
Tissue Sodium Accumulation: Pathophysiology and Clinical Implications


 ,
, 
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Nonosmotic Tissue Sodium Storage
2.1. The Function of Skin Interstitium
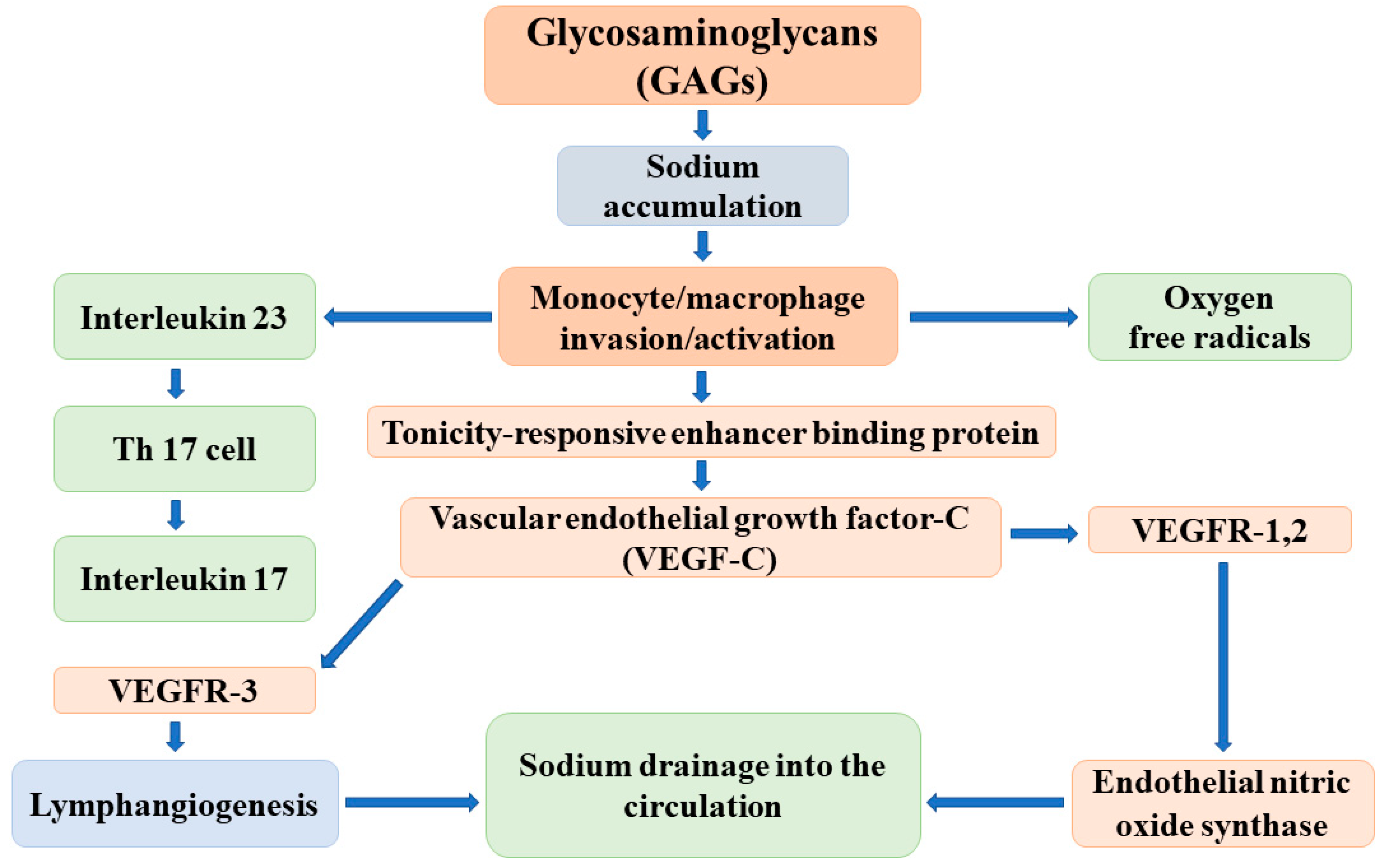
2.2. Endothelial Glycocalyx
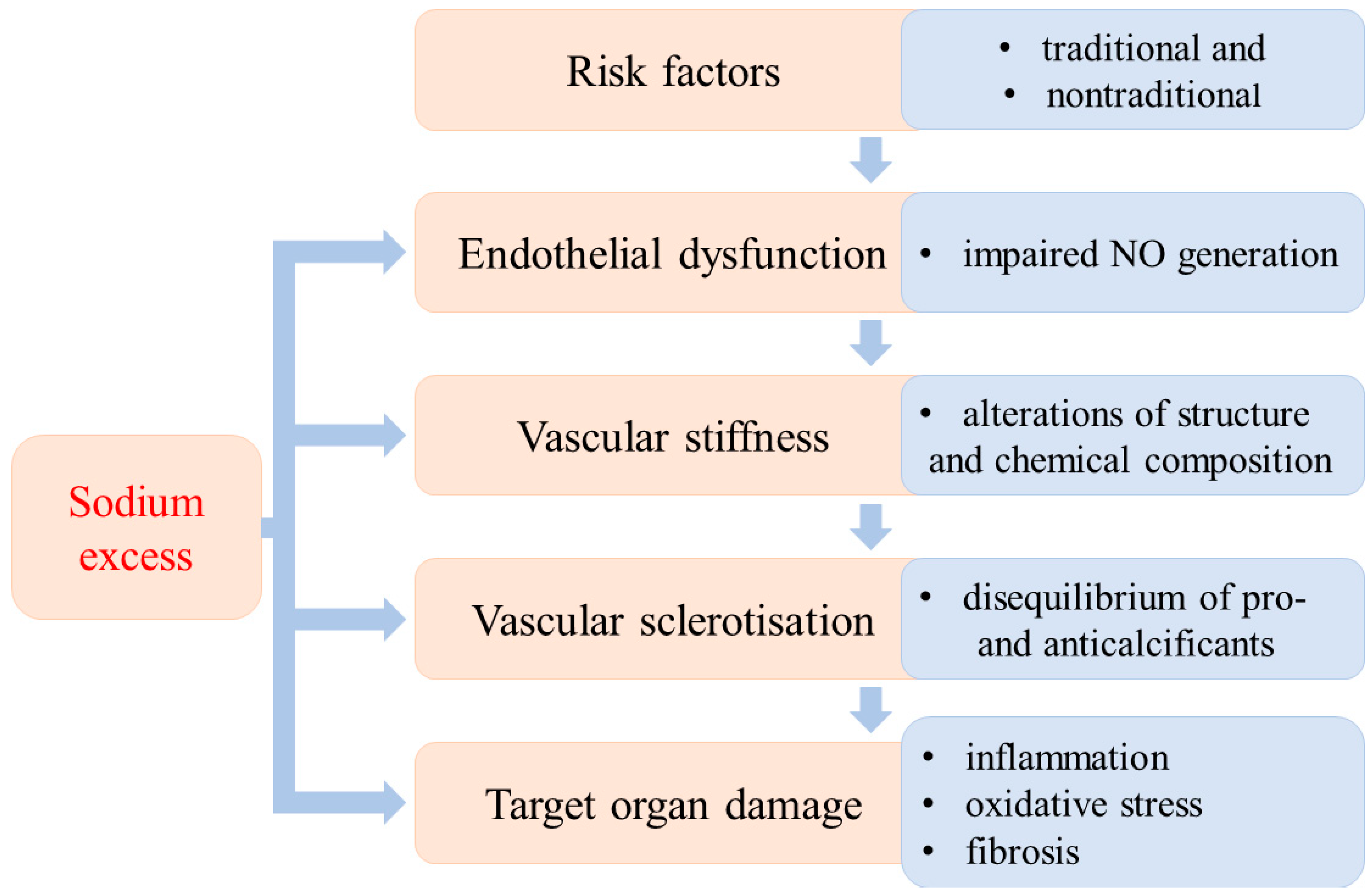
3. Sodium Loading and Oxidative Tissue Damage
3.1. Kidney
3.2. Myocardium
4. Perinatal Pathologies Associated with Sodium Intake and OS
4.1. Preeclamptic Pregnancy
4.2. Prenatal High Sodium Intake and Vascular Programming
4.3. Perinatal Adaptation and Oxidative Stress
5. Conclusions
- How and where the chloride, the accompanying anion of sodium, is stored and released.
- Water-free sodium release cannot meet the volume requirements; therefore, bound sodium and bound water should be released simultaneously.
- How the sodium reservoir functions are regulated in the given tissue.
- How we can prevent/attenuate the sodium-induced tissue damage.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO Regional Office for Europe. European Health for All Database 2009 (HFA-DB). Available online: http//dasta.euro.who.int (accessed on 20 January 2010).
