1. Introduction
According to the WHO’s Global Health Estimates Report of 2019, stroke represents the second largest cause of death worldwide [
1], with the ischemic subtype accounting for approximately 80–85% of registered cases [
1]. Brain ischemia is usually caused by a thrombus occluding a blood vessel, or by a systemic hypoperfusion, and is characterized by a decrease in the blood supply to the brain, which triggers a set of processes known as ischemic cascade within the cerebral tissue [
2]. Depending on the severity of the ischemic insult (i.e., level and duration of blood reduction), and the particular area of the brain affected, an ischemic insult may lead to the loss of cell function and, eventually, cell death [
2]. Thus, the first therapeutic approach must be aimed at recovering normal blood flow levels to the tissue affected. Currently, mechanical thrombectomy or pharmacological thrombolysis (e.g., with alteplase or tenecteplase) may afford recanalization in eligible patients [
3]. However, despite being essential to stop further cell death, tissue reperfusion does not ensure a total recovery of cell function. In fact, reperfusion may also exacerbate the damage produced within the ischemic penumbra in what is called “ischemia-reperfusion damage” [
4].
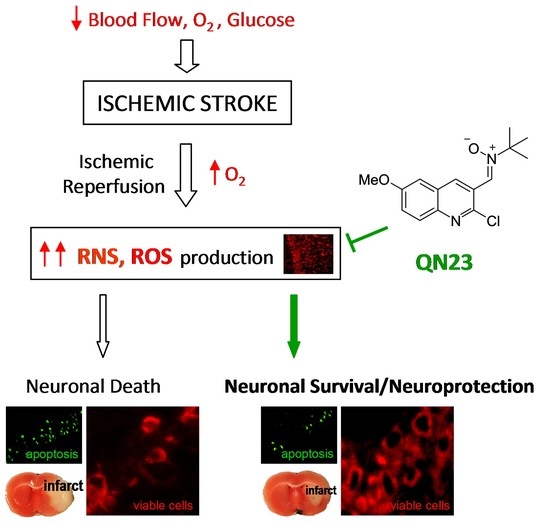
One of the main actors playing a role in ischemic tissue recovery following reperfusion is oxidative stress. As a consequence of the massive influx of oxygen upon the previously compromised tissue of the penumbra, radical species production overwhelms the endogenous antioxidant systems and exerts structural and functional damage, hindering tissue recovery and inducing cell death [
5,
6]. Therefore, antioxidant therapies able to reduce the impact of this damage have been highly sought after. The nitrone functional group (i.e.,
N-oxide of an imine) has been a widely studied chemical group in the search for drugs aimed at reducing oxidative stress. Long described as radical traps due to their radical species scavenging activity [
7], nitrones were proposed early on as potentially therapeutic antioxidant compounds [
8,
9]. In particular for ischemic stroke treatment, the widely known nitrone NXY-059 (Cerovive, developed by AstraZeneca) served for many years as the antioxidant nitrone paradigm. As a more soluble and potent derivative of the original
N-
tert-Butyl-α-phenylnitrone (PBN), NXY-059 was reported to reduce lesion volume and motor impairment in preclinical phases, including several animal species and experimental stroke models [
10]. Phase I and II clinical trials reported a good pharmacokinetic profile [
11,
12,
13], but a lower brain permeability than original PBN was reported, even though it was increased as a consequence of the ischemic episode [
14]. These results, along with no remarkable safety concerns, promoted NXY-059 to Phase III clinical trials, SAINT I and SAINT II, whose pooled results eventually revealed that NXY-059 did not afford a significant improvement in stroke patients when compared with placebo [
15]. Posterior reviews and meta-analysis pointed out an overestimation of efficacy in preclinical trials, which presented several drawbacks that avoided the translation of the preclinical results of NXY-059 to clinical practice [
10], apart from unresolved enquiries regarding NXY-059’s true mechanism of action [
16]. Currently, numerous guidelines highlight the need for a rigorous preclinical study including relevant aspects in the clinical practice treatment of particular diseases and their outcomes early in the development of new neuroprotectant drugs [
17].
In spite of it, NXY-059 served to make clear the therapeutic potential of nitrones as candidates for the treatment of different diseases, especially ischemic stroke. For many years, our group has been developing new nitrone-derived compounds based on diverse scaffolds as potential drug candidates for the treatment of the ischemic neuropathology. Apart from the cholesterol-derived nitrone candidate ISQ-201 [
18], our group identified a set of nitrones bearing a quinoline core as interesting neuroprotective compounds in oxygen and glucose deprivation (OGD) injury on primary neuronal cultures [
19]. One of these quinolyl nitrones, named RP19, was identified as the most potent of the set, with a promising neuroprotective effect against OGD insult, and thus prompted its study on in vivo stroke experimental models, confirming its neuroprotective effect [
20]. Further exploration of the quinoline core and screening of more than 40 quinolyl-derived compounds in our phenotypical OGD model has led to new interesting candidates, with the one identified as QN23 being our current lead quinolyl nitrone [
21,
22]. QN23 presented higher protection and tolerability than previous RP19, and higher potency than NXY-059, on primary neuronal cultures subjected to OGD. Furthermore, QN23 had a remarkable antioxidant profile, being particularly strong against hydroxyl radical, which was complemented by studies in primary neuronal cultures in which QN23 reduced reactive oxygen species (ROS) and lipid peroxidation levels [
22]. The in vivo study of its neuroprotective activity revealed decreased levels of neuronal death and apoptosis and an improvement in neurological function, after intraperitoneal injection of QN23 at the onset of post-ischemic reperfusion [
22]. In addition, the study of the anti-inflammatory activity of QN23 in the carrageenan-induced rat paw edema experimental model showed anti-inflammatory properties [
22]. In our search for new therapeutic compounds for the treatment of ischemic stroke,
Scheme 1 shows QN23 development as the lead compound among the new quinolyl drug candidates. In order to keep with its development, new evidence of antioxidant activity and relevant preclinical characterization studies of QN23 are described in this report.
In particular, in the present study we assess the effectivity of QN23 when administered intravenously, in contrast to previous studies based on intraperitoneal administration. We also report the dose–response study of QN23 in order to establish its tolerability and efficacy in vivo, and we evaluate the infarct size and neurofunctional impairment reductions by QN23 treatment in an alternative transient middle cerebral artery occlusion (tMCAO) experimental stroke model performed by an independent laboratory. Another aspect of importance in the development of new therapeutic candidates is the assessment of a clinically useful therapeutic window [
3]. In this report, a therapeutic window study of QN23 was also included. Finally, and considering the dramatic increase in stroke incidence in younger individuals (i.e., younger than 55 years) being reported recently [
23], an assessment of the effect of any potential treatment on functional sequelae later in time after the ischemic episode is required. These adults may eventually suffer from compromising neurofunctional or neuropsychiatric outcomes, long after the ischemic episode but still early in life [
24]. Due to this, the assessment of a long-term effect should also be considered early in preclinical phases of drug development [
17]. Therefore, long-term efficacy of QN23 was assessed 3 months after the ischemic insult.
2. Materials and Methods
2.1. Compounds
Quinolyl nitrone QN23 was synthesized as described [
22]. NXY-059 was supplied by TargetMol (Boston, MA, USA) (T6201).
2.2. Animal Model of Global Cerebral Ischemia, Experimental Design, and Treatment
Transient forebrain ischemia was induced in adult male Wistar rats (10–12 weeks, Charles River, L’Arbresle, France) by the standard four-vessel occlusion (4VO) model previously described [
25,
26]. Briefly, animals were anesthetized by intraperitoneal injection of a combination of ketamine, diazepam, and atropine (62.5, 5.0, and 0.25 mg/kg, respectively); positioned in a stereotactic device; and both vertebral arteries were cauterized. The next day, anesthesia was induced with 4% isoflurane and maintained at 2–2.5% isoflurane for carotid artery dissection. After anesthesia withdrawal, the carotid arteries were occluded with an atraumatic vascular clamp to induce brain ischemia. During the entire surgical procedure, temperature was monitored at 2 min intervals and kept at 37 °C. After 15 min of ischemia, the carotid arteries were unclamped for brain reperfusion. The animals were studied after 5 days of reperfusion (R5d), or after 11 weeks of reperfusion (long-term studies), and then they were euthanized under deep anesthesia. Animals from the sham control group were handled in the same way, but without occlusion of the carotid arteries. Ischemic animals were treated with vehicle solution (ethanol/polyethylene glycol 400/saline, 1:300:200
v/v), or with nitrones QN23 or NXY-059 in vehicle solution, as a single injection through the tail vein at the onset of reperfusion. For the therapeutic window experiments, the animals were also treated at reperfusion times 1, 3, and 6 h after ischemia. The treatments were prepared by an independent researcher and administered with concealed allocation. Treatments were administered in a randomized order obtained by a computer-based randomization program. To determine the sample size, a power analysis (
http://www.biomath.info/power/ttest.htm, accessed on 12 March 2020) with significance level set at 0.05 and statistical power set at 0.8 (80%) rendered a sample size of <6 subjects per group. A total of 120 ischemic animals (30 animals treated with vehicle, 60 treated with QN23, and 30 treated with NXY-059) and 9 sham control animals were included in these studies. Additionally, 13 animals were excluded from the study: 6 animals were not ischemic and 7 animals died within the 24 h after the surgical procedure (5 vehicles, 1 QN23-treated at 1.0 mg/kg, and 1 QN23-treated at 2.0 mg/kg). The Animal Care Ethics Committee of the Hospital Universitario Ramón y Cajal (Madrid, Spain) authorized all the procedures involving the animal experimentation reported here (authorization number 04/2020). These experiments were performed according to ARRIVE guidelines and the legislation on the protection of animals used for scientific purposes in Spain (RD 53/2013) and the EU (Directive 2010/63/EU). The study was designed and conducted according to the STAIR guidelines [
3,
17] regarding physiological monitoring, sample randomization, predefined exclusion criteria, allocation concealment, blinded assessment of outcomes, dose–response definition, and a conflict-of-interest statement.
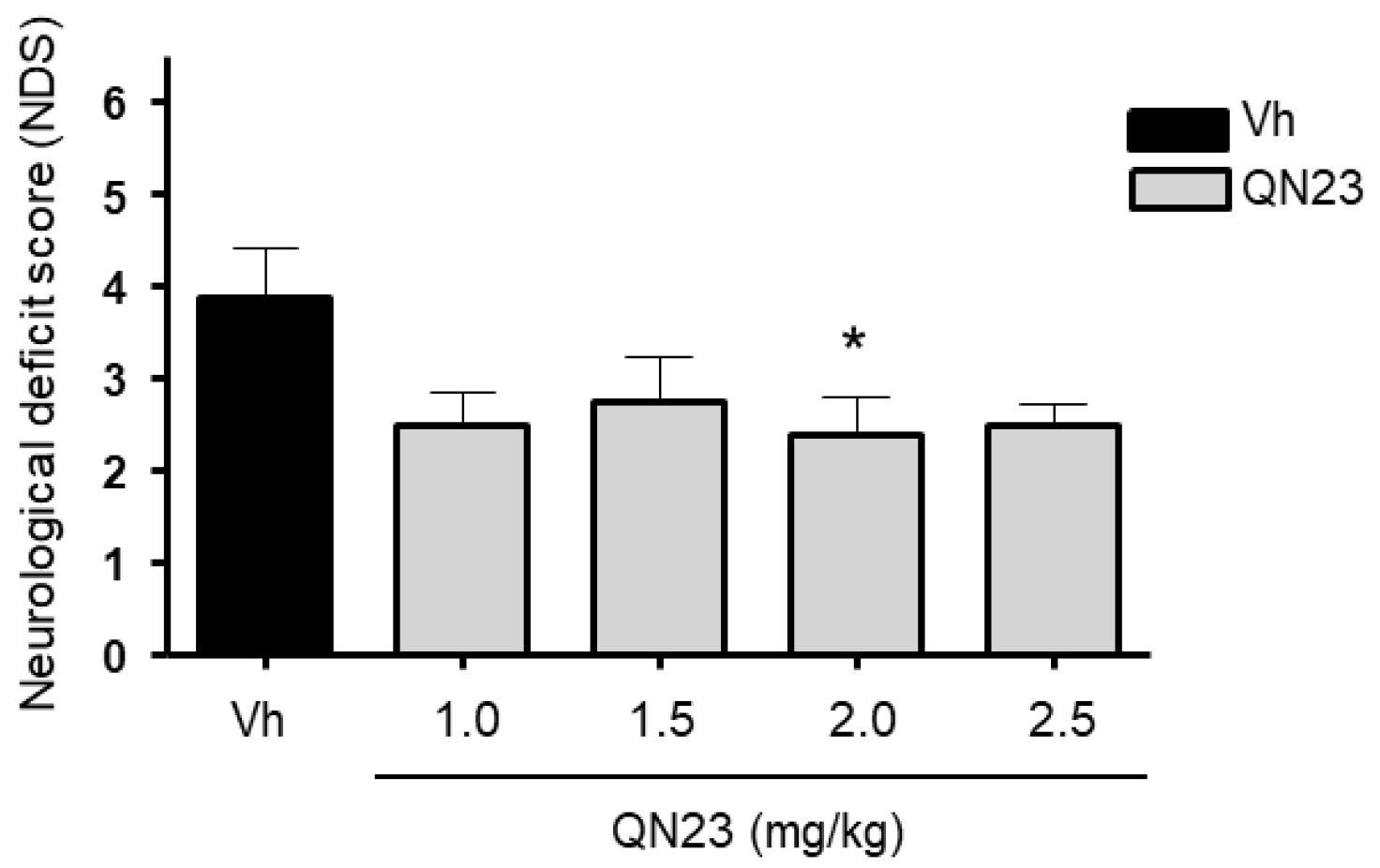
2.3. Evaluation of Neurological Deficits
After 5 days of post-ischemic reperfusion (R5d), neurological deficits were evaluated in the animals by a researcher blind to the treatment allocation. General status and mobility impairments were quantified according to a scale previously validated in an entire cohort of R5d animals (
n = 20) [
27]. This scale ranges from 0 (best, non-operated control animal) to 10 (animal with a high neurofunctional deficit).
2.4. Behavioral Tests
Long-term assessment of neurological damage after the ischemic episode was performed by the evaluation of the spontaneous exploratory activity (spatial recognition) and spatial memory skills in the Y maze [
28].
For the assessment of the exploratory activity and spatial recognition, control animals and ischemic animals treated with vehicle, QN23, or NXY-059 were placed in the center of the maze and allowed free exploration for 8 min. An independent researcher blind to the treatment allocation registered the total number of arms that the animal entered indistinctly (arm entries), and the times that the animal explored the three different arms of the maze consecutively (alternations or triads).
For the assessment of spatial memory, experiments consisted of two trials. For the first trial (training), control and ischemic animals treated with vehicle or QN23 were placed into one of the arms (start arm) and allowed to explore the Y maze for 10 min with one of the arms closed. One hour after the training trial, the animals were returned to the Y maze and the second trial was conducted. The animals were placed in the same start arm as in the training trial, but now with the remaining two arms of the maze available for exploration for 3 min. A researcher blind to the treatment allocation registered the first choice of arm for entry (novel or old).
