

Mitochondrial Oxidative Stress and Mitophagy Activation Contribute to TNF-Dependent Impairment of Myogenesis
 ,
,  ,
,  and
and
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
Materials
3. Results
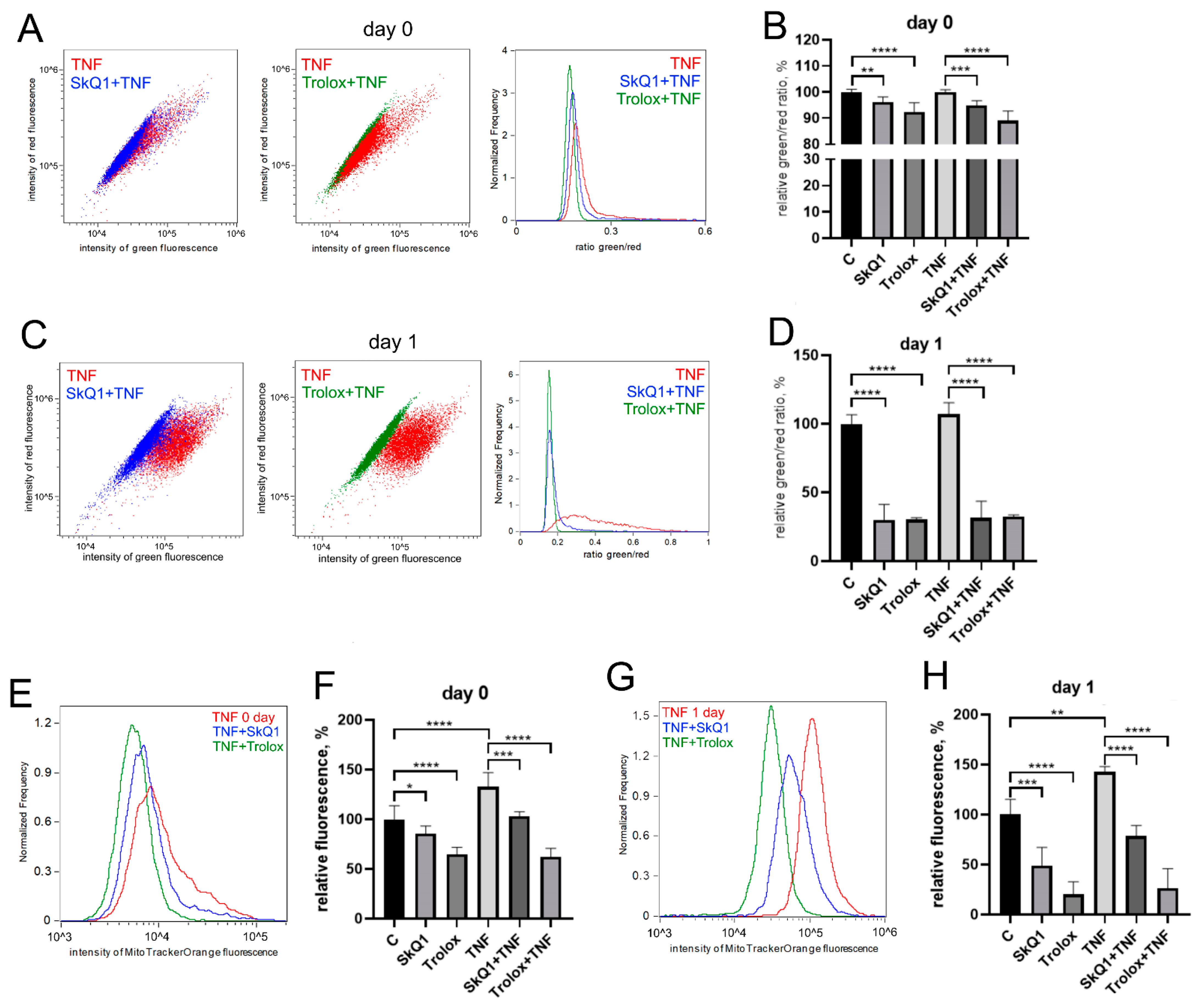
3.1. TNF Suppresses Myogenic Differentiation of MB135 Myoblasts and Stimulates Mitochondrial Oxidative Processes and Mitophagy
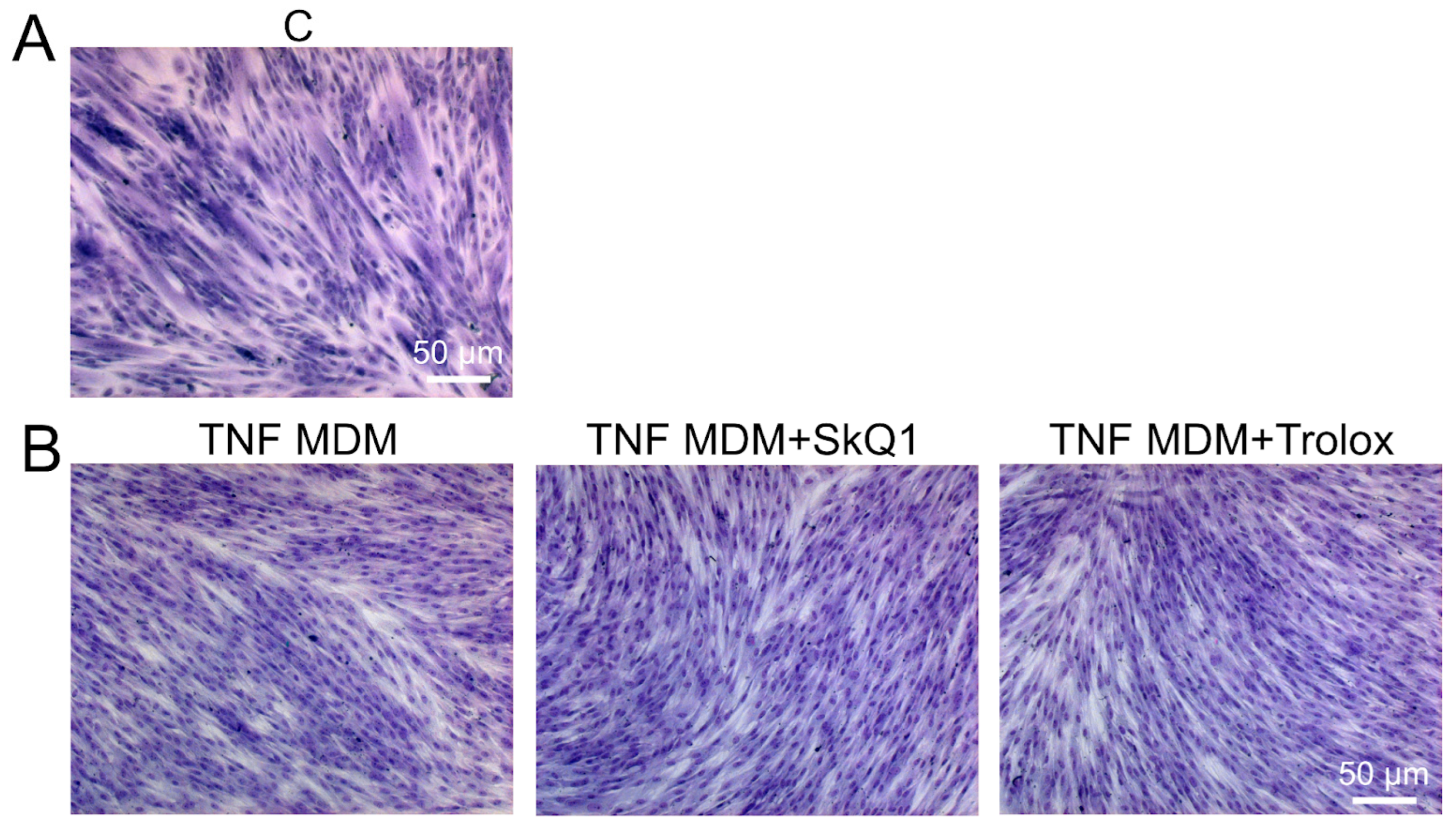
3.2. Antioxidants Partially Restore Differentiation Impaired by TNF
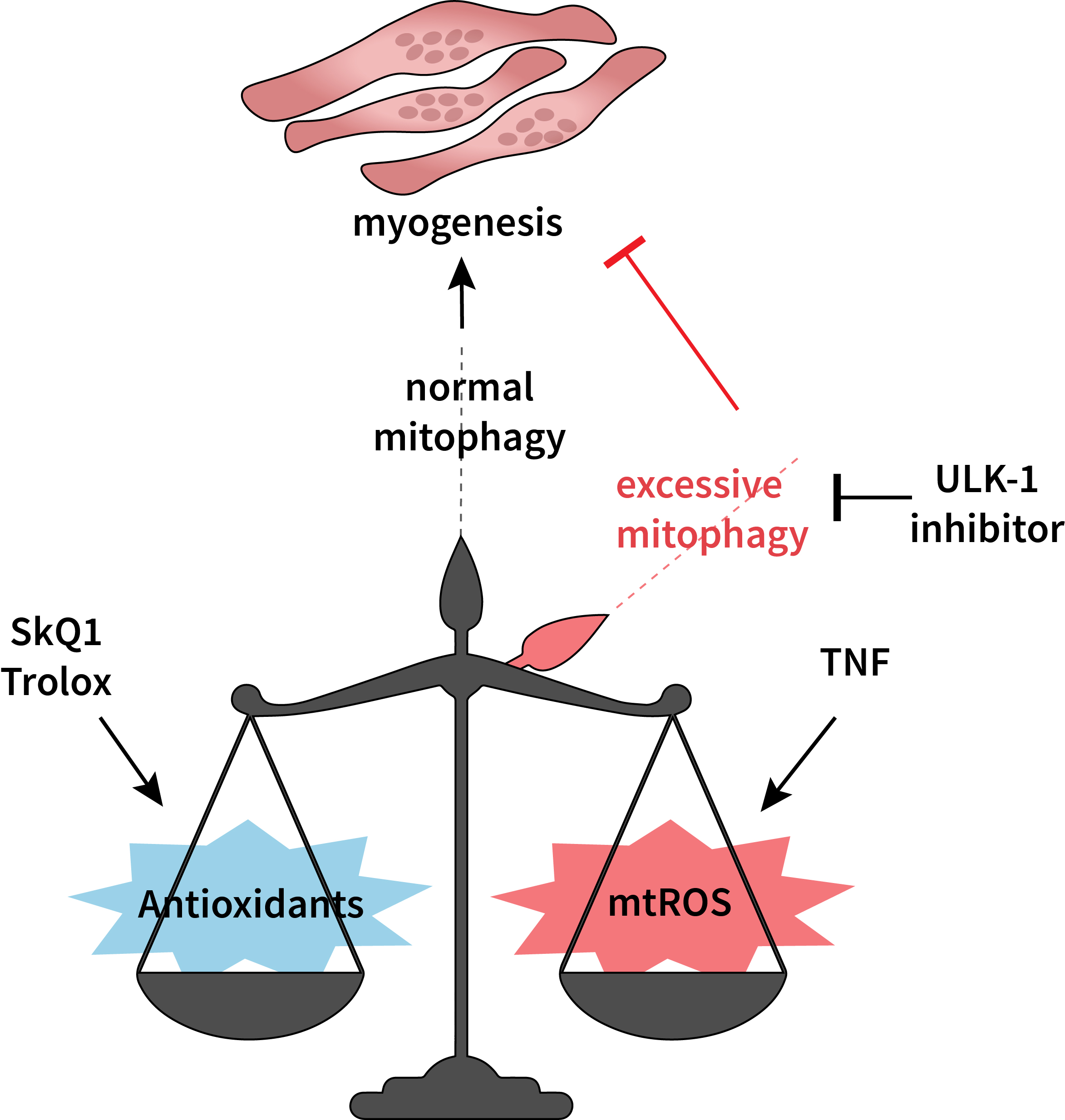
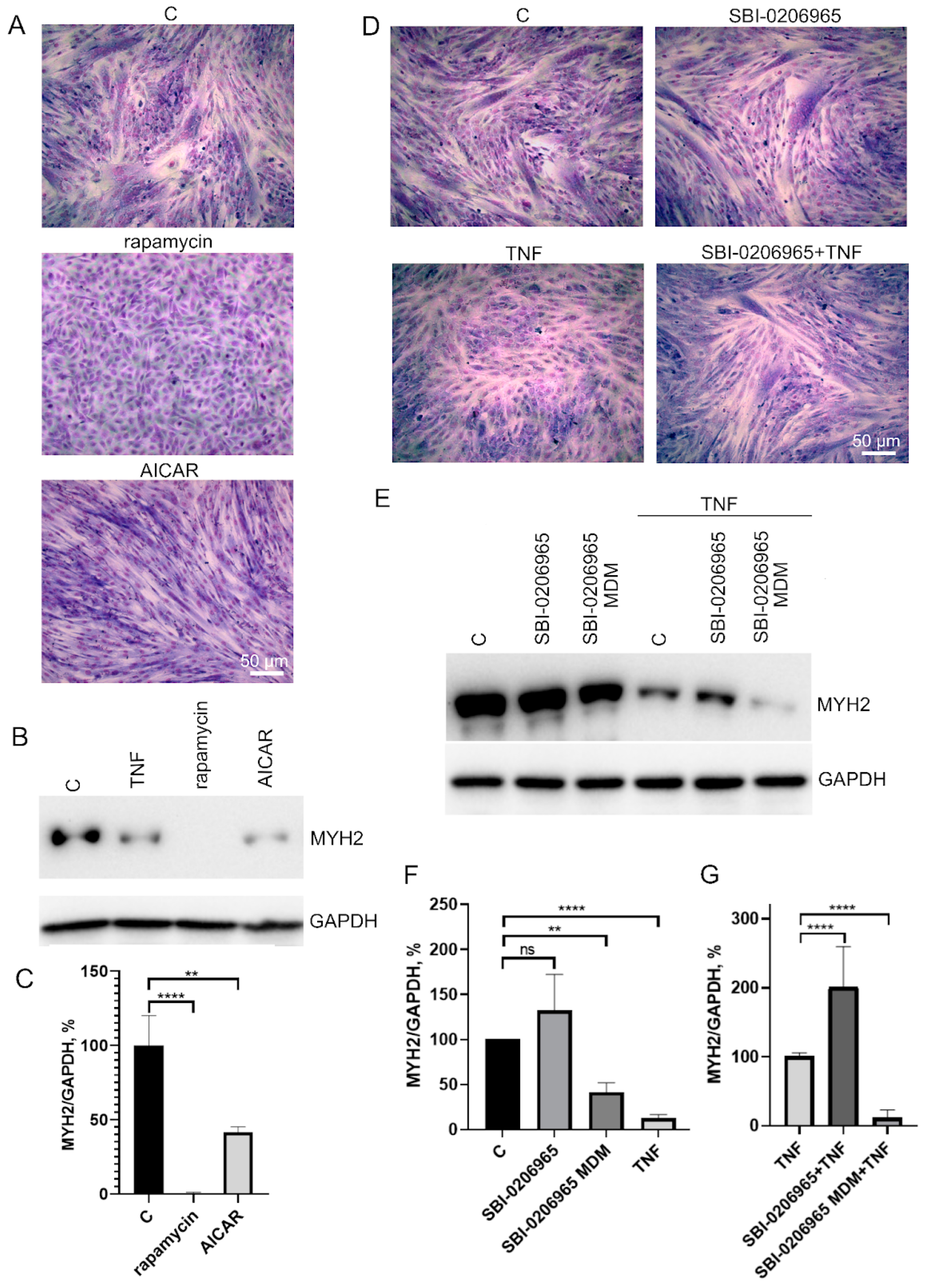
3.3. Antioxidants Restore TNF-Impaired Myogenesis, in Part by Suppressing Excessive Mitophagy
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A



References
- Tulangekar, A.; Sztal, T.E. Inflammation in Duchenne Muscular Dystrophy-Exploring the Role of Neutrophils in Muscle Damage and Regeneration. Biomedicines 2021, 9, 1366. [Google Scholar] [CrossRef]
- Tidball, J.G.; Flores, I.; Welc, S.S.; Wehling-Henricks, M.; Ochi, E. Aging of the Immune System and Impaired Muscle Regeneration: A Failure of Immunomodulation of Adult Myogenesis. Exp. Gerontol. 2021, 145, 111200. [Google Scholar] [CrossRef]
- Mosca, N.; Petrillo, S.; Bortolani, S.; Monforte, M.; Ricci, E.; Piemonte, F.; Tasca, G. Redox Homeostasis in Muscular Dystrophies. Cells 2021, 10, 1364. [Google Scholar] [CrossRef]
- Yang, W.; Hu, P. Skeletal Muscle Regeneration Is Modulated by Inflammation. J. Orthop. Translat. 2018, 13, 25–32. [Google Scholar] [CrossRef]
- Coletti, D.; Moresi, V.; Adamo, S.; Molinaro, M.; Sassoon, D. Tumor Necrosis Factor-α Gene Transfer Induces Cachexia and Inhibits Muscle Regeneration. Genesis 2005, 43, 120–128. [Google Scholar] [CrossRef]
- Langen, R.C.J.; Van Der Velden, J.L.J.; Schols, A.M.W.J.; Kelders, M.C.J.M.; Wouters, E.F.M.; Janssen-Heininger, Y.M.W. Tumor Necrosis Factor-Alpha Inhibits Myogenic Differentiation through MyoD Protein Destabilization. FASEB J. 2004, 18, 227–237. [Google Scholar] [CrossRef]
- Stewart, C.E.H.; Newcomb, P.V.; Holly, J.M.P. Multifaceted Roles of TNF-Alpha in Myoblast Destruction: A Multitude of Signal Transduction Pathways. J. Cell. Physiol. 2004, 198, 237–247. [Google Scholar] [CrossRef]
- Alter, J.; Rozentzweig, D.; Bengal, E. Inhibition of Myoblast Differentiation by Tumor Necrosis Factor Alpha Is Mediated by c-Jun N-Terminal Kinase 1 and Leukemia Inhibitory Factor. J. Biol. Chem. 2008, 283, 23224–23234. [Google Scholar] [CrossRef]
- Langen, R.C.J.; Schols, A.M.W.J.; Kelders, M.C.J.M.; Van Der Velden, J.L.J.; Wouters, E.F.M.; Janssen-Heininger, Y.M.W. Tumor Necrosis Factor-Alpha Inhibits Myogenesis through Redox-Dependent and -Independent Pathways. Am. J. Physiol. Cell Physiol. 2002, 283, C714–C721. [Google Scholar] [CrossRef]
- Li, Y.P.; Reid, M.B. NF-kappaB Mediates the Protein Loss Induced by TNF-Alpha in Differentiated Skeletal Muscle Myotubes. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2000, 279, R1165–R1170. [Google Scholar] [CrossRef]
- Li, Y.-P.; Chen, Y.; John, J.; Moylan, J.; Jin, B.; Mann, D.L.; Reid, M.B. TNF-α Acts via p38 MAPK to Stimulate Expression of the Ubiquitin Ligase atrogin1/MAFbx in Skeletal Muscle. FASEB J. 2005, 19, 362–370. [Google Scholar] [CrossRef] [PubMed]
- Tumor Necrosis Factor Alpha (TNF-α) Inactivates the PI3-kinase/PKB Pathway and Induces Atrophy and Apoptosis in L6 Myotubes. Cytokine 2011, 54, 173–184. [CrossRef] [PubMed]
- Sen, C.K.; Khanna, S.; Reznick, A.Z.; Roy, S.; Packer, L. Glutathione Regulation of Tumor Necrosis Factor-Alpha-Induced NF-Kappa B Activation in Skeletal Muscle-Derived L6 Cells. Biochem. Biophys. Res. Commun. 1997, 237, 645–649. [Google Scholar] [CrossRef] [PubMed]
- Goichberg, P.; Shtutman, M.; Ben-Ze’ev, A.; Geiger, B. Recruitment of Beta-Catenin to Cadherin-Mediated Intercellular Adhesions Is Involved in Myogenic Induction. J. Cell Sci. 2001, 114, 1309–1319. [Google Scholar] [CrossRef]
- Barbieri, E.; Sestili, P. Reactive Oxygen Species in Skeletal Muscle Signaling. J. Signal Transduct. 2012, 2012, 982794. [Google Scholar] [CrossRef]
- Chen, M.-M.; Li, Y.; Deng, S.-L.; Zhao, Y.; Lian, Z.-X.; Yu, K. Mitochondrial Function and Reactive Oxygen/Nitrogen Species in Skeletal Muscle. Front. Cell Dev. Biol. 2022, 10, 826981. [Google Scholar] [CrossRef]
- Le Moal, E.; Pialoux, V.; Juban, G.; Groussard, C.; Zouhal, H.; Chazaud, B.; Mounier, R. Redox Control of Skeletal Muscle Regeneration. Antioxid. Redox Signal. 2017, 27, 276–310. [Google Scholar] [CrossRef]
- Malinska, D.; Kudin, A.P.; Bejtka, M.; Kunz, W.S. Changes in Mitochondrial Reactive Oxygen Species Synthesis during Differentiation of Skeletal Muscle Cells. Mitochondrion 2012, 12, 144–148. [Google Scholar] [CrossRef]
- Kozakowska, M.; Pietraszek-Gremplewicz, K.; Jozkowicz, A.; Dulak, J. The Role of Oxidative Stress in Skeletal Muscle Injury and Regeneration: Focus on Antioxidant Enzymes. J. Muscle Res. Cell Motil. 2016, 36, 377–393. [Google Scholar] [CrossRef]
- Youm, T.H.; Woo, S.-H.; Kwon, E.-S.; Park, S.S. NADPH Oxidase 4 Contributes to Myoblast Fusion and Skeletal Muscle Regeneration. Oxid. Med. Cell. Longev. 2019, 2019, 3585390. [Google Scholar] [CrossRef]
- Lee, S.; Tak, E.; Lee, J.; Rashid, M.A.; Murphy, M.P.; Ha, J.; Kim, S.S. Mitochondrial H2O2 Generated from Electron Transport Chain Complex I Stimulates Muscle Differentiation. Cell Res. 2011, 21, 817–834. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-H.; Choi, T.G.; Park, S.; Yun, H.R.; Nguyen, N.N.Y.; Jo, Y.H.; Jang, M.; Kim, J.; Kim, J.; Kang, I.; et al. Mitochondrial ROS-Derived PTEN Oxidation Activates PI3K Pathway for mTOR-Induced Myogenic Autophagy. Cell Death Differ. 2018, 25, 1921–1937. [Google Scholar] [CrossRef] [PubMed]
- Lian, D.; Chen, M.-M.; Wu, H.; Deng, S.; Hu, X. The Role of Oxidative Stress in Skeletal Muscle Myogenesis and Muscle Disease. Antioxidants 2022, 11, 755. [Google Scholar] [CrossRef] [PubMed]
- Franco, A.A.; Odom, R.S.; Rando, T.A. Regulation of Antioxidant Enzyme Gene Expression in Response to Oxidative Stress and during Differentiation of Mouse Skeletal Muscle. Free Radic. Biol. Med. 1999, 27, 1122–1132. [Google Scholar] [CrossRef] [PubMed]
- Shelar, S.B.; Narasimhan, M.; Shanmugam, G.; Litovsky, S.H.; Gounder, S.S.; Karan, G.; Arulvasu, C.; Kensler, T.W.; Hoidal, J.R.; Darley-Usmar, V.M.; et al. Disruption of Nuclear Factor (erythroid-Derived-2)-like 2 Antioxidant Signaling: A Mechanism for Impaired Activation of Stem Cells and Delayed Regeneration of Skeletal Muscle. FASEB J. 2016, 30, 1865–1879. [Google Scholar] [CrossRef] [PubMed]
- Broome, S.C.; Woodhead, J.S.T.; Merry, T.L. Mitochondria-Targeted Antioxidants and Skeletal Muscle Function. Antioxidants 2018, 7, 107. [Google Scholar] [CrossRef] [PubMed]
