
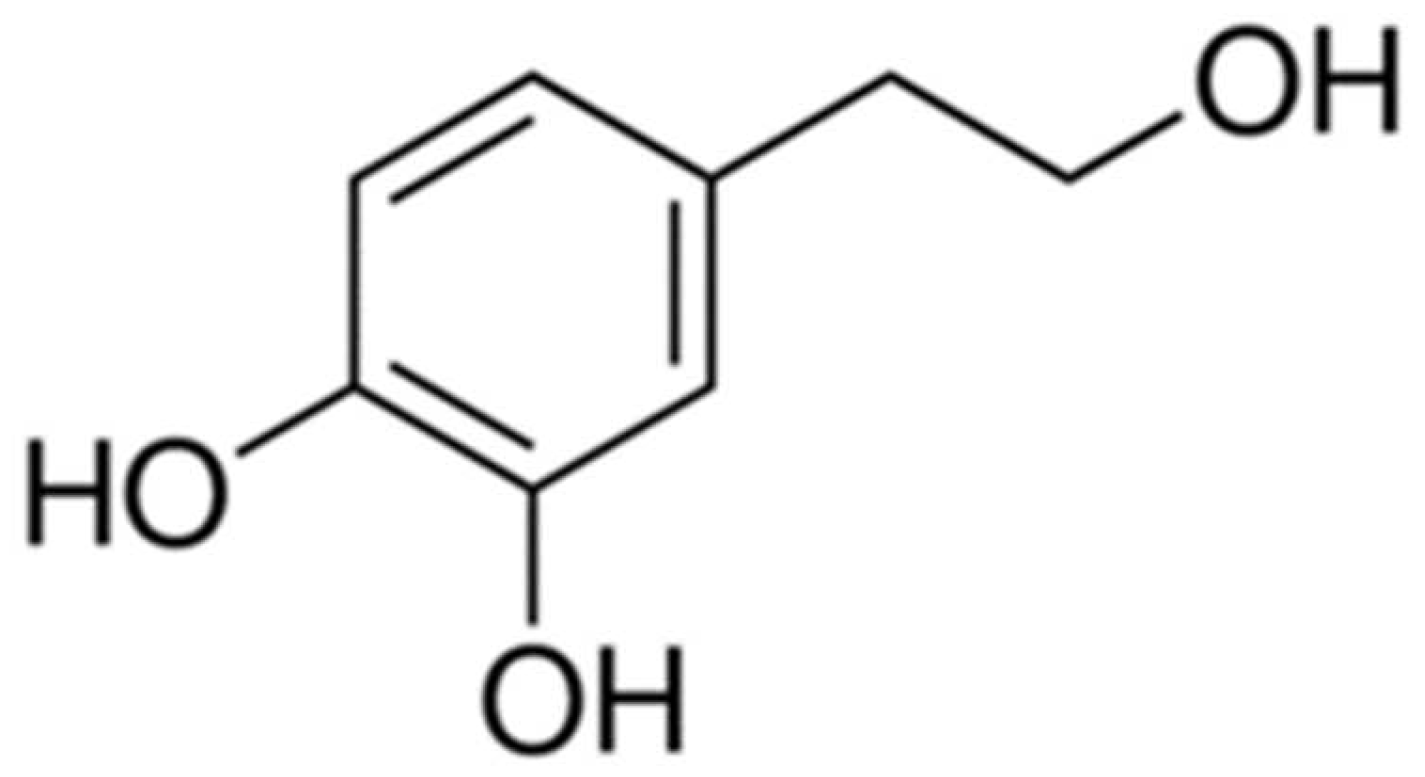
Hydroxytyrosol Prevents Doxorubicin-Induced Oxidative Stress and Apoptosis in Cardiomyocytes


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Cultures and Treatments
2.3. MTT Assay
2.4. Detection of Intracellular ROS
2.5. Immunoblotting
2.6. Trypan Blue Assay
2.7. Statistical Analysis
3. Results
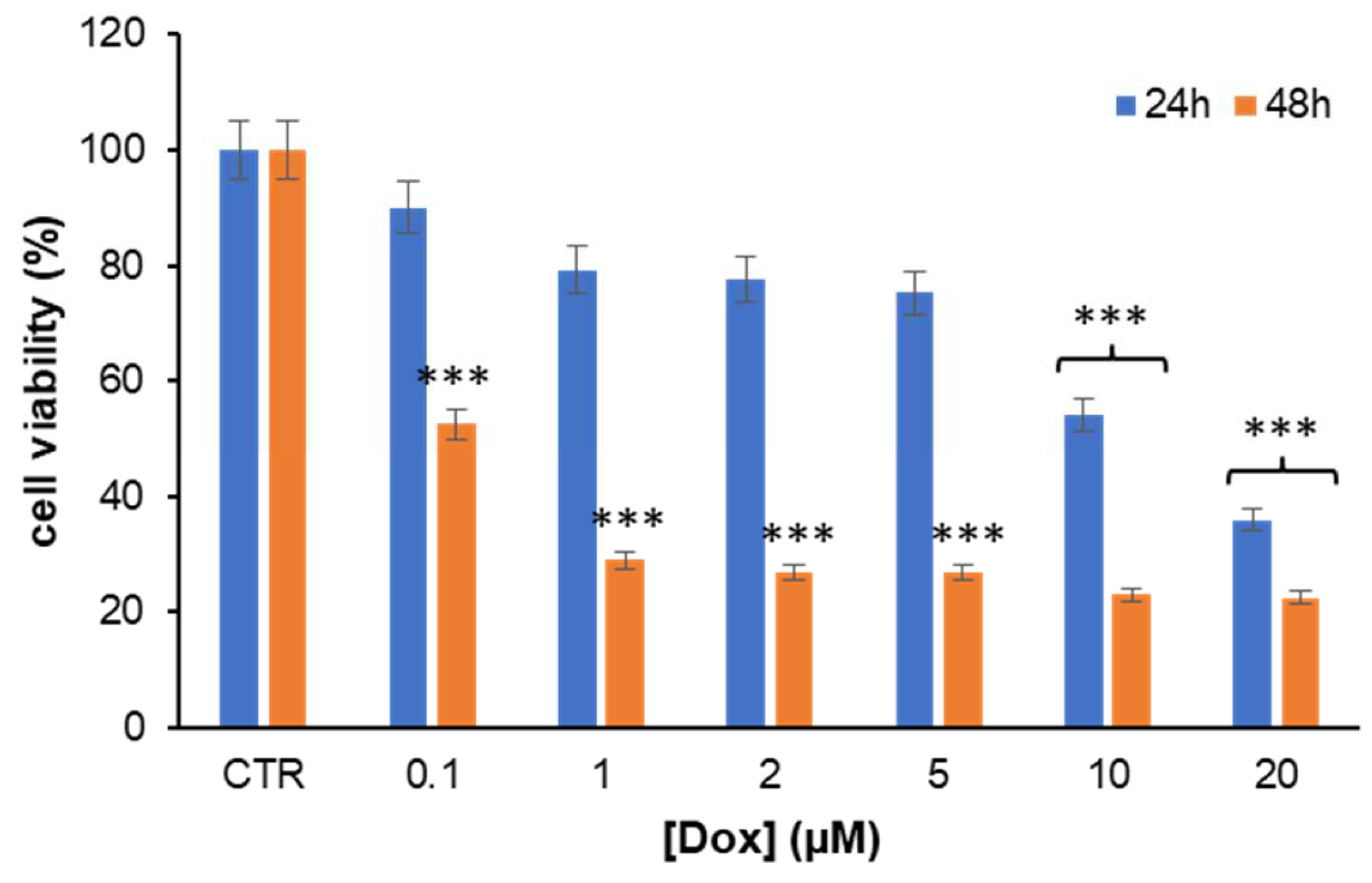
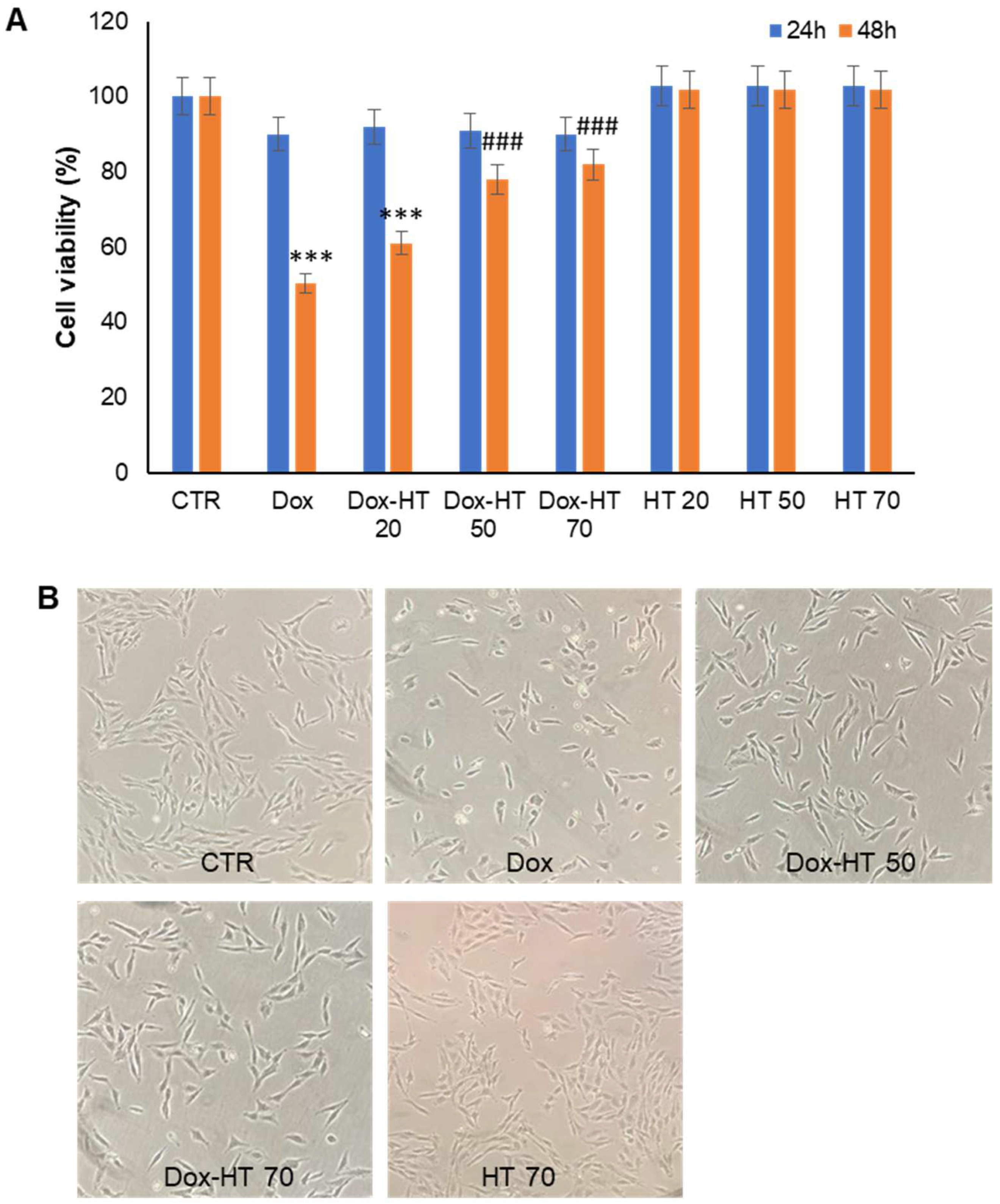
3.1. HT Markedly Reduces Dox-Induced Toxicity in H9c2 Cells
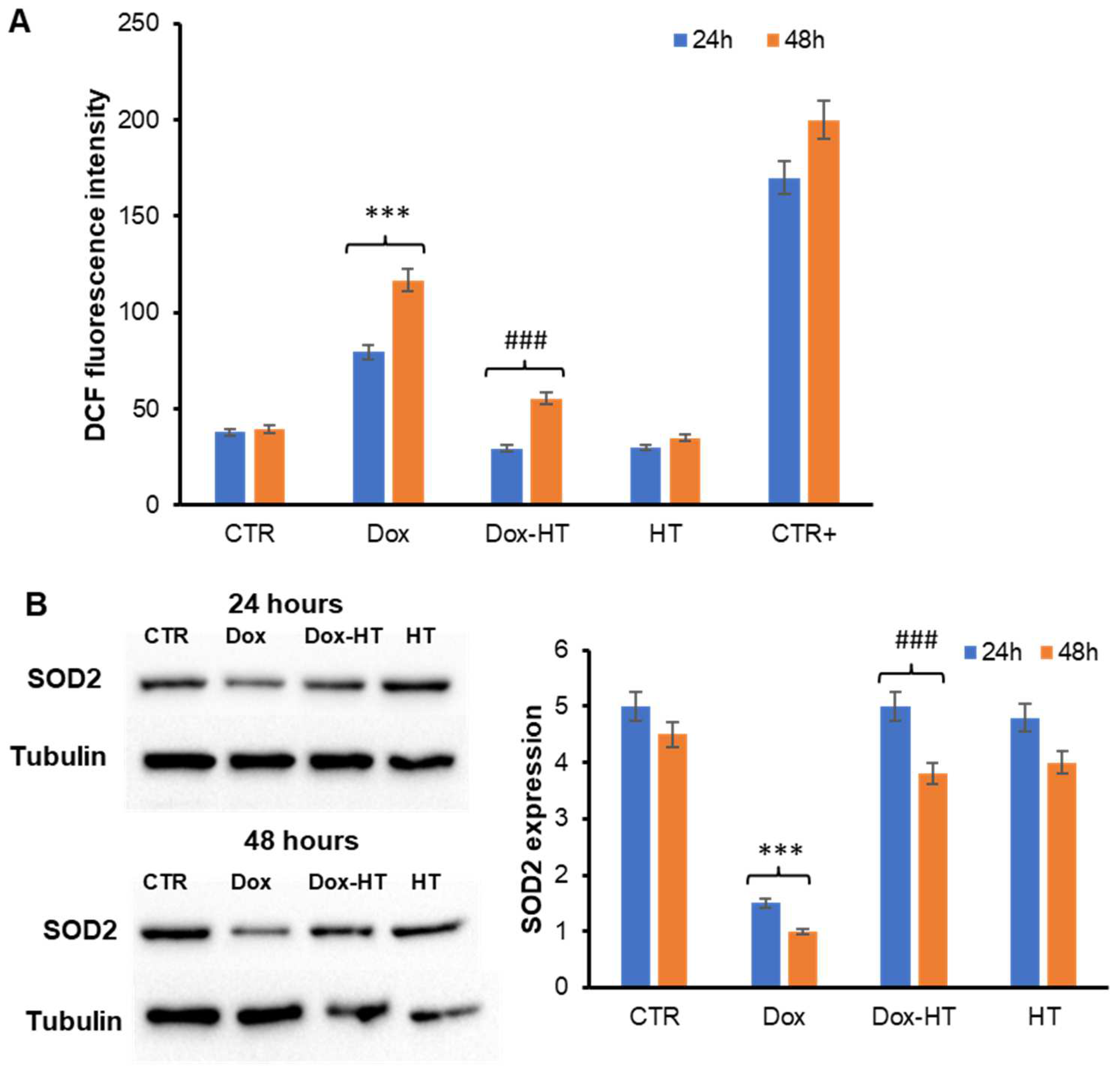
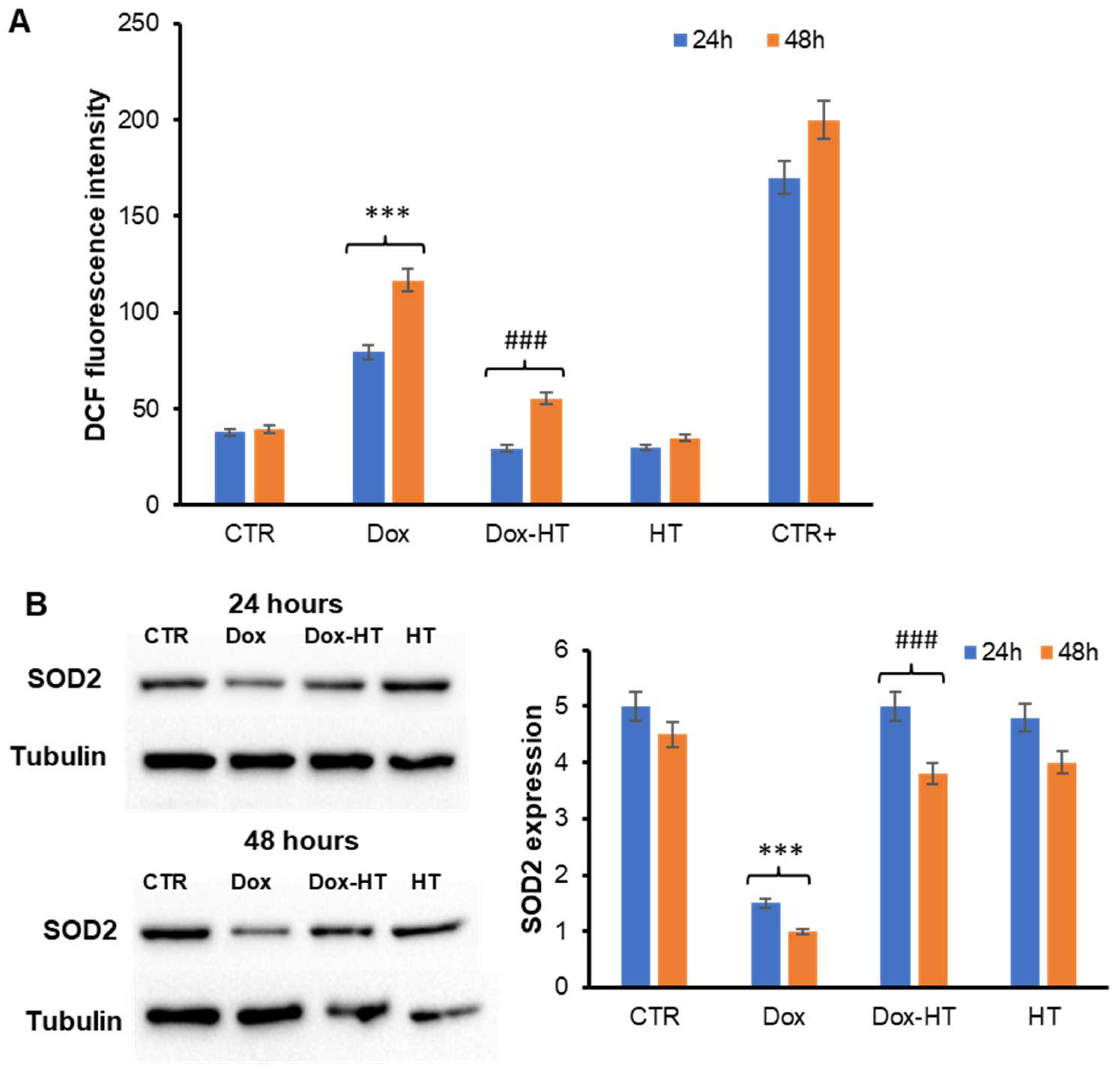
3.2. HT Prevents Dox-Induced ROS Production and SOD2 Activation in H9c2 Cells
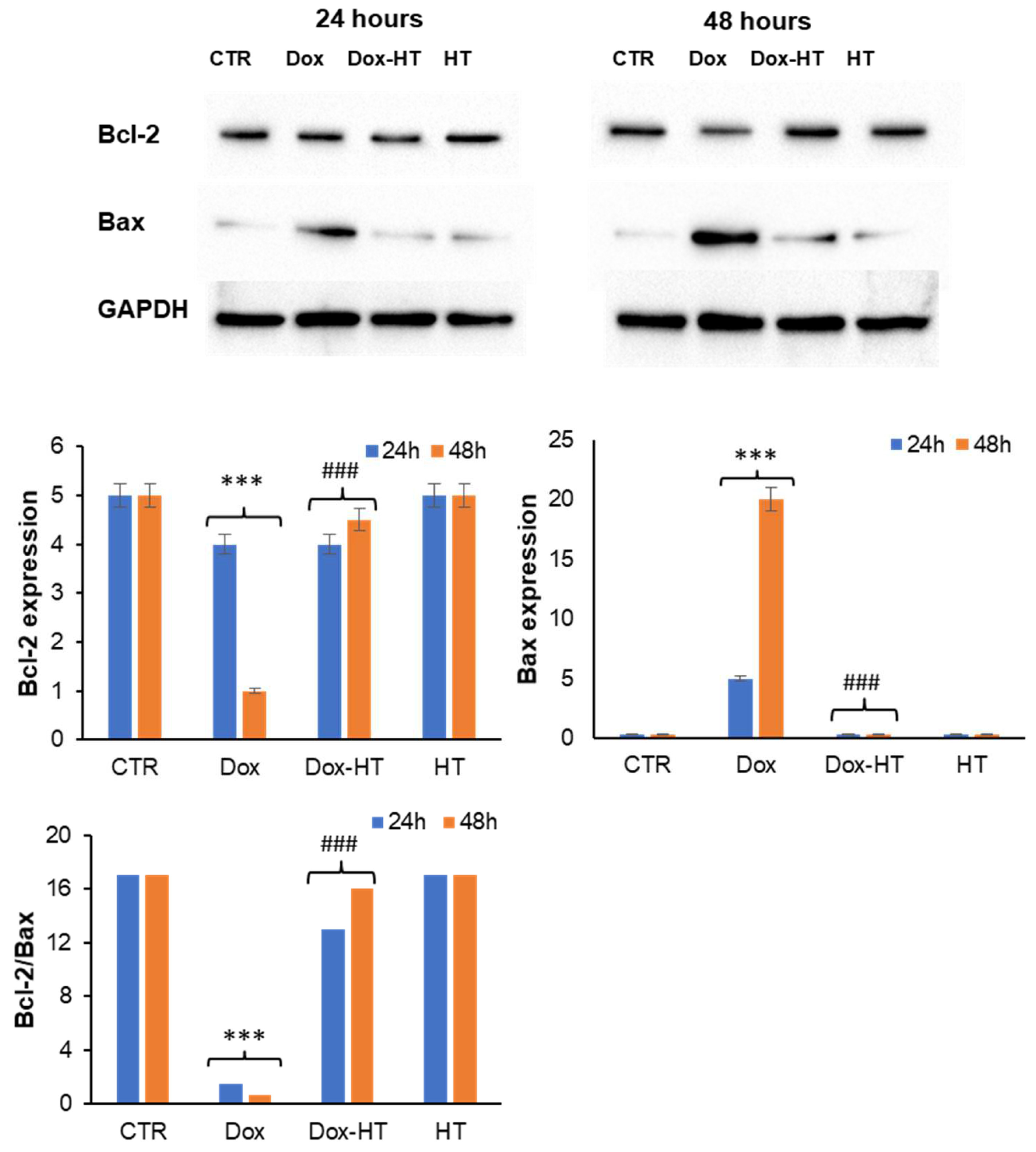
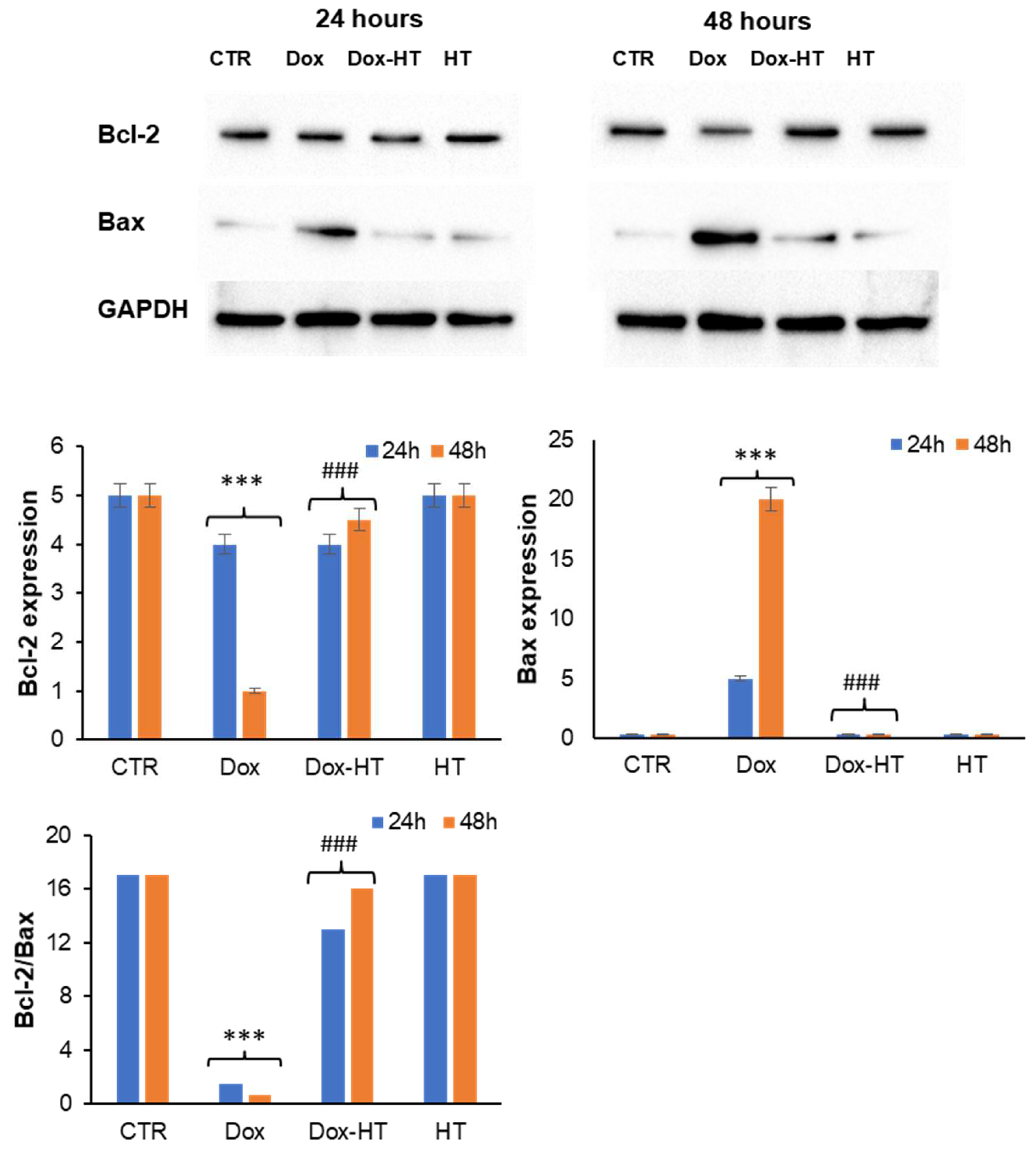
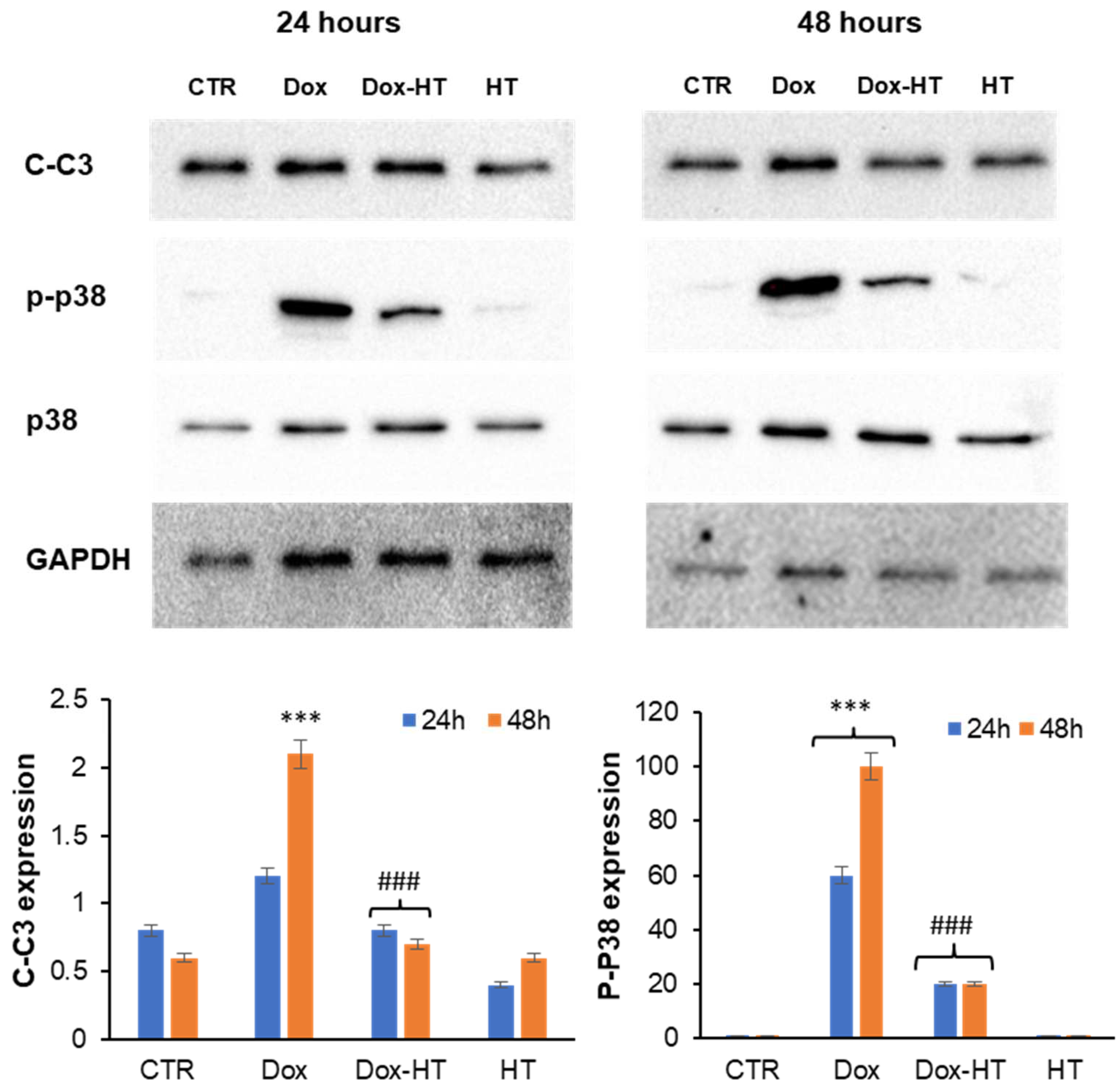
3.3. HT Protects H9c2 Cells by the Dox-Induced Apoptosis
3.4. HT Effect in DNA Damage Dox-Induced in H9c2 Cells
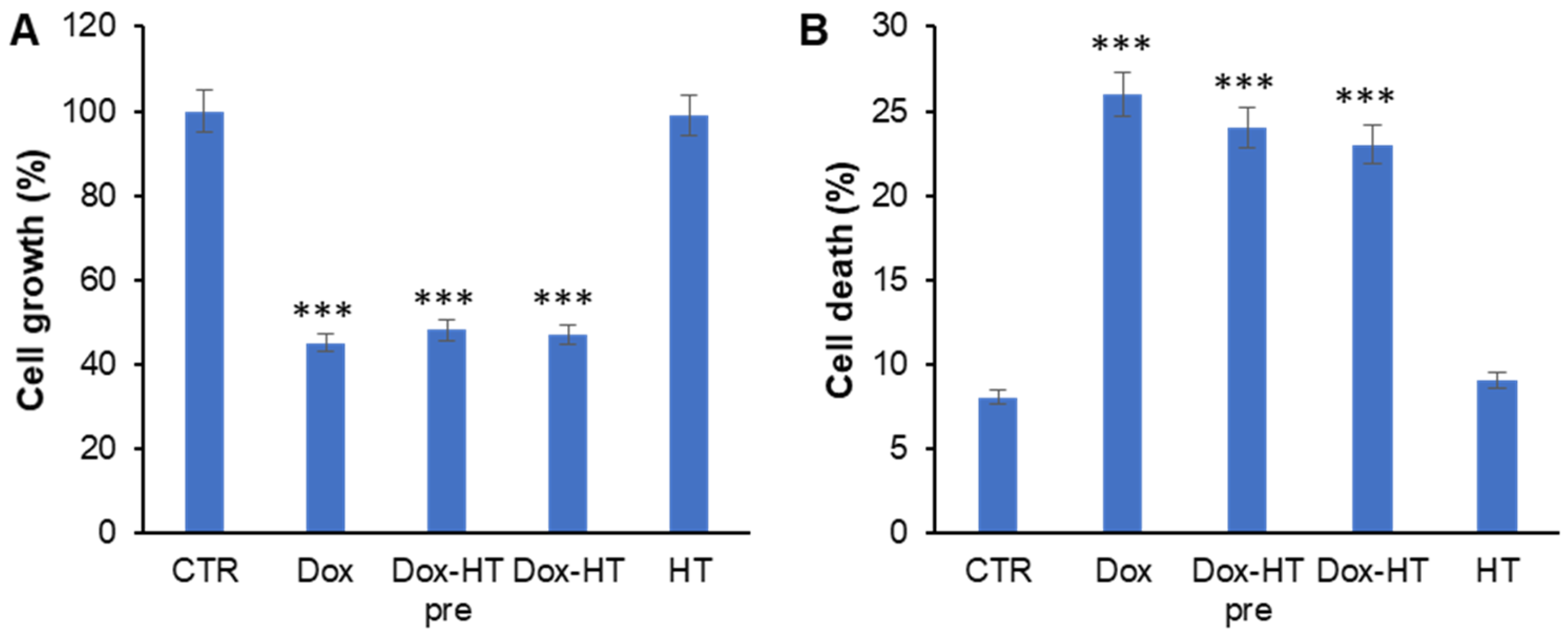
3.5. HT Does Not Impair Antitumoral Dox Activity in Osteosarcoma Cells
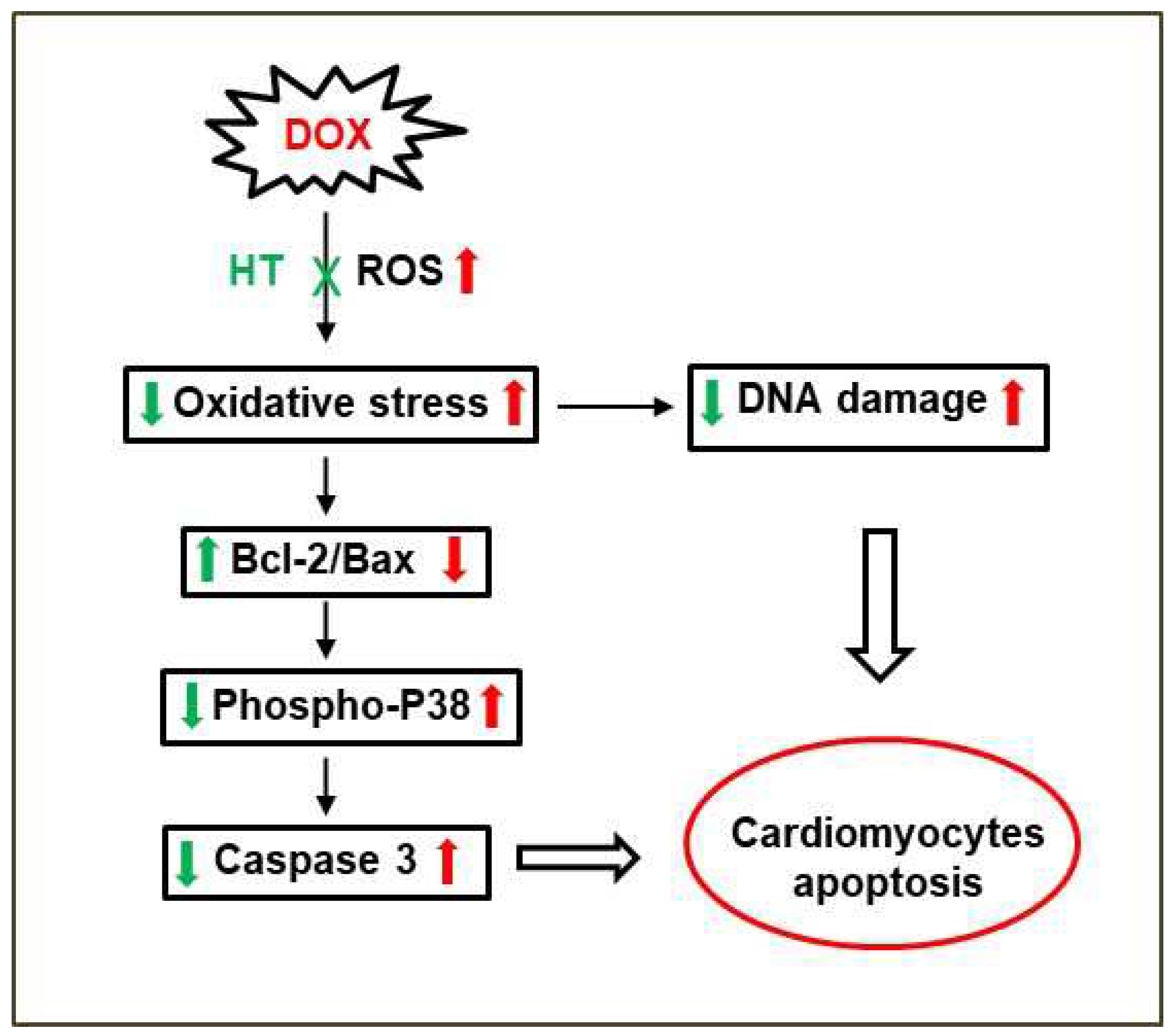
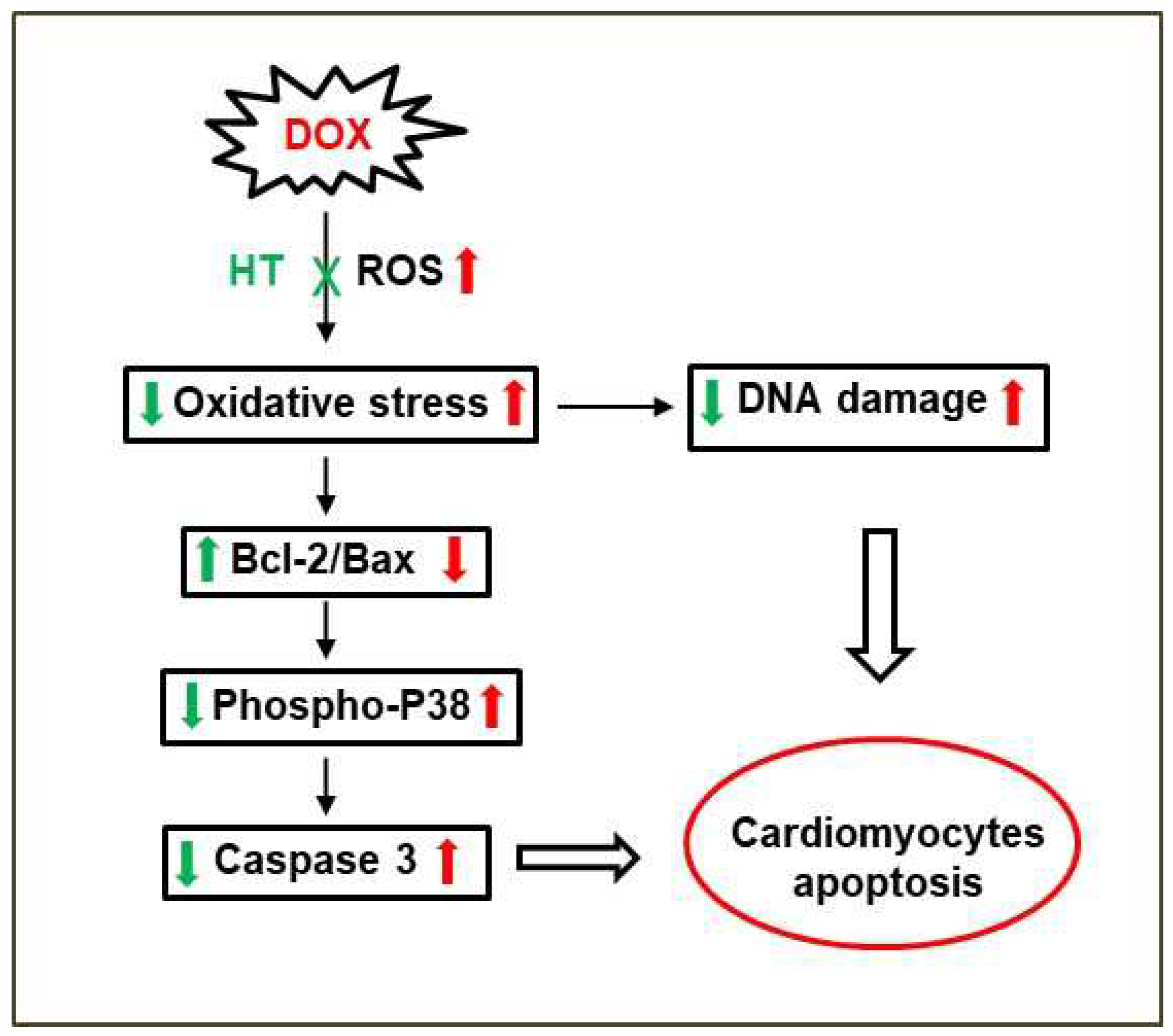
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mordente, A.; Meucci, E.; Martorana, G.E.; Tavian, D.; Silvestrini, A. Topoisomerases and Anthracyclines: Recent Advances and Perspectives in Anticancer Therapy and Prevention of Cardiotoxicity. Curr. Med. Chem. 2017, 24, 1607–1626. [Google Scholar] [CrossRef] [PubMed]
- Marinello, J.; Delcuratolo, M.; Capranico, G. Anthracyclines as Topoisomerase II Poisons: From Early Studies to New Perspectives. Int. J. Mol. Sci. 2018, 19, 3480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Songbo, M.; Lang, H.; Xinyong, C.; Bin, X.; Ping, Z.; Liang, S. Oxidative stress injury in doxorubicin-induced cardiotoxicity. Toxicol. Lett. 2019, 307, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Prudowsky, Z.D.; Yustein, J.T. Recent Insights into Therapy Resistance in Osteosarcoma. Cancers 2020, 13, 83. [Google Scholar] [CrossRef] [PubMed]
- Podyacheva, E.Y.; Kushnareva, E.A.; Karpov, A.A.; Toropova, Y.G. Analysis of Models of Doxorubicin-Induced Cardiomyopathy in Rats and Mice. A Modern View from the Perspective of the Pathophysiologist and the Clinician. Front. Pharmacol. 2021, 12, 670479. [Google Scholar] [CrossRef]
- Rawat, P.S.; Jaiswal, A.; Khurana, A.; Bhatti, J.S.; Navik, U. Doxorubicin-induced cardiotoxicity: An update on the molecular mechanism and novel therapeutic strategies for effective management. Biomed. Pharmacother. 2021, 139, 111708. [Google Scholar] [CrossRef]
- Kalyanaraman, B. Teaching the basics of the mechanism of doxorubicin-induced cardiotoxicity: Have we been barking up the wrong tree? Redox Biol. 2020, 29, 101394. [Google Scholar] [CrossRef]
- Cunha-Oliveira, T.; Ferreira, L.L.; Coelho, A.R.; Deus, C.M.; Oliveira, P.J. Doxorubicin triggers bioenergetic failure and p53 activation in mouse stem cell-derived cardiomyocytes. Toxicol. Appl. Pharmacol. 2018, 348, 1–13. [Google Scholar] [CrossRef]
- Wenningmann, N.; Knapp, M.; Ande, A.; Vaidya, T.R.; Ait-Oudhia, S. Insights into Doxorubicin-induced Cardiotoxicity: Molecular Mechanisms, Preventive Strategies, and Early Monitoring. Mol. Pharmacol. 2019, 96, 219–232. [Google Scholar] [CrossRef]
- Adhikari, A.; Asdaq, S.M.B.; Al Hawaj, M.A.; Chakraborty, M.; Thapa, G.; Bhuyan, N.R.; Imran, M.; Alshammari, M.K.; Alshehri, M.M.; Harshan, A.A.; et al. Anticancer Drug-Induced Cardiotoxicity: Insights and Pharmacogenetics. Pharmaceuticals 2021, 14, 970. [Google Scholar] [CrossRef]
- Nicoletto, R.E.; Ofner, C.M., 3rd. Cytotoxic mechanisms of doxorubicin at clinically relevant concentrations in breast cancer cells. Cancer Chemother. Pharmacol. 2022, 89, 285–311. [Google Scholar] [CrossRef]
- Cappetta, D.; De Angelis, A.; Sapio, L.; Prezioso, L.; Illiano, M.; Quaini, F.; Rossi, F.; Berrino, L.; Naviglio, S.; Urbanek, K. Oxidative Stress and Cellular Response to Doxorubicin: A Common Factor in the Complex Milieu of Anthracycline Cardiotoxicity. Oxid. Med. Cell. Longev. 2017, 2017, 1521020. [Google Scholar] [CrossRef]
- Li, T.; Singal, P.K. Adriamycin-induced early changes in myocardial antioxidant enzymes and their modulation by probucol. Circulation 2000, 102, 2105–2110. [Google Scholar] [CrossRef]
- Members, T.F.; Piepoli, M.F.; Hoes, A.W.; Agewall, S.; Albus, C.; Brotons, C.; Catapano, A.L.; Cooney, M.T.; Corrà, U.; Cosyns, B.; et al. European guidelines on cardiovascular disease prevention in clinical practice: The Sixth Joint Task Force of the European Society of Cardiology and Other Societies on Cardiovascular Disease Prevention in Clinical Practice (constituted by representatives of 10 societies and by invited experts) developed with the special contribution of the European Association for Cardiovascular Prevention & Rehabilitation (EACPR). Eur. Heart J. 2016, 37, 2315–2381. [Google Scholar] [CrossRef]
- Chang, H.M.; Moudgil, R.; Scarabelli, T.; Okwuosa, T.M.; Yeh, E.T.H. Cardiovascular Complications of Cancer Therapy: Best Practices in Diagnosis, Prevention, and Management: Part 1. J. Am. Coll. Cardiol. 2017, 70, 2536–2551. [Google Scholar] [CrossRef]
- Korzeniowska, K.; Jankowski, J.; Cieślewicz, A.; Jabłecka, A. Is it possible to prevent chemotherapy-induced heart failure with cardiovascular drugs—The review of the current clinical evidence. Ther. Clin. Risk Manag. 2019, 15, 1095–1110. [Google Scholar] [CrossRef] [Green Version]
- Bikiewicz, A.; Banach, M.; von Haehling, S.; Maciejewski, M.; Bielecka-Dabrowa, A. Adjuvant breast cancer treatments cardiotoxicity and modern methods of detection and prevention of cardiac complications. ESC Heart Fail. 2021, 8, 2397–2418. [Google Scholar] [CrossRef]
- Avila, M.S.; Ayub-Ferreira, S.M.; de Barros Wanderley, M.R.; das Dores Cruz, F., Jr.; Gonçalves Brandão, S.M.; Rigaud, V.O.C.; Higuchi-Dos-Santos, M.H.; Hajjar, L.A.; Kalil Filho, R.; Hoff, P.M.; et al. Carvedilol for Prevention of Chemotherapy-Related Cardiotoxicity: The CECCY Trial. J. Am. Coll. Cardiol. 2018, 71, 2281–2290. [Google Scholar] [CrossRef]
- Huang, S.; Zhao, Q.; Yang, Z.G.; Diao, K.Y.; He, Y.; Shi, K.; Shen, M.T.; Fu, H.; Guo, Y.K. Protective role of beta-blockers in chemotherapy-induced cardiotoxicity-a systematic review and meta-analysis of carvedilol. Heart Fail. Rev. 2019, 24, 325–333. [Google Scholar] [CrossRef] [Green Version]
- Padegimas, A.; Clasen, S.; Ky, B. Cardioprotective strategies to prevent breast cancer therapy-induced cardiotoxicity. Trends Cardiovasc. Med. 2020, 30, 22–28. [Google Scholar] [CrossRef]
- Mecinaj, A.; Gulati, G.; Heck, S.L.; Holte, E.; Fagerland, M.W.; Larsen, A.I.; Blix, E.S.; Geisler, J.; Wethal, T.; Omland, T. Rationale and design of the PRevention of cArdiac Dysfunction during Adjuvant breast cancer therapy (PRADA II) trial: A randomized, placebo-controlled, multicenter trial. Cardiooncology 2021, 7, 33. [Google Scholar] [CrossRef] [PubMed]
- Tebbi, C.K.; London, W.B.; Friedman, D.; Villaluna, D.; De Alarcon, P.A.; Constine, L.S.; Mendenhall, N.P.; Sposto, R.; Chauvenet, A.; Schwartz, C.L. Dexrazoxane-associated risk for acute myeloid leukemia/myelodysplastic syndrome and other secondary malignancies in pediatric Hodgkin’s disease. J. Clin. Oncol. 2007, 25, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Ruan, Y.; Shen, T.; Qiu, Q.; Yan, M.; Sun, S.; Dou, L.; Huang, X.; Wang, Q.; Zhang, X.; et al. Dexrazoxane Protects Cardiomyocyte from Doxorubicin-Induced Apoptosis by Modulating miR-17-5p. Biomed. Res. Int. 2020, 2020, 5107193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chow, E.J.; Aplenc, R.; Vrooman, L.M.; Doody, D.R.; Huang, Y.V.; Aggarwal, S.; Armenian, S.H.; Baker, K.S.; Bhatia, S.; Constine, L.S.; et al. Late health outcomes after dexrazoxane treatment: A report from the Children’s Oncology Group. Cancer 2022, 128, 788–796. [Google Scholar] [CrossRef]
- Eneh, C.; Lekkala, M.R. Dexrazoxane. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Abushouk, A.I.; Ismail, A.; Salem, A.M.A.; Afifi, A.M.; Abdel-Daim, M.M. Cardioprotective mechanisms of phytochemicals against doxorubicin-induced cardiotoxicity. Biomed. Pharmacother. 2017, 90, 935–946. [Google Scholar] [CrossRef]
- Shabalala, S.; Muller, C.J.F.; Louw, J.; Johnson, R. Polyphenols, autophagy and doxorubicin-induced cardiotoxicity. Life Sci. 2017, 180, 160–170. [Google Scholar] [CrossRef]
- Lin, H.; Zhang, J.; Ni, T.; Lin, N.; Meng, L.; Gao, F.; Luo, H.; Liu, X.; Chi, J.; Guo, H. Yellow Wine Polyphenolic Compounds prevents Doxorubicin-induced cardiotoxicity through activation of the Nrf2 signalling pathway. J. Cell. Mol. Med. 2019, 23, 6034–6047. [Google Scholar] [CrossRef] [Green Version]
- Sirangelo, I.; Sapio, L.; Ragone, A.; Naviglio, S.; Iannuzzi, C.; Barone, D.; Giordano, A.; Borriello, M. Vanillin Prevents Doxorubicin-Induced Apoptosis and Oxidative Stress in Rat H9c2 Cardiomyocytes. Nutrients 2020, 12, 2317. [Google Scholar] [CrossRef]
- Liu, C.; Ma, X.; Zhuang, J.; Liu, L.; Sun, C. Cardiotoxicity of doxorubicin-based cancer treatment: What is the protective cognition that phytochemicals provide us? Pharmacol. Res. 2020, 160, 105062. [Google Scholar] [CrossRef]
- Koss-Mikołajczyk, I.; Todorovic, V.; Sobajic, S.; Mahajna, J.; Gerić, M.; Tur, J.A.; Bartoszek, A. Natural Products Counteracting Cardiotoxicity during Cancer Chemotherapy: The Special Case of Doxorubicin, a Comprehensive Review. Int. J. Mol. Sci. 2021, 22, 37. [Google Scholar] [CrossRef]
- Syahputra, R.A.; Harahap, U.; Dalimunthe, A.; Nasution, M.P.; Satria, D. The Role of Flavonoids as a Cardioprotective Strategy against Doxorubicin-Induced Cardiotoxicity: A Review. Molecules 2022, 27, 1320. [Google Scholar] [CrossRef]
- Rigacci, S.; Stefani, M. Nutraceutical Properties of Olive Oil Polyphenols. An Itinerary from Cultured Cells through Animal Models to Humans. Int. J. Mol. Sci. 2016, 17, 843. [Google Scholar] [CrossRef] [Green Version]
- Karković Marković, A.; Torić, J.; Barbarić, M.; Jakobušić Brala, C. Hydroxytyrosol, Tyrosol and Derivatives and Their Potential effects on Human Health. Molecules 2019, 24, 2001. [Google Scholar] [CrossRef] [Green Version]
- Leri, M.; Scuto, M.; Ontario, M.L.; Calabrese, V.; Calabrese, E.J.; Bucciantini, M.; Stefani, M. Healthy Effects of Plant Polyphenols: Molecular Mechanisms. Int. J. Mol. Sci. 2020, 21, 1250. [Google Scholar] [CrossRef] [Green Version]
- Bucciantini, M.; Leri, M.; Nardiello, P.; Casamenti, F.; Stefani, M. Olive Polyphenols: Antioxidant and Anti-Inflammatory Properties. Antioxidants 2021, 10, 1044. [Google Scholar] [CrossRef]
- Sirangelo, I.; Borriello, M.; Liccardo, M.; Scafuro, M.; Russo, P.; Iannuzzi, C. Hydroxytyrosol Selectively Affects Non-Enzymatic Glycation in Human Insulin and Protects by AGEs Cytotoxicity. Antioxidants 2021, 10, 1127. [Google Scholar] [CrossRef]
- Sirangelo, I.; Borriello, M.; Vilasi, S.; Iannuzzi, C. Hydroxytyrosol Inhibits Protein Oligomerization and Amyloid Aggregation in Human Insulin. Int. J. Mol. Sci. 2020, 21, 4636. [Google Scholar] [CrossRef]
- Granados-Principal, S.; El-Azem, N.; Pamplona, R.; Ramirez-Tortosa, C.; Pulido-Moran, M.; Vera-Ramirez, L.; Quiles, J.L.; Sanchez-Rovira, P.; Naudí, A.; Portero-Otin, M.; et al. Hydroxytyrosol ameliorates oxidative stress and mitochondrial dysfunction in doxorubicin-induced cardiotoxicity in rats with breast cancer. Biochem. Pharmacol. 2014, 90, 25–33. [Google Scholar] [CrossRef]
- Kumar, P.; Nagarajan, A.; Uchil, P.D. Analysis of Cell Viability by the MTT Assay. Cold Spring Harb. Protoc. 2018, 2018(6), 469–471. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Sirangelo, I.; Vella, F.M.; Irace, G.; Manco, G.; Iannuzzi, C. Glycation in Demetalated Superoxide Dismutase 1 Prevents Amyloid Aggregation and Produces Cytotoxic Ages Adducts. Front. Mol. Biosci. 2016, 3, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LeBel, C.P.; Ischiropoulos, H.; Bondy, S.C. Evaluation of the probe 2′,7′-dichlorofluorescin as an indicator of reactive oxygen species formation and oxidative stress. Chem. Res. Toxicol. 1992, 5, 227–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallace, K.B. Doxorubicin-induced cardiac mitochondrionopathy. Pharmacol. Toxicol. 2003, 93, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Octavia, Y.; Tocchetti, C.G.; Gabrielson, K.L.; Janssens, S.; Crijns, H.J.; Moens, A.L. Doxorubicin-induced cardiomyopathy: From molecular mechanisms to therapeutic strategies. J. Mol. Cell. Cardiol. 2012, 52, 1213–1225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angsutararux, P.; Luanpitpong, S.; Issaragrisil, S. Chemotherapy-Induced Cardiotoxicity: Overview of the Roles of Oxidative Stress. Oxid. Med. Cell. Longev. 2015, 2015, 795602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotamraju, S.; Konorev, E.A.; Joseph, J.; Kalyanaraman, B. Doxorubicin-induced apoptosis in endothelial cells and cardiomyocytes is ameliorated by nitrone spin traps and ebselen. Role of reactive oxygen and nitrogen species. J. Biol. Chem. 2000, 275, 33585–33592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsang, W.P.; Chau, S.P.; Kong, S.K.; Fung, K.P.; Kwok, T.T. Reactive oxygen species mediate doxorubicin induced p53-independent apoptosis. Life Sci. 2003, 73, 2047–2058. [Google Scholar] [CrossRef]
- Crompton, M. Bax, Bid and the permeabilization of the mitochondrial outer membrane in apoptosis. Curr. Opin. Cell. Biol. 2000, 12, 414–419. [Google Scholar] [CrossRef]
- Tsujimoto, Y. Role of Bcl-2 family proteins in apoptosis: Apoptosomes or mitochondria? Genes Cells 1998, 3, 697–707. [Google Scholar] [CrossRef]
- Saelens, X.; Festjens, N.; Vande Walle, L.; van Gurp, M.; van Loo, G.; Vandenabeele, P. Toxic proteins released from mitochondria in cell death. Oncogene 2004, 23, 2861–2874. [Google Scholar] [CrossRef] [Green Version]
- Oh, J.; Lee, B.S.; Lim, G.; Lim, H.; Lee, C.J.; Park, S.; Lee, S.H.; Chung, J.H.; Kang, S.M. Atorvastatin protects cardiomyocyte from doxorubicin toxicity by modulating survivin expression through FOXO1 inhibition. J. Mol. Cell. Cardiol. 2020, 138, 244–255. [Google Scholar] [CrossRef]
- Parrish, A.B.; Freel, C.D.; Kornbluth, S. Cellular mechanisms controlling caspase activation and function. Cold Spring Harb. Perspect. Biol. 2013, 5, a008672. [Google Scholar] [CrossRef]
- Spallarossa, P.; Garibaldi, S.; Altieri, P.; Fabbi, P.; Manca, V.; Nasti, S.; Rossettin, P.; Ghigliotti, G.; Ballestrero, A.; Patrone, F.; et al. Carvedilol prevents doxorubicin-induced free radical release and apoptosis in cardiomyocytes in vitro. J. Mol. Cell. Cardiol. 2004, 37, 837–846. [Google Scholar] [CrossRef]
- Lyu, Y.L.; Kerrigan, J.E.; Lin, C.P.; Azarova, A.M.; Tsai, Y.C.; Ban, Y.; Liu, L.F. Topoisomerase IIbeta mediated DNA double-strand breaks: Implications in doxorubicin cardiotoxicity and prevention by dexrazoxane. Cancer Res. 2007, 67, 8839–8846. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Liu, X.; Bawa-Khalfe, T.; Lu, L.S.; Lyu, Y.L.; Liu, L.F.; Yeh, E.T. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat. Med. 2012, 18, 1639–1642. [Google Scholar] [CrossRef]
