

The Role of Lonp1 on Mitochondrial Functions during Cardiovascular and Muscular Diseases
 , and
, and 
Abstract
1. Introduction
2. General Features and Functions of Lonp1 and Its Implication in Diseases

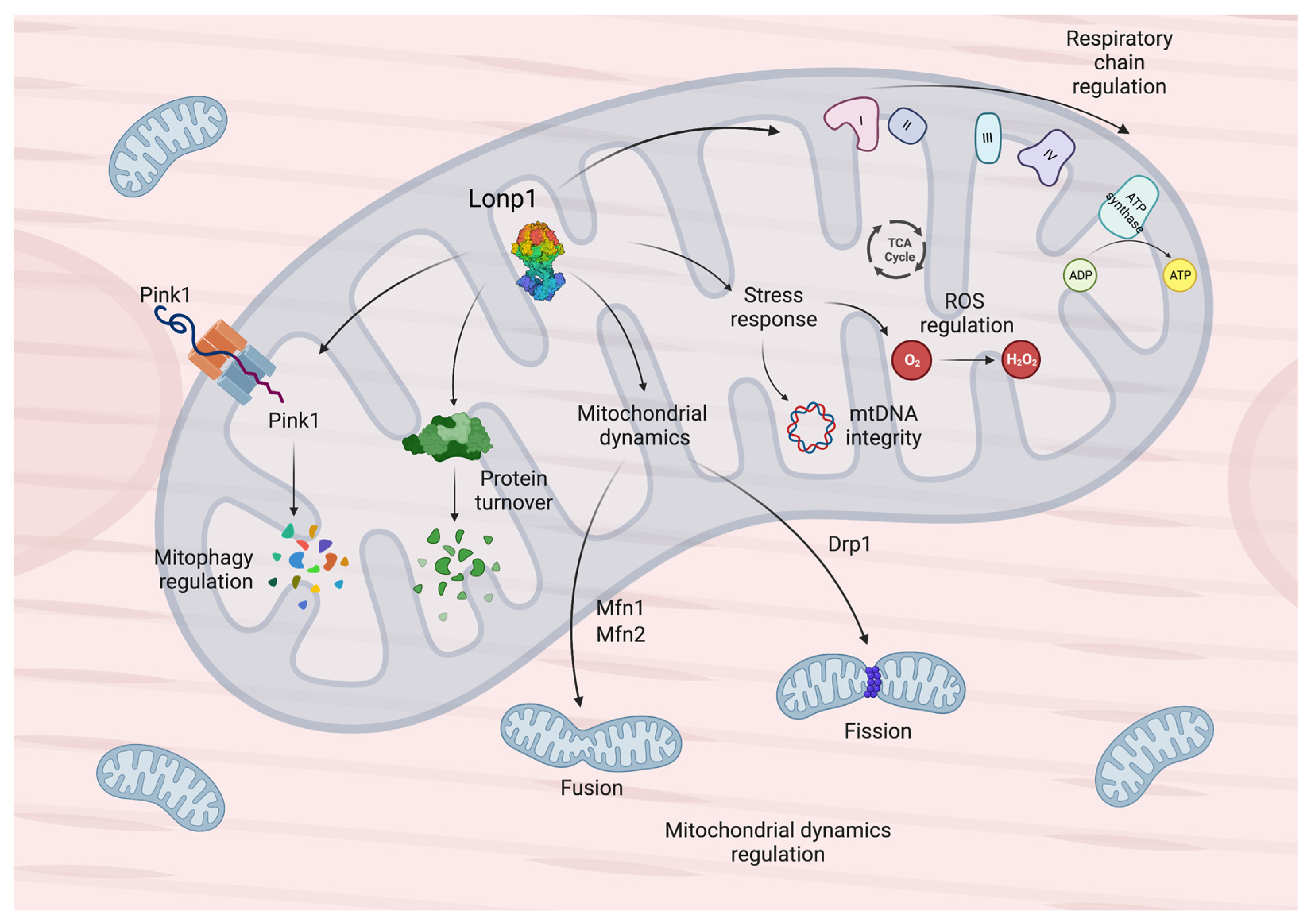{kind=link}
{kind=link}
| Lonp1 Functions | References |
|---|---|
| Selectively degrades oxidized and damaged proteins, including aconitase | [23] |
| Maintains normal cell viability in response to acute stress stimuli when upregulated | [24,25] |
| Works with mitochondrial HSP70 chaperone system to promote protein folding in mitochondria | [27] |
| Supports incoming proteins folding through its chaperone-like function and degrades abnormal imported proteins | [28] |
| Contributes to the solubilization of mitochondrial proteins | [28] |
| Promotes cell proliferation, apoptotic resistance to stresses | [25,29,30] |
| Involved in the regulation of mitochondrial dynamics and mitophagy | [16] |
| Implicated in the proteolytic system responsible for PINK1 degradation | [33,34,35] |
| Can bind mitochondrial DNA | [18] |
| Involved in the regulation of mitochondrial DNA copy number, through direct and indirect mechanisms | [18,40,41,42,43] |
3. The Role of Lonp1 in Heart Homeostasis and Response to Stress
4. The Functions of Lonp1 in the Skeletal Muscle
5. Lonp1 and Aging: A Role in Sarcopenia
6. Conclusions and Future Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Galluzzi, L.; Kepp, O.; Trojel-Hansen, C.; Kroemer, G. Mitochondrial control of cellular life, stress, and death. Circ. Res. 2012, 111, 1198–1207. [Google Scholar] [CrossRef] [PubMed]
- Delbridge, L.M.D.; Mellor, K.M.; Taylor, D.J.; Gottlieb, R.A. Myocardial stress and autophagy: Mechanisms and potential therapies. Nat. Rev. Cardiol. 2017, 14, 412–425. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, C.; Marchi, S.; Pinton, P. The machineries, regulation and cellular functions of mitochondrial calcium. Nat. Rev. Mol. Cell Biol. 2018, 19, 713–730. [Google Scholar] [CrossRef] [PubMed]
- Bonora, M.; Wieckowski, M.R.; Sinclair, D.A.; Kroemer, G.; Pinton, P.; Galluzzi, L. Targeting mitochondria for cardiovascular disorders: Therapeutic potential and obstacles. Nat. Rev. Cardiol. 2019, 16, 33–55. [Google Scholar] [CrossRef]
- Murphy, E.; Ardehali, H.; Balaban, R.S.; DiLisa, F.; Dorn, G.W., 2nd; Kitsis, R.N.; Otsu, K.; Ping, P.; Rizzuto, R.; Sack, M.N.; et al. Mitochondrial Function, Biology, and Role in Disease: A Scientific Statement From the American Heart Association. Circ. Res. 2016, 118, 1960–1991. [Google Scholar] [CrossRef]
- Lopez-Crisosto, C.; Pennanen, C.; Vasquez-Trincado, C.; Morales, P.E.; Bravo-Sagua, R.; Quest, A.F.G.; Chiong, M.; Lavandero, S. Sarcoplasmic reticulum-mitochondria communication in cardiovascular pathophysiology. Nat. Rev. Cardiol. 2017, 14, 342–360. [Google Scholar] [CrossRef]
- Bravo-San Pedro, J.M.; Kroemer, G.; Galluzzi, L. Autophagy and Mitophagy in Cardiovascular Disease. Circ. Res. 2017, 120, 1812–1824. [Google Scholar] [CrossRef]
- Terman, A.; Brunk, U.T. Myocyte aging and mitochondrial turnover. Exp. Gerontol. 2004, 39, 701–705. [Google Scholar] [CrossRef]
- Pinti, M.; Gibellini, L.; Nasi, M.; De Biasi, S.; Bortolotti, C.A.; Iannone, A.; Cossarizza, A. Emerging role of Lon protease as a master regulator of mitochondrial functions. Biochim. Biophys. Acta 2016, 1857, 1300–1306. [Google Scholar] [CrossRef]
- Wang, N.; Gottesman, S.; Willingham, M.C.; Gottesman, M.M.; Maurizi, M.R. A human mitochondrial ATP-dependent protease that is highly homologous to bacterial Lon protease. Proc. Natl. Acad. Sci. USA 1993, 90, 11247–11251. [Google Scholar] [CrossRef]
- Suzuki, C.K.; Rep, M.; van Dijl, J.M.; Suda, K.; Grivell, L.A.; Schatz, G. ATP-dependent proteases that also chaperone protein biogenesis. Trends Biochem. Sci. 1997, 22, 118–123. [Google Scholar] [CrossRef]
- Fukui, T.; Eguchi, T.; Atomi, H.; Imanaka, T. A membrane-bound archaeal Lon protease displays ATP-independent proteolytic activity towards unfolded proteins and ATP-dependent activity for folded proteins. J. Bacteriol. 2002, 184, 3689–3698. [Google Scholar] [CrossRef] [PubMed]
- Polo, M.; Alegre, F.; Moragrega, A.B.; Gibellini, L.; Marti-Rodrigo, A.; Blas-Garcia, A.; Esplugues, J.V.; Apostolova, N. Lon protease: A novel mitochondrial matrix protein in the interconnection between drug-induced mitochondrial dysfunction and endoplasmic reticulum stress. Br. J. Pharmacol. 2017, 174, 4409–4429. [Google Scholar] [CrossRef] [PubMed]
- Gibellini, L.; Borella, R.; De Gaetano, A.; Zanini, G.; Tartaro, D.L.; Carnevale, G.; Beretti, F.; Losi, L.; De Biasi, S.; Nasi, M.; et al. Evidence for mitochondrial Lonp1 expression in the nucleus. Sci. Rep. 2022, 12, 10877. [Google Scholar] [CrossRef]
- Venkatesh, S.; Lee, J.; Singh, K.; Lee, I.; Suzuki, C.K. Multitasking in the mitochondrion by the ATP-dependent Lon protease. Biochim. Biophys. Acta 2012, 1823, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Gibellini, L.; De Gaetano, A.; Mandrioli, M.; Van Tongeren, E.; Bortolotti, C.A.; Cossarizza, A.; Pinti, M. The biology of Lonp1: More than a mitochondrial protease. Int. Rev. Cell Mol. Biol. 2020, 354, 1–61. [Google Scholar] [CrossRef]
- Quiros, P.M.; Barcena, C.; Lopez-Otin, C. Lon protease: A key enzyme controlling mitochondrial bioenergetics in cancer. Mol. Cell. Oncol. 2014, 1, e968505. [Google Scholar] [CrossRef]
- Lu, B.; Lee, J.; Nie, X.; Li, M.; Morozov, Y.I.; Venkatesh, S.; Bogenhagen, D.F.; Temiakov, D.; Suzuki, C.K. Phosphorylation of human TFAM in mitochondria impairs DNA binding and promotes degradation by the AAA+ Lon protease. Mol. Cell 2013, 49, 121–132. [Google Scholar] [CrossRef]
