’s Disease Patients
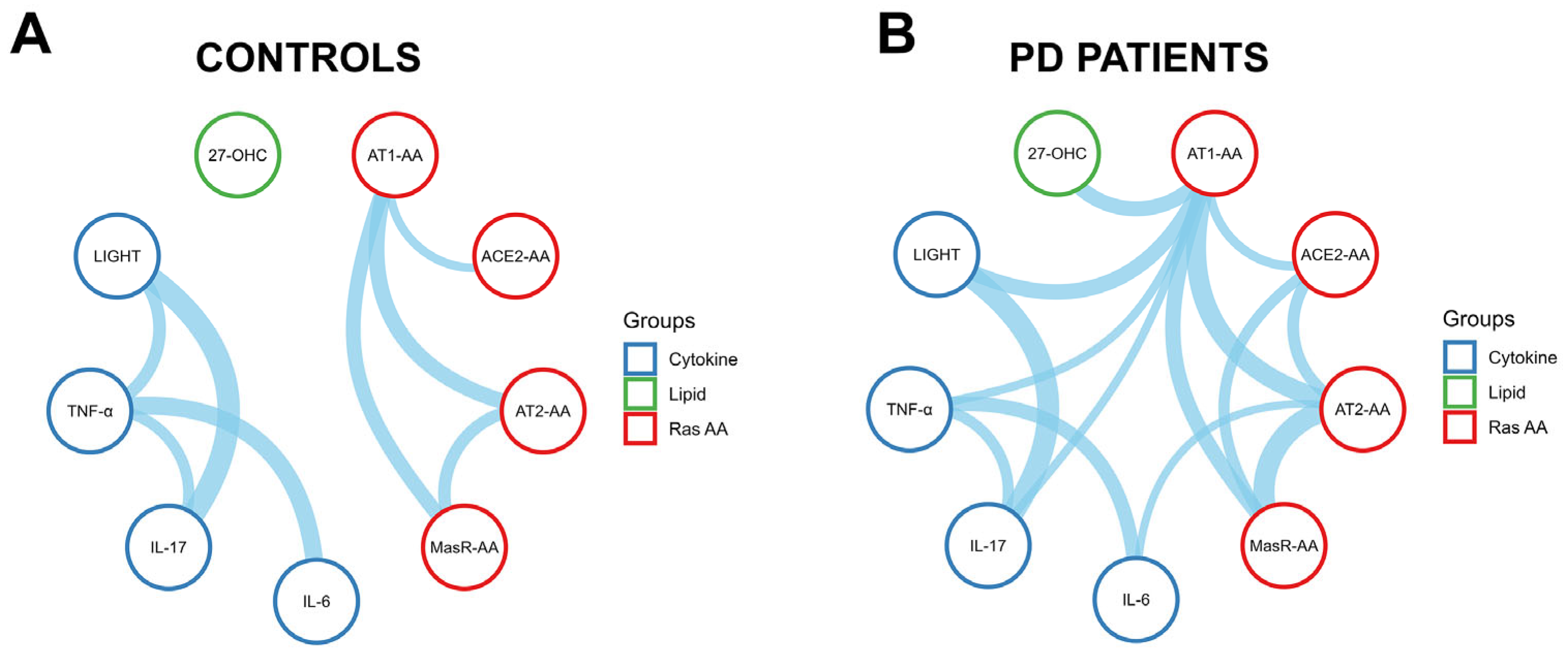
3.6. Immune and Oxidative Crosstalk with the RAS Autoantibody Network in Parkinson’s Disease Patients
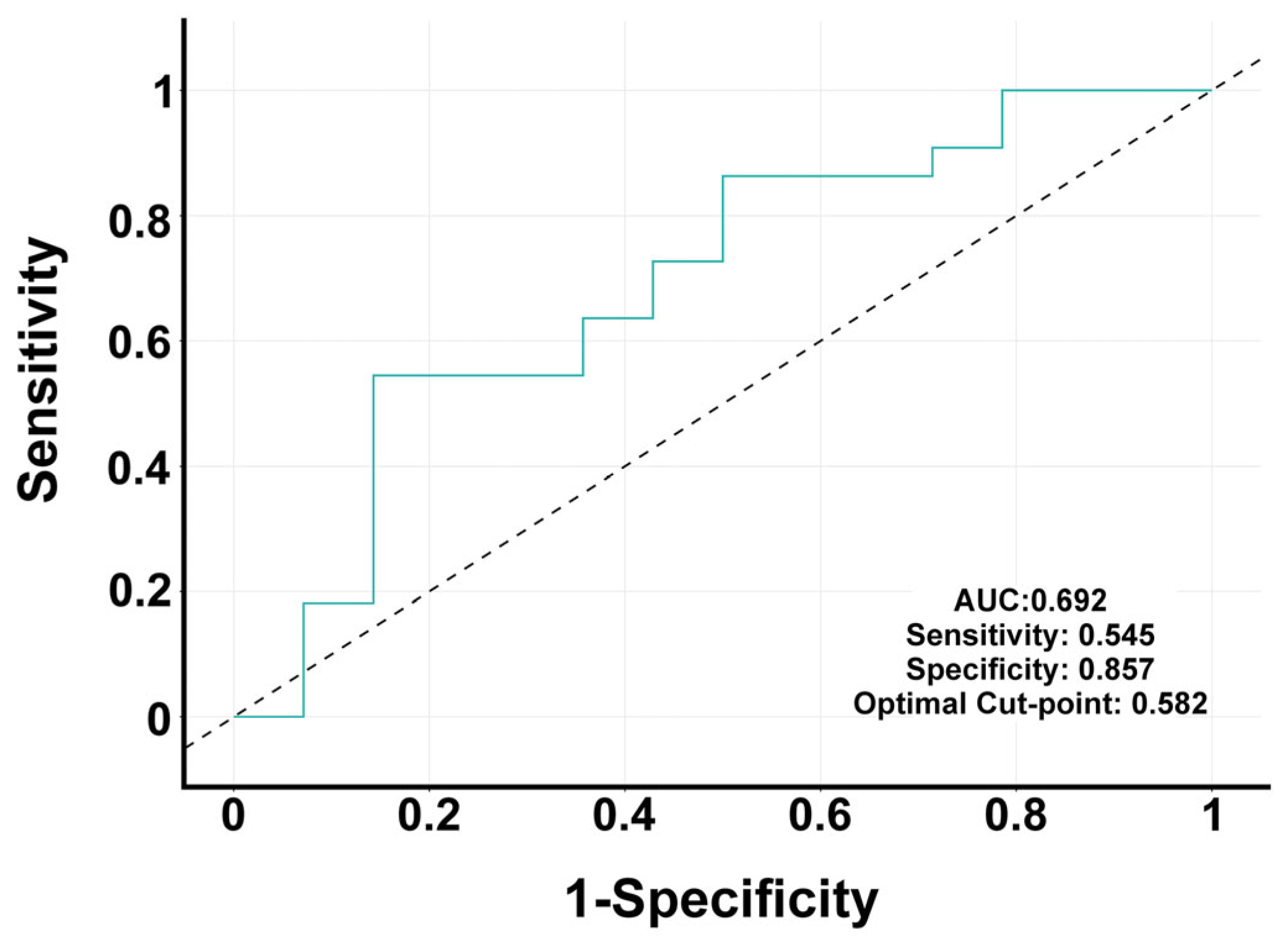
3.7. Multivariate Analysis Identifies Predictors of PD Status
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 27-OHC | 27-hydroxycholesterol |
| 6-OHDA | 6-hydroxydopamine |
| ACE2 | Angiotensin-converting enzyme 2 |
| ACE2-AAs | ACE2 autoantibodies |
| AIC | Akaike Information Criterion |
| Ang II | Angiotensin II |
| Ang1-7 | Angiotensin-(1-7) |
| AUC | Area under curve |
| AT1R | AT1 receptors |
| AT2-AAs | AT2 autoantibodies |
| AT2R | AT2 receptors |
| AAs | Autoantibodies |
| AT1-AA | Autoantibodies for AT1 receptors |
| BBB | Blood–brain barrier |
| DALT | Dural-associated lymphoid tissues |
| ELISA | Enzyme-linked immunosorbent assay |
| 4PL | Four-parameter logistic |
| GPCR | G protein-coupled receptors |
| CEIm-G | Galician Drug Research Ethics Committee |
| CSF | Cerebrospinal fluid |
| IL-17 | Interleukin-17 |
| IL-6 | Interleukin-6 |
| IQR | Interquartile ranges |
| MasR | Mas receptors |
| MasR-AAs | MasR autoantibodies |
| PD | Parkinson’s disease |
| ROC | Receiver operating characteristic |
| RAS | Renin–angiotensin system |
| TG2 | Transglutaminase 2 |
| TNFSF14, LIGHT | Tumor necrosis factor superfamily member 14 |
| TNF-α | Tumor necrosis factor-alpha |
References
- Jackson, L.; Eldahshan, W.; Fagan, S.C.; Ergul, A. Within the Brain: The Renin Angiotensin System. Int. J. Mol. Sci. 2018, 19, 876. [Google Scholar] [CrossRef] [PubMed]
- Labandeira-Garcia, J.L.; Labandeira, C.M.; Guerra, M.J.; Rodriguez-Perez, A.I. The role of the brain renin-angiotensin system in Parkinson’s disease. Transl. Neurodegener. 2024, 13, 22. [Google Scholar] [CrossRef] [PubMed]
- Costa-Besada, M.A.; Valenzuela, R.; Garrido-Gil, P.; Villar-Cheda, B.; Parga, J.A.; Lanciego, J.L.; Labandeira-Garcia, J.L. Paracrine and Intracrine Angiotensin 1-7/Mas Receptor Axis in the Substantia Nigra of Rodents, Monkeys, and Humans. Mol. Neurobiol. 2018, 55, 5847–5867. [Google Scholar] [CrossRef]