2.5. Brain Sections
After 5 days of reperfusion (R5d), or at 11 weeks of reperfusion in long-term studies, animals were euthanized by transcardiac perfusion via the left ventricle under deep anesthesia. First, blood was washed out with 200 mL of ice-cold saline medium, followed by fixation with 4% (w/v) paraformaldehyde solution in PBS for brain preservation. Then, the brains were carefully removed from the cranial cavity and placed in 4% (w/v) paraformaldehyde solution in PBS overnight at 4 °C. The next day, the brains were sequentially placed in 10%, 20%, and 30% (w/v) sucrose in PBS until sunken, and finally embedded in Tissue-Tek O.C.T. (Sakura Finetek, Torrance, CA, USA) before freezing at −80 °C. Brain coronal sections containing the hippocampus region were obtained by cryostat sectioning at the level of interaural line +5.7 ± 0.2 mm on Real Capillary Gap microscope slides (Agilent Dako, Santa Clara, CA, USA).
2.6. Neuronal Death Evaluation
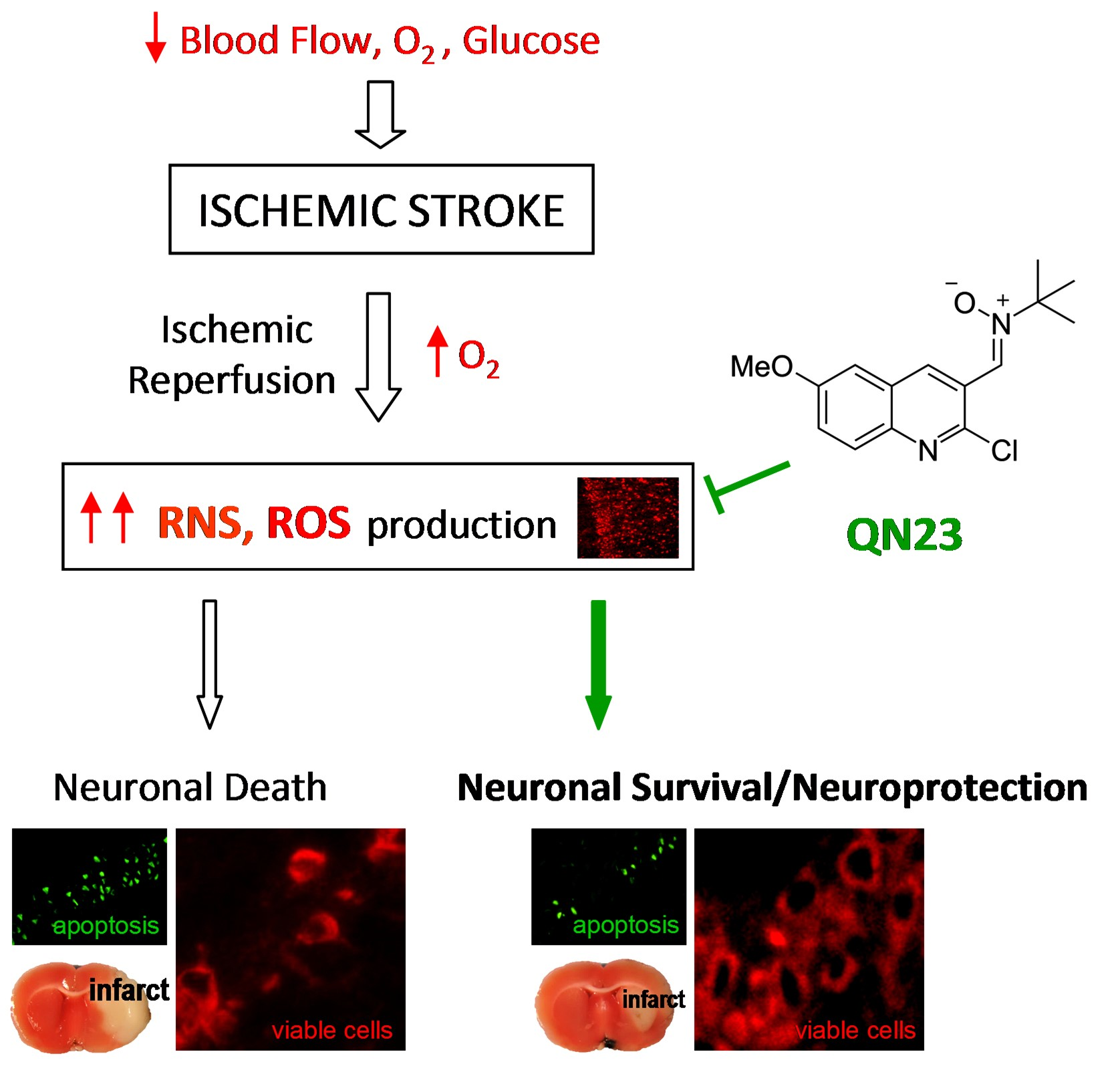
Neuronal death was determined by Fluoro-Jade B staining [
25] of brain cryosections (10 µm thick) obtained from ischemic animals after 5 days of post-ischemic reperfusion and fixed as described above. The cerebral and lateral cortex fields and the hippocampal CA1 subfield from a given section were analyzed with fluorescence microscopy (40× objective) and dead cells (in green) were counted using a grid of 330 × 220 µm, with a total area of 1.017 mm
2 per section analyzed and two sections per brain sample. Fluorescence images were digitized with a color CCD camera (1280 × 960 pixel resolution). Data analysis was independently carried out by two researchers blind to the treatment allocation.
2.7. TUNEL Assay
Neuronal apoptosis was determined in brain sections from ischemic animals by the Terminal deoxynucleotidyl transferase-mediated dUTP Nick-End Labeling (TUNEL) assay (Promega, Madison, WI, USA), following the supplier protocol with little modifications. Briefly, 5 µm-thick coronal brain sections obtained as described above were post-fixed with 4% (w/v) paraformaldehyde solution in PBS (5 min, at room temperature). After washing in PBS (3×, 5 min), sections were permeabilized with 0.1 M sodium citrate (pH 6.0) at 95 °C for 1 min, quickly cooled in ice, and washed in PBS at room temperature. Then, brain sections were blocked (0.1 M Tris-HCl, pH 7.5, 20% fetal bovine serum and 3% bovine serum albumin), washed in PBS, and fixed again with 4% (w/v) paraformaldehyde solution in PBS (5 min, at room temperature). After washing in PBS (3×, 5 min), sections were incubated with the kit buffer for 10 min, followed by incubation with terminal deoxynucleotidyl transferase (TdT) and fluorescein-12-dUTP (1.5 h, 37 °C). The reaction was stopped by extensive washing in saline–sodium citrate buffer (30 mM sodium citrate in 0.3 M sodium chloride) at room temperature, followed by washing in PBS (3×, 5 min). Finally, sections were mounted with coverslips in antifade buffered glycerol solution containing 4.6 mM p-phenylenediamine and 30 µM bisbenzimide (Hoechst 33342) for nuclear staining.
Green-labeled apoptosis-positive cells were counted as in Fluoro-Jade B staining (see above). Data analysis was carried out by two independent researchers blinded to the treatment allocation.