- Harrison, D.J.; Geller, D.S.; Gill, J.D.; Lewis, V.O.; Gorlick, R. Current and future therapeutic approaches for osteosarcoma. Expert. Rev. Anticancer Ther. 2018, 18, 39–50. [Google Scholar] [CrossRef]
- Cai, F.; Luis, M.A.F.; Lin, X.; Wang, M.; Cai, L.; Cen, C.; Biskup, E. Anthracycline-induced cardiotoxicity in the chemotherapy treatment of breast cancer: Preventive strategies and treatment. Mol. Clin. Oncol. 2019, 11, 15–23. [Google Scholar] [CrossRef]
- Mohseny, A.B.; Machado, I.; Cai, Y.; Schaefer, K.L.; Serra, M.; Hogendoorn, P.C.; Llombart-Bosch, A.; Cleton-Jansen, A.M. Functional characterization of osteosarcoma cell lines provides representative models to study the human disease. Lab. Investig. 2011, 91, 1195–1205. [Google Scholar] [CrossRef]
- Menna, P.; Paz, O.G.; Chello, M.; Covino, E.; Salvatorelli, E.; Minotti, G. Anthracycline cardiotoxicity. Expert Opin. Drug Saf. 2012, 11 (Suppl. 1), S21–S36. [Google Scholar] [CrossRef]
- Ojha, S.; Al Taee, H.; Goyal, S.; Mahajan, U.B.; Patil, C.R.; Arya, D.S.; Rajesh, M. Cardioprotective potentials of plant-derived small molecules against doxorubicin associated cardiotoxicity. Oxid. Med. Cell. Longev. 2016, 2016, 5724973. [Google Scholar] [CrossRef] [Green Version]
- Squillaro, T.; Schettino, C.; Sampaolo, S.; Galderisi, U.; Di Iorio, G.; Giordano, A.; Melone, M. Adult-onset Brain Tumors and Neurodegeneration: Are Polyphenols Protective? J. Cell. Physiol. 2018, 233, 3955–3967. [Google Scholar] [CrossRef] [PubMed]
- Damiano, S.; Iovane, V.; Squillacioti, C.; Mirabella, N.; Prisco, F.; Ariano, A.; Amenta, M.; Giordano, A.; Florio, S.; Ciarcia, R. Red Orange and Lemon Extract Prevents the Renal Toxicity Induced by Ochratoxin A in Rats. J. Cell. Physiol. 2020, 235, 5386–5393. [Google Scholar] [CrossRef] [PubMed]
- Dong, Q.; Chen, L.; Lu, Q.; Sharma, S.; Li, L.; Morimoto, S.; Wang, G. Quercetin attenuates doxorubicin cardiotoxicity by modulating Bmi-1 expression. Br. J. Pharmacol. 2014, 171, 4440–4454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imbaby, S.; Ewais, M.; Essawy, S.; Farag, N. Cardioprotective effects of curcumin and nebivolol against doxorubicin-induced cardiac toxicity in rats. Hum. Exp. Toxicol. 2014, 33, 800–813. [Google Scholar] [CrossRef]
- Nayak, P.G.; Paul, P.; Bansal, P.; Kutty, N.G.; Pai, K.S. Sesamol prevents doxorubicin-induced oxidative damage and toxicity on H9c2 cardiomyoblasts. J. Pharm. Pharmacol. 2013, 65, 1083–1093. [Google Scholar] [CrossRef]