- Valenzuela, R.; Rodriguez-Perez, A.I.; Costa-Besada, M.A.; Rivas-Santisteban, R.; Garrido-Gil, P.; Lopez-Lopez, A.; Navarro, G.; Lanciego, J.L.; Franco, R.; Labandeira-Garcia, J.L. An ACE2/Mas-related receptor MrgE axis in dopaminergic neuron mitochondria. Redox Biol. 2021, 46, 102078. [Google Scholar] [CrossRef]
- Bild, W.; Vasincu, A.; Rusu, R.N.; Ababei, D.C.; Stana, A.B.; Stanciu, G.D.; Savu, B.; Bild, V. Impact of the Renin-Angiotensin System on the Pathogeny and Pharmacotherapeutics of Neurodegenerative Diseases. Biomolecules 2022, 12, 1429. [Google Scholar] [CrossRef]
- Kamath, T.; Abdulraouf, A.; Burris, S.J.; Langlieb, J.; Gazestani, V.; Nadaf, N.M.; Balderrama, K.; Vanderburg, C.; Macosko, E.Z. Single-cell genomic profiling of human dopamine neurons identifies a population that selectively degenerates in Parkinson’s disease. Nat. Neurosci. 2022, 25, 588–595. [Google Scholar] [CrossRef]
- Lee, A.J.; Kim, C.; Park, S.; Joo, J.; Choi, B.; Yang, D.; Jun, K.; Eom, J.; Lee, S.J.; Chung, S.J.; et al. Characterization of altered molecular mechanisms in Parkinson’s disease through cell type-resolved multiomics analyses. Sci. Adv. 2023, 9, eabo2467. [Google Scholar] [CrossRef]
- Martirosyan, A.; Ansari, R.; Pestana, F.; Hebestreit, K.; Gasparyan, H.; Aleksanyan, R.; Hnatova, S.; Poovathingal, S.; Marneffe, C.; Thal, D.R.; et al. Unravelling cell type-specific responses to Parkinson’s Disease at single cell resolution. Mol. Neurodegener. 2024, 19, 7. [Google Scholar] [CrossRef]
- Jo, Y.; Kim, S.; Ye, B.S.; Lee, E.; Yu, Y.M. Protective Effect of Renin-Angiotensin System Inhibitors on Parkinson’s Disease: A Nationwide Cohort Study. Front. Pharmacol. 2022, 13, 837890. [Google Scholar] [CrossRef]
- Lin, H.C.; Tseng, Y.F.; Shen, A.L.; Chao, J.C.; Hsu, C.Y.; Lin, H.L. Association of Angiotensin Receptor Blockers with Incident Parkinson Disease in Patients with Hypertension: A Retrospective Cohort Study. Am. J. Med. 2022, 135, 1001–1007. [Google Scholar] [CrossRef]