- Brenner, D.; Labreuche, J.; Touboul, P.J.; Schmidt-Petersen, K.; Poirier, O.; Perret, C.; Schönfelder, C.; Combadiere, C.; Lathrop, M.; Cambien, F.; et al. Cytokine polymorphism associated with carotid intima-media thickness in stroke patients. Stroke 2006, 37, 1691–1696. [Google Scholar] [CrossRef] [PubMed]
- Sulyok, E. New aspects of the pathomechanism of salt-sensitive hypertension. Orv. Hetil. 2019, 160, 43–49. (In Hungarian) [Google Scholar] [CrossRef] [PubMed]
- Intersalt Cooperative Research Group. Intersalt: An international study of electrolyte excretion and blood pressure. Results for 24 hour urinary sodium and potassium excetion. Br. Med. J. 1988, 297, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Forte, J.G.; Miguel, J.M.; Miguel, M.J.; de Pádua, F.; Rose, G. Salt and blood pressure, a community trial. Hum. Hypertens. 1989, 3, 179–184. [Google Scholar]
- Denton, D.; Weisinger, R.; Mundy, N.I.; Wickings, N.J.; Dixson, A.; Moisson, P.; Pingard, M.A.; Shade, R.; Carey, D.; Ardaillou, R.; et al. The effect of increased salt intake on blood pressure of chimpanzees. Nat. Med. 1995, 1, 1009–1016. [Google Scholar] [CrossRef]
- Adrogné, H.J.; Madias, N.E. Sodium and potassium in the pathogenesis of hypertension. N. Engl. J. Med. 2007, 356, 1966–1978. [Google Scholar] [CrossRef]
- He, F.J.; MacGregor, G.A. Effect of modest salt reduction on blood pressure: A meta-analysis of randomized trials. Implications for public health. J. Hum. Hypertens. 2002, 16, 761–770. [Google Scholar] [CrossRef]
- Cook, N.R.; Cutler, J.A.; Obarzanek, E.; Buring, J.E.; Rexrode, K.M.; Kumanyika, S.K.; Appel, L.J.; Whelton, P.K. Long term effects of dietary sodium reduction on cardiovascular disease outcomes: Observational follow-up of the trials of hypertension prevention (TOHP). Br. Med. J. 2007, 334, 885–888. [Google Scholar] [CrossRef]
- Guyton, A.C.; Coleman, T.G.; Cowley, A.V., Jr.; Scheel, K.W.; Manning, R.D., Jr.; Norman, R.A., Jr. Arterial pressure regulation. Overriding dominance of the kidney in long-term regulation and hypertension. Am. J. Med. 1972, 52, 584–594. [Google Scholar] [CrossRef]
- Grassi, G.; Arenare, F.; Pieruzzi, F.; Brambilla, G.; Mancia, G. Sympathetic activation in cardiovascular and renal disease. J. Nephrol. 2008, 22, 190–195. [Google Scholar]
- Esler, M. The 2009 Carl Ludwig Lecture: Pathophysiology of the human sympathetic nervous system in cardiovascular diseases: The transition from mechanisms to medical management. J. Appl. Physiol. 2010, 108, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Malpas, S.C. Sympathetic nervous system overactivity and its role on the development of cardiovascular disease. Physiol. Rev. 2010, 90, 513–557. [Google Scholar] [CrossRef] [PubMed]
- Ralph, A.F.; Grenier, C.; Costello, H.M.; Stewart, K.; Ivy, J.R.; Dhaun, N.; Baile, A.M. Activation of sympathetic nervous system promotes blood pressure salt-sensitivity in C57BL 6/J. mice. Hypertension 2021, 77, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Guild, S.-J.; McBryde, F.D.; Malpas, S.C.; Barrett, C.J. High dietary salt and angiotensin II chronically increase renal sympathetic nerve activation. Hypertension 2012, 59, 614–620. [Google Scholar] [CrossRef]
- Brands, M.W.; Manhiani, M. Sodium-retaining effect of insulin in diabetes. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2012, 303, R1101–R1109. [Google Scholar] [CrossRef]
- Weidmann, P.; Ferrari, P. Central role of sodium in hypertension in diabetic subject. Diabetes Care 1991, 14, 220–232. [Google Scholar] [CrossRef]
- DeFronzo, R.A.; Ferranini, E. Insulin resistance. A multifaceted syndrome responsible for NIDDM, obesity, hypertension, dyslipidemia, and artherosclerotin cardiovascular disease. Diabetes Care 1991, 14, 173–194. [Google Scholar] [CrossRef]
- Muniyappa, R.; Montagnani, M.; Koh, K.K.; Quon, M.J. Cardiovascular actions of insulin. Endocr. Rev. 2007, 28, 463–491. [Google Scholar] [CrossRef]
- Muniyappa, R.; Sowers, J.R. Role of insulin resistance in endothelial dysfunction. Rev. Endocr. Metab. Disord. 2013, 14, 5–12. [Google Scholar] [CrossRef]
- Janus, A.; Szahidewitz-Krupska, E.; Mauer, G.; Doroszko, A. Insulin resistance and endothelial dysfunction constitute a common therapeutic target in cardiometabolic disorders. Mediat. Inflamm. 2016, 2016, 3634948. [Google Scholar] [CrossRef]
- Titze, J.; Maillet, A.; Lang, R.; Gunga, H.C.; Johannes, B.; Gauquelin-Koch, G.; Kihm, E.; Larina, I.; Gharib, C.; Kirsch, K.A. Long-term sodium balance in humans in a terrestrial space station simulation study. Am. J. Kidney Dis. 2002, 40, 508–516. [Google Scholar] [CrossRef] [PubMed]
- Heer, M.; Basich, F.; Kropp, J.; Gerzer, R.; Drummer, C. High dietary sodium chloride consumption may not induce body fluid retention in humans. Am. J. Physiol. Ren. Physiol. 2000, 278, F585. [Google Scholar] [CrossRef] [PubMed]
- Qi, N.; Rapp, J.P.; Brand, P.H.; Metting, P.J.; Britton, S.L. Body fluid expansion is not essential for salt-induced hypertension in SS/Jr rats. Am. J. Physiol. 1999, 277, R1392–R1400. [Google Scholar] [CrossRef] [PubMed]
- Titze, J.; Shakinacibaei, M.; Schaffhuber, M.; Schulze-Tanzil, G.; Porst, M.; Schwind, K.H.; Dietsch, P.; Hilgers, K.F. Glycosaminoglycan polymerization may enable osmotically inactive Na+ storage in the skin. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H203–H208. [Google Scholar] [CrossRef]
- Machnik, A.; Neuhofer, W.; Jantsch, J.; Dahlmann, A.; Tammela, T.; Machura, K.; Park, J.K.; Beck, F.X.; Müller, D.N.; Derer, W.; et al. Macrophages regulate salt-dependent volume and blood pressure by a vascular endothelial growth factor-C-dependent buffering mechanisms. Nat. Med. 2009, 15, 545–552. [Google Scholar] [CrossRef]
- Titze, J.; Krause, H.; Hecht, H.; Dietsch, P.; Rittweger, J.; Lang, R.; Kirsch, K.A.; Hilgers, K.F. Reduced osmotically inactive Na storage capacity and hypertension in the Dahl model. Am. J. Physiol. Renal. Physiol. 2002, 283, F134–F141. [Google Scholar] [CrossRef]
- Wiig, H.; Schröder, A.; Neuhofer, W.; Jantsch, J.; Kopp, C.; Karlsen, T.V.; Boschmann, M.; Goss, J.; Bry, M.; Rakova, N.; et al. Immune cells control skin lymphatic electrolyte homeostasis and blood pressure. J. Clin. Investig. 2013, 123, 2803–2815. [Google Scholar] [CrossRef]
- Slagman, M.C.; Kwakernaak, A.; Yazdani, S.; Laverman, G.D.; van den Born, J.; Titze, J.; Navis, G. Vascular endothelial growth factor C levels are modulated by dietary salt intake in proteinuric chronic kidney disease patients and in healthy subjects. Nephrol. Dial. Transpl. 2012, 27, 978–982. [Google Scholar] [CrossRef]
- Soria, J.C.; DeBraud, F.; Bahleda, R.; Adamo, B.; Andre, F.; Dientsmann, R.; Delmonte, A.; Cereda, R.; Isaacson, J.; Litten, J.; et al. Phase I/IIa study evaluating the safety, efficacy, pharmatokinetics, and pharmacodynamics of lucitanib in advanced solid tumors. Ann. Oncol. 2014, 25, 2244–2251. [Google Scholar] [CrossRef]
- Lankhorst, S.; Severs, D.; Markó, I.; Rakova, N.; Titze, J.; Müller, D.N.; Jan Danser, A.H.; van den Meiracker, A.H. Salt sensitivity of angiogenesis inhibition-induced blood pressure rise: Role of interstitial sodium inhibition? Hypertension 2017, 69, 919–926. [Google Scholar] [CrossRef]
- Wahlgren, V.; Magnus, R. Über die Bedeutung der Gewebe als Chlordepots. Arch. Exp. Pathol. Pharmakol. 1909, 71, 97–112. [Google Scholar] [CrossRef]
- Sulyok, E. Physical water compartments: A revised concept of perinatal water metabolism. Physiol. Res. 2006, 55, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Sulyok, E.; Nyul, Z. Hyaluronan–related limited concentration by the immature kidney. Med. Hypotheses 2005, 65, 1058–1065. [Google Scholar] [CrossRef] [PubMed]