- Carresi, C.; Musolino, V.; Gliozzi, M.; Maiuolo, J.; Mollace, R.; Nucera, S.; Maretta, A.; Sergi, D.; Muscoli, S.; Gratteri, S.; et al. Anti-oxidant effect of bergamot polyphenolic fraction counteracts doxorubicin-induced cardiomyopathy: Role of autophagy and c-kitposCD45negCD31neg cardiac stem cell activation. J. Mol. Cell. Cardiol. 2018, 119, 10–18. [Google Scholar] [CrossRef]
- Abdel-Raheem, I.T.; Abdel-Ghany, A.A. Hesperidin alleviates doxorubicin-induced cardiotoxicity in rats. J. Egypt Natl. Cancer Inst. 2009, 21, 175–184. [Google Scholar]
- Razavi-Azarkhiavi, K.; Iranshahy, M.; Sahebkar, A.; Shirani, K.; Karimi, G. The Protective Role of Phenolic Compounds against Doxorubicin-induced Cardiotoxicity: A Comprehensive Review. Nutr. Cancer 2016, 68, 892–917. [Google Scholar] [CrossRef]
- Kitsati, N.; Mantzaris, M.D.; Galaris, D. Hydroxytyrosol inhibits hydrogen peroxide-induced apoptotic signaling via labile iron chelation. Redox Biol. 2016, 10, 233–242. [Google Scholar] [CrossRef] [Green Version]
- May, P.M.; Williams, G.K.; Williams, D.R. Solution Chemistry Studies of Adriamycin-Iron Complexes Present In Vivo. Eur. J. Cancer 1980, 16, 1275–1276. [Google Scholar] [CrossRef]
- Nitobe, J.; Yamaguchi, S.; Okuyama, M.; Nozaki, N.; Sata, M.; Miyamoto, T.; Takeishi, Y.; Kubota, I.; Tomoike, H. Reactive Oxygen Species Regulate FLICE Inhibitory Protein (FLIP) and Susceptibility to Fas-Mediated Apoptosis in Cardiac Myocytes. Cardiovasc. Res. 2003, 57, 119–128. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sirangelo, I.; Liccardo, M.; Iannuzzi, C. Hydroxytyrosol Prevents Doxorubicin-Induced Oxidative Stress and Apoptosis in Cardiomyocytes. Antioxidants 2022, 11, 1087. https://doi.org/10.3390/antiox11061087
Sirangelo I, Liccardo M, Iannuzzi C. Hydroxytyrosol Prevents Doxorubicin-Induced Oxidative Stress and Apoptosis in Cardiomyocytes. Antioxidants. 2022; 11(6):1087. https://doi.org/10.3390/antiox11061087
Chicago/Turabian StyleSirangelo, Ivana, Maria Liccardo, and Clara Iannuzzi. 2022. "Hydroxytyrosol Prevents Doxorubicin-Induced Oxidative Stress and Apoptosis in Cardiomyocytes" Antioxidants 11, no. 6: 1087. https://doi.org/10.3390/antiox11061087
APA StyleSirangelo, I., Liccardo, M., & Iannuzzi, C. (2022). Hydroxytyrosol Prevents Doxorubicin-Induced Oxidative Stress and Apoptosis in Cardiomyocytes. Antioxidants, 11(6), 1087. https://doi.org/10.3390/antiox11061087