- Romanowska, J.; Bjornevik, K.; Cortese, M.; Tuominen, J.A.; Solheim, M.; Abolpour Mofrad, A.; Igland, J.; Scherzer, C.R.; Riise, T. Association Between Use of Any of the Drugs Prescribed in Norway and the Subsequent Risk of Parkinson Disease: A Drug-wide Association Study. Neurology 2023, 101, e2068–e2077. [Google Scholar] [CrossRef] [PubMed]
- Tansey, M.G.; Wallings, R.L.; Houser, M.C.; Herrick, M.K.; Keating, C.E.; Joers, V. Inflammation and immune dysfunction in Parkinson disease. Nat. Rev. Immunol. 2022, 22, 657–673. [Google Scholar] [CrossRef] [PubMed]
- Labandeira, C.M.; Pedrosa, M.A.; Quijano, A.; Valenzuela, R.; Garrido-Gil, P.; Sanchez-Andrade, M.; Suarez-Quintanilla, J.A.; Rodriguez-Perez, A.I.; Labandeira-Garcia, J.L. Angiotensin type-1 receptor and ACE2 autoantibodies in Parkinson’s disease. NPJ Park. Dis. 2022, 8, 76. [Google Scholar] [CrossRef] [PubMed]
- Pedrosa, M.A.; Labandeira, C.M.; Valenzuela, R.; Quijano, A.; Sanchez-Andrade, M.; Suarez-Quintanilla, J.A.; Lanciego, J.L.; Labandeira-Garcia, J.L.; Rodriguez-Perez, A.I. AT1 receptor autoantibodies mediate effects of metabolic syndrome on dopaminergic vulnerability. Brain Behav. Immun. 2023, 108, 255–268. [Google Scholar] [CrossRef]
- Giral, M.; Foucher, Y.; Dufay, A.; Duong Van Huyen, J.P.; Renaudin, K.; Moreau, A.; Philippe, A.; Hegner, B.; Dechend, R.; Heidecke, H.; et al. Pretransplant sensitization against angiotensin II type 1 receptor is a risk factor for acute rejection and graft loss. Am. J. Transplant. 2013, 13, 2567–2576. [Google Scholar] [CrossRef]
- Lamarca, B.; Speed, J.; Ray, L.F.; Cockrell, K.; Wallukat, G.; Dechend, R.; Granger, J. Hypertension in response to IL-6 during pregnancy: Role of AT1-receptor activation. Int. J. Interferon Cytokine Mediat. Res. 2011, 2011, 65–70. [Google Scholar] [CrossRef]
- Riemekasten, G.; Philippe, A.; Nather, M.; Slowinski, T.; Muller, D.N.; Heidecke, H.; Matucci-Cerinic, M.; Czirjak, L.; Lukitsch, I.; Becker, M.; et al. Involvement of functional autoantibodies against vascular receptors in systemic sclerosis. Ann. Rheum. Dis. 2011, 70, 530–536. [Google Scholar] [CrossRef]