- Crewe, C.; Schafer, C.; Lee, I.; Kinter, M.; Szweda, L.I. Regulation of Pyruvate Dehydrogenase Kinase 4 in the Heart through Degradation by the Lon Protease in Response to Mitochondrial Substrate Availability. J. Biol. Chem. 2017, 292, 305–312. [Google Scholar] [CrossRef]
- Lee, Y.G.; Kim, H.W.; Nam, Y.; Shin, K.J.; Lee, Y.J.; Park, D.H.; Rhee, H.W.; Seo, J.K.; Chae, Y.C. LONP1 and ClpP cooperatively regulate mitochondrial proteostasis for cancer cell survival. Oncogenesis 2021, 10, 18. [Google Scholar] [CrossRef]
- Pinti, M.; Gibellini, L.; Liu, Y.; Xu, S.; Lu, B.; Cossarizza, A. Mitochondrial Lon protease at the crossroads of oxidative stress, ageing and cancer. Cell. Mol. Life. Sci. 2015, 72, 4807–4824. [Google Scholar] [CrossRef] [PubMed]
- Teng, H.; Wu, B.; Zhao, K.; Yang, G.; Wu, L.; Wang, R. Oxygen-sensitive mitochondrial accumulation of cystathionine beta-synthase mediated by Lon protease. Proc. Natl. Acad. Sci. USA 2013, 110, 12679–12684. [Google Scholar] [CrossRef] [PubMed]
- Bota, D.A.; Davies, K.J. Lon protease preferentially degrades oxidized mitochondrial aconitase by an ATP-stimulated mechanism. Nat. Cell Biol. 2002, 4, 674–680. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, R.; Zhang, H.; Kim, J.W.; Shimoda, L.; Dang, C.V.; Semenza, G.L. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell 2007, 129, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Quiros, P.M.; Espanol, Y.; Acin-Perez, R.; Rodriguez, F.; Barcena, C.; Watanabe, K.; Calvo, E.; Loureiro, M.; Fernandez-Garcia, M.S.; Fueyo, A.; et al. ATP-dependent Lon protease controls tumor bioenergetics by reprogramming mitochondrial activity. Cell Rep. 2014, 8, 542–556. [Google Scholar] [CrossRef]
- Kuo, C.Y.; Chiu, Y.C.; Lee, A.Y.; Hwang, T.L. Mitochondrial Lon protease controls ROS-dependent apoptosis in cardiomyocyte under hypoxia. Mitochondrion 2015, 23, 7–16. [Google Scholar] [CrossRef]
- Shin, C.S.; Meng, S.; Garbis, S.D.; Moradian, A.; Taylor, R.W.; Sweredoski, M.J.; Lomenick, B.; Chan, D.C. LONP1 and mtHSP70 cooperate to promote mitochondrial protein folding. Nat. Commun. 2021, 12, 265. [Google Scholar] [CrossRef]
- Matsushima, Y.; Takahashi, K.; Yue, S.; Fujiyoshi, Y.; Yoshioka, H.; Aihara, M.; Setoyama, D.; Uchiumi, T.; Fukuchi, S.; Kang, D. Mitochondrial Lon protease is a gatekeeper for proteins newly imported into the matrix. Commun. Biol. 2021, 4, 974. [Google Scholar] [CrossRef]
- Cheng, C.W.; Kuo, C.Y.; Fan, C.C.; Fang, W.C.; Jiang, S.S.; Lo, Y.K.; Wang, T.Y.; Kao, M.C.; Lee, A.Y. Overexpression of Lon contributes to survival and aggressive phenotype of cancer cells through mitochondrial complex I-mediated generation of reactive oxygen species. Cell Death Dis. 2013, 4, e681. [Google Scholar] [CrossRef]
- Zanini, G.; Selleri, V.; De Gaetano, A.; Gibellini, L.; Malerba, M.; Mattioli, A.V.; Nasi, M.; Apostolova, N.; Pinti, M. Differential Expression of Lonp1 Isoforms in Cancer Cells. Cells 2022, 11, 3940. [Google Scholar] [CrossRef]
- Galluzzi, L.; Kepp, O.; Kroemer, G. Mitochondria: Master regulators of danger signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 780–788. [Google Scholar] [CrossRef] [PubMed]
- Koyano, F.; Okatsu, K.; Kosako, H.; Tamura, Y.; Go, E.; Kimura, M.; Kimura, Y.; Tsuchiya, H.; Yoshihara, H.; Hirokawa, T.; et al. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 2014, 510, 162–166. [Google Scholar] [CrossRef] [PubMed]
- Thomas, R.E.; Andrews, L.A.; Burman, J.L.; Lin, W.Y.; Pallanck, L.J. PINK1-Parkin pathway activity is regulated by degradation of PINK1 in the mitochondrial matrix. PLoS Genet. 2014, 10, e1004279. [Google Scholar] [CrossRef] [PubMed]
- Greene, A.W.; Grenier, K.; Aguileta, M.A.; Muise, S.; Farazifard, R.; Haque, M.E.; McBride, H.M.; Park, D.S.; Fon, E.A. Mitochondrial processing peptidase regulates PINK1 processing, import and Parkin recruitment. EMBO Rep. 2012, 13, 378–385. [Google Scholar] [CrossRef]
- Wang, X.; Xu, X.; Zhao, Y.; Qi, L.; Ge, H. Inhibition of Lonp1 induces mitochondrial remodeling and autophagy suppression in cervical cancer cells. Acta Histochem. 2023, 125, 151986. [Google Scholar] [CrossRef]
- Drummond, M.J.; Addison, O.; Brunker, L.; Hopkins, P.N.; McClain, D.A.; LaStayo, P.C.; Marcus, R.L. Downregulation of E3 ubiquitin ligases and mitophagy-related genes in skeletal muscle of physically inactive, frail older women: A cross-sectional comparison. J. Gerontol. A Biol. Sci. Med. Sci. 2014, 69, 1040–1048. [Google Scholar] [CrossRef]
- Leduc-Gaudet, J.P.; Hussain, S.N.A.; Barreiro, E.; Gouspillou, G. Mitochondrial Dynamics and Mitophagy in Skeletal Muscle Health and Aging. Int. J. Mol. Sci. 2021, 22, 8179. [Google Scholar] [CrossRef]
- Kao, T.Y.; Chiu, Y.C.; Fang, W.C.; Cheng, C.W.; Kuo, C.Y.; Juan, H.F.; Wu, S.H.; Lee, A.Y. Mitochondrial Lon regulates apoptosis through the association with Hsp60-mtHsp70 complex. Cell Death Dis. 2015, 6, e1642. [Google Scholar] [CrossRef]
- He, L.; Nair, M.K.M.; Chen, Y.; Liu, X.; Zhang, M.; Hazlett, K.R.O.; Deng, H.; Zhang, J.R. The Protease Locus of Francisella tularensis LVS Is Required for Stress Tolerance and Infection in the Mammalian Host. Infect. Immun. 2016, 84, 1387–1402. [Google Scholar] [CrossRef]
- Chen, S.H.; Suzuki, C.K.; Wu, S.H. Thermodynamic characterization of specific interactions between the human Lon protease and G-quartet DNA. Nucleic Acids Res. 2008, 36, 1273–1287. [Google Scholar] [CrossRef]
- Lu, B.; Yadav, S.; Shah, P.G.; Liu, T.; Tian, B.; Pukszta, S.; Villaluna, N.; Kutejova, E.; Newlon, C.S.; Santos, J.H.; et al. Roles for the human ATP-dependent Lon protease in mitochondrial DNA maintenance. J. Biol. Chem. 2007, 282, 17363–17374. [Google Scholar] [CrossRef] [PubMed]