- Rügheimer, L.; Johnsson, C.; Maric, C.; Hansell, P. Hormonal regulation of renomedullary hyaluronan. Acta Physiol. 2008, 193, 195–198. [Google Scholar] [CrossRef]
- Stridh, S.; Palm, F.; Hansell, P. Renal Interstital hyaluronan: Functional aspects during normal and pathological conditions. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2012, 302, R1235–R1249. [Google Scholar] [CrossRef]
- Stridh, S.; Palm, F.; Takahashi, T.; Ikegami-Kawai, M.; Friederich-Persson, M.; Hansell, P. Hyluronan production by renomedullary interstitial cells: Influence of endothelin, angiotensin II and vasopressin. Int. J. Mol. Sci. 2017, 18, 2701. [Google Scholar] [CrossRef]
- Rossitto, G.; Mary, S.; Chen, J.Y.; Boder, P.; Chew, K.S.; Neves, K.B.; Alves, R.L.; Montezano, A.C.; Welsh, P.; Petrie, M.C.; et al. Tissue sodium excess is not hypertonic and reflects EC volume expansion. Nat. Commun. 2020, 11, 4222. [Google Scholar] [CrossRef]
- Selvarajah, V.; Connolly, K.; McEniery, C.; Wilkinson, I. Skin sodium and hypertension: A paradigm shift? Curr. Hypertens. Rep. 2018, 20, 94. [Google Scholar] [CrossRef]
- Olde Engberink, R.H.; Rorije, N.M.; Homan van der Heide, J.J.; van den Born, B.J.H.; Vogt, L. Role of the vascular wall in sodium homeostasis and salt sensitivity. J. Am. Soc. Nephrol. 2015, 26, 777–783. [Google Scholar] [CrossRef]
- Oberleihtner, H.; Peters, W.; Kusche-Vihrog, K.; Korte, S.; Schillers, H.; Kliche, K.; Oberleithner, K. Salt overload damages the glycocalyx sodium barrier of vascular endothelium. Pflug. Arch. 2011, 462, 519–528. [Google Scholar] [CrossRef]
- Kusche-Vihrog, K.; Jeggle, P.; Oberleithner, H. The role of NaCl in vascular endothelium. Pflug. Arch. 2014, 466, 851–859. [Google Scholar] [CrossRef] [PubMed]
- Dragovich, M.A.; Chester, D.; Fu, B.M.; Wu, C.; Xu, Y.; Goligorsky, M.S.; Zhang, X.F. Mechanotransduction of the endothelial glycocalyx mediates nitric oxide production through activation of TRP channels. Am. J. Physiol. Cell Physiol. 2016, 311, C846–C853. [Google Scholar] [CrossRef] [PubMed]
- Nader, H.B.; Buonassisi, V.; Colburn, P.; Dietrich, C.P. Heparin stimulates the synthesis and modifies the sulfation pattern of heparan sulfate proteoglycan from endothelial cells. J. Cell Physiol. 1989, 140, 305–310. [Google Scholar] [CrossRef] [PubMed]
- Broekhuizen, L.N.; Lemkes, B.A.; Mooij, H.L.; Meuwese, M.C.; Verberne, H.; Holleman, F.; Schlingemann, R.O.; Nieuwdorp, M.; Stroes, E.S.; Vink, H. Effect of sulodexide on endothelial glycocolyx and vascular permeability in patients with type 2 diabetes mellitus. Diabetologia 2010, 53, 2646–2655. [Google Scholar] [CrossRef]
- Liu, M.; Deng, M.; Luo, Q.; Dou, X.; Jia, Z. High salt loading downregulates Nrf2 expression in a sodium-dependent manner in renal connecting duct cells. Front. Physiol. 2019, 10, 1565. [Google Scholar] [CrossRef] [PubMed]
- Kitiyakara, C.; Chabrashinvili, T.; Chen, Y.; Blam, J.; Karbera, A.; Aslam, S.; Welch, W.J.; Wilcox, C.S. Salt intake, oxidative stress and renal expression of NADPH oxidase and superoxide dismutase. J. Am. Soc. Nephrol. 2003, 14, 2775–2782. [Google Scholar] [CrossRef]
- Vaziri, N.D.; Dicus, M.; Ho, N.D.; Boroujerdi-Rad, L.; Sindhu, R.K. Oxidative stress and dysregulation of superoxide dismutase and NADPH oxidase in renal insufficiency. Kidney Int. 2003, 62, 179–185. [Google Scholar] [CrossRef]
- John, E.J.; Shau, G.H.; Nessy, B.; O’Neill, S.; Lane, B.; Healy, V. Impact of elevated dietary sodium intake on NAD(P)H oxidase and SOD in the cortex and medulla in rat kidney. Am. J. Physiol. Regul. Integr. Physiol. 2010, 299, R234–R240. [Google Scholar] [CrossRef]
- Kleinewietfeld, M.; Manzel, A.; Titze, J.; Kvakan, H.; Yosef, N.; Linker, R.A.; Muller, D.N.; Hafler, D.A. Sodium chloride drives autoimmune disease by the induction of pathogenic TH17 cells. Nature 2013, 946, 518–522. [Google Scholar] [CrossRef]