2.8. Immunohistochemistry for Neuronal Viability Evaluation
Brain cryosections from ischemic animals after 11 weeks of reperfusion were used after fixation to detect neuronal viability by immunohistochemistry. Sections (10-µm thick) were subsequently post-fixed with 4% (w/v) paraformaldehyde solution in PBS for 5 min at room temperature, washed in PBS (3×, 5 min), and permeabilized with 10 mM sodium citrate (pH 6.0) for 3 min at 95 °C, followed by cooling at room temperature for 20 min and washing in PBS (3×, 5 min). Then, brain sections were blocked (5% heat-inactivated donkey serum, 0.1% Triton X-100 in PBS) for 1 h at room temperature and incubated with mouse monoclonal anti-S6 protein antibody (Cell Signaling Technology, Danvers, MA, USA) overnight at 4 °C. After washing in PBS (3×, 5 min), sections were incubated with X Red-conjugated anti-mouse antibody (Jackson ImmunoResearch, West Grove, PA, USA) for 1 h at room temperature and washed again in PBS. Sections were mounted with coverslips in antifade buffered glycerol solution containing 4.6 mM p-phenylenediamine and 30 µM bisbenzimide (Hoechst 33342, Thermo Fisher Scientific, formerly Invitrogen, Waltham, MA, USA) for nuclear staining. The fields of the hippocampal CA1 region of a given section were examined by fluorescence microscopy. Images of more than 6 different fields within a given section were acquired with a 40× objective, and Image-J software (version 1.53, National Institute of Health, Bethesda, Maryland, USA) was used for quantitative analysis of the number of S6-labeled cells per field in the area of interest.
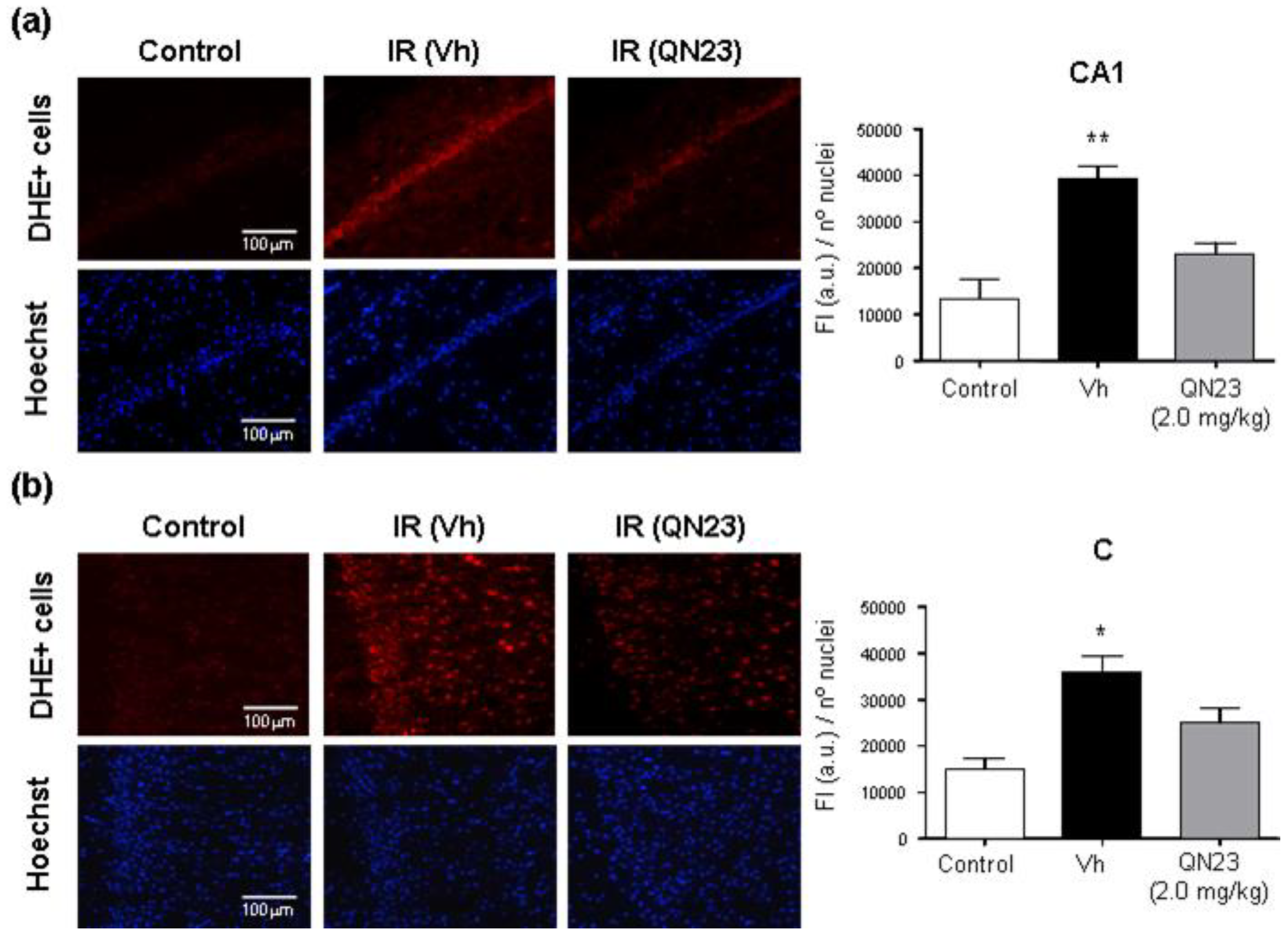
2.9. Imaging of Cerebral ROS in Brain Ischemia
In order to evaluate the antioxidant capacity of QN23 in vivo, the fluorescent probe dihydroethidium (DHE) was used for superoxide and hydrogen peroxide detection according to the experimental procedure described in [
29], with minor modifications. Animals subjected to global cerebral ischemia (Wistar male rats of 280–300 g) were treated with vehicle or QN23 at the onset of the reperfusion period, and after 2 h of reperfusion DHE (3.0 mg/kg; 0.9 mg/mL in DMSO/polyethylene glycol 400/saline, 2.5:50:47.5
v/v) was intravenously injected (1 mL). DHE (Sigma-Aldrich, Merck KGaA, Darmstadt, Germany) was freshly weighted and dissolved just before the injection to avoid undesired air-driven oxidation. At 6 h of reperfusion after ischemia (4 h after DHE injection), the animals were euthanized and perfused with saline and 4% (
w/v) paraformaldehyde in PBS. The untreated control animals were also injected with DHE and euthanized and perfused in the same way at 4 h after DHE injection.
Rat brains were excised and kept in the same solution in the dark at 4 °C for 2 h, after which paraformaldehyde solution was replaced by 30% sucrose and kept in the dark at 4 °C until brain sinking. Two days after the surgery, the brains were embedded in Tissue-Tek O.C.T. (Sakura Finetek), frozen at −80 °C, and cryo-sectioned at the level of interaural line +5.7 ± 0.2 mm on Real Capillary Gap microscope slides (Dako). Ten µm-thick brain sections were mounted with coverslips in Fluoroshield® with DAPI (Sigma-Aldrich, Merck KGaA, Darmstadt, Germany).
Hippocampal CA1 subfields and cortical fields from a given brain section were randomly selected and analyzed with fluorescence microscopy (20× objective), and the selected images (425 × 340 μm) were digitized with a color CCD camera (1280 × 1024 pixel resolution). Fluorescence intensity (in red) of ROS-reacted DHE was evaluated and normalized to the number of DAPI-stained nuclei by using an image analysis software (Image-J 1.53). At least four images per brain sample, and 3 to 6 brains per experimental group, were evaluated and averaged. Treatment information was kept concealed throughout the study.
2.10. Animal Model of Transient Focal Cerebral Ischemia
For the transient focal cerebral ischemia experimental model we used 12-week-old male Wistar rats (300–350 g, Charles River), housed under standard conditions with food and water ad libitum. The animals were anesthetized by an intraperitoneal injection of a mixture of 100 mg/kg ketamine, 5 mg/kg diazepam, and 0.3 mg/kg atropine. Anesthesia was maintained during the surgical procedure by inhalation of 1–1.5% sevoflurane in 80% medicinal air plus 20% O
2. For the transient occlusion of the right middle cerebral artery (tMCAO, 60 min), we followed the intraluminal suture procedure as originally described [
30], adapted to our experimental setup [
31]. The procedure included continuous monitoring of cortical perfusion (CP, by Laser-Doppler flowmetry), arterial blood pressure, and core temperature, and discontinuous measurement of glycemia levels during preischemia (basal), ischemia, and reperfusion stages. Buprenorphine (subcutaneous, 0.05 mg/kg) was used as an analgesic. Forty-eight hours after the ischemic insult, the animals were euthanized by intracardiac injection of potassium chloride (200 mg/kg) under deep anesthesia, and the brains were obtained.
A power analysis (see above) provided a sample size of 6 subjects per group. A total of 35 animals was used in this study. The animals were randomly assigned to four experimental tMCAO groups, treated with (1) vehicle; (2) QN23, 1.5 mg/kg; (3) QN23, 2.5 mg/kg; and (4) QN23, 4 mg/kg. QN23 was dissolved in vehicle and injected through the femoral vein 20 min after the end of tMCAO. Nine animals were excluded from the study, based on predefined criteria, because of death within the first 24 h after the procedure (1 vehicle, 1 QN23-treated at 2.5 mg/kg, and 4 QN23-treated at 4.0 mg/kg) or due to no infarct after the procedure (1 vehicle, 1 QN23-treated at 1.5 mg/kg, and 1 QN23-treated at 2.5 mg/kg). Experiments were conducted in compliance with the legislation on the protection of animals used for scientific purposes in Spain (RD 53/2013) and the EU (Directive 2010/63/EU). The protocols were approved by the Animal Experimentation Ethics Committee from IIS La Fe (Valencia, Spain, authorization number 2020/VSC/PEA/0076). The study was designed and conducted according to the STAIR/RIGOR guidelines [
3,
17] regarding physiological monitoring, simple randomization, predefined exclusion criteria, allocation concealment, blinded assessment of outcomes, dose response definition, and a conflict-of-interest statement.
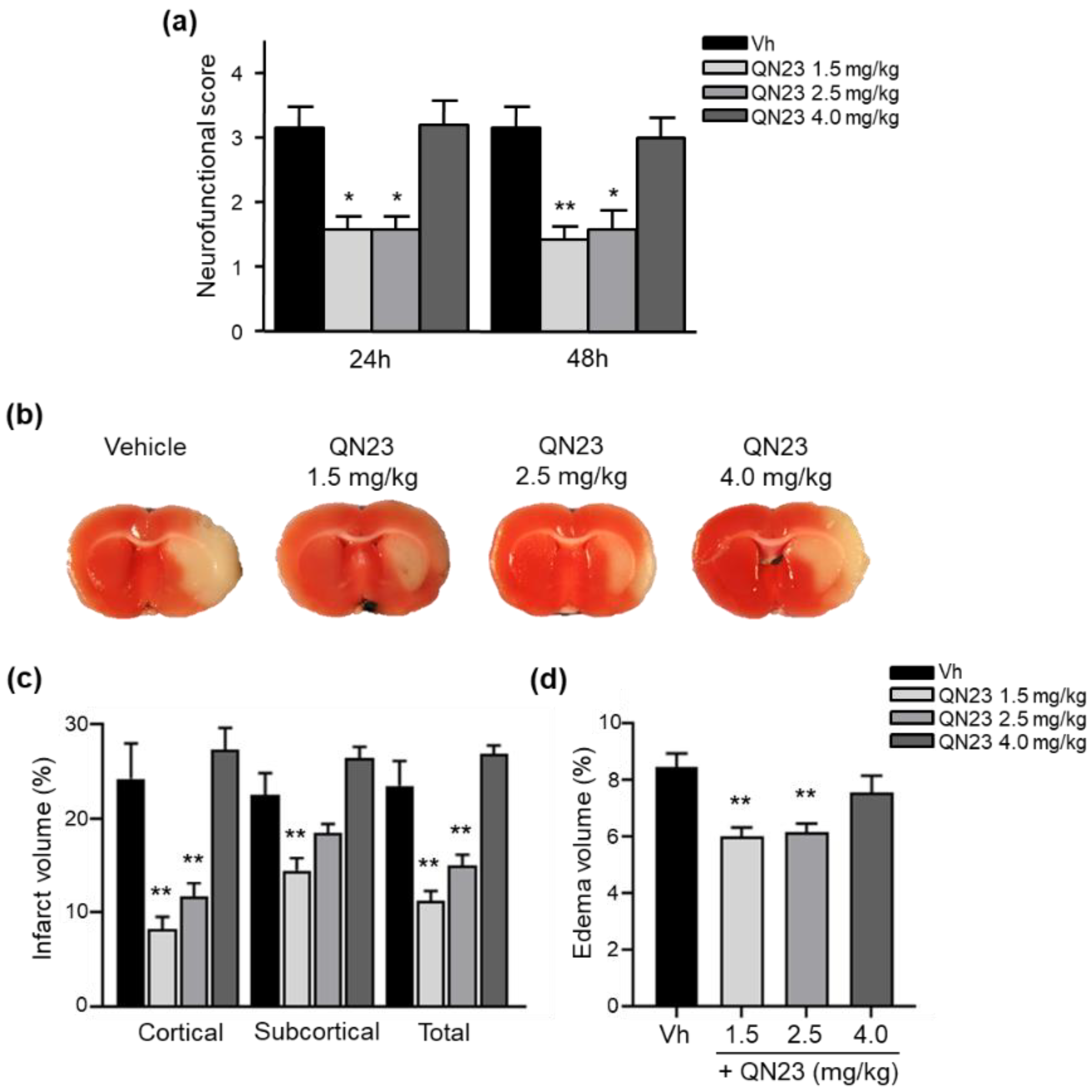
2.11. Evaluation of Neurofunctional Score in Transient Focal Cerebral Ischemia
Twenty-four and 48 hours after the ischemic insult, the rats’ neurofunctional status was evaluated following the procedure previously reported [
32]. The severity of functional deficits was determined with the sum of the score of four tests assessing (a) spontaneous activity (moving/exploring = 0, no moving or moving only when pushed = 1), (b) circling to the left (none = 0, circling when elevated by the tail and pushed = 1, circling without displacement, spinning = 2), (c) parachute reflex: protective abduction of forelimbs (symmetrical = 0, asymmetrical, contralateral forelimb retracted = 1), and (d) resistance to left forepaw stretching (not allowed = 0, allowed = 1). The total score ranged from 0 (no neurological deficits) to 5 (highest neurological deficits) or 6 (death). Neurofunctional evaluation was performed by an independent researcher blind to the treatment allocation.
2.12. Evaluation of Infarct Volume in Transient Focal Cerebral Ischemia
Brain infarct volume was determined by the 2,3,5-triphenyltetrazolium chloride (TTC; Sigma-Aldrich, Merck KGaA, Darmstadt, Germany) vital staining method [
33], followed by morphometric analysis [
31] by a researcher blind to the treatment allocation. Briefly, rats were euthanized and the brain was sliced in seven 2 mm-thick coronal sections, which were immersed in a 2% solution of TTC in saline solution at 37 °C for 10 min and fixed in 10% phosphate-buffered formalin (pH 7.4) overnight. Digital photographs were taken (Olympus CAMEDIA C-5050, Tokyo, Japan) and the border between infarcted and non-infarcted tissue was outlined with an image analysis system (Image-J software). In order to correct for the influence of edema, the infarcted area was calculated as follows: corrected infarct area = infarct area × (contralateral hemisphere area / ipsilateral hemisphere area). The corrected infarct area was measured on both the anterior and the posterior sides of each slice and averaged; the corrected infarct volume was calculated by multiplying the average corrected infarct area by the thickness of the slice (2 mm), and the total corrected infarct volume by adding up the corrected infarct volume of the seven slices. The operational sequence was applied separately to the cortex and striatum as well as to the sum of both. The subtraction between the raw and corrected infarcted hemisphere equaled the edema volume.
2.13. Pharmacokinetic and Toxicity Studies
For the toxicokinetics study of QN23, female Sprague Dawley rats (9–10 weeks, Envigo, Barcelona, Spain) were divided into three groups (n = 6 per group) and treated intravenously with different concentrations of QN23 (2.0, 6.0, and 18 mg/kg), in vehicle solution (ethanol/polyethylene glycol 400/saline, 1:200:60 v/v), as a single bolus of 1 mL injection at 3 mL/min. Samples of plasma were taken at 0.25, 0.5, 1, 2, 4, and 8 h after the first administration (on day 1) and QN23 concentration was determined by liquid chromatography–mass spectrometry in tandem (LC-MS/MS) analysis for pharmacokinetic study. For the determination of the toxicokinetic parameters, a noncompartmental analysis was performed using the Phoenix 64 WinNonlin software (version 8.0, Certara, Sant Louis, MO, USA). This study was carried out by Swiss BioQuant AG (Reinach, Switzerland). For the toxicity study of QN23, rats were divided into four groups (n = 5 per group) and treated intravenously with vehicle (control group) or different concentrations of QN23 (2.0, 6.0, and 18 mg/kg) as a single bolus injection in a 4-day repeated dose as described above. Pathology analyses, comprising hematology, clinical biochemistry, and coagulation, were performed, and heart, kidney, liver, and spleen necropsies were collected for macroscopic analysis. This study was carried out by Vivotecnia Research S.L (Madrid, Spain).
2.14. Statistical Analysis
Data from each animal and experimental condition were independently analyzed, and their averaged values were used for statistical analysis. Data are represented as mean ± SE. For comparison between multiple concentrations or experimental groups, an analysis of variance (ANOVA) or non-parametric Kruskal–Wallis test (for neurological scoring variables) was performed, followed by a post-hoc test (Dunnett’s or Dunn’s test, respectively) to compare with control group means by pairs when ANOVA or Kruskal–Wallis tests were significant. The statistical significance level was set at α = 0.05, and the Prism statistical package (version 5.0, GraphPad Software, San Diego, CA, USA) was used.
4. Discussion
In the current clinical practice, there is a direct need for therapies for the treatment of ischemic stroke. Recanalization strategies, either mechanical (thrombectomy) or pharmacological (e.g., rtPA), are the only therapies approved by the Food and Drug Administration or European Medicines Agency for the treatment of this pathology and must be combined with new approaches in order to effectively improve the patient’s recovery and decrease damage associated with ischemic reperfusion injury. In this paper, we updated the assessment of the biological effect of QN23 as a preclinical candidate for the treatment of ischemic stroke by reporting, based on different animal experimental models, a dose–response study, in addition to a therapeutic window screening and an evaluation of the long-term efficacy. Remarkably, we include here the in vivo antioxidant activity and also report the results of a pharmacokinetics study of QN23.
In this report, the administration of QN23 was determined to be feasible, safe, and effective as an intravenous single injection, and therefore, potentially useful for the treatment of pathologies such as acute ischemic stroke (AIS). The solubility of the compound required of an in-house designed vehicle was composed of ethanol, polyethyleneglycol 400, and saline, which was proven safe in healthy animals (data not shown). With this approach, doses ranging from 1.0 to 2.5 mg/kg was assayed on a transient four-vessel occlusion (4VO) model, and the effect, based on the evaluation of neuronal death and apoptosis induction, concluded 2.0 mg/kg as the most effective dose. An independent laboratory performed the study of the neuroprotective effect of QN23 on a transient middle cerebral artery occlusion (tMCAO) model by intraluminal suture procedure, a preclinical relevant model of focal ischemia. Dose–response analysis included 1.5, 2.5, and 4.0 mg/kg. Evaluation of the functional outcomes was in accordance with the infarct and edema volumes measured on the ischemic brains of the animals and reported the range of 1.5–2.5 mg/kg as the most effective. These results agree with the optimal dose found for the global ischemia model. The aforementioned limited solubility of QN23 in aqueous solutions may explain the lower effect with high doses of QN23, since it is feasible that with QN23 at the high doses used, part of the compound may have precipitated as an insoluble compound after the injection, and therefore been ineffective. This effect could be more intense at the highest dose used. Of note, four animals treated with QN23 4.0 mg/kg died after the administration within the next 24 h, in contrast with one animal death in the group treated with vehicle and QN23 2.5 mg/kg. Given that the toxicokinetic study tested higher doses of QN23 (6.0 and 18 mg/kg) without finding apparent effects of toxicity, the deaths may have been caused by a poor solubility and/or precipitation of the compound in a vehicle solution with lower polyethylene glycol 400 concentration than in the toxicokinetic study, causing the obstruction of a vessel and a lack of effectiveness.
As radical traps, nitrones such as QN23 have long been thought to exert their neuroprotective mechanism of action by stabilizing highly reactive radical species and avoiding further damage to the cell structures [
9]. Previous studies of the antioxidant activity of QN23 revealed a very potent scavenging activity against hydroxyl radical, the most reactive and harmful radical species produced during ischemia–reperfusion damage [
22]. Furthermore, QN23 was reported to significantly reduce ROS and lipid peroxidation levels in primary neuronal cultures subjected to OGD to comparable levels of undamaged cells [
22]. However, could this activity be translated to in vivo? There has been controversy about the real effect of radical scavengers in vivo, raised also for nitrones such as PBN or NXY-059, mainly due to the unachievable high concentrations required in cerebral tissue in order to effectively act as radical traps [
9]. In this report we describe for the first time evidence of the antioxidant effect of QN23 in vivo, using dihydroethidium (DHE) as a molecular probe for oxidative stress [
29,
35]. The obtained results show a remarkable effect of QN23 on the suppression of the fluorescence intensity of ROS-reacted DHE, which accounts for a decreased production of radical species and meaning that QN23 may indeed work as an antioxidant nitrone. Based on the extensive literature of nitrones working as radical traps, it seems coherent to attribute this effect to radical-trapping activity. However, other mechanisms different to radical trapping, such as an activation of endogenous antioxidant systems, may also participate in this reduction in ROS-reacted DHE fluorescence intensity [
36]. In any case, the results reported here shed light on a controversial field such as the in vivo effect of an antioxidant, which was proven effective in this report.
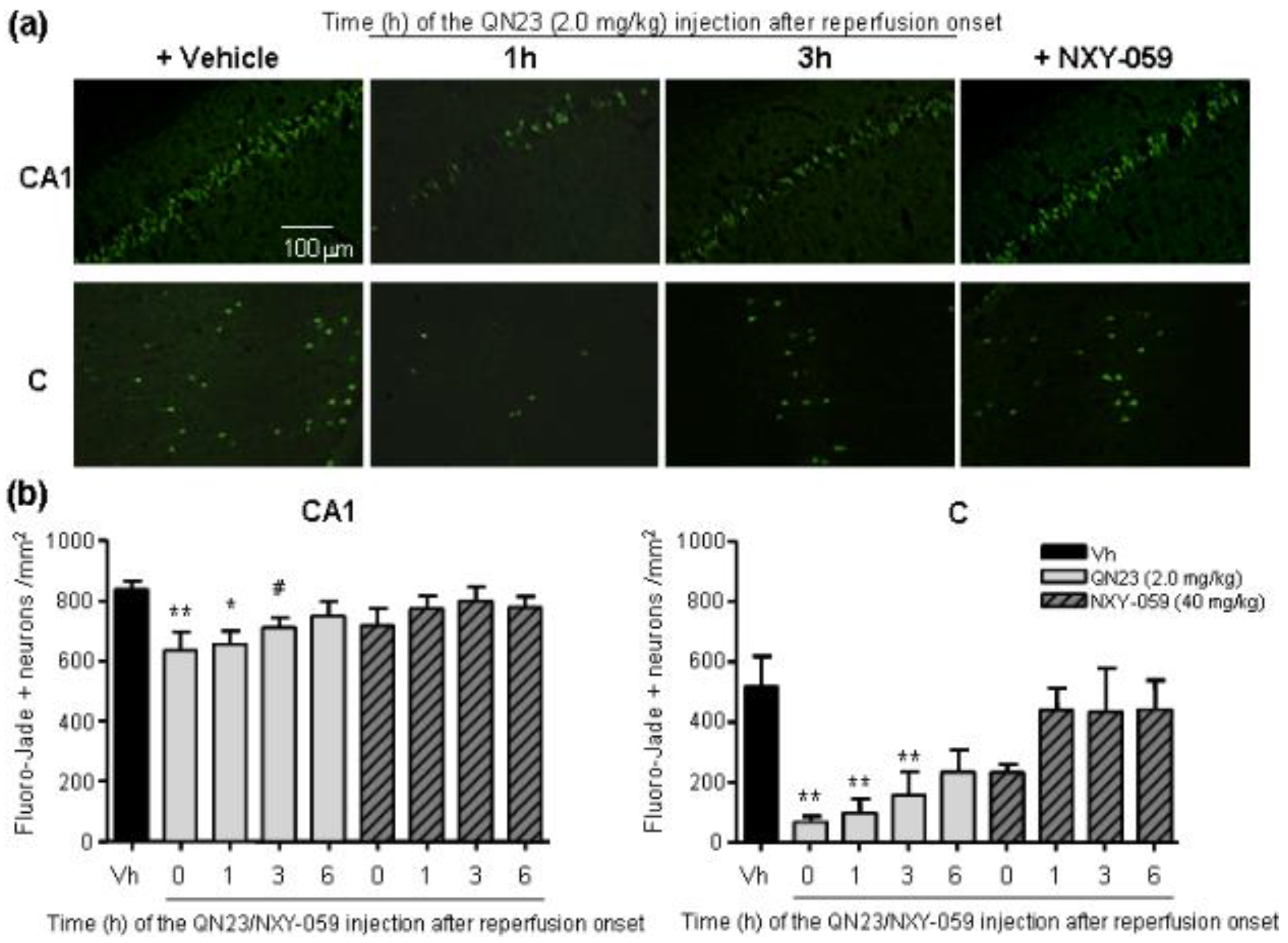
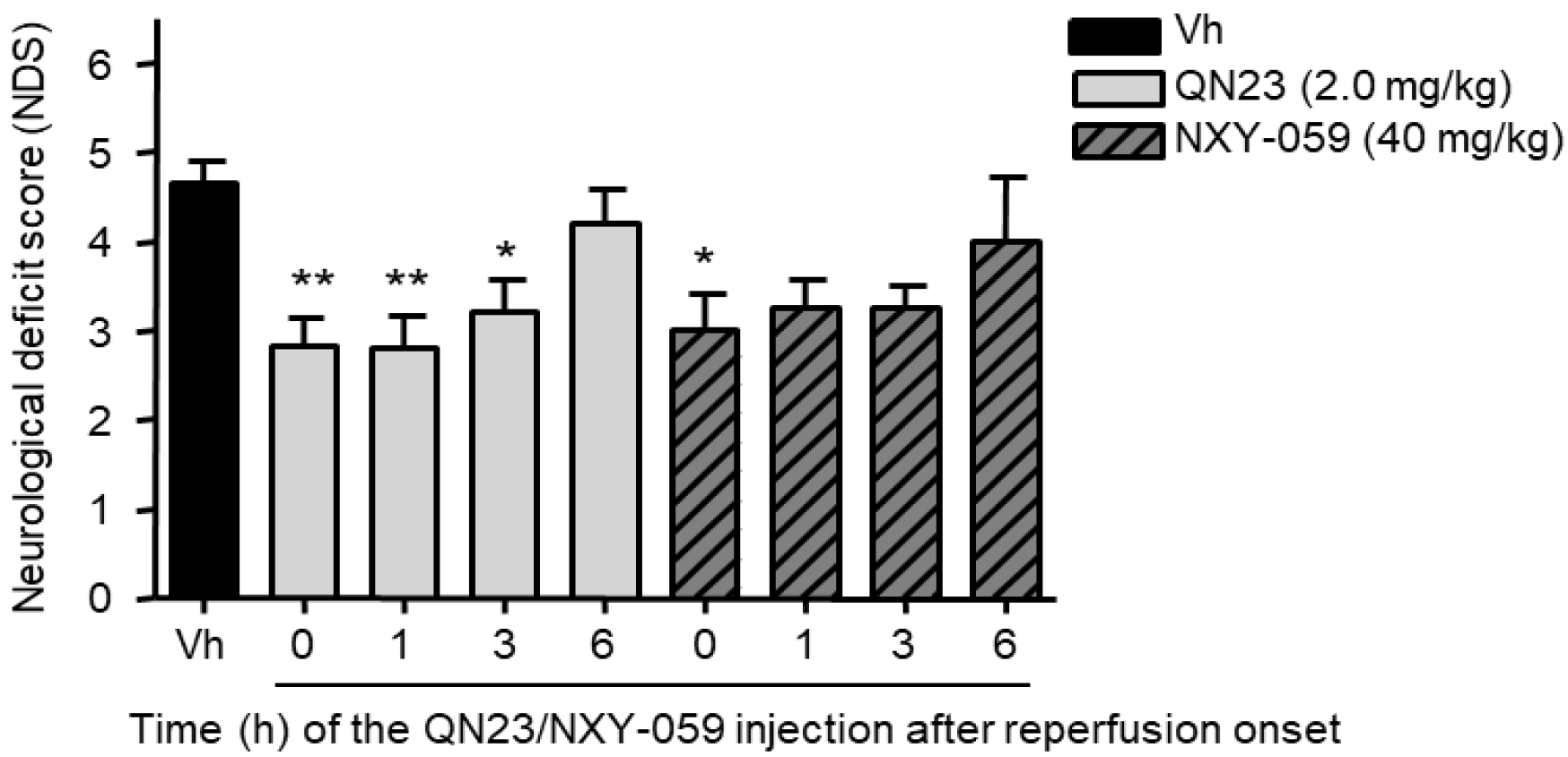
All of the previous studies were carried out with QN23 being administered at the onset of reperfusion. This served as a way to find the most effective dose of QN23 in our experimental model and to determine an antioxidant mechanism of action of QN23, but this injection schedule is less likely to occur in real clinical practice. After the first signs of an acute ischemic attack, the management, diagnosis, and treatment of a stroke patient may take several hours and, therefore, any drug candidate must be proven safe and effective also at administration times with relatively long delays from symptom onset and/or recanalization therapy, mirroring clinical practice in stroke patients [
3]. In this report we explored a delayed time of injection ranging from 1 to 6 h post ischemia. Significant reduction of damage outcomes were observed with QN23 (2.0 mg/kg) injection up until 3 h (R3h) in our preclinical model, with an effect comparable to the one observed when treatment was administered just after the ischemic period. Remarkably, treatment with NXY-059 (40 mg/kg) also produced a slight improvement, although not significant, with the same administration schedule. Whether this extended time of administration might be applicable to humans and compatible with current recanalization approaches must still be assessed, but it represents an interesting starting point into the development of more clinically useful therapies.
Related to this, the influence of an extended therapeutic window on the mechanism of action of QN23 is an aspect that still needs to be assessed. Considering the antioxidant activity of QN23 as the responsible mechanism for the neuroprotective effect found, an extended administration time, later than the peak of radical species usually produced right after the recovery of normal oxygen levels in the compromised tissue, may explain the decrease in the effectivity in our experimental model. In other words, we can consider that the later QN23 is injected, the less effective it is at decreasing radical species levels, since they have already been generated and damage to cellular structures has already been produced. This may justify that the antioxidant activity of QN23 is its main beneficious role, and that further mechanisms of action—i.e., binding to biological targets, modulating signaling pathways, etc.—that could take place at later times (i.e., at 6 h) would not be expected to have a crucial influence on the final outcome.
Long-term sequelae of ischemic stroke, manifested normally as psychiatric or neurological disorders, represent an aspect that is gaining importance in the current clinical practice and social security systems due to an increased incidence of stroke episodes in younger adults. The evaluation of a potential protective effect longer term after an ischemic episode is, therefore, of crucial relevance. The use of preclinical models to assess this kind of outcome may be problematic since life span comparison between species is hard to evaluate. Some literature has mirrored a human life period of five to seven years to a period of three months in adult rats [
37]. Regardless of the exact equivalence between the two species, this time period allows for the determination of outcomes, either functional or biochemical, once the brain-remodeling action after the ischemic–reperfusion injury carried out by endogenous neurorepair processes has been completed [
38]. The immunofluorescence detection of ribosomal protein S6 after 11 weeks (three months) of reperfusion allowed for the observation of the anatomical disruption produced within the hippocampal CA1 region as a consequence of the ischemic-reperfusion injury. These results were confirmed by behavioral tests, which detected an impaired performance in the spontaneous spatial recognition and spatial memory tests in those untreated ischemic animals. Remarkably, treatment with QN23 decreased the long-term functional impairment and preserved neuronal viability at a three-month-long period, which adds to the short-term effect described previously and contributes to the preclinical confirmation of QN23 as a promising candidate for the treatment of ischemic stroke.
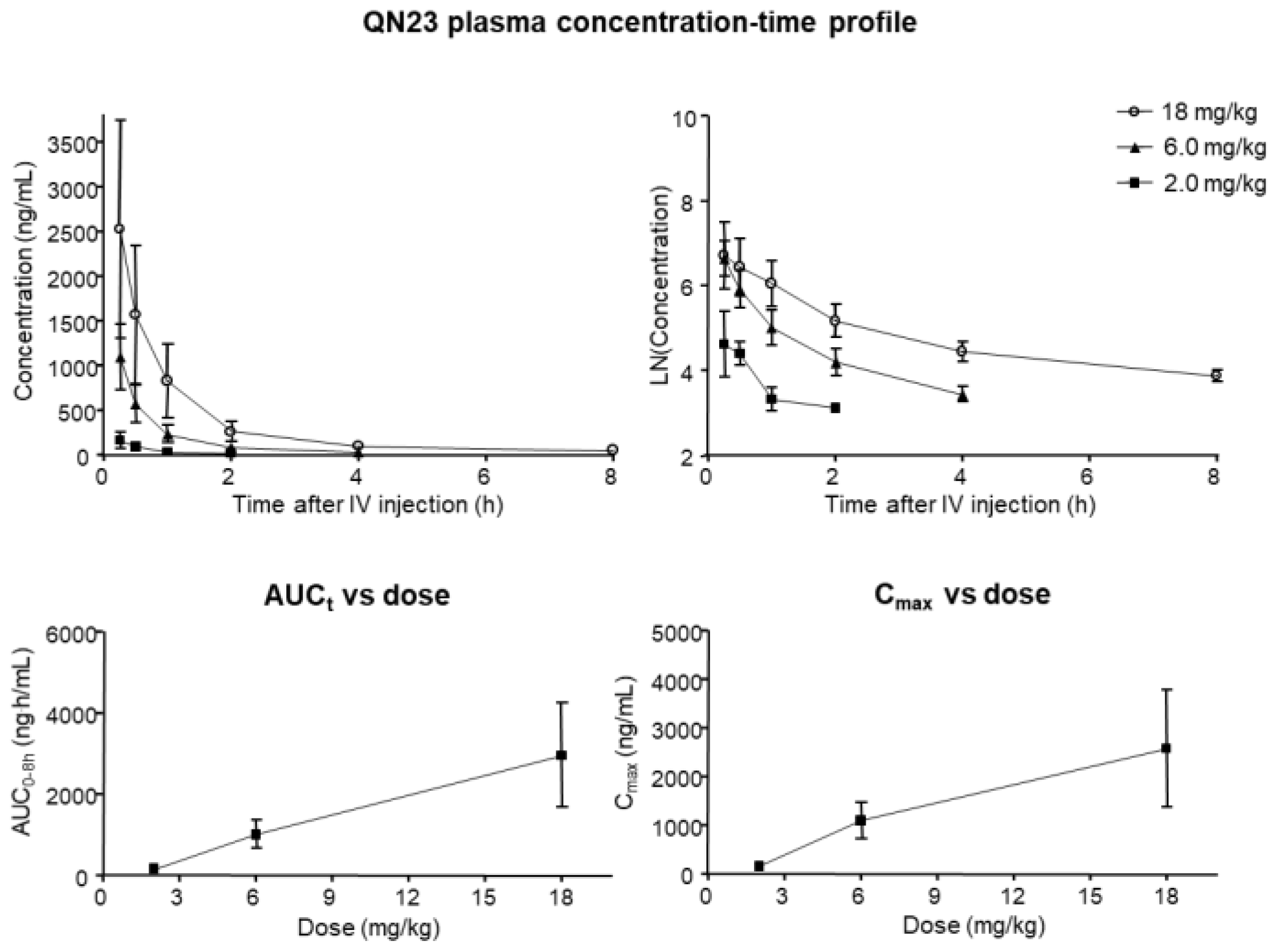
As its name indicates, acute ischemic stroke is characterized by a sudden appearance of symptoms, and the therapeutic window, spanning only several hours, requires a rapid and efficient route of administration. A single bolus intravenous injection allows for a fast delivery of drugs to the systemic circulation, rapidly reaching the target tissue, i.e., the brain. Determination of the characteristics of this type of administration for QN23 in terms of the achieved concentration, time of elimination or accumulation, and potential toxicity, allows for the optimization of a dose regime for further development. In this report, we analyzed the pharmacokinetics parameters of different doses of QN23 (2.0, 6.0, and 18 mg/kg) when injected intravenously as a single bolus in healthy animals in a period of 8 h after administration. Non-compartmental analysis of the obtained concentration–time profile showed a very fast and gradual decrease in concentration in blood in the time studied, with very short half-lives ranging from 0.16 (2.0 mg/kg) to 0.24 h (6.0 mg/kg) or 1.13 h (18 mg/kg), and rapid elimination due to high clearance rates. This fast elimination of QN23 from the organism may indicate a very fast mechanism of action able to produce the effect described in this report in a period of minutes but, at the same time, with consequences long after the injection, as described in the long-term-effect assessment. Regarding the relevance of oxidative stress in the course of tissue recovery after ischemia–reperfusion, this fast mechanism of action of QN23 agrees with the evidence demonstrated here that the antioxidant activity of QN23 would be its main target of action.
Finally, this study was designed and conducted with predefined exclusion criteria, physiological monitoring, sample randomization, allocation concealment, blinded assessment of outcomes, dose–response definition, and a conflict-of-interest statement, in accordance with STAIR and RIGOR guidelines. In addition, we assessed preclinical relevant aspects of the potential use of QN23 as a treatment, such as its therapeutic window, its long-term effect, and its pharmacokinetics, including preliminary results on its antioxidant mechanism of action [
3,
17]. For these studies we used a four-vessel occlusion (4VO) global ischemia and a transient middle cerebral artery occlusion (tMCAO) focal ischemia as preclinical relevant models for preclinical studies. As a first preclinical characterization, we decided to use young-adult male rats in order to assess important features of QN23 performance and the mechanism of action in more standardized systems. The results obtained in these models are indispensable in preliminary steps of preclinical characterization, and these results are required for design optimization in more advanced preclinical stages.


 ,
, 













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}