- Winkler, J.; Seybert, A.; Konig, L.; Pruggnaller, S.; Haselmann, U.; Sourjik, V.; Weiss, M.; Frangakis, A.S.; Mogk, A.; Bukau, B. Quantitative and spatio-temporal features of protein aggregation in Escherichia coli and consequences on protein quality control and cellular ageing. EMBO J. 2010, 29, 910–923. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Lu, B.; Lee, I.; Ondrovicova, G.; Kutejova, E.; Suzuki, C.K. DNA and RNA binding by the mitochondrial lon protease is regulated by nucleotide and protein substrate. J. Biol. Chem. 2004, 279, 13902–13910. [Google Scholar] [CrossRef] [PubMed]
- Dikoglu, E.; Alfaiz, A.; Gorna, M.; Bertola, D.; Chae, J.H.; Cho, T.J.; Derbent, M.; Alanay, Y.; Guran, T.; Kim, O.H.; et al. Mutations in LONP1, a mitochondrial matrix protease, cause CODAS syndrome. Am. J. Med. Genet. A 2015, 167, 1501–1509. [Google Scholar] [CrossRef]
- Strauss, K.A.; Jinks, R.N.; Puffenberger, E.G.; Venkatesh, S.; Singh, K.; Cheng, I.; Mikita, N.; Thilagavathi, J.; Lee, J.; Sarafianos, S.; et al. CODAS syndrome is associated with mutations of LONP1, encoding mitochondrial AAA+ Lon protease. Am. J. Hum. Genet. 2015, 96, 121–135. [Google Scholar] [CrossRef]
- Tang, Y.; Liu, Y.X.; Sheng, Y.; Fan, L.L.; Zhang, A.Q.; Zheng, Z.F. The first case report of CODAS syndrome in Chinese population caused by two LONP1 pathogenic mutations. Front. Genet. 2022, 13, 1031856. [Google Scholar] [CrossRef]
- Marlin, S.; Ducou Le Pointe, H.; Le Merrer, M.; Portnoi, M.F.; Chantot, S.; Jonard, L.; Mantel-Guiochon, A.; Siffroi, J.P.; Garabedian, E.N.; Denoyelle, F. Fourth case of cerebral, ocular, dental, auricular, skeletal syndrome (CODAS), description of new features and molecular analysis. Am. J. Med. Genet. A 2010, 152A, 1510–1514. [Google Scholar] [CrossRef]
- Nimmo, G.A.M.; Venkatesh, S.; Pandey, A.K.; Marshall, C.R.; Hazrati, L.N.; Blaser, S.; Ahmed, S.; Cameron, J.; Singh, K.; Ray, P.N.; et al. Bi-allelic mutations of LONP1 encoding the mitochondrial LonP1 protease cause pyruvate dehydrogenase deficiency and profound neurodegeneration with progressive cerebellar atrophy. Hum. Mol. Genet. 2019, 28, 290–306. [Google Scholar] [CrossRef]
- Qiao, L.; Xu, L.; Yu, L.; Wynn, J.; Hernan, R.; Zhou, X.; Farkouh-Karoleski, C.; Krishnan, U.S.; Khlevner, J.; De, A.; et al. Rare and de novo variants in 827 congenital diaphragmatic hernia probands implicate LONP1 as candidate risk gene. Am. J. Hum. Genet. 2021, 108, 1964–1980. [Google Scholar] [CrossRef]
- Welle, S.; Glueck, S.B. In for the long run: Focus on “Lifelong voluntary exercise in the mouse prevents age-related alterations in gene expression in the heart”. Physiol. Genomics 2003, 12, 71–72. [Google Scholar] [CrossRef]
- Chandel, N.S. Mitochondria as signaling organelles. BMC Biol. 2014, 12, 34. [Google Scholar] [CrossRef]
- Henning, R.J. Type-2 diabetes mellitus and cardiovascular disease. Future. Cardiol. 2018, 14, 491–509. [Google Scholar] [CrossRef]
- Borghetti, G.; von Lewinski, D.; Eaton, D.M.; Sourij, H.; Houser, S.R.; Wallner, M. Diabetic Cardiomyopathy: Current and Future Therapies. Beyond Glycemic Control. Front. Physiol. 2018, 9, 1514. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, D.; Montecucco, F.; Dallegri, F.; Carbone, F. Impact of different ectopic fat depots on cardiovascular and metabolic diseases. J. Cell. Physiol. 2019, 234, 21630–21641. [Google Scholar] [CrossRef] [PubMed]
- Paolisso, P.; Bergamaschi, L.; Saturi, G.; D’Angelo, E.C.; Magnani, I.; Toniolo, S.; Stefanizzi, A.; Rinaldi, A.; Bartoli, L.; Angeli, F.; et al. Secondary Prevention Medical Therapy and Outcomes in Patients With Myocardial Infarction With Non-Obstructive Coronary Artery Disease. Front. Pharmacol. 2019, 10, 1606. [Google Scholar] [CrossRef] [PubMed]
- Mattioli, A.V.; Coppi, F.; Manenti, A.; Farinetti, A. Subclinical Vascular Damage: Current Insights and Future Potential. Vasc. Health Risk Manag. 2021, 17, 729–738. [Google Scholar] [CrossRef]
- Ilkun, O.; Boudina, S. Cardiac dysfunction and oxidative stress in the metabolic syndrome: An update on antioxidant therapies. Curr. Pharm. Des. 2013, 19, 4806–4817. [Google Scholar] [CrossRef]
- Schilling, J.D. The mitochondria in diabetic heart failure: From pathogenesis to therapeutic promise. Antioxid. Redox. Signal. 2015, 22, 1515–1526. [Google Scholar] [CrossRef]
- Loor, G.; Schumacker, P.T. Role of hypoxia-inducible factor in cell survival during myocardial ischemia-reperfusion. Cell Death Differ. 2008, 15, 686–690. [Google Scholar] [CrossRef]
- Circu, M.L.; Aw, T.Y. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic. Biol. Med. 2010, 48, 749–762. [Google Scholar] [CrossRef]
- Venkatesh, S.; Li, M.; Saito, T.; Tong, M.; Rashed, E.; Mareedu, S.; Zhai, P.; Barcena, C.; Lopez-Otin, C.; Yehia, G.; et al. Mitochondrial LonP1 protects cardiomyocytes from ischemia/reperfusion injury in vivo. J. Mol. Cell. Cardiol. 2019, 128, 38–50. [Google Scholar] [CrossRef] [PubMed]
- Bruneau, B.G. The developing heart: From The Wizard of Oz to congenital heart disease. Development 2020, 147, 194233. [Google Scholar] [CrossRef]
- Menendez-Montes, I.; Escobar, B.; Palacios, B.; Gomez, M.J.; Izquierdo-Garcia, J.L.; Flores, L.; Jimenez-Borreguero, L.J.; Aragones, J.; Ruiz-Cabello, J.; Torres, M.; et al. Myocardial VHL-HIF Signaling Controls an Embryonic Metabolic Switch Essential for Cardiac Maturation. Dev. Cell 2016, 39, 724–739. [Google Scholar] [CrossRef] [PubMed]
- Maroli, G.; Braun, T. The long and winding road of cardiomyocyte maturation. Cardiovasc. Res. 2021, 117, 712–726. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Sun, Q.; Zhou, L.; Liu, K.; Jiao, K. Complex Regulation of Mitochondrial Function During Cardiac Development. J. Am. Heart. Assoc. 2019, 8, e012731. [Google Scholar] [CrossRef] [PubMed]
- De Gaetano, A.; Gibellini, L.; Bianchini, E.; Borella, R.; De Biasi, S.; Nasi, M.; Boraldi, F.; Cossarizza, A.; Pinti, M. Impaired Mitochondrial Morphology and Functionality in Lonp1(wt/-) Mice. J. Clin. Med. 2020, 9, 1783. [Google Scholar] [CrossRef] [PubMed]
- Zhao, K.; Huang, X.; Zhao, W.; Lu, B.; Yang, Z. LONP1-mediated mitochondrial quality control safeguards metabolic shifts in heart development. Development 2022, 149, 200458. [Google Scholar] [CrossRef]
- Crewe, C.; Kinter, M.; Szweda, L.I. Rapid inhibition of pyruvate dehydrogenase: An initiating event in high dietary fat-induced loss of metabolic flexibility in the heart. PLoS ONE 2013, 8, e77280. [Google Scholar] [CrossRef]
- Onat, U.I.; Yildirim, A.D.; Tufanli, O.; Cimen, I.; Kocaturk, B.; Veli, Z.; Hamid, S.M.; Shimada, K.; Chen, S.; Sin, J.; et al. Intercepting the Lipid-Induced Integrated Stress Response Reduces Atherosclerosis. J. Am. Coll. Cardiol. 2019, 73, 1149–1169. [Google Scholar] [CrossRef]
- Hoshino, A.; Okawa, Y.; Ariyoshi, M.; Kaimoto, S.; Uchihashi, M.; Fukai, K.; Iwai-Kanai, E.; Matoba, S. Oxidative post-translational modifications develop LONP1 dysfunction in pressure overload heart failure. Circ. Heart Fail. 2014, 7, 500–509. [Google Scholar] [CrossRef]
- Cocchi, C.; Coppi, F.; Farinetti, A.; Mattioli, A.V. Cardiovascular disease prevention and therapy in women with Type 2 diabetes. Future Cardiol. 2021, 17, 487–496. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Stanyer, L.; Jorgensen, W.; Hori, O.; Clark, J.B.; Heales, S.J. Inactivation of brain mitochondrial Lon protease by peroxynitrite precedes electron transport chain dysfunction. Neurochem. Int. 2008, 53, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Chou, H.C.; Chen, Y.W.; Lee, T.R.; Wu, F.S.; Chan, H.T.; Lyu, P.C.; Timms, J.F.; Chan, H.L. Proteomics study of oxidative stress and Src kinase inhibition in H9C2 cardiomyocytes: A cell model of heart ischemia-reperfusion injury and treatment. Free Radic. Biol. Med. 2010, 49, 96–108. [Google Scholar] [CrossRef]
- Hsieh, S.R.; Hsu, C.S.; Lu, C.H.; Chen, W.C.; Chiu, C.H.; Liou, Y.M. Epigallocatechin-3-gallate-mediated cardioprotection by Akt/GSK-3beta/caveolin signalling in H9c2 rat cardiomyoblasts. J. Biomed. Sci. 2013, 20, 86. [Google Scholar] [CrossRef]
- Baines, C.P. The cardiac mitochondrion: Nexus of stress. Annu. Rev. Physiol. 2010, 72, 61–80. [Google Scholar] [CrossRef]
- Whelan, R.S.; Kaplinskiy, V.; Kitsis, R.N. Cell death in the pathogenesis of heart disease: Mechanisms and significance. Annu. Rev. Physiol. 2010, 72, 19–44. [Google Scholar] [CrossRef]
- Takahashi, R.; Kawawa, A.; Kubota, S. Short time exposure to hypoxia promotes H9c2 cell growth. Biochim. Biophys. Acta. 2006, 1760, 1293–1297. [Google Scholar] [CrossRef]
- Gurusamy, N.; Lekli, I.; Gorbunov, N.V.; Gherghiceanu, M.; Popescu, L.M.; Das, D.K. Cardioprotection by adaptation to ischaemia augments autophagy in association with BAG-1 protein. J. Cell. Mol. Med. 2009, 13, 373–387. [Google Scholar] [CrossRef]
- Gurusamy, N.; Lekli, I.; Mukherjee, S.; Ray, D.; Ahsan, M.K.; Gherghiceanu, M.; Popescu, L.M.; Das, D.K. Cardioprotection by resveratrol: A novel mechanism via autophagy involving the mTORC2 pathway. Cardiovasc. Res. 2010, 86, 103–112. [Google Scholar] [CrossRef]
- Ristow, M.; Schmeisser, S. Extending life span by increasing oxidative stress. Free Radic. Biol. Med. 2011, 51, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Ristow, M.; Zarse, K. How increased oxidative stress promotes longevity and metabolic health: The concept of mitochondrial hormesis (mitohormesis). Exp. Gerontol. 2010, 45, 410–418. [Google Scholar] [CrossRef] [PubMed]
- Pasupathy, S.; Homer-Vanniasinkam, S. Ischaemic preconditioning protects against ischaemia/reperfusion injury: Emerging concepts. Eur. J. Vasc. Endovasc. Surg. 2005, 29, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Venkatesh, S.; Suzuki, C.K. Cell stress management by the mitochondrial LonP1 protease—Insights into mitigating developmental, oncogenic and cardiac stress. Mitochondrion 2020, 51, 46–61. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Zhong, H.; Bosch-Marce, M.; Fox-Talbot, K.; Wang, L.; Wei, C.; Trush, M.A.; Semenza, G.L. Complete loss of ischaemic preconditioning-induced cardioprotection in mice with partial deficiency of HIF-1 alpha. Cardiovasc. Res. 2008, 77, 463–470. [Google Scholar] [CrossRef]
- Eckle, T.; Kohler, D.; Lehmann, R.; El Kasmi, K.; Eltzschig, H.K. Hypoxia-inducible factor-1 is central to cardioprotection: A new paradigm for ischemic preconditioning. Circulation 2008, 118, 166–175. [Google Scholar] [CrossRef]
- Pinti, M.; Gibellini, L.; De Biasi, S.; Nasi, M.; Roat, E.; O’Connor, J.E.; Cossarizza, A. Functional characterization of the promoter of the human Lon protease gene. Mitochondrion 2011, 11, 200–206. [Google Scholar] [CrossRef]
- Sepuri, N.B.V.; Angireddy, R.; Srinivasan, S.; Guha, M.; Spear, J.; Lu, B.; Anandatheerthavarada, H.K.; Suzuki, C.K.; Avadhani, N.G. Mitochondrial LON protease-dependent degradation of cytochrome c oxidase subunits under hypoxia and myocardial ischemia. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 519–528. [Google Scholar] [CrossRef]
- Guillon, B.; Bulteau, A.L.; Wattenhofer-Donze, M.; Schmucker, S.; Friguet, B.; Puccio, H.; Drapier, J.C.; Bouton, C. Frataxin deficiency causes upregulation of mitochondrial Lon and ClpP proteases and severe loss of mitochondrial Fe-S proteins. FEBS J. 2009, 276, 1036–1047. [Google Scholar] [CrossRef]
- Prabu, S.K.; Anandatheerthavarada, H.K.; Raza, H.; Srinivasan, S.; Spear, J.F.; Avadhani, N.G. Protein kinase A-mediated phosphorylation modulates cytochrome c oxidase function and augments hypoxia and myocardial ischemia-related injury. J. Biol. Chem. 2006, 281, 2061–2070. [Google Scholar] [CrossRef]
- Vijayasarathy, C.; Damle, S.; Prabu, S.K.; Otto, C.M.; Avadhani, N.G. Adaptive changes in the expression of nuclear and mitochondrial encoded subunits of cytochrome c oxidase and the catalytic activity during hypoxia. Eur. J. Biochem. 2003, 270, 871–879. [Google Scholar] [CrossRef] [PubMed]
- Kadenbach, B.; Ramzan, R.; Moosdorf, R.; Vogt, S. The role of mitochondrial membrane potential in ischemic heart failure. Mitochondrion 2011, 11, 700–706. [Google Scholar] [CrossRef] [PubMed]
- Sedlic, F.; Muravyeva, M.Y.; Sepac, A.; Sedlic, M.; Williams, A.M.; Yang, M.; Bai, X.; Bosnjak, Z.J. Targeted Modification of Mitochondrial ROS Production Converts High Glucose-Induced Cytotoxicity to Cytoprotection: Effects on Anesthetic Preconditioning. J. Cell. Physiol. 2017, 232, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Bulteau, A.L.; Lundberg, K.C.; Ikeda-Saito, M.; Isaya, G.; Szweda, L.I. Reversible redox-dependent modulation of mitochondrial aconitase and proteolytic activity during in vivo cardiac ischemia/reperfusion. Proc. Natl. Acad. Sci. USA 2005, 102, 5987–5991. [Google Scholar] [CrossRef]
- Cantu, D.; Schaack, J.; Patel, M. Oxidative inactivation of mitochondrial aconitase results in iron and H2O2-mediated neurotoxicity in rat primary mesencephalic cultures. PLoS ONE 2009, 4, e7095. [Google Scholar] [CrossRef]
- Lopaschuk, G.D.; Jaswal, J.S. Energy metabolic phenotype of the cardiomyocyte during development, differentiation, and postnatal maturation. J. Cardiovasc. Pharmacol. 2010, 56, 130–140. [Google Scholar] [CrossRef]
- Lopaschuk, G.D.; Ussher, J.R.; Folmes, C.D.; Jaswal, J.S.; Stanley, W.C. Myocardial fatty acid metabolism in health and disease. Physiol. Rev. 2010, 90, 207–258. [Google Scholar] [CrossRef]
- Stanley, W.C.; Recchia, F.A.; Lopaschuk, G.D. Myocardial substrate metabolism in the normal and failing heart. Physiol. Rev. 2005, 85, 1093–1129. [Google Scholar] [CrossRef]
- Fragasso, G.; Salerno, A.; Lattuada, G.; Cuko, A.; Calori, G.; Scollo, A.; Ragogna, F.; Arioli, F.; Bassanelli, G.; Spoladore, R.; et al. Effect of partial inhibition of fatty acid oxidation by trimetazidine on whole body energy metabolism in patients with chronic heart failure. Heart 2011, 97, 1495–1500. [Google Scholar] [CrossRef]
- Masoud, W.G.; Ussher, J.R.; Wang, W.; Jaswal, J.S.; Wagg, C.S.; Dyck, J.R.; Lygate, C.A.; Neubauer, S.; Clanachan, A.S.; Lopaschuk, G.D. Failing mouse hearts utilize energy inefficiently and benefit from improved coupling of glycolysis and glucose oxidation. Cardiovasc. Res. 2014, 101, 30–38. [Google Scholar] [CrossRef]
- Xu, Z.; Fu, T.; Guo, Q.; Zhou, D.; Sun, W.; Zhou, Z.; Chen, X.; Zhang, J.; Liu, L.; Xiao, L.; et al. Disuse-associated loss of the protease LONP1 in muscle impairs mitochondrial function and causes reduced skeletal muscle mass and strength. Nat. Commun. 2022, 13, 894. [Google Scholar] [CrossRef]
- Cordeiro, A.V.; Bricola, R.S.; Braga, R.R.; Lenhare, L.; Silva, V.R.R.; Anaruma, C.P.; Katashima, C.K.; Crisol, B.M.; Simabuco, F.M.; Silva, A.S.R.; et al. Aerobic Exercise Training Induces the Mitonuclear Imbalance and UPRmt in the Skeletal Muscle of Aged Mice. J. Gerontol. A Biol. Sci. Med. Sci. 2020, 75, 2258–2261. [Google Scholar] [CrossRef] [PubMed]
- Grotewiel, M.S.; Martin, I.; Bhandari, P.; Cook-Wiens, E. Functional senescence in Drosophila melanogaster. Ageing Res. Rev. 2005, 4, 372–397. [Google Scholar] [CrossRef] [PubMed]
- Martinez, V.G.; Javadi, C.S.; Ngo, E.; Ngo, L.; Lagow, R.D.; Zhang, B. Age-related changes in climbing behavior and neural circuit physiology in Drosophila. Dev. Neurobiol. 2007, 67, 778–791. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.S.; Lekkas, P.; Braddock, J.M.; Farman, G.P.; Ballif, B.A.; Irving, T.C.; Maughan, D.W.; Vigoreaux, J.O. Aging enhances indirect flight muscle fiber performance yet decreases flight ability in Drosophila. Biophys. J. 2008, 95, 2391–2401. [Google Scholar] [CrossRef]
- Baggio, F.; Bratic, A.; Mourier, A.; Kauppila, T.E.; Tain, L.S.; Kukat, C.; Habermann, B.; Partridge, L.; Larsson, N.G. Drosophila melanogaster LRPPRC2 is involved in coordination of mitochondrial translation. Nucleic Acids Res. 2014, 42, 13920–13938. [Google Scholar] [CrossRef]
- Bratic, A.; Wredenberg, A.; Gronke, S.; Stewart, J.B.; Mourier, A.; Ruzzenente, B.; Kukat, C.; Wibom, R.; Habermann, B.; Partridge, L.; et al. The bicoid stability factor controls polyadenylation and expression of specific mitochondrial mRNAs in Drosophila melanogaster. PLoS Genet. 2011, 7, e1002324. [Google Scholar] [CrossRef]
- Rackham, O.; Busch, J.D.; Matic, S.; Siira, S.J.; Kuznetsova, I.; Atanassov, I.; Ermer, J.A.; Shearwood, A.M.; Richman, T.R.; Stewart, J.B.; et al. Hierarchical RNA Processing Is Required for Mitochondrial Ribosome Assembly. Cell Rep. 2016, 16, 1874–1890. [Google Scholar] [CrossRef]
- Pareek, G.; Thomas, R.E.; Vincow, E.S.; Morris, D.R.; Pallanck, L.J. Lon protease inactivation in Drosophila causes unfolded protein stress and inhibition of mitochondrial translation. Cell Death. Discov. 2018, 4, 51. [Google Scholar] [CrossRef]
- Nouri, K.; Feng, Y.; Schimmer, A.D. Mitochondrial ClpP serine protease-biological function and emerging target for cancer therapy. Cell Death Dis. 2020, 11, 841. [Google Scholar] [CrossRef]
- Inui, T.; Anzai, M.; Takezawa, Y.; Endo, W.; Kakisaka, Y.; Kikuchi, A.; Onuma, A.; Kure, S.; Nishino, I.; Ohba, C.; et al. A novel mutation in the proteolytic domain of LONP1 causes atypical CODAS syndrome. J. Hum. Genet. 2017, 62, 653–655. [Google Scholar] [CrossRef] [PubMed]
- Hannah-Shmouni, F.; MacNeil, L.; Brady, L.; Nilsson, M.I.; Tarnopolsky, M. Expanding the Clinical Spectrum of LONP1-Related Mitochondrial Cytopathy. Front. Neurol. 2019, 10, 981. [Google Scholar] [CrossRef] [PubMed]
- Masiero, E.; Agatea, L.; Mammucari, C.; Blaauw, B.; Loro, E.; Komatsu, M.; Metzger, D.; Reggiani, C.; Schiaffino, S.; Sandri, M. Autophagy is required to maintain muscle mass. Cell. Metab. 2009, 10, 507–515. [Google Scholar] [CrossRef]
- Neel, B.A.; Lin, Y.; Pessin, J.E. Skeletal muscle autophagy: A new metabolic regulator. Trends. Endocrinol. Metab. 2013, 24, 635–643. [Google Scholar] [CrossRef]
- Mammucari, C.; Milan, G.; Romanello, V.; Masiero, E.; Rudolf, R.; Del Piccolo, P.; Burden, S.J.; Di Lisi, R.; Sandri, C.; Zhao, J.; et al. FoxO3 controls autophagy in skeletal muscle in vivo. Cell. Metab. 2007, 6, 458–471. [Google Scholar] [CrossRef]
- O’Leary, M.F.; Vainshtein, A.; Iqbal, S.; Ostojic, O.; Hood, D.A. Adaptive plasticity of autophagic proteins to denervation in aging skeletal muscle. Am. J. Physiol. Cell Physiol. 2013, 304, C422–C430. [Google Scholar] [CrossRef]
- Vainshtein, A.; Desjardins, E.M.; Armani, A.; Sandri, M.; Hood, D.A. PGC-1alpha modulates denervation-induced mitophagy in skeletal muscle. Skelet. Muscle 2015, 5, 9. [Google Scholar] [CrossRef]
- Huang, S.; Wang, X.; Yu, J.; Tian, Y.; Yang, C.; Chen, Y.; Chen, H.; Ge, H. LonP1 regulates mitochondrial network remodeling through the PINK1/Parkin pathway during myoblast differentiation. Am. J. Physiol. Cell Physiol. 2020, 319, C1020–C1028. [Google Scholar] [CrossRef]
- Barbieri, E.; Battistelli, M.; Casadei, L.; Vallorani, L.; Piccoli, G.; Guescini, M.; Gioacchini, A.M.; Polidori, E.; Zeppa, S.; Ceccaroli, P.; et al. Morphofunctional and Biochemical Approaches for Studying Mitochondrial Changes during Myoblasts Differentiation. J. Aging Res. 2011, 2011, 845379. [Google Scholar] [CrossRef]
- Huang, S.; Wang, X.; Wu, X.; Yu, J.; Li, J.; Huang, X.; Zhu, C.; Ge, H. Yap regulates mitochondrial structural remodeling during myoblast differentiation. Am. J. Physiol. Cell Physiol. 2018, 315, C474–C484. [Google Scholar] [CrossRef] [PubMed]
- Pownall, M.E.; Gustafsson, M.K.; Emerson, C.P., Jr. Myogenic regulatory factors and the specification of muscle progenitors in vertebrate embryos. Annu. Rev. Cell Dev. Biol. 2002, 18, 747–783. [Google Scholar] [CrossRef] [PubMed]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010, 8, e1000298. [Google Scholar] [CrossRef] [PubMed]
- Baechler, B.L.; Bloemberg, D.; Quadrilatero, J. Mitophagy regulates mitochondrial network signaling, oxidative stress, and apoptosis during myoblast differentiation. Autophagy 2019, 15, 1606–1619. [Google Scholar] [CrossRef]
- Sin, J.; Andres, A.M.; Taylor, D.J.; Weston, T.; Hiraumi, Y.; Stotland, A.; Kim, B.J.; Huang, C.; Doran, K.S.; Gottlieb, R.A. Mitophagy is required for mitochondrial biogenesis and myogenic differentiation of C2C12 myoblasts. Autophagy 2016, 12, 369–380. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Dorn, G.W., 2nd. PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science 2013, 340, 471–475. [Google Scholar] [CrossRef]
- Gegg, M.E.; Cooper, J.M.; Chau, K.Y.; Rojo, M.; Schapira, A.H.; Taanman, J.W. Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin-dependent manner upon induction of mitophagy. Hum. Mol. Genet. 2010, 19, 4861–4870. [Google Scholar] [CrossRef]
- Lutz, A.K.; Exner, N.; Fett, M.E.; Schlehe, J.S.; Kloos, K.; Lammermann, K.; Brunner, B.; Kurz-Drexler, A.; Vogel, F.; Reichert, A.S.; et al. Loss of parkin or PINK1 function increases Drp1-dependent mitochondrial fragmentation. J. Biol. Chem. 2009, 284, 22938–22951. [Google Scholar] [CrossRef]
- Sebastian, D.; Sorianello, E.; Segales, J.; Irazoki, A.; Ruiz-Bonilla, V.; Sala, D.; Planet, E.; Berenguer-Llergo, A.; Munoz, J.P.; Sanchez-Feutrie, M.; et al. Mfn2 deficiency links age-related sarcopenia and impaired autophagy to activation of an adaptive mitophagy pathway. EMBO J. 2016, 35, 1677–1693. [Google Scholar] [CrossRef]
- De Gaetano, A.; Gibellini, L.; Zanini, G.; Nasi, M.; Cossarizza, A.; Pinti, M. Mitophagy and Oxidative Stress: The Role of Aging. Antioxidants 2021, 10, 794. [Google Scholar] [CrossRef]
- Bodine, S.C.; Latres, E.; Baumhueter, S.; Lai, V.K.; Nunez, L.; Clarke, B.A.; Poueymirou, W.T.; Panaro, F.J.; Na, E.; Dharmarajan, K.; et al. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science 2001, 294, 1704–1708. [Google Scholar] [CrossRef]
- Gomes, M.D.; Lecker, S.H.; Jagoe, R.T.; Navon, A.; Goldberg, A.L. Atrogin-1, a muscle-specific F-box protein highly expressed during muscle atrophy. Proc. Natl. Acad. Sci. USA 2001, 98, 14440–14445. [Google Scholar] [CrossRef] [PubMed]
- Ruegg, M.A.; Glass, D.J. Molecular mechanisms and treatment options for muscle wasting diseases. Annu. Rev. Pharmacol. Toxicol. 2011, 51, 373–395. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.; Nathan, J.A.; Goldberg, A.L. Muscle wasting in disease: Molecular mechanisms and promising therapies. Nat. Rev. Drug Discov. 2015, 14, 58–74. [Google Scholar] [CrossRef] [PubMed]
- Egerman, M.A.; Glass, D.J. Signaling pathways controlling skeletal muscle mass. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 59–68. [Google Scholar] [CrossRef]
- Guo, Q.; Xu, Z.; Zhou, D.; Fu, T.; Wang, W.; Sun, W.; Xiao, L.; Liu, L.; Ding, C.; Yin, Y.; et al. Mitochondrial proteostasis stress in muscle drives a long-range protective response to alleviate dietary obesity independently of ATF4. Sci. Adv. 2022, 8, eabo0340. [Google Scholar] [CrossRef]
- Wang, D.; Day, E.A.; Townsend, L.K.; Djordjevic, D.; Jorgensen, S.B.; Steinberg, G.R. GDF15: Emerging biology and therapeutic applications for obesity and cardiometabolic disease. Nat. Rev. Endocrinol. 2021, 17, 592–607. [Google Scholar] [CrossRef]
- Keipert, S.; Ost, M. Stress-induced FGF21 and GDF15 in obesity and obesity resistance. Trends Endocrinol. Metab. 2021, 32, 904–915. [Google Scholar] [CrossRef]
- Kharitonenkov, A.; Shiyanova, T.L.; Koester, A.; Ford, A.M.; Micanovic, R.; Galbreath, E.J.; Sandusky, G.E.; Hammond, L.J.; Moyers, J.S.; Owens, R.A.; et al. FGF-21 as a novel metabolic regulator. J. Clin. Investig. 2005, 115, 1627–1635. [Google Scholar] [CrossRef]
- Inagaki, T.; Dutchak, P.; Zhao, G.; Ding, X.; Gautron, L.; Parameswara, V.; Li, Y.; Goetz, R.; Mohammadi, M.; Esser, V.; et al. Endocrine regulation of the fasting response by PPARalpha-mediated induction of fibroblast growth factor 21. Cell Metab. 2007, 5, 415–425. [Google Scholar] [CrossRef]
- Owen, B.M.; Ding, X.; Morgan, D.A.; Coate, K.C.; Bookout, A.L.; Rahmouni, K.; Kliewer, S.A.; Mangelsdorf, D.J. FGF21 acts centrally to induce sympathetic nerve activity, energy expenditure, and weight loss. Cell Metab. 2014, 20, 670–677. [Google Scholar] [CrossRef]
- Quiros, P.M.; Mottis, A.; Auwerx, J. Mitonuclear communication in homeostasis and stress. Nat. Rev. Mol. Cell Biol. 2016, 17, 213–226. [Google Scholar] [CrossRef] [PubMed]
- Bar-Ziv, R.; Bolas, T.; Dillin, A. Systemic effects of mitochondrial stress. EMBO Rep. 2020, 21, e50094. [Google Scholar] [CrossRef] [PubMed]
- Shpilka, T.; Haynes, C.M. The mitochondrial UPR: Mechanisms, physiological functions and implications in ageing. Nat. Rev. Mol. Cell Biol. 2018, 19, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Anderson, N.S.; Haynes, C.M. Folding the Mitochondrial UPR into the Integrated Stress Response. Trends. Cell Biol. 2020, 30, 428–439. [Google Scholar] [CrossRef] [PubMed]
- Kaspar, S.; Oertlin, C.; Szczepanowska, K.; Kukat, A.; Senft, K.; Lucas, C.; Brodesser, S.; Hatzoglou, M.; Larsson, O.; Topisirovic, I.; et al. Adaptation to mitochondrial stress requires CHOP-directed tuning of ISR. Sci. Adv. 2021, 7, eabf0971. [Google Scholar] [CrossRef]
- Quiros, P.M.; Prado, M.A.; Zamboni, N.; D’Amico, D.; Williams, R.W.; Finley, D.; Gygi, S.P.; Auwerx, J. Multi-omics analysis identifies ATF4 as a key regulator of the mitochondrial stress response in mammals. J. Cell Biol. 2017, 216, 2027–2045. [Google Scholar] [CrossRef] [PubMed]
- Delaval, E.; Perichon, M.; Friguet, B. Age-related impairment of mitochondrial matrix aconitase and ATP-stimulated protease in rat liver and heart. Eur. J. Biochem. 2004, 271, 4559–4564. [Google Scholar] [CrossRef]
- Bakala, H.; Delaval, E.; Hamelin, M.; Bismuth, J.; Borot-Laloi, C.; Corman, B.; Friguet, B. Changes in rat liver mitochondria with aging. Lon protease-like reactivity and N(epsilon)-carboxymethyllysine accumulation in the matrix. Eur. J. Biochem. 2003, 270, 2295–2302. [Google Scholar] [CrossRef]
- Ngo, J.K.; Davies, K.J. Importance of the lon protease in mitochondrial maintenance and the significance of declining lon in aging. Ann. N. Y. Acad. Sci. 2007, 1119, 78–87. [Google Scholar] [CrossRef]
- Erjavec, N.; Bayot, A.; Gareil, M.; Camougrand, N.; Nystrom, T.; Friguet, B.; Bulteau, A.L. Deletion of the mitochondrial Pim1/Lon protease in yeast results in accelerated aging and impairment of the proteasome. Free Radic. Biol. Med. 2013, 56, 9–16. [Google Scholar] [CrossRef]
- Taouktsi, E.; Kyriakou, E.; Smyrniotis, S.; Borbolis, F.; Bondi, L.; Avgeris, S.; Trigazis, E.; Rigas, S.; Voutsinas, G.E.; Syntichaki, P. Organismal and Cellular Stress Responses upon Disruption of Mitochondrial Lonp1 Protease. Cells 2022, 11, 1363. [Google Scholar] [CrossRef]
- Luce, K.; Osiewacz, H.D. Increasing organismal healthspan by enhancing mitochondrial protein quality control. Nat. Cell Biol. 2009, 11, 852–858. [Google Scholar] [CrossRef] [PubMed]
- Pareek, G.; Pallanck, L.J. Inactivation of Lon protease reveals a link between mitochondrial unfolded protein stress and mitochondrial translation inhibition. Cell Death Dis. 2018, 9, 1168. [Google Scholar] [CrossRef] [PubMed]
- Pomatto, L.C.D.; Carney, C.; Shen, B.; Wong, S.; Halaszynski, K.; Salomon, M.P.; Davies, K.J.A.; Tower, J. The Mitochondrial Lon Protease Is Required for Age-Specific and Sex-Specific Adaptation to Oxidative Stress. Curr. Biol. 2017, 27, 1–15. [Google Scholar] [CrossRef]
- Kurihara, Y.; Kanki, T.; Aoki, Y.; Hirota, Y.; Saigusa, T.; Uchiumi, T.; Kang, D. Mitophagy plays an essential role in reducing mitochondrial production of reactive oxygen species and mutation of mitochondrial DNA by maintaining mitochondrial quantity and quality in yeast. J. Biol. Chem. 2012, 287, 3265–3272. [Google Scholar] [CrossRef] [PubMed]
- Hepple, R.T.; Baker, D.J.; Kaczor, J.J.; Krause, D.J. Long-term caloric restriction abrogates the age-related decline in skeletal muscle aerobic function. FASEB J. 2005, 19, 1320–1322. [Google Scholar] [CrossRef] [PubMed]
- Lanza, I.R.; Short, D.K.; Short, K.R.; Raghavakaimal, S.; Basu, R.; Joyner, M.J.; McConnell, J.P.; Nair, K.S. Endurance exercise as a countermeasure for aging. Diabetes 2008, 57, 2933–2942. [Google Scholar] [CrossRef]
- Stadtman, E.R.; Starke-Reed, P.E.; Oliver, C.N.; Carney, J.M.; Floyd, R.A. Protein modification in aging. EXS 1992, 62, 64–72. [Google Scholar] [CrossRef]
- LaFrance, R.; Brustovetsky, N.; Sherburne, C.; Delong, D.; Dubinsky, J.M. Age-related changes in regional brain mitochondria from Fischer 344 rats. Aging. Cell. 2005, 4, 139–145. [Google Scholar] [CrossRef]
- Sachs, H.G.; Colgan, J.A.; Lazarus, M.L. Ultrastructure of the aging myocardium: A morphometric approach. Am. J. Anat. 1977, 150, 63–71. [Google Scholar] [CrossRef]
- Richter, C. Oxidative damage to mitochondrial DNA and its relationship to ageing. Int. J. Biochem. Cell Biol. 1995, 27, 647–653. [Google Scholar] [CrossRef] [PubMed]