- Zhang, W.C.; Zheng, X.J.; Du, L.J.; Sun, J.Y.; Shen, Z.X.; Shi, C.; Sun, S.; Zhang, Z.; Chen, X.O.; Qin, M.; et al. High salt primes a specific activation state of macrophages, M(Na). Cell Res. 2015, 25, 893–910. [Google Scholar] [CrossRef]
- Yi, B.; Titze, J.; Rykova, M.; Feuerecker, M.; Vassilieva, G.; Nichiporuk, I.; Schelling, G.; Morukov, B.; Choukèr, A. Effects of dietary salt levels on monocyte cells and immune responses in healthy human subjects: A longitudinal study. Transl. Res. 2015, 166, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Iwakura, Y.; Ishigame, H. The IL-23/IL-17 axis in inflammation. J. Clin. Investig. 2006, 116, 1218–1222. [Google Scholar] [CrossRef] [PubMed]
- Fehrenbach, D.; Mattson, D.L. Inflammatory macrophages in the kidney contribute to salt-sensitive hypertension. Am. J. Physiol. Ren. Physiol. 2020, 318, F544–F548. [Google Scholar] [CrossRef] [PubMed]
- Sakata, F.; Ito, Y.; Mizuno, M.; Sawai, A.; Suzuki, Y.; Tomita, T.; Tawada, M.; Tanaka, A.; Hirayama, A.; Sagara, A.; et al. Sodium chloride promotes tissue inflammation via osmotic stimuli in subtotal-nephrectomized mice. Lab. Investig. 2017, 97, 432–446. [Google Scholar] [CrossRef]
- Washino, S.; Hosohata, K.; Jin, D.; Takai, S.; Miyagawa, T. Early urinary biomarkers of renal tubular damage by a high-salt intake independent of blood pressure in normotensive rats. Clin. Exp. Pharm. Psyhol. 2018, 45, 261–268. [Google Scholar] [CrossRef]
- Kayamba, V.; Kelly, P. Estimated 24-hour urinary sodium excretion as risk factor for oxidative stress in Zambian adults. A cross-sectional study. PLoS ONE 2020, 15, e0242144. [Google Scholar] [CrossRef]
- Dimitrieva, N.I.; Cui, K.; Kitchaev, D.A.; Zhao, K.; Burg, M.B. DNA double-strand breaks induced by high NaCl occur predominantly in gene deserts. Proc. Natl. Acad. Sci. USA 2011, 108, 20796–20801. [Google Scholar] [CrossRef]
- Huang, P.; Shen, Z.; Liu, J.; Huang, Y.; Chen, S.; Yu, W.; Wang, S.; Ren, Y.; Li, X.; Tang, C.; et al. Hydrogen sulphide inhibits high-salt diet renal oxidative stress and kidney injury in Dahl rats. Oxid. Med. Cell Longev. 2016, 2016, 2807490. [Google Scholar] [CrossRef]
- Tian, N.; Moore, R.S.; Brandy, S.; Rose, R.A.; Gu, J.W.; Hughson, M.D.; Manning, R.D., Jr. Interactions between oxidative stress and inflammation in salt-sensitive hypertension. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H3388–H3395. [Google Scholar] [CrossRef]
- Yang, G.H.; Zhou, X.; Ji, E.J.; Liu, J.X.; Sun, J.; Dong, Y.; Jiang, T.M.; Li, Y.M. VEGF-C mediated cardiac lymphangiogenesis in high salt intake accelerated progression of left ventricular remodelling in spontaneously hypertensive rats. Clin. Exp. Hypertens. 2017, 39, 740–747. [Google Scholar] [CrossRef]
- Varagic, J.; Frohlich, E.D.; Dies, J.; Susic, D.; Ahn, J.; González, A.; López, B. Myocardial fibrosis, impaired coronary hemodynamics and ventriicular dysfunction in salt-loaded SHR. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H1503–H1509. [Google Scholar] [CrossRef] [PubMed]
- Selvajar, S.D.; Djoussel, L.; Agular, F.G.; Martinez, E.E.; Polsinelli, V.B.; Irvin, M.R.; Arnett, D.K.; Shah, S.J. Association of estimated sodium intake with adverse cardiac structure and function from the HyperGEN study. J. Am. Cell Cardiol. 2017, 70, 715–724. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Shen, Z.; Yu, W.; Huang, Y.; Tang, C.; Du, J.; Jin, H. Hydrogen sulphide inhibits high-salt diet-induced myocardial oxidative stress and myocardial hypertrophy in Dahl rats. Front. Pharmacol. 2017, 8, 128. [Google Scholar] [CrossRef] [PubMed]