- Zinovkin, R.A.; Romaschenko, V.P.; Galkin, I.I.; Zakharova, V.V.; Pletjushkina, O.Y.; Chernyak, B.V.; Popova, E.N. Role of Mitochondrial Reactive Oxygen Species in Age-Related Inflammatory Activation of Endothelium. Aging 2014, 6, 661–674. [Google Scholar] [CrossRef]
- Zakharova, V.V.; Pletjushkina, O.Y.; Galkin, I.I.; Zinovkin, R.A.; Chernyak, B.V.; Krysko, D.V.; Bachert, C.; Krysko, O.; Skulachev, V.P.; Popova, E.N. Low Concentration of Uncouplers of Oxidative Phosphorylation Decreases the TNF-Induced Endothelial Permeability and Lethality in Mice. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 968–977. [Google Scholar] [CrossRef]
- Romaschenko, V.P.; Zinovkin, R.A.; Galkin, I.I.; Zakharova, V.V.; Panteleeva, A.A.; Tokarchuk, A.V.; Lyamzaev, K.G.; Pletjushkina, O.Y.; Chernyak, B.V.; Popova, E.N. Low Concentrations of Uncouplers of Oxidative Phosphorylation Prevent Inflammatory Activation of Endothelial Cells by Tumor Necrosis Factor. Biochemistry 2015, 80, 610–619. [Google Scholar] [CrossRef]
- Li, Y.P.; Atkins, C.M.; Sweatt, J.D.; Reid, M.B. Mitochondria Mediate Tumor Necrosis Factor-alpha/NF-kappaB Signaling in Skeletal Muscle Myotubes. Antioxid. Redox Signal. 1999, 1, 97–104. [Google Scholar] [CrossRef]
- Karpukhina, A.; Galkin, I.; Ma, Y.; Dib, C.; Zinovkin, R.; Pletjushkina, O.; Chernyak, B.; Popova, E.; Vassetzky, Y. Analysis of Genes Regulated by DUX4 via Oxidative Stress Reveals Potential Therapeutic Targets for Treatment of Facioscapulohumeral Dystrophy. Redox Biol. 2021, 43, 102008. [Google Scholar] [CrossRef] [PubMed]
- Acharya, S.; Peters, A.M.; Norton, A.S.; Murdoch, G.K.; Hill, R.A. Change in Nox4 Expression Is Accompanied by Changes in Myogenic Marker Expression in Differentiating C2C12 Myoblasts. Pflügers Arc.—Eur. J. Physiol. 2013, 465, 1181–1196. [Google Scholar] [CrossRef]
- Piao, Y.J.; Seo, Y.H.; Hong, F.; Kim, J.H.; Kim, Y.-J.; Kang, M.H.; Kim, B.S.; Jo, S.A.; Jo, I.; Jue, D.-M.; et al. Nox 2 Stimulates Muscle Differentiation via NF-kappaB/iNOS Pathway. Free Radic. Biol. Med. 2005, 38, 989–1001. [Google Scholar] [CrossRef] [PubMed]
- Doughan, A.K.; Harrison, D.G.; Dikalov, S.I. Molecular Mechanisms of Angiotensin II-Mediated Mitochondrial Dysfunction: Linking Mitochondrial Oxidative Damage and Vascular Endothelial Dysfunction. Circ. Res. 2008, 102, 488–496. [Google Scholar] [CrossRef] [PubMed]
- Nazarewicz, R.R.; Dikalova, A.E.; Bikineyeva, A.; Dikalov, S.I. Nox2 as a Potential Target of Mitochondrial Superoxide and Its Role in Endothelial Oxidative Stress. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H1131–H1140. [Google Scholar] [CrossRef] [PubMed]
- Kröller-Schön, S.; Steven, S.; Kossmann, S.; Scholz, A.; Daub, S.; Oelze, M.; Xia, N.; Hausding, M.; Mikhed, Y.; Zinssius, E.; et al. Molecular Mechanisms of the Crosstalk between Mitochondria and NADPH Oxidase through Reactive Oxygen Species-Studies in White Blood Cells and in Animal Models. Antioxid. Redox Signal. 2014, 20, 247–266. [Google Scholar] [CrossRef]
- Vorobjeva, N.; Prikhodko, A.; Galkin, I.; Pletjushkina, O.; Zinovkin, R.; Sud’ina, G.; Chernyak, B.; Pinegin, B. Mitochondrial Reactive Oxygen Species Are Involved in Chemoattractant-Induced Oxidative Burst and Degranulation of Human Neutrophils in Vitro. Eur. J. Cell Biol. 2017, 96, 254–265. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial Reactive Oxygen Species (ROS) and ROS-Induced ROS Release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef]
- Sebastián, D.; Zorzano, A. Self-Eating for Muscle Fitness: Autophagy in the Control of Energy Metabolism. Dev. Cell 2020, 54, 268–281. [Google Scholar] [CrossRef]
- Chen, W.; Chen, Y.; Liu, Y.; Wang, X. Autophagy in Muscle Regeneration: Potential Therapies for Myopathies. J. Cachexia Sarcopenia Muscle 2022, 13, 1673–1685. [Google Scholar] [CrossRef]
- Sin, J.; Andres, A.M.; Taylor, D.J.R.; Weston, T.; Hiraumi, Y.; Stotland, A.; Kim, B.J.; Huang, C.; Doran, K.S.; Gottlieb, R.A. Mitophagy Is Required for Mitochondrial Biogenesis and Myogenic Differentiation of C2C12 Myoblasts. Autophagy 2016, 12, 369–380. [Google Scholar] [CrossRef] [PubMed]
- Ashrafi, G.; Schwarz, T.L. The Pathways of Mitophagy for Quality Control and Clearance of Mitochondria. Cell Death Differ. 2013, 20, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Nichenko, A.S.; Southern, W.M.; Atuan, M.; Luan, J.; Peissig, K.B.; Foltz, S.J.; Beedle, A.M.; Warren, G.L.; Call, J.A. Mitochondrial Maintenance via Autophagy Contributes to Functional Skeletal Muscle Regeneration and Remodeling. Am. J. Physiol. Cell Physiol. 2016, 311, C190–C200. [Google Scholar] [CrossRef]
- Esteca, M.V.; Severino, M.B.; Silvestre, J.G.; Palmeira Dos Santos, G.; Tamborlin, L.; Luchessi, A.D.; Moriscot, A.S.; Gustafsson, Å.B.; Baptista, I.L. Loss of Parkin Results in Altered Muscle Stem Cell Differentiation during Regeneration. Int. J. Mol. Sci. 2020, 21, 8007. [Google Scholar] [CrossRef]
- Baldelli, S.; Aquilano, K.; Ciriolo, M.R. PGC-1α Buffers ROS-Mediated Removal of Mitochondria during Myogenesis. Cell Death Dis. 2014, 5, e1515. [Google Scholar] [CrossRef]
- Shu, L.; Houghton, P.J. The mTORC2 Complex Regulates Terminal Differentiation of C2C12 Myoblasts. Mol. Cell. Biol. 2009, 29, 4691–4700. [Google Scholar] [CrossRef]
- Yoo, Y.-M.; Jung, E.-M.; Jeung, E.-B. Rapamycin-Induced Autophagy Decreases Myf5 and MyoD Proteins in C2C12 Myoblast Cells. Toxicol. In Vitro 2019, 58, 132–141. [Google Scholar] [CrossRef]
- Thomson, D.M. The Role of AMPK in the Regulation of Skeletal Muscle Size, Hypertrophy, and Regeneration. Int. J. Mol. Sci. 2018, 19, 3125. [Google Scholar] [CrossRef]
- Togliatto, G.; Trombetta, A.; Dentelli, P.; Cotogni, P.; Rosso, A.; Tschöp, M.H.; Granata, R.; Ghigo, E.; Brizzi, M.F. Unacylated Ghrelin Promotes Skeletal Muscle Regeneration Following Hindlimb Ischemia via SOD-2-Mediated miR-221/222 Expression. J. Am. Heart Assoc. 2013, 2, e000376. [Google Scholar] [CrossRef]
- Hidalgo, M.; Marchant, D.; Quidu, P.; Youcef-Ali, K.; Richalet, J.P.; Beaudry, M.; Besse, S.; Launay, T. Oxygen Modulates the Glutathione Peroxidase Activity during the L6 Myoblast Early Differentiation Process. Cell. Physiol. Biochem. 2014, 33, 67–77. [Google Scholar] [CrossRef]
- Dmitriev, P.; Bou Saada, Y.; Dib, C.; Ansseau, E.; Barat, A.; Hamade, A.; Dessen, P.; Robert, T.; Lazar, V.; Louzada, R.A.N.; et al. DUX4-Induced Constitutive DNA Damage and Oxidative Stress Contribute to Aberrant Differentiation of Myoblasts from FSHD Patients. Free Radic. Biol. Med. 2016, 99, 244–258. [Google Scholar] [PubMed]
- Vermeij, W.P.; Hoeijmakers, J.H.J.; Pothof, J. Genome Integrity in Aging: Human Syndromes, Mouse Models, and Therapeutic Options. Annu. Rev. Pharmacol. Toxicol. 2016, 56, 427–445. [Google Scholar] [CrossRef] [PubMed]
- Bosutti, A.; Degens, H. The Impact of Resveratrol and Hydrogen Peroxide on Muscle Cell Plasticity Shows a Dose-Dependent Interaction. Sci. Rep. 2015, 5, 8093. [Google Scholar]
- Bou Saada, Y.; Zakharova, V.; Chernyak, B.; Dib, C.; Carnac, G.; Dokudovskaya, S.; Vassetzky, Y.S. Control of DNA Integrity in Skeletal Muscle under Physiological and Pathological Conditions. Cell. Mol. Life Sci. 2017, 74, 3439–3449. [Google Scholar] [CrossRef] [PubMed]
- Call, J.A.; Wilson, R.J.; Laker, R.C.; Zhang, M.; Kundu, M.; Yan, Z. Ulk1-Mediated Autophagy Plays an Essential Role in Mitochondrial Remodeling and Functional Regeneration of Skeletal Muscle. Am. J. Physiol. Cell Physiol. 2017, 312, C724–C732. [Google Scholar] [PubMed]
- Laker, R.C.; Drake, J.C.; Wilson, R.J.; Lira, V.A.; Lewellen, B.M.; Ryall, K.A.; Fisher, C.C.; Zhang, M.; Saucerman, J.J.; Goodyear, L.J.; et al. Ampk Phosphorylation of Ulk1 Is Required for Targeting of Mitochondria to Lysosomes in Exercise-Induced Mitophagy. Nat. Commun. 2017, 8, 548. [Google Scholar] [PubMed]
- Jagannathan, S.; Shadle, S.C.; Resnick, R.; Snider, L.; Tawil, R.N.; van der Maarel, S.M.; Bradley, R.K.; Tapscott, S.J. Model Systems of DUX4 Expression Recapitulate the Transcriptional Profile of FSHD Cells. Hum. Mol. Genet. 2016, 25, 4419–4431. [Google Scholar] [CrossRef] [PubMed]
- Allen, G.F.G.; Toth, R.; James, J.; Ganley, I.G. Loss of Iron Triggers PINK1/Parkin-Independent Mitophagy. EMBO Rep. 2013, 14, 1127–1135. [Google Scholar] [CrossRef]
- Lyamzaev, K.G.; Panteleeva, A.A.; Karpukhina, A.A.; Galkin, I.I.; Popova, E.N.; Pletjushkina, O.Y.; Rieger, B.; Busch, K.B.; Mulkidjanian, A.Y.; Chernyak, B.V. Novel Fluorescent Mitochondria-Targeted Probe MitoCLox Reports Lipid Peroxidation in Response to Oxidative Stress. Oxid. Med. Cell. Longev. 2020, 2020, 3631272. [Google Scholar] [CrossRef]
- Lyamzaev, K.G.; Sumbatyan, N.V.; Nesterenko, A.M.; Kholina, E.G.; Voskoboynikova, N.; Steinhoff, H.-J.; Mulkidjanian, A.Y.; Chernyak, B.V. MitoCLox: A Novel Mitochondria-Targeted Fluorescent Probe for Tracing Lipid Peroxidation. Oxid. Med. Cell. Longev. 2019, 2019, 9710208. [Google Scholar] [CrossRef]
- Skulachev, V.P.; Antonenko, Y.N.; Cherepanov, D.A.; Chernyak, B.V.; Izyumov, D.S.; Khailova, L.S.; Klishin, S.S.; Korshunova, G.A.; Lyamzaev, K.G.; Pletjushkina, O.Y.; et al. Prevention of Cardiolipin Oxidation and Fatty Acid Cycling as Two Antioxidant Mechanisms of Cationic Derivatives of Plastoquinone (SkQs). Biochim. Biophys. Acta 2010, 1797, 878–889. [Google Scholar] [CrossRef]
- Bhattacharya, D.; Scimè, A. Mitochondrial Function in Muscle Stem Cell Fates. Front. Cell Dev. Biol. 2020, 8, 480. [Google Scholar] [CrossRef]
- Schofield, J.H.; Schafer, Z.T. Mitochondrial Reactive Oxygen Species and Mitophagy: A Complex and Nuanced Relationship. Antioxid. Redox Signal. 2021, 34, 517–530. [Google Scholar] [CrossRef]
- Emodin Attenuates TNF-α-Induced Apoptosis and Autophagy in Mouse C2C12 Myoblasts Though the Phosphorylation of Akt. Int. Immunopharmacol. 2016, 34, 107–113. [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chernyavskij, D.A.; Pletjushkina, O.Y.; Kashtanova, A.V.; Galkin, I.I.; Karpukhina, A.; Chernyak, B.V.; Vassetzky, Y.S.; Popova, E.N. Mitochondrial Oxidative Stress and Mitophagy Activation Contribute to TNF-Dependent Impairment of Myogenesis. Antioxidants 2023, 12, 602. https://doi.org/10.3390/antiox12030602
Chernyavskij DA, Pletjushkina OY, Kashtanova AV, Galkin II, Karpukhina A, Chernyak BV, Vassetzky YS, Popova EN. Mitochondrial Oxidative Stress and Mitophagy Activation Contribute to TNF-Dependent Impairment of Myogenesis. Antioxidants. 2023; 12(3):602. https://doi.org/10.3390/antiox12030602
Chicago/Turabian StyleChernyavskij, Daniil A., Olga Yu. Pletjushkina, Anastasia V. Kashtanova, Ivan I. Galkin, Anna Karpukhina, Boris V. Chernyak, Yegor S. Vassetzky, and Ekaterina N. Popova. 2023. "Mitochondrial Oxidative Stress and Mitophagy Activation Contribute to TNF-Dependent Impairment of Myogenesis" Antioxidants 12, no. 3: 602. https://doi.org/10.3390/antiox12030602
APA StyleChernyavskij, D. A., Pletjushkina, O. Y., Kashtanova, A. V., Galkin, I. I., Karpukhina, A., Chernyak, B. V., Vassetzky, Y. S., & Popova, E. N. (2023). Mitochondrial Oxidative Stress and Mitophagy Activation Contribute to TNF-Dependent Impairment of Myogenesis. Antioxidants, 12(3), 602. https://doi.org/10.3390/antiox12030602