- Cadenas, E.; Davies, K.J. Mitochondrial free radical generation, oxidative stress, and aging. Free Radic. Biol. Med. 2000, 29, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Khrapko, K.; Bodyak, N.; Thilly, W.G.; van Orsouw, N.J.; Zhang, X.; Coller, H.A.; Perls, T.T.; Upton, M.; Vijg, J.; Wei, J.Y. Cell-by-cell scanning of whole mitochondrial genomes in aged human heart reveals a significant fraction of myocytes with clonally expanded deletions. Nucleic Acids Res. 1999, 27, 2434–2441. [Google Scholar] [CrossRef] [PubMed]
- Aiken, J.; Bua, E.; Cao, Z.; Lopez, M.; Wanagat, J.; McKenzie, D.; McKiernan, S. Mitochondrial DNA deletion mutations and sarcopenia. Ann. N. Y. Acad. Sci. 2002, 959, 412–423. [Google Scholar] [CrossRef] [PubMed]
- Ermini, M. Ageing changes in mammalian skeletal muscle: Biochemical studies. Gerontology 1976, 22, 301–316. [Google Scholar] [CrossRef]
- Koltai, E.; Hart, N.; Taylor, A.W.; Goto, S.; Ngo, J.K.; Davies, K.J.; Radak, Z. Age-associated declines in mitochondrial biogenesis and protein quality control factors are minimized by exercise training. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2012, 303, R127–R134. [Google Scholar] [CrossRef]
- Bota, D.A.; Davies, K.J. Mitochondrial Lon protease in human disease and aging: Including an etiologic classification of Lon-related diseases and disorders. Free Radic. Biol. Med. 2016, 100, 188–198. [Google Scholar] [CrossRef]
- Gibellini, L.; Pinti, M.; Beretti, F.; Pierri, C.L.; Onofrio, A.; Riccio, M.; Carnevale, G.; De Biasi, S.; Nasi, M.; Torelli, F.; et al. Sirtuin 3 interacts with Lon protease and regulates its acetylation status. Mitochondrion 2014, 18, 76–81. [Google Scholar] [CrossRef]
- Granot, Z.; Kobiler, O.; Melamed-Book, N.; Eimerl, S.; Bahat, A.; Lu, B.; Braun, S.; Maurizi, M.R.; Suzuki, C.K.; Oppenheim, A.B.; et al. Turnover of mitochondrial steroidogenic acute regulatory (StAR) protein by Lon protease: The unexpected effect of proteasome inhibitors. Mol. Endocrinol. 2007, 21, 2164–2177. [Google Scholar] [CrossRef]


| Organ | Experimental Model | Lonp1 Levels | Effects | References |
|---|---|---|---|---|
| Heart | Mouse cardiomyocytes | Normal expression | Glucose, FAO enzymes and PDH levels modulation in maturing cardiomyocytes | [48] |
| Rat cardiomyocyte H9c2 cells | Overexpression | Apoptosis under normoxic conditions | [74,75] | |
| Downregulation | Mitigation of cell death induced by hypoxia | [74,75] | ||
| Mouse | Knock out | Severe defective heart development; embryonic lethality; Reduction nof cardioprotective effect of IPC; Increment in myocardial infarct size | [66,67,74,75,84] | |
| Downregulation | fragmentation of mitochondria; cardiomyocytes aberrant metabolic reprogramming; cardiomyopathy; HF | [67] | ||
| Normal expression | Reduction of cardiac stress and injury by reprogramming energy metabolism, thorough the regulation of PDH activity and OXPHOS complexes | [19,24,48,83] | ||
| Upregulation | Reduction of oxidative damage; preservation of redox state of mitochondria; reprograming of mitochondrial bioenergetics; reduction of complex I activity, ROS production, and cardiac cell death | [61] | ||
| Overexpression | Reduction of protein carbonylation and lipid peroxidation during ischemia and early reperfusion | [61,94] | ||
| Skeletal muscle | Drosophila | Knockout | Locomotion defects; alteration of respiratory chain function; accumulation of unfolded and oxidized mitochondrial proteins; reduction of OXPHOS capacity and ATP production; stimulation of UPRmt response | [106,107,108,109] |
| Mouse C2C12 cells | Knockout | Suppression of PINK1/Parkin pathway; alterations of mitochondrial dynamics; accumulation of damaged mitochondria | [128,129] | |
| Immortalized mouse myoblasts | Downregulation | Block of autophagy, only at the late stage of myoblast differentiation | [118] | |
| Mouse myotubes | Knock out | Reduction of fully assembled respiratory complexes IV; alterations of mitochondrial respiration | [101] | |
| Mouse | Knock out | Alteration of mitochondrial ultrastructure and organelle functions; hypotonia, mild to moderate motor delay; Release of myokines; Reduction of lean and fat mass and lower body weight | [16,25,66,101] | |
| Mouse under high-fat diet (HFD) | Knock out | Activation of UPRmt; alterations of mitochondrial protein turnover; improvement of insulin resistance; reduction of liver steatosis; prevention of high-fat diet HFD–induced obesity | [135] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zanini, G.; Selleri, V.; Malerba, M.; Solodka, K.; Sinigaglia, G.; Nasi, M.; Mattioli, A.V.; Pinti, M. The Role of Lonp1 on Mitochondrial Functions during Cardiovascular and Muscular Diseases. Antioxidants 2023, 12, 598. https://doi.org/10.3390/antiox12030598
Zanini G, Selleri V, Malerba M, Solodka K, Sinigaglia G, Nasi M, Mattioli AV, Pinti M. The Role of Lonp1 on Mitochondrial Functions during Cardiovascular and Muscular Diseases. Antioxidants. 2023; 12(3):598. https://doi.org/10.3390/antiox12030598
Chicago/Turabian StyleZanini, Giada, Valentina Selleri, Mara Malerba, Kateryna Solodka, Giorgia Sinigaglia, Milena Nasi, Anna Vittoria Mattioli, and Marcello Pinti. 2023. "The Role of Lonp1 on Mitochondrial Functions during Cardiovascular and Muscular Diseases" Antioxidants 12, no. 3: 598. https://doi.org/10.3390/antiox12030598
APA StyleZanini, G., Selleri, V., Malerba, M., Solodka, K., Sinigaglia, G., Nasi, M., Mattioli, A. V., & Pinti, M. (2023). The Role of Lonp1 on Mitochondrial Functions during Cardiovascular and Muscular Diseases. Antioxidants, 12(3), 598. https://doi.org/10.3390/antiox12030598