- Nilsen, L.H.; Ovesen, P.; Hansen, M.R.; Brantlov, S.; Jespersen, B.; Bie, P.; Jensen, B.L. Changes in the renin-angiotensin-aldosterone system in response to dietary salt intake in normal and hypertensive pregnancy. A randomised trial. Am. J. Soc. Hypertens. 2016, 10, e4–e890. [Google Scholar]
- Braun, M.A.; Gallery, E.D.; Ross, M.R.; Esber, R.P. Sodium excretion in normal and hypertensive pregnancy: A prospective study. Am. J. Obs. Gynecol. 1988, 159, 297–307. [Google Scholar]
- Birukov, A.; Andersen, L.B.; Herse, F.; Rakova, N.; Kitlen, G.; Kyhl, H.B.; Golic, M.; Haase, N.; Kräker, K.; Müller, D.N.; et al. Aldosterone, salt and potassium intakes as predictors of pregnancy outcome including preeclampsia. Hypertension 2019, 74, 391–398. [Google Scholar] [CrossRef]
- Escher, G.; Cristiano, M.; Causevic, M.; Baumann, M.; Frey, F.J.; Surbek, D.; Mohaupt, M.G. High aldosterone-to-renin variants of CYP11B2 and pregnancy outcome. Nephrol. Dial. Transpl. 2009, 24, 1870–1875. [Google Scholar] [CrossRef]
- Todkar, A.; Di Chiara, M.; Loffing-Cueni, D.; Bettoni, C.; Mohaupt, M.; Loffing, J.; Wagner, C.A. Aldosterone deficiency adversely affects pregnancy outcome in mice. Pflug. Arch. 2012, 464, 331–343. [Google Scholar] [CrossRef]
- Scaife, P.J.; Mohaupt, M.G. Salt, aldosterone and extrarenal Na+-sensitive responses in pregnancy. Placenta 2017, 65, 53–58. [Google Scholar] [CrossRef]
- Achur, M.; Valiyaveettil, A.; Alkahalil, A.; Gowda, D.C. Characterization of proteoglycans of human placenta and identification of unique chondroitin sulfate proteoglycans of the intervillous spaces that mediate the adherence of Plasmodium falciparum- infected erythrocytes to the placenta. J. Biol. Chem. 2000, 27551, 40344–40356. [Google Scholar] [CrossRef]
- Chui, P.; Murthi, S.P.; Brennecke, V.; Ignjatovic, V.; Monagle, P.T.; Said, J.M. The expression of placental proteoglycans in pre-eclampsia. Gynecol. Obstet. Investig. 2012, 13, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Williams, P.J.; Bulmer, J.N.; Searle, R.F.; Innes, B.A.; Robson, S.C. Altered decidnal leucocyte population in the placental bed in pre-eclampsia and foetal growth restriction: A comparison with late normal pregnancy. Reproduction 2009, 138, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Yosef, N.; Thalhamer, T.; Zhu, C.; Xiao, S.; Kishi, Y.; Regev, A.; Kuchroo, V. Induction of pathogenic TH17 cells by inducible salt-sensing kinase SGK1. Nature 2013, 496, 513–517. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M. Salt in pregnancy. Lancet 1958, 1, 168–185. [Google Scholar]
- Barker, D.J.P. In utero programming of chronic disease. Clin. Sci. 1998, 95, 115–128. [Google Scholar] [CrossRef]
- Barker, D.J.P.; Bagby, S.P.; Hanson, M.A. Mechanisms of disease: In utero programming in the pathogenesis of hypertension. Nat. Clin. Pract. Nephrol. 2007, 2, 700–707. [Google Scholar] [CrossRef]
- Porter, J.P.; King, S.H.; Honeycutt, A.D. Prenatal high-salt diet in the Sprague-Dawley rat programs blood pressure in adult female offspring. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 293, R334–R342. [Google Scholar] [CrossRef]
- Piecha, G.; Koleganova, Í.N.; Ritz, E.; Müller, A.; Fedorova, O.V.; Bagrov, A.Y.; Lutz, D.; Schirmacher, P.; Gross-Weissmann, M.L. High salt intake causes adverse fetal programming-vascular effects beyond blood pressure. Nephrol. Dial. Transpl. 2012, 27, 3464–3476. [Google Scholar] [CrossRef]
- Rotmans, J.I.; Babelink, T.J. Antenatal excessive sodium intake induces adverse vascular remodelling in offspring. Nephrol. Dial. Tranplant. 2012, 27, 3379–3381. [Google Scholar] [CrossRef][Green Version]
- McCarty, M.F. Endothelial membrane potential regulates production of both nitric oxide and superoxide- a fundamental determinant of vascular health. Med. Hypotheses 1999, 53, 277–289. [Google Scholar] [CrossRef]
- Gescha, S.; Kretschner, A.; Sharkovska, Y.; Evgenov, O.V.; Lawrenz, B.; Hucke, A.; Hocher, B.; Stasch, J.P. Solube guanylate cyclase stimulation prevents fibrotic tissue remodelling and improves survival in salt-sensitive Dahl rats. PLoS ONE 2011, 6, e21853. [Google Scholar]
- Vida, G.; Sulyok, E.; Lakatos, O.; Ertl, T.; Martens-Lobenhoffer, J.; Bode-Böger, S.M. Plasma levels of asymmetric dimethyarginine in premature neonates: Its possible involvement in developmental programming of chronic diseases. Acta Pediatr. 2009, 98, 437–441. [Google Scholar] [CrossRef] [PubMed]
- Worgall, S.; Rascher, W.; Gyódi, G.; Nyul, Z.; Baranyai, Z.; Sulyok, E. Urinary excretion of endogenous ouabain-like substance is reduced in NaCl supplemented premature infants. Biol. Neonate 1997, 72, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Nagy, G.; Gaszner, B.; Lányi, É.; Markó, L.; Fehér, E.; Cseh, J.; Kőszegi, T.; Betlehem, J.; Sulyok, E.; Cziráki, A.; et al. Selective association of endogenous ouabain with subclinical organ damage in treated hypertensive patients. J. Hum. Hypertens. 2011, 25, 122–129. [Google Scholar] [CrossRef][Green Version]
- Lembo, C.; Buonocore, G.; Perrone, S. Oxidative stress in preterm newborns. Antioxidants 2021, 10, 1672. [Google Scholar] [CrossRef]
- Jung, E.; Romero, R.; Yeo, L.; Diaz-Primera, R.; Marin-Concha, J.; Para, R.; Lopez, A.M.; Pacora, P.; Gomez-Lopez, N.; Yoon, B.H.; et al. The fetal inflammatory response syndrome: The origin of concepts, pathophysiology, diagnosis, and obstetrical implications. Semin. Fetal Neonatal Med. 2020, 25, 101146. [Google Scholar] [CrossRef]
- Tammela, O.K.; Koivisto, M.E. Fluid restriction for preventing bronchopulmonary dysplasia? Reduced fluid intake during the first week of life improves the outcome of low-birth-weight infant. Acta Paediatr. 1992, 81, 207–212. [Google Scholar] [CrossRef]
- Costarino, A.T.; Gruskay, J.A.; Corcoran, L.; Polin, R.A.; Baumgart, S. Sodium restriction versus daily maintenance replacement in very low birth weight premature neonates: A randomized blind therapeutic trial. J. Pediatr. 1992, 120, 99–106. [Google Scholar] [CrossRef]
- Hartnoll, G.; Bétrémieux, P.; Modi, N. Randomised controlled trial of postnatal sodium supplementationin infants of 25–30 weeks gestational age: Effects on cardiopulmonary adaptation. Arch. Dis. Child. Fetal Neonatal Ed. 2001, 85, 29–32. [Google Scholar] [CrossRef]
- OH, W.; Pondexter, B.B.; Perritt, R.; Lemons, J.A.; Bauer, C.R.; Ehrenkranz, R.A.; Stoll, B.J.; Poole, K.; Wright, L.L. Association between fluid intake and weight loss during the first ten days of life and risk of bronchopulmonary dysplasia in extremely low birth weight infants. J. Pediatr. 2005, 147, 786–790. [Google Scholar] [CrossRef]
- Hartnoll, G.; Bétrémieux, P.; Modi, N. Randomised controlled trial of postnatal sodium supplementation on oxygen dependency and body weight in 25–30 week gestational age infants. Arch. Dis. Child. Fetal Neonatal Ed. 2000, 82, 19–23. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sulyok, E.; Farkas, B.; Nagy, B.; Várnagy, Á.; Kovács, K.; Bódis, J. Tissue Sodium Accumulation: Pathophysiology and Clinical Implications. Antioxidants 2022, 11, 750. https://doi.org/10.3390/antiox11040750
Sulyok E, Farkas B, Nagy B, Várnagy Á, Kovács K, Bódis J. Tissue Sodium Accumulation: Pathophysiology and Clinical Implications. Antioxidants. 2022; 11(4):750. https://doi.org/10.3390/antiox11040750
Chicago/Turabian StyleSulyok, Endre, Bálint Farkas, Bernadett Nagy, Ákos Várnagy, Kálmán Kovács, and József Bódis. 2022. "Tissue Sodium Accumulation: Pathophysiology and Clinical Implications" Antioxidants 11, no. 4: 750. https://doi.org/10.3390/antiox11040750
APA StyleSulyok, E., Farkas, B., Nagy, B., Várnagy, Á., Kovács, K., & Bódis, J. (2022). Tissue Sodium Accumulation: Pathophysiology and Clinical Implications. Antioxidants, 11(4), 750. https://doi.org/10.3390/antiox11040750