- Wallukat, G.; Homuth, V.; Fischer, T.; Lindschau, C.; Horstkamp, B.; Jupner, A.; Baur, E.; Nissen, E.; Vetter, K.; Neichel, D.; et al. Patients with preeclampsia develop agonistic autoantibodies against the angiotensin AT1 receptor. J. Clin. Investig. 1999, 103, 945–952. [Google Scholar] [CrossRef]
- Lage, L.; Rodriguez-Perez, A.I.; Labandeira-Garcia, J.L.; Dominguez-Meijide, A. Angiotensin type-1 receptor autoantibodies promote alpha-synuclein aggregation in dopaminergic neurons. Front. Immunol. 2024, 15, 1457459. [Google Scholar] [CrossRef]
- Arthur, J.M.; Forrest, J.C.; Boehme, K.W.; Kennedy, J.L.; Owens, S.; Herzog, C.; Liu, J.; Harville, T.O. Development of ACE2 autoantibodies after SARS-CoV-2 infection. PLoS ONE 2021, 16, e0257016. [Google Scholar] [CrossRef]
- Takahashi, Y.; Haga, S.; Ishizaka, Y.; Mimori, A. Autoantibodies to angiotensin-converting enzyme 2 in patients with connective tissue diseases. Arthritis Res. Ther. 2010, 12, R85. [Google Scholar] [CrossRef] [PubMed]
- Wallukat, G.; Wernike, K.; Bachamanda Somesh, D.; Mettenleiter, T.C.; Muller, J. Animals Experimentally Infected with SARS-CoV-2 Generate Functional Autoantibodies against G-Protein-Coupled Receptors. Biomedicines 2023, 11, 2668. [Google Scholar] [CrossRef] [PubMed]
- Liles, C.; Li, H.; Veitla, V.; Liles, J.T.; Murphy, T.A.; Cunningham, M.W.; Yu, X.; Kem, D.C. AT2R autoantibodies block angiotensin II and AT1R autoantibody-induced vasoconstriction. Hypertension 2015, 66, 830–835. [Google Scholar] [CrossRef]
- Skiba, M.A.; Kruse, A.C. Autoantibodies as Endogenous Modulators of GPCR Signaling. Trends Pharmacol. Sci. 2021, 42, 135–150. [Google Scholar] [CrossRef]
- Cabral-Marques, O.; Carvalho-Marques, A.H.; Schimke, L.F.; Heidecke, H.; Riemekasten, G. Loss of balance in normal GPCR-mediated cell trafficking. Front. Biosci. (Landmark Ed.) 2019, 24, 18–34. [Google Scholar] [CrossRef]
- Cabral-Marques, O.; Marques, A.; Giil, L.M.; De Vito, R.; Rademacher, J.; Gunther, J.; Lange, T.; Humrich, J.Y.; Klapa, S.; Schinke, S.; et al. GPCR-specific autoantibody signatures are associated with physiological and pathological immune homeostasis. Nat. Commun. 2018, 9, 5224. [Google Scholar] [CrossRef]
- Riemekasten, G.; Petersen, F.; Heidecke, H. What Makes Antibodies Against G Protein-Coupled Receptors so Special? A Novel Concept to Understand Chronic Diseases. Front. Immunol. 2020, 11, 564526. [Google Scholar] [CrossRef]
- Dai, L.; Wang, J.; Zhang, X.; Yan, M.; Zhou, L.; Zhang, G.; Meng, L.; Chen, L.; Cao, X.; Zhang, Z.; et al. 27-Hydroxycholesterol Drives the Spread of alpha-Synuclein Pathology in Parkinson’s Disease. Mov. Disord. 2023, 38, 2005–2018. [Google Scholar] [CrossRef]
- Durrenberger, P.F.; Grunblatt, E.; Fernando, F.S.; Monoranu, C.M.; Evans, J.; Riederer, P.; Reynolds, R.; Dexter, D.T. Inflammatory Pathways in Parkinson’s Disease; A BNE Microarray Study. Park. Dis. 2012, 2012, 214714. [Google Scholar] [CrossRef]
- Karpenko, M.N.; Vasilishina, A.A.; Gromova, E.A.; Muruzheva, Z.M.; Miliukhina, I.V.; Bernadotte, A. Interleukin-1beta, interleukin-1 receptor antagonist, interleukin-6, interleukin-10, and tumor necrosis factor-alpha levels in CSF and serum in relation to the clinical diversity of Parkinson’s disease. Cell Immunol. 2018, 327, 77–82. [Google Scholar] [CrossRef]
- Liu, Z.; Qiu, A.W.; Huang, Y.; Yang, Y.; Chen, J.N.; Gu, T.T.; Cao, B.B.; Qiu, Y.H.; Peng, Y.P. IL-17A exacerbates neuroinflammation and neurodegeneration by activating microglia in rodent models of Parkinson’s disease. Brain Behav. Immun. 2019, 81, 630–645. [Google Scholar] [CrossRef] [PubMed]
- Hughes, A.J.; Daniel, S.E.; Kilford, L.; Lees, A.J. Accuracy of clinical diagnosis of idiopathic Parkinson’s disease: A clinico-pathological study of 100 cases. J. Neurol. Neurosurg. Psychiatry 1992, 55, 181–184. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J. Statistical Power Analysis for the Behavioral Sciences, 2nd ed.; Lawrence Erlbaum Associates: Hillsdale, NJ, USA, 1988. [Google Scholar]
- R Core Team. R: A Language and Environment for Statistical Computing. Available online: https://www.R-project.org/ (accessed on 3 April 2025).
- Bian, J.; Lei, J.; Yin, X.; Wang, P.; Wu, Y.; Yang, X.; Wang, L.; Zhang, S.; Liu, H.; Fu, M.L.X. Limited AT1 Receptor Internalization Is a Novel Mechanism Underlying Sustained Vasoconstriction Induced by AT1 Receptor Autoantibody from Preeclampsia. J. Am. Heart Assoc. 2019, 8, e011179. [Google Scholar] [CrossRef]
- Liu, C.; Luo, R.; Elliott, S.E.; Wang, W.; Parchim, N.F.; Iriyama, T.; Daugherty, P.S.; Blackwell, S.C.; Sibai, B.M.; Kellems, R.E.; et al. Elevated Transglutaminase Activity Triggers Angiotensin Receptor Activating Autoantibody Production and Pathophysiology of Preeclampsia. J. Am. Heart Assoc. 2015, 4, e002323. [Google Scholar] [CrossRef]
- Liu, C.; Luo, R.; Wang, W.; Peng, Z.; Johnson, G.V.W.; Kellems, R.E.; Xia, Y. Tissue Transglutaminase-Mediated AT1 Receptor Sensitization Underlies Pro-inflammatory Cytokine LIGHT-Induced Hypertension. Am. J. Hypertens. 2019, 32, 476–485. [Google Scholar] [CrossRef]
- Liu, C.; Wang, W.; Parchim, N.; Irani, R.A.; Blackwell, S.C.; Sibai, B.; Jin, J.; Kellems, R.E.; Xia, Y. Tissue transglutaminase contributes to the pathogenesis of preeclampsia and stabilizes placental angiotensin receptor type 1 by ubiquitination-preventing isopeptide modification. Hypertension 2014, 63, 353–361. [Google Scholar] [CrossRef]
- Hein, L.; Meinel, L.; Pratt, R.E.; Dzau, V.J.; Kobilka, B.K. Intracellular trafficking of angiotensin II and its AT1 and AT2 receptors: Evidence for selective sorting of receptor and ligand. Mol. Endocrinol. 1997, 11, 1266–1277. [Google Scholar] [CrossRef]
- Villar-Cheda, B.; Costa-Besada, M.A.; Valenzuela, R.; Perez-Costas, E.; Melendez-Ferro, M.; Labandeira-Garcia, J.L. The intracellular angiotensin system buffers deleterious effects of the extracellular paracrine system. Cell Death Dis. 2017, 8, e3044. [Google Scholar] [CrossRef]
- Kotas, M.E.; Medzhitov, R. Homeostasis, inflammation, and disease susceptibility. Cell 2015, 160, 816–827. [Google Scholar] [CrossRef]
- Labandeira-Garcia, J.L.; Rodriguez-Perez, A.I.; Valenzuela, R.; Costa-Besada, M.A.; Guerra, M.J. Menopause and Parkinson’s disease. Interaction between estrogens and brain renin-angiotensin system in dopaminergic degeneration. Front. Neuroendocrinol. 2016, 43, 44–59. [Google Scholar] [CrossRef]
- Villar-Cheda, B.; Dominguez-Meijide, A.; Valenzuela, R.; Granado, N.; Moratalla, R.; Labandeira-Garcia, J.L. Aging-related dysregulation of dopamine and angiotensin receptor interaction. Neurobiol. Aging 2014, 35, 1726–1738. [Google Scholar] [CrossRef] [PubMed]
- Villar-Cheda, B.; Rodriguez-Pallares, J.; Valenzuela, R.; Munoz, A.; Guerra, M.J.; Baltatu, O.C.; Labandeira-Garcia, J.L. Nigral and striatal regulation of angiotensin receptor expression by dopamine and angiotensin in rodents: Implications for progression of Parkinson’s disease. Eur. J. Neurosci. 2010, 32, 1695–1706. [Google Scholar] [CrossRef] [PubMed]
- Irani, R.A.; Zhang, Y.; Zhou, C.C.; Blackwell, S.C.; Hicks, M.J.; Ramin, S.M.; Kellems, R.E.; Xia, Y. Autoantibody-mediated angiotensin receptor activation contributes to preeclampsia through tumor necrosis factor-alpha signaling. Hypertension 2010, 55, 1246–1253. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Kellems, R.E.; Xia, Y. Inflammation, Autoimmunity, and Hypertension: The Essential Role of Tissue Transglutaminase. Am. J. Hypertens. 2017, 30, 756–764. [Google Scholar] [CrossRef]
- Labandeira, C.M.; Pedrosa, M.A.; Suarez-Quintanilla, J.A.; Cortes-Ayaso, M.; Labandeira-Garcia, J.L.; Rodriguez-Perez, A.I. Angiotensin System Autoantibodies Correlate with Routine Prognostic Indicators for COVID-19 Severity. Front. Med. 2022, 9, 840662. [Google Scholar] [CrossRef]
- Debnath, M.; Berk, M.; Maes, M. Translational evidence for the Inflammatory Response System (IRS)/Compensatory Immune Response System (CIRS) and neuroprogression theory of major depression. Prog. Neuropsychopharmacol. Biol. Psychiatry 2021, 111, 110343. [Google Scholar] [CrossRef]
- Woiciechowsky, C.; Schoning, B.; Lanksch, W.R.; Volk, H.D.; Docke, W.D. Mechanisms of brain-mediated systemic anti-inflammatory syndrome causing immunodepression. J. Mol. Med. 1999, 77, 769–780. [Google Scholar] [CrossRef]
- Rodriguez-Perez, A.I.; Garrido-Gil, P.; Pedrosa, M.A.; Garcia-Garrote, M.; Valenzuela, R.; Navarro, G.; Franco, R.; Labandeira-Garcia, J.L. Angiotensin type 2 receptors: Role in aging and neuroinflammation in the substantia nigra. Brain Behav. Immun. 2020, 87, 256–271. [Google Scholar] [CrossRef]
- Sun, Y.; Li, Y.; Wang, M.; Yue, M.; Bai, L.; Bian, J.; Hao, W.; Sun, J.; Zhang, S.; Liu, H. Increased AT(2)R expression is induced by AT(1)R autoantibody via two axes, Klf-5/IRF-1 and circErbB4/miR-29a-5p, to promote VSMC migration. Cell Death Dis. 2020, 11, 432. [Google Scholar] [CrossRef]
- Cabral-Marques, O.; Riemekasten, G. Functional autoantibodies targeting G protein-coupled receptors in rheumatic diseases. Nat. Rev. Rheumatol. 2017, 13, 648–656. [Google Scholar] [CrossRef]
- Dhande, I.; Ali, Q.; Hussain, T. Proximal tubule angiotensin AT2 receptors mediate an anti-inflammatory response via interleukin-10: Role in renoprotection in obese rats. Hypertension 2013, 61, 1218–1226. [Google Scholar] [CrossRef] [PubMed]
- Garrido-Gil, P.; Pedrosa, M.A.; Garcia-Garrote, M.; Pequeno-Valtierra, A.; Rodriguez-Castro, J.; Garcia-Souto, D.; Rodriguez-Perez, A.I.; Labandeira-Garcia, J.L. Microglial angiotensin type 2 receptors mediate sex-specific expression of inflammatory cytokines independently of circulating estrogen. Glia 2022, 70, 2348–2360. [Google Scholar] [CrossRef] [PubMed]
- Quijano, A.; Rodriguez-Perez, A.I.; Costa-Besada, M.A.; Lopez-Lopez, A.; Guerra, M.J.; Labandeira-Garcia, J.L.; Valenzuela, R. Modulation of Mitochondrial Dynamics by the Angiotensin System in Dopaminergic Neurons and Microglia. Aging Dis. 2024. [Google Scholar] [CrossRef]
- Sommer, A.; Marxreiter, F.; Krach, F.; Fadler, T.; Grosch, J.; Maroni, M.; Graef, D.; Eberhardt, E.; Riemenschneider, M.J.; Yeo, G.W.; et al. Th17 Lymphocytes Induce Neuronal Cell Death in a Human iPSC-Based Model of Parkinson’s Disease. Cell Stem Cell 2018, 23, 123–131.e126. [Google Scholar] [CrossRef]
- Rodriguez-Perez, A.I.; Serrano-Heras, G.; Labandeira, C.M.; Camacho-Meno, L.; Castro-Robles, B.; Suarez-Quintanilla, J.A.; Munoz-Lopez, M.; Piqueras-Landete, P.; Guerra, M.J.; Segura, T.; et al. Serum angiotensin type-1 receptor autoantibodies and neurofilament light chain as markers of neuroaxonal damage in post-COVID patients. Front. Immunol. 2025, 16, 1571027. [Google Scholar] [CrossRef]
- Fleegal-DeMotta, M.A.; Doghu, S.; Banks, W.A. Angiotensin II modulates BBB permeability via activation of the AT(1) receptor in brain endothelial cells. J. Cereb. Blood Flow Metab. 2009, 29, 640–647. [Google Scholar] [CrossRef]
- Setiadi, A.; Korim, W.S.; Elsaafien, K.; Yao, S.T. The role of the blood-brain barrier in hypertension. Exp. Physiol. 2018, 103, 337–342. [Google Scholar] [CrossRef]
- Yasar, S.; Moored, K.D.; Adam, A.; Zabel, F.; Chuang, Y.F.; Varma, V.R.; Carlson, M.C. Angiotensin II Blood Levels Are Associated with Smaller Hippocampal and Cortical Volumes in Cognitively Normal Older Adults. J. Alzheimers Dis. 2020, 75, 521–529. [Google Scholar] [CrossRef]
- Fitzpatrick, Z.; Ghabdan Zanluqui, N.; Rosenblum, J.S.; Tuong, Z.K.; Lee, C.Y.C.; Chandrashekhar, V.; Negro-Demontel, M.L.; Stewart, A.P.; Posner, D.A.; Buckley, M.; et al. Venous-plexus-associated lymphoid hubs support meningeal humoral immunity. Nature 2024, 628, 612–619. [Google Scholar] [CrossRef]
- Negi, N.; Das, B.K. Decoding intrathecal immunoglobulins and B cells in the CNS: Their synthesis, function, and regulation. Int. Rev. Immunol. 2020, 39, 67–79. [Google Scholar] [CrossRef]
- Sun, B.; Ramberger, M.; O’Connor, K.C.; Bashford-Rogers, R.J.M.; Irani, S.R. The B cell immunobiology that underlies CNS autoantibody-mediated diseases. Nat. Rev. Neurol. 2020, 16, 481–492. [Google Scholar] [CrossRef] [PubMed]
- Nager, A.R.; Goldstein, J.S.; Herranz-Perez, V.; Portran, D.; Ye, F.; Garcia-Verdugo, J.M.; Nachury, M.V. An Actin Network Dispatches Ciliary GPCRs into Extracellular Vesicles to Modulate Signaling. Cell 2017, 168, 252–263.e214. [Google Scholar] [CrossRef] [PubMed]
- Pedrosa, M.A.; Labandeira, C.M.; Lago-Baameiro, N.; Valenzuela, R.; Pardo, M.; Labandeira-Garcia, J.L.; Rodriguez-Perez, A.I. Extracellular Vesicles and Their Renin-Angiotensin Cargo as a Link between Metabolic Syndrome and Parkinson’s Disease. Antioxidants 2023, 12, 2045. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Robles, C.; Athauda, D.; Barber, T.R.; Barker, R.A.; Dexter, D.T.; Duty, S.; Ellis-Doyle, R.; Gandhi, S.; Handley, J.; Jabbari, E.; et al. Treatment Selection and Prioritization for the EJS ACT-PD MAMS Trial Platform. Mov. Disord. 2025. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Labandeira, C.M.; Camacho-Meño, L.; Aracil-Pastor, P.; Suárez-Quintanilla, J.A.; Labandeira-García, J.L.; Rodríguez-Pérez, A.I. Renin–Angiotensin System Autoantibody Network in Parkinson’s Disease Patients. Antioxidants 2025, 14, 706. https://doi.org/10.3390/antiox14060706
Labandeira CM, Camacho-Meño L, Aracil-Pastor P, Suárez-Quintanilla JA, Labandeira-García JL, Rodríguez-Pérez AI. Renin–Angiotensin System Autoantibody Network in Parkinson’s Disease Patients. Antioxidants. 2025; 14(6):706. https://doi.org/10.3390/antiox14060706
Chicago/Turabian StyleLabandeira, Carmen M., Laura Camacho-Meño, Paula Aracil-Pastor, Juan A. Suárez-Quintanilla, Jose L. Labandeira-García, and Ana I. Rodríguez-Pérez. 2025. "Renin–Angiotensin System Autoantibody Network in Parkinson’s Disease Patients" Antioxidants 14, no. 6: 706. https://doi.org/10.3390/antiox14060706
APA StyleLabandeira, C. M., Camacho-Meño, L., Aracil-Pastor, P., Suárez-Quintanilla, J. A., Labandeira-García, J. L., & Rodríguez-Pérez, A. I. (2025). Renin–Angiotensin System Autoantibody Network in Parkinson’s Disease Patients. Antioxidants, 14(6), 706. https://doi.org/10.3390/antiox14060706