
Oxidative Stress Induced by High Salt Diet—Possible Implications for Development and Clinical Manifestation of Cutaneous Inflammation and Endothelial Dysfunction in Psoriasis vulgaris



Abstract
1. Introduction
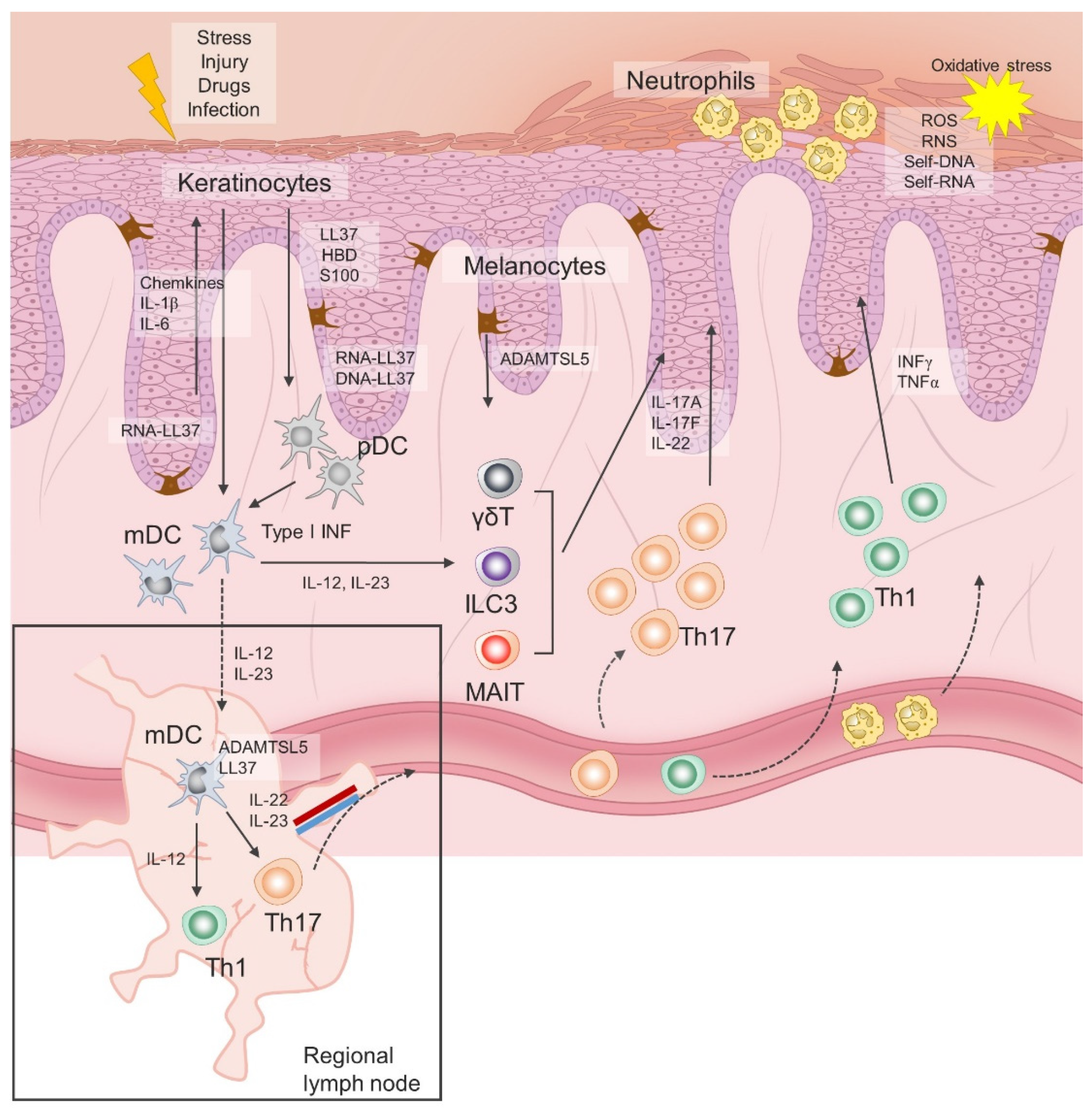
2. Immunopathogenesis of Psoriasis vulgaris
3. Increased Dietary Salt Intake Impairs Redox System Mechanism, Leading to Increased Oxidative Stress—Implications for Endothelial Dysfunction
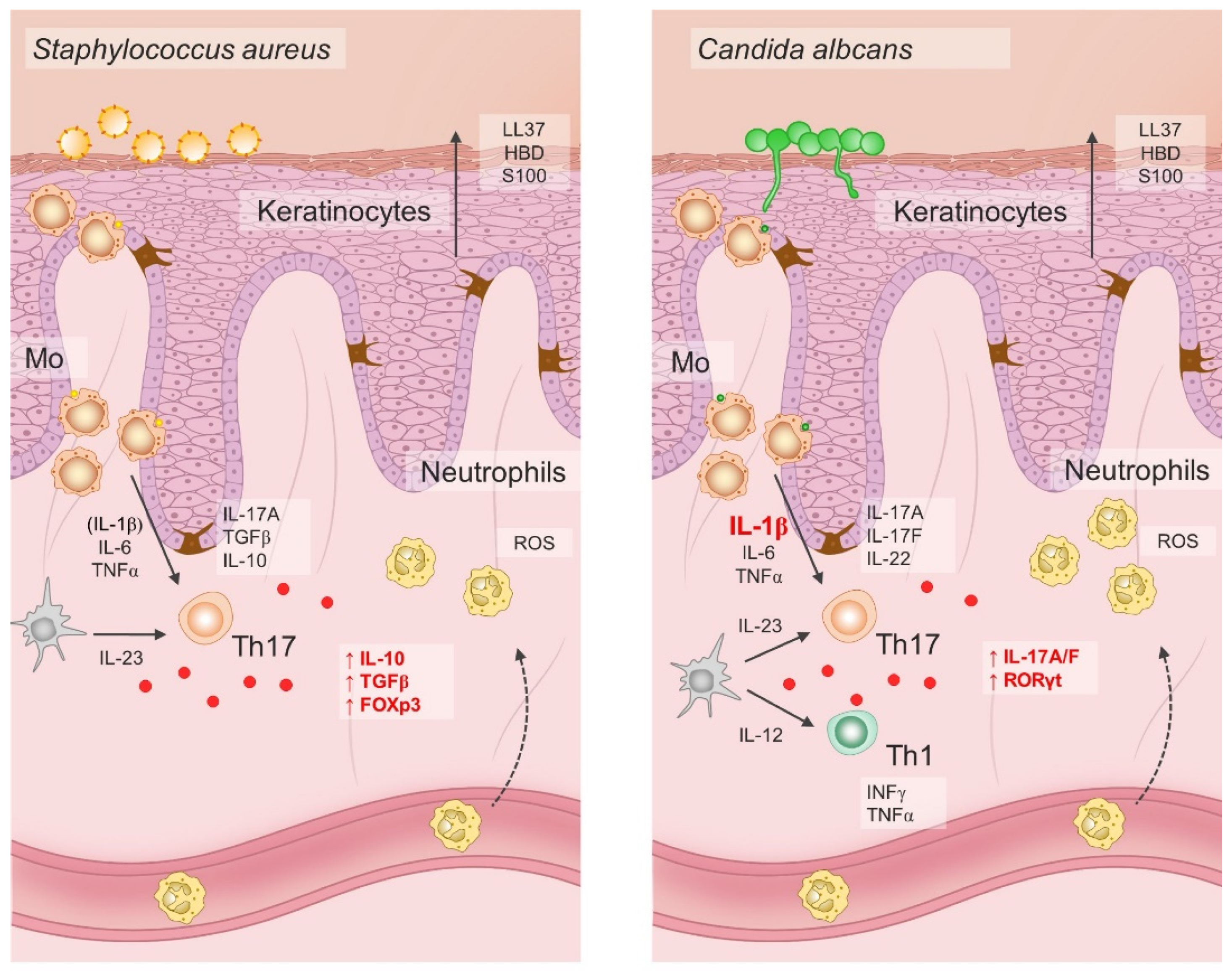
4. Role of Oxidative Stress in Immunopathogenesis of Psoriasis vulgaris
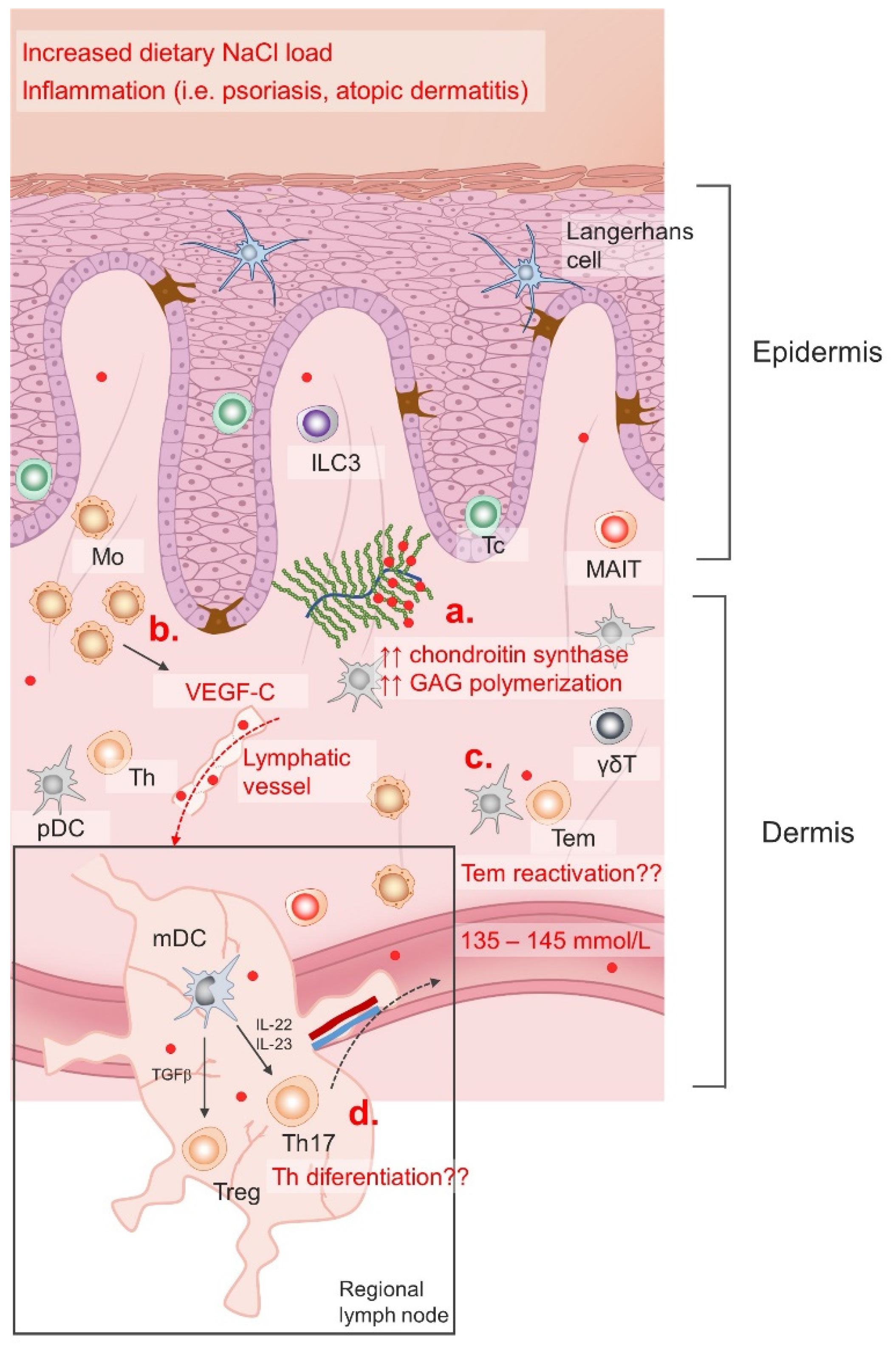
5. Evidence for NaCl-Mediated Modulation of Type-3 Inflammation—Dichotomous, Context-Dependent Effect of NaCl on the Th17 and Treg Phenotype
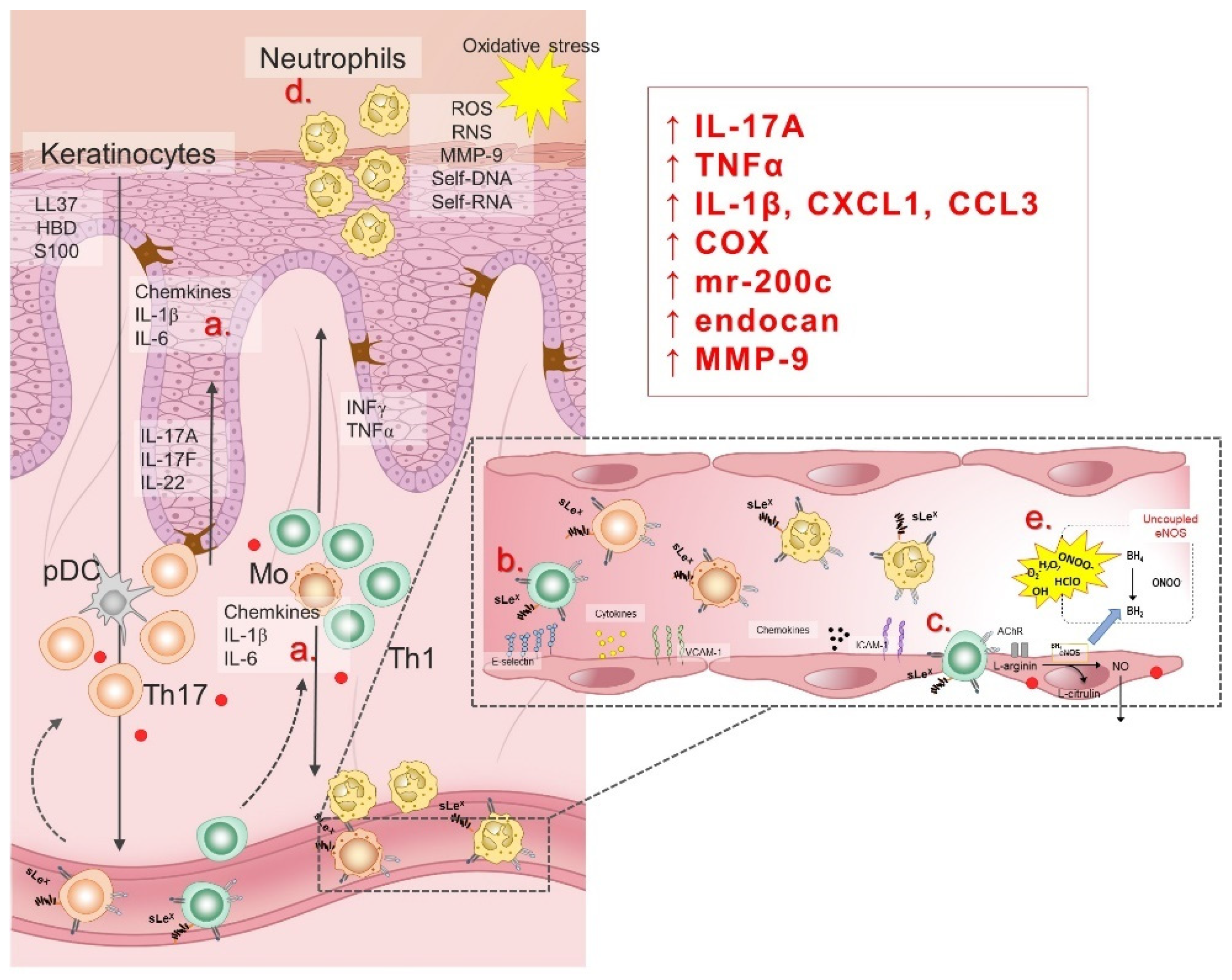
6. Deleterious Effects of Increased Systemic Oxidative Stress and Low-Grade Inflammation on Endothelial Function in Psoriasis Patients

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| First Author, Year, [Ref.] | Country | Study Design | Study Groups | Psoriasis Severity/PASI Inclusion Criteria | PASI Mean ± SD/[SEM] or Median (IQR) | Disease Duration (Years, Mean ± SD or [SEM]) | Systemic Antipsoriatic Therapy | Assessment Method/ Occlusion Site | Vessel, Measurement Site | Measurements | Effect on Measured Inflammation and Oxidative Stress Parameters | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Psoriasis | Controls | |||||||||||
| Jensen, 2011, [208] | Denmark | Case-control Study | 30 | 30 | Mild to moderate psoriasis/PASI < 10 | 7.3 ± 3.8 | 21.3 ± 17.0 | None | PAT (reactive hyperemia index, RHI; augmentation index, %)/Brachial artery (occlusion) | both index fingers | ↔ RHI; ↔ AI% | ↑ hsCRP (p = 0.011) |
| Mallbris, 2008, [223] | Sweden | Case-control Study | 20 | 20 | Severe psoriasis/PASI > 12 | 14.3 ± 4.8 | 0.4 ± 0.3 | None | FMD, NMD (absolute value vessel dilatation (B2-B1); vessel dilatation as the % of baseline value, %)/Forearm cuff (occlusion) | BA, above the elbow | ↔ B2-B1 ↔ %FMD ↔ %NMD | ↑ hsCRP (p < 0.05) |
| Martyn-Simmons, 2011, [205] | United Kingdom | Prospective Cohort Study | 60 | 117 | Moderate to severe psoriasis/PASI > 10 | 9.15 ± [0.91] | 31 ± [1.6] | Standard systemic therapy: MTX (n = 16, 26.7%); Acitretin (n = 5, 8.3%); ciclosporin (n = 3, 5%), fumaric acid esters (n = 5, 8.3%); Biologics: anti TNF-therapy (n = 13, 21.7%) | FMD, NMD (vessel dilatation as the % of baseline value, %)/Forearm cuff (occlusion) | BA, above the elbow | ↔ %FMD ↔ %NMD FMD associated with ciclosporin (β = 0.29, p < 0.04) | ↑ hsCRP (p < 0.05) |
| Gisondi, 2009, [224] | Italy | Case-control Study | 39 | 38 | Moderate to severe psoriasis/PASI > 10 | 12.4 ± 4.7 | 14.8 ± 12.7 | None (at least 2 months before inclusion) | cfPWV; no occlusion site crPWV(m/s); no occlusion site | cfPWV—sensor on CA and FA; crPWV—sensors on CA and RA | ↑ cfPWV (p = 0.001); positive correlation with disease duration (p = 0.0001), not with PASI. ↔ PWVcr | ↔ CRP |
| Balci, 2008, [207] | Turkey | Case-control Study | 43 | 43 | All PASI included | 6.5 ± 4.4 | 13.26 ± 10.55 | None (n = 32, 74%) Standard systemic treatment: acitretin (n = 10, 23%) Biologics: etanercept (n = 1, 2.3%) | cIMT (mm), no occlusion site FMD, NMD (vessel dilatation as the % of baseline value, %)/Forearm cuff (occlusion) | cIMT—left and right CCA FMD-BA, above the elbow | ↑ cIMT (p = 0.003) ↓ FMD% (p = 0.002), correlating with disease duration (β = −0.259, p < 0.05) ↓ NMD% (p = 0.013) No association with systemic therapies found. | / |
| Ulusoy, 2010, [202] | Turkey | Case-control Study | 28 | 28 | Mild to moderate/PASI 0.1–49.9 | 13 ± 8 | 4 ± 3 | None | FMD, NMD (vessel dilatation as the % of baseline value, %)/3–4 cm proximal to the section of the brachial artery (occlusion) | BA, above the elbow | ↓ FMD% (p < 0.001) ↔ NMD% | / |
| Von Stebut, 2019, [220] | Germany | Randomized Controlled Trial | 151 (35 + PsA) | 44 | Moderate to severe/PASI > 10 | A. 19.3 ± 7.9 B. 21.7 ± 10.5 C. 17.5 ± 4.2 D. 19.5 ± 6.1 | A. 20.6 ± 12.7 B. 20.8 ± 13.3 C. 18.9 ± 11.7 D. 20.3 ± 11.7 | A. secukinumab 300 mg from baseline to week 52 (n = 48) B. secukinumab 150 mg from baseline to week 52 (n = 54) C. placebo until week 12, then secukinumab 300 mg until week 52 (n = 26) D. placebo until week 12, then secukinumab 150 mg until week 52 (n = 23) | FMD (vessel dilatation as the % of baseline value, %)/5 cm distal to the measurement site (occlusion) PWVcf (distance/Δtime [m/s]), AI [%], no occlusion site | FMD—BA, 5–10 cm proximal to the antecubital fossa PWVcf—on CA and FA | Psoriasis patients compared to healthy controls: ↓ FMD% (at baseline), (p < 0.01) Group A and B compared to 3 and 4 at 12 weeks: ↔ FMD% Group A compared to baseline: ↑ FMD% (p < 0.002) Group B compared to baseline: ↑ FMD% (p = 0.0034) Group D compared to baseline: ↔ FMD%, ↔ PWVcf | A compared with C + D at week 12: ↓ S100B (mean −0.02, 95%CI −0.03 to 0.01) |
| de Simone, 2011, [201] | Italy | Case-control Study | 32 | 31 | Not specified/all PASI included | 17.9 ± 10.9 | 12.6 ± 10.2 | None (at least 3 months prior) | FMD, NMD (vessel dilatation as the % of baseline value, %)/forearm (occlusion) | Right BA, 2 to 15 cm proximal to the antecubital fossa | ↓ FMD% (p = 0.012), no correlation found with PASI or disease duration ↔NMD% | ↔ CRP ↔ ESR |
| Erfan, 2005, [225] | Turkey | Case-control Study | 60 | 30 | Moderate to severe/PASI ≥ 5 | Pso-ED 10.9 (5–24.6) Pso + ED 10.3 (5–26.9) | Pso-ED 7.8 (1–30) Pso + ED 15.5 (1–50) | none | FMD, NMD (vessel dilatation as the % of baseline value, %)/not specified (occlusion) | BA | ↓ FMD (p < 0.05) | ↑ YKL-40 (p < 0.05) ↑ CRP (p < 0.05) Pso + ED vs. controls + ED: ↑ YKL-40 (p < 0.05) |
| Haberka, 2018, [226] | Poland | Case-control Study | 80 | 39 | Mild to moderate | 18.6 ± 10.5 | 15.3 ± 11.2 | none | cfPWV (m/s), no occlusion site, FMD (vessel dilatation as the % of baseline value, %)/proximal portion of the arm (occlusion) cIMT (mm), no occlusion site | cfPWV—sensors (CCA and CFA) FMD- BA, above the antecubital fossa cIMT—CCA | ↑ cIMT(mm) (p < 0.05) ↓ FMD% (p < 0.001) ↔ PWV m/s | ↑ AOPPs (p < 0.001), sign. assoc. with IMT (r = 0.3), FMD (r = -0.25) ↑ visfatin (p < 0.001) ↔ osteoprogerin, ↔ nesfatin |
| Holzer, 2021, [227] | Austria | Randomized Controlled Trial | 65 | Moderate to severe/PASI ≥ 10 | Adalimumab group: 16.3 ± 5.8 FAE group:16.4 ± 5.9 | Adalimumab group: 11.9 ± 11.3 FAE group:10.1 ± 8.8 | Intervention with: Adalimumab (n = 33, 50.8%) FAE (n = 32, 49.2%) + NB-UVB (for non-responders) | FMD, NMD (vessel dilatation as the % of baseline value, %)/not specified (occlusion) cIMT (mm), no occlusion site | FMD—BA, above the antecubital fossa cIMT—1st cm of the CCA | Adalimumab group: ↑ FMD% after intervention (p = 0.048) FAE group: ↔FMD% Both groups: ↔ NMD%, ↔cIMT (mm) | Adalimumab a.i.:↓hsCRP (p = 0.022); FAE a.i.: ↔ hsCRP; ↓ p-selectin (p = 0.034) Both groups a.i.: ↓E-selectin (FAE: p = 0.041; adalimumab: p = 0.001) | |
| Erturan, 2014, [228] | Turkey | Case-control Study | 56 | 53 | Mild to moderate/PASI 0.1–49.9 | 3 (range 0.6–27) | 5.5 (range 0.5–50) | None (at least 3 months prior) | FMD (vessel dilatation as the % of baseline value, %)/forearm, bottom of the cuff on the wrist (occlusion) IMT(mm), no occlusion site | FMD—BA, 2–5 cm proximal to the antecubital fossa cIMT—previous segment of the bifurcation of the CA | ↓ FMD % (p = 0.0001) ↔ cIMT | ↑ sCD40L (p = 0.012) ↔ homocysteine ↔ ESR ↔ hsCRP |
| Karadag, 2010, [204] | Turkey | Case-control Study | 75 (24 + PsA) | 50 | All PASI included | 4.4 (1.8–34) | No data provided | No data obtained | FMD (vessel dilatation as the % of baseline value, %)/proximal forearm (occlusion) | BA | Pso vs. controls: ↓ FMD% (p < 0.001), no correlation with PASI PsA vs. Pso: ↓ FMD (p = 0.096) | ↑ ESR (p = 0.006) |
| Białecka, 2021, [229] | Poland | Case-control Study | 62 (6 + PsA) | 42 | All PASI included | 14.92 ± 6.99 | Assessed, data not provided | Data obtained on past use of systemic therapy (systemic treatment was used in n = 39; 62.9%) | cIMT (mm), no occlusion site cardiac CT: calcium score according to the Agatston scale (CS); mass of calcifications (CM, mg); the volume of calcifications in coronary arteries (CV, mm3) | cIMT—Both CCA, 2 cm from their bifurcation | ↑ cIMT (p < 0.0001); no correlation with PASI or CRP ↑ amount of calcification | ↑ CRP (p < 0.0001) |
| Bańska-Kisiel, 2016, [230] | Poland | Cross-sectional Study | 74 | none | Mild to moderate/PASI ≤ 50 | 18.7 ± 10.6 | 17.1 ± 11.2 | Biologics (n = 5; 7%) | cIMT (mm), no occlusion site | Both CCA, distal segments | Association between cIMT and PASI (r = 0.33; p = 0.007) | / |
| Troitzsch, 2012, [231] | Germany | Cross-sectional Study | 72 | 1955 | No data | No data | No data | No data provided | cIMT (mm), no occlusion site | Both CCA (10 consecutive measurement points, in 1 mm steps, from the bulb of both sides) | ↑ cIMT (p = 0.001) ↔ carotid plaque prevalence | ↑ hsCRP (p = 0.003) |
| de Oliveira, 2019, [232] | Brazil | Case-control Study | 11 | 33 | Severe/PASI > 10 | No data | No data | MTX (n = 2, 18%) | PWV (m/s), AIx, arm (occlusion) cIMT(mm), no occlusion site | PWV—not specified cIMT—1 cm from the posterior wall of the CCA | ↑ PWV (p = 0.033) ↑ IMT (left CCA) above the 75th centile (p = 0.045) | ↑ CRP (p < 0.001) |
| Fabi, 2022, [233] | Italy | Case-control Study | 20 * age < 18 | 20 | Not specified | 2.64 ± 2.6 | 1.84 ± 1.18 | Cyclosporine (n = 3, 15%), 2 switched to guselkumab | cIMT(mm), no occlusion site | Both CCA, at least 5 mm below its end | ↑ cIMT (right, p = 0.001; left, p = 0.00), positively correlating with disease duration | / |
| Awad, 2017, [234] | Egypt | Case-control Study | 45 | 45 | Not specified/all PASI included | 10.18 ± 4.6 | cIMT < 1 mm: 10.27 ± 14.07 cIMT > 1 mm: 11.33 ± 6.98 | none | cIMT(mm), no occlusion site | Both CCA, distal portion of the CCA (10–20 mm proximal to the carotid bulb) | ↑ cIMT (p < 0.001), positively correlating with PASI (r = 0.78, p < 0.001), serum psoriasin (r = 0.48, p > 0.01) and serum koebnerisin (r = 0.48, p < 0.01), but not with disease duration | ↑ psoriasin (p < 0.001) ↑ koebnerisin (p = 0.001), higher levels in patients with subclinical atherosclerosis (p = 0.04) |
| Liu, 2015, [235] | China | Case-control Study | 35 | 20 | BSA > 10% | 15.5 ± 12.7 | 14.0 ± 7.2 | MTX (n = 13, 37.1%) Retinoids (n = 2, 5.7%) | haPWV (m/s), no occlusion site, cIMT(mm), no occlusion site AI | haPWV—precordium and both posterior PA cIMT—max. thickness point along a 1-cm section of the CCA proximal to the carotid bulb, both sides | CD34 + EPC was independently predictive of increased haPWV | ↓ CD34 + EPC (p = 0.02); neg. correlating with haPWV (r = -0.43, p = 0.01) ↔ CD34/KDR + EPC, ↔ CD133/KDR + EPC and ↔ CD133 + EPC |
| El-Mongy, 2009, [236] | Egypt | Case-control Study | 80 (25 + PsA) | 50 | Not specified/all PASI included | 29.1 ± 16 | 12.6 ± 9.5 | No data provided (patients treated with cyclosporine or retinoid were excluded) | cIMT(mm), no occlusion site | right CCA, 1 cm distal to the carotid bifurcation in the posterior wall | ↑ cIMT (p < 0.001), positively correlating with age (r = 0.6, p ≤ 0.001), duration of the disease (r = 0.4, p = 0.001) and PASI (r= 0.5, p ≤ 0.001) | ↑ CRP (p ≤ 0.001) ↑ ESR (p = 0.004) |
| Martinez-Lopez, 2018, [237] | Spain | Prospective Cohort Study | 53 (21 + PsA) | Self-controlled, 8 m | PASI ≥ 5 | 9.46 ± 3.62 | 17.33 ± 10.78 | Systemic therapy (n = 30, 56.6%) Cyclosporine (n = 10, 18.8%), MTX (n = 10, 18.8%), acitretin (n = 10, 18.8%); Biologics (n = 23, 43.4%); TNF-α inhibitor (etanercept, infliximab, adalimumab), (n = 13, 24.5%); anti-IL12/23 (ustekinumab), (n = 10, 18.8%) Wash out period of 3 months before baseline | cIMT (mm), no occlusion site | cIMT—left CCA, 1 cm from the carotid bifurcation (6 measurements) | All patients: Decreasing tendency IMT (p = 0.086) MTX a.i.: ↓ IMT (p = 0.045) ustekinumab a.i.: ↓ IMT (p = 0.010) | / |
| Piros, 2021, [238] | Hungary | Prospective Cohort Study | 31 (17 + PsA) | Self-controlled, 6 m | Severe psoriasis/PASI > 10 | 18 (14–24) | 24 (16–28) | anti-IL-17 therapy- intervention: secukinumab (n = 20, 64.5%), ixekizumab (n = 11, 35.5%) | cIMT (mm) bIMT (mm) fIMT (mm); no occlusion site | cIMT—CCA; bIMT—middle third of the BA fIMT—middle third of the CFA * on both sides | 6 months after baseline, a.i. ↓ cIMT, ↓ bIMT, ↓ fIMT (p < 0.001 for all)—the improvement was more significant in non-calcified arteries than in calcified arteries | / |
| Jokai, 2013, [239] | Hungary | Prospective Cohort Study | 16 | Self-controlled, 6 m | Severe psoriasis/PASI > 15 | Baseline: 25.64 (21.2–32.4); ↓ of PASI after 6 months for 1.04 (0–8.8) | 16.8 (4–40) | No biologic therapy at baseline; intervention with TNF-α inhibitors: etanercept (n = 3, 18.8%), infliximab (n = 7, 43.8%), adalimumab (n = 6, 37.5%) during 6 months | cIMT (mm), bIMT (mm), no occlusion site | cIMT—carotid bifurcation bIMT—middle third of the BA | Group 1— no apparent atherosclerosis (n = 13) ↓ after intervention cIMT(mm) (p = 0.011) ↓ after intervention bIMT(mm) (p = 0.006) Group 2—atherosclerosis present (n = 3) ↔ cIMT, ↔ bIMT (but increasing tendency) | / |
| Ikonomidis, 2015, [240] | Greece | Case-control Study | 59 | 59 CAD patients; 40 healthy controls | All PASI included | 11.5 ± 8 | 5.1 ± 1.25 | Ciclosporine (n = 59, 100%) | cfPWV (m/s), augmentation index (CAI, %), no occlusion site, FMD (vessel dilatation as the % of baseline value, %)/occlusion site not specified cIMT(mm), no occlusion site CFR (ratio of peak diastolic velocity after adenosine infusion to peak diastolic velocity at rest), no occlusion site | cfPWV—sensors (CCA and CFA FMD—BA cIMT—CCA, bulb, ICA; on both sides CFR—color Doppler on LAD | Compared to healthy controls: ↑ cfPWV; ↑ CAI, ↑ IMT (p < 0.05 for all); IMT values correlating with PASI (r = 0.67, p < 0.01) ↓ FMD, ↓ CFR Compared to CAD patients: ↔ cfPWV; ↔ CAI, ↔ IMT ↔ FMD, ↔ CFR | Compared to healthy controls: ↑ MDA, ↑ IL-6 (p < 0.05 for both), correlating with cIMT (r = 0.35, p = 0.01 and r = 0.58, p < 0.001) Compared to CAD patients: ↔ MDA, ↔ IL-6 |
| Robati, 2014, [241] | Iran | Case-control Study | 60 | 60 | All PASI included | 23.45 (14.92–33.18) | 10 (4–16.5) | None (exclusion criteria was systemic therapy within the last 6 months) | cIMT (mm), no occlusion site | Right CCA, 1 cm proximal to the bifurcation (at least 3 measurements) | ↑ cIMT (p < 0.0001) | ↑ leptin, ↑ resistin (p < 0.0001) |
| Antonucci, 2014, [242] | Italy | Case-control Study | 40 | 40 | Moderate to severe/PASI > 10 | 16.1 ± ? | Not assessed | Exclusion criteria were: cyclosporine, oral retinoids, systemic steroids; no other data on therapy available | cIMT (mm), no occlusion site | cIMT—CCA, 1 cm proximal to the bifurcation | ↑ IMT (p < 0.001), positively correlating with PASI (r = 0.515, p < 0.01), not with BMI | / |
| Marovt, 2020, [243] | Slovenia | Prospective Cohort Study | 15 (4 + PsA) | Self-controlled | Moderate to severe/PASI > 10 | PASI 16.78 (11.0–19.8) BSA 12.62 (8–20) | 20.9 (range 3–52) | Intervention with anti-IL-23/IL-17: ustekinumab (n = 4, 26.67%); secukinumab (n = 10, 66.67%); ixekizumab (n = 1, 6.67%) | cfPWV (m/s), no occlusion site, aortic AIx, cIMT (mm), no occlusion site | CfPWV—CA, FA cIMT—CA, bifurcation level, both sides | ↔ cfPWV ↔ cIMT ↑ central aortic diastolic pressure (mmHg) (p = 0.03) | / |
| Elsheikh, 2013, [244] | Egypt | Case-control Study | 60 | 20 | Mild, moderate, severe/all PASI included | 18.49 ± 11.29 | 11.25 ± 6.95 | None (at least 6 weeks prior to cIMT) | cIMT (mm; internal diameter—ID; arterial wall mass index—AWMI), no occlusion site | Both sides at three points: - CCA (10 mm before the bulb) - Bulb (5–10 mm cranially to the start of the bulb - Internal carotid artery column after the flow divider | ↑ cIMT (p = 0.001); ↑ AWMI (p = 0.010) ↓ ID (p = 0.001) - independent predictor of cIMT: duration of disease (r = 0.425, p = 0.008); age (r = 0.362, p = 0.021), PASI score (r = 0.326, p = 0.014); BMI (r = 0.243, p = 0.019) | / |
| Yiu, 2010, [206] | China | Case-control Study | 52 | 50 | BSA > 10 | 14.7 ± 12.1 | 15.4 ± 7.1 | Methotrexate (n = 26, 50%) | baPWV (m/s), no occlusion, PAT (index), proximal forearm of the studied hand (occlusion) | baPWV—ATP and BA; PAT—tip of both middle fingers | Psoriasis vs. controls: ↑ baPWV (p < 0.01) ↔ PAT index Psoriasis patients on MTX vs. without MTX: ↔ baPWV ↔ PAT index no correlation between baPWV and PAT index (r = 0.09, p = 0.40) | ↑ hsCRP (p < 0.01)—correlating with baPWV (r = 0.51, p < 0.01) and with PASI (r = 0.48, p < 0.01) |
| Kim, 2015, [245] | South Korea | Case-control Study | 54 | 60 | Mild and moderate to severe/all PASI included | 10.7 + 7.0 | 10.4 + 9.7 | Data on previous systemic treatment obtained: n = 49, 90.7% had received systemic treatment at some point | BSI (β) cIMT (mm), no occlusion site | BSI—region 2 cm from the carotid bifurcation toward the center of the body cIMT—1 cm distal to the far wall of each CCA | ↑ BSI (p < 0.001), correlating with PASI ↔ cIMT (intended to be ↑, no significance) | / |
| Patschan, 2018, [246] | Germany | Case-control Study | 30 | 26 | Not specified | 10.2 ± 2.0 | 18.3 ± 2.7 | Past/present treatment with biological drug (n = 10, 33%) | cfPWV (m/s), augmentation index, AI; no occlusion site | Sensors on CA and FA | ↔ PWV (m/s) | ↑CRP ↔ CD133+/KDR+ (EPC) cells |
| Pina, 2016, [219] | Spain | Prospective Cohort Study | 29 | Self-controlled | Moderate to severe psoriasis | 18.9 ± 7.8 | 18.2 ± 12.1 | Anti-TNF-α (intervention): adalimumab Washout period from other systemic therapies of 4 weeks | FMD (vessel dilatation as the % of baseline value, %), forearm (occlusion); PWV | FMD—BA, 2–12 cm proximal to the antecubital fossa PWV—right CCA | A.i. vs. baseline: ↑ FMD%(p = 0.008) ↓ PWV (p = 0.03) | hsCRP? |
| Balta, 2014, [247] | Turkey | Case-control Study | 32 | 35 | All PASI included | Assessed, but values not presented in paper | Assessed, but values not presented in paper | No data provided | PWV (m/s), augmentation index (AIx), BA (occlusion) | Distance between jugular notch and symphysis pubis | ↑ PWV (m/s), (p = 0.01), no correlation with disease duration or PASI | ↑ hsCRP (p = 0.01) |
| Dregan, 2018, [248] | United Kingdom | Cross-sectional Study | 2091 | 165 149 | Presence of psoriasis diagnosis/all included | Not assessed | Not assessed | Corticosteroids (n = 166, 8% DMARDs (n = 168, 8%) | photoplethysmography (arterial stiffness index, SI, m/s), no occlusion | Index finger of the dominant hand | ↑ SI (p = 0.016) | / |
| Choi, 2016, [249] | South Korea | Case-control Study | 103 | 103 | All PASI included | 8.7 + 5.5 | 3 (0.5–10) | No data provided | CAVI, right brachial, right ankle (occlusion) PWV(m/s), cAIx | PWV—between aortic valve and ankle cAIx—BA, RA | ↑ CAVI (p = 0.03) cAIx, correlating with disease duration (r = 0.319, p = 0.001), not with PASI | ↑ CRP (p = 0.025) |
| Hansen, 2018, [250] | Denmark | Cross-sectional Study | 254 | 4431 | Self-reported psoriasis/all included | Not assessed | Not assessed | Not assessed | photoplethysmography (arterial stiffness index, SI), no occlusion | Index finger of the non-dominant hand | ↑ SI (p = 0.04) | ↑ hsCRP |
| Jensen, 2014, [251] | Denmark | Randomized Controlled Trial | 30 Pso, low energy diet | 30 Pso, normal diet | All PASI included | 4.8 (3.8–8.2) intervention group; 5.5 (3.6–6.8) controls | Not assessed | Not assessed | PAT (reactive hyperemia index, RHI)/Brachial artery (occlusion on the upper arm) | Both index fingers | ↔ RHI | ↔ hsCRP ↔ VCAM ↔ ICAM |
| Nakao, 2018, [222] | Japan | Cohort Study | 15 (7 + PsA) | Self-controlled | Not specified | 5.7 (3.2–12.8) | Mean 18.7 | Intervention with anti TNF-α: infliximab | RH-PAT (RHI), arm opposite to the dominant arm (occlusion) | Fingers of each hand | 6 weeks: ↔ RHI in responders ↓ trend RHI in non-responders (p = 0.09) | ↓ CRP, ↓ ESR (p = 0.016) |
| Sunbul, 2015, [252] | Turkey | Case-control Study | 50 | 50 | Not specified | 13.7 ± 8.9 | 13.5 ± 10.7 | No data (data obtained about previous medication) | PWV(m/s), AIx | ↑ PWV (p = 0.001) ↑ AIx (p = 0.001), no correlation with PASI observed | ↑ NLR (p = 0.002) | |
| Altekin, 2012, [253] | Turkey | Case-control Study | 57 | 60 | Not specified/all PASI included | 7.8 ± 7.4 | 11.3 ± 8.5 | None (no systemic immunosuppressive therapy at least 6 months prior) | cfPWV(m/s), cIMT (mm), no occlusion site | cfPWV—CA, FA cIMT—1 cm segment of both CCA, 2–3 cm distal to the bulb | ↑ cfPWV (p < 0.001), positively correlating with PASI (r = 0.417, p = 0.001) ↑ max cIMT (p < 0.001) ↑ mean cIMT (p < 0.001) | / |
| Enany, 2011, [254] | Egypt | Case-control Study | 50 | 10 | All PASI included | 20.99 ± 16.67 | 6.50 ± 2.95 | None (at least 6 months prior) | cIMT(mm), no occlusion site | CCA, 1 cm proximal to the carotid bulb; bulb; ICA, 1 cm distal to the carotid bifurcation | ↑ cIMT (p < 0.05), correlating with age, disease duration, BMI, PASI score, systolic blood pressure, diastolic blood pressure, leptin levels, LDL levels and triglyceride levels | ↑ leptin (p < 0.05) |
| Divito, 2018, [255] | Italy | Case-control Study | ↓ CV R 34; ↑ CVR 23 | ↓ CVR 39; ↑ CVR15 | Severe/PASI not specified | No data provided | No data provided | No data provided | PWV (m/s) | Not specified | Low CV risk, Pso vs. controls: ↔ PWV High CV risk, Pso vs. controls: ↑ PWV (p = 0.037) | / |
| Asha, 2014, [256] | India | Case-control Study | 80 | 80 | Not specified/all PASI included | 15.60 ± 10.79 | 3.42 ± 2.56 | No data provided | cIMT(mm), no occlusion site | Both CA | ↑ mean cIMT (p < 0.001), significant cumulative association with leptin and apoB/apoA-I | ↑ leptin (p < 0.001) |
| Usta, 2011, [203] | Turkey | Case-control Study | 29 | 25 | Not specified/all PASI included | 4.6 ± 3.8 | 13 ± 10 | None (at least 1 month prior) | FMD, NMD (vessel dilatation as the % of baseline value, %), upper arm proximal to the imaged artery segment (occlusion); | BA, 2–4 cm above the antecubital fossa | ↔ FMD ↔ NMD | ↑ CRP (p = 0.011) ↑ fibrinogen (p = 0.011) ↔ ADMA |
| Arias-Santiago, 2012, [257] | Spain | Case-control Study | 72 (25 +PsA) | 61 | Severe/PASI > 10 | mean 19.25 | mean 17.64 | None (at least 2 months prior) | cIMT(mm), no occlusion site | Distal portion of the both CCA, 1 to 2 cm proximal to the carotid bulb | ↑ right cIMT (p = 0.013) ↑ left cIMT (p = 0.042) ↑ presence of carotid atheroma plaques (p < 0.001) | ↑ fibrinogen, ↑ CRP, ↑ ESR, ↑ D-dimer, ↑ homocysteine |
| Alba, 2018, [210] | USA | Case-control Study | 9 (1+ PsA) | 9 | ≥5% BSA | 16 ± 2 BSA | No data provided | None | LDF (NO-dependent vasodilatation—ΔCVClocal heating and CVCpost-l-NAME SNP-induced vasodilatation (flux/mmHg); vascular adrenergic responsiveness, logEC50) | Cutaneous microcirculation, forearm skin | ↓ NO-dependent vasodilation (p < 0.01), correlating with BSA (r = 0.54, p = 0.04) ↔ SNP-induced vasodilatation After ascorbate infusion: ↔ NO-dependent vasodilation ↔ vascular adrenergic responsiveness ↔ max NE-induced vasoconstriction | / |
7. Increased CVD Risk in PV Patients Due to Impaired Endothelial Function
8. Rationale for Reducing Dietary Salt Intake and Antioxidant Supplementation in Patients Suffering from Psoriasis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yamazaki, F. Psoriasis: Comorbidities. J. Dermatol. 2021, 48, 732–740. [Google Scholar] [CrossRef] [PubMed]
- Sahi, F.M.; Masood, A.; Danawar, N.A.; Mekaiel, A.; Malik, B.H. Association Between Psoriasis and Depression: A Traditional Review. Cureus 2022, 12, e9708. [Google Scholar] [CrossRef] [PubMed]
- Tohid, H.; Aleem, D.; Jackson, C. Major Depression and Psoriasis: A Psychodermatological Phenomenon. SPP Skin Pharmacol. Physiol. 2016, 29, 220–230. [Google Scholar] [CrossRef]
- Damiani, G.; Bragazzi, N.L.; Karimkhani Aksut, C.; Wu, D.; Alicandro, G.; McGonagle, D.; Guo, C.; Dellavalle, R.; Grada, A.; Wong, P.; et al. The Global, Regional, and National Burden of Psoriasis: Results and Insights from the Global Burden of Disease 2019 Study. Front. Med. 2021, 8, 743180. [Google Scholar] [CrossRef] [PubMed]
- Salomon, J.A.; Wang, H.; Freeman, M.K.; Vos, T.; Flaxman, A.D.; Lopez, A.D.; Murray, C.J. Healthy life expectancy for 187 countries, 1990–2010, a systematic analysis for the Global Burden Disease Study 2010. Lancet 2012, 380, 2144–2162. [Google Scholar] [CrossRef]
- Merola, J.F.; Qureshi, A.; Husni, M.E. Underdiagnosed and undertreated psoriasis: Nuances of treating psoriasis affecting the scalp, face, intertriginous areas, genitals, hands, feet, and nails. Dermatol. Ther. 2018, 31, e12589. [Google Scholar] [CrossRef]
- Ten Bergen, L.L.; Petrovic, A.; Aarebrot, A.K.; Appel, S. Current knowledge on autoantigens and autoantibodies in psoriasis. Scand. J. Immunol. 2020, 92, e12945. [Google Scholar] [CrossRef]
- Dave, R.; Alkeswani, A. An Overview of Biologics for Psoriasis. JDD 2021, 20, 1246–1247. [Google Scholar] [CrossRef]
- Dand, N.; Mahil, S.; Capon, F.; Smith, C.; Simpson, M.; Barker, J. Psoriasis and Genetics. Acta. Derm. Venerol. 2020, 100, 55–65. [Google Scholar] [CrossRef]
- Rendon, A.; Schäkel, K. Psoriasis Pathogenesis and Treatment. Int. J. Mol. Sci. 2019, 20, 1475. [Google Scholar] [CrossRef]
- Armstrong, A.W.; Read, C. Pathophysiology, Clinical Presentation, and Treatment of Psoriasis: A Review. JAMA 2020, 323, 1945. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, C.E.M.; Armstrong, A.W.; Gudjonsson, J.E.; Barker, J.N.W.N. Psoriasis. Lancet 2021, 397, 1301–1315. [Google Scholar] [CrossRef]
- Su, W.; Zhao, Y.; Wei, Y.; Zhang, X.; Ji, J.; Yang, S. Exploring the Pathogenesis of Psoriasis Complicated with Atherosclerosis via Microarray Data Analysis. Front. Immunol. 2021, 12, 667690. Available online: https://www.frontiersin.org/article/10.3389/fimmu.2021.667690 (accessed on 4 April 2022). [CrossRef] [PubMed]
- Patik, J.C.; Lennon, S.L.; Farquhar, W.B.; Edwards, D.G. Mechanisms of dietary sodium-induced impairments in endothelial function and potential countermeasures. Nutrients 2021, 13, 270. [Google Scholar] [CrossRef]
- Boegehold, M.A. The Effect of High Salt Intake on Endothelial Function: Reduced Vascular Nitric Oxide in the Absence of Hypertension. JVR J. Vasc. Res. 2013, 50, 458–467. [Google Scholar] [CrossRef]
- Maifeld, A.; Wild, J.; Karlsen, T.V.; Rakova, N.; Wistorf, E.; Linz, P.; Jung, R.; Birukov, A.; Gimenez-Rivera, V.A.; Wilck, N.; et al. Skin Sodium Accumulates in Psoriasis and Reflects Disease Severity. J. Investig. Dermatol. 2022, 142, 166–178.e8. [Google Scholar] [CrossRef]
- Kleinewietfeld, M.; Manzel, A.; Titze, J.; Kvakan, H.; Yosef, N.; Linker, R.A.; Muller, D.N.; Hafler, D.A. Sodium chloride drives autoimmune disease by the induction of pathogenic TH 17 cells. Nature 2013, 496, 518–522. [Google Scholar] [CrossRef]
- Wu, C.; Yosef, N.; Thalhamer, T.; Zhu, C.; Xiao, S.; Kishi, Y.; Regev, A.; Kuchroo, V.K. Induction of pathogenic TH17 cells by inducible salt-sensing kinase SGK1. Nature 2013, 496, 513–517. [Google Scholar] [CrossRef]
- Balan, Y.; Packirisamy, R.M.; Mohanraj, P.S. High dietary salt intake activates inflammatory cascades via Th17 immune cells: Impact on health and diseases. Arch. Med. Sci. 2022, 18, 459. [Google Scholar] [CrossRef]
- Liang, Y.; Sarkar, M.K.; Tsoi, L.C.; Gudjonsson, J.E. Psoriasis: A mixed autoimmune and autoinflammatory disease. Curr. Opin. Immunol. 2017, 49, 1–8. [Google Scholar] [CrossRef]
- Murthy, A.S.; Leslie, K. Autoinflammatory Skin Disease: A Review of Concepts and Applications to General Dermatology. Dermatology 2016, 232, 534–540. [Google Scholar] [CrossRef] [PubMed]
- Fanoni, D.; Venegoni, L.; Vergani, B.; Tavecchio, S.; Cattaneo, A.; Leone, B.E.; Berti, E.; Marzano, A.V. Evidence for a role of autoinflammation in early-phase psoriasis. Clin. Exp. Immunol. 2019, 198, 283–291. [Google Scholar] [CrossRef]
- Li, B.; Huang, L.; Lv, P.; Li, X.; Liu, G.; Chen, Y.; Wang, Z.; Qian, X.; Shen, Y.; Li, Y.; et al. The role of Th17 cells in psoriasis. Immunol. Res. 2020, 68, 296–309. [Google Scholar] [CrossRef] [PubMed]
- Furue, K.; Ito, T.; Tsuji, G.; Kadono, T.; Nakahara, T.; Furue, M. Autoimmunity and autoimmune co-morbidities in psoriasis. Immunology 2018, 154, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.-Y.; Shao, S.; Wang, G. Antimicrobial peptides: Bridging innate and adaptive immunity in the pathogenesis of psoriasis. Chin. Med. J. 2020, 133, 2966–2975. [Google Scholar] [CrossRef]
- Takahashi, T.; Yamasaki, K. Psoriasis and Antimicrobial Peptides. Int. J. Mol. Sci. 2020, 21, 6791. [Google Scholar] [CrossRef]
- Polese, B.; Zhang, H.; Thurairajah, B.; King, I.L. Innate Lymphocytes in Psoriasis. Front. Immunol. 2020, 11, 242. [Google Scholar] [CrossRef]
- Sato, Y.; Ogawa, E.; Okuyama, R. Role of Innate Immune Cells in Psoriasis. Int. J. Mol. Sci. 2020, 21, 6604. [Google Scholar] [CrossRef]
- Gambichler, T.; Skrygan, M.; Tomi, N.S.; Othlinghaus, N.; Brockmeyer, N.H.; Altmeyer, P.; Kreuter, A. Differential mRNA expression of antimicrobial peptides and proteins in atopic dermatitis as compared to Psoriasis vulgaris and healthy skin. Int. Arch. Allergy Immunol. 2008, 147, 17–24. [Google Scholar] [CrossRef]
- Lande, R.; Chamilos, G.; Ganguly, D.; Demaria, O.; Frasca, L.; Durr, S.; Conrad, C.; Schröder, J.; Gilliet, M. Cationic antimicrobial peptides in psoriatic skin cooperate to break innate tolerance to self-DNA. Eur. J. Immunol. 2015, 45, 203–213. [Google Scholar] [CrossRef]
- Nestle, F.O.; Conrad, C.; Tun-Kyi, A.; Homey, B.; Gombert, M.; Boyman, O.; Burg, G.; Liu, Y.-J.; Gilliet, M. Plasmacytoid predendritic cells initiate psoriasis through interferon-alpha production. J. Exp. Med. 2005, 202, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Shelley, J.R.; Davidson, D.J.; Dorin, J.R. The Dichotomous Responses Driven by β-Defensins. Front. Immunol. 2020, 11, 1176. [Google Scholar] [CrossRef] [PubMed]
- Morizane, S.; Yamasaki, K.; Mühleisen, B.; Kotol, P.F.; Murakami, M.; Aoyama, Y.; Iwatsuki, K.; Hata, T.; Gallo, R.L. Cathelicidin antimicrobial peptide LL-37 in psoriasis enables keratinocyte reactivity against TLR9 ligands. J. Investig. Dermatol. 2012, 132, 135–143. [Google Scholar] [CrossRef]
- Akiyama, T.; Niyonsaba, F.; Kiatsurayanon, C.; Nguyen, T.T.; Ushio, H.; Fujimura, T.; Ueno, T.; Okumura, K.; Ogawa, H.; Ikeda, S. The human cathelicidin LL-37 host defense peptide upregulates tight junction-related proteins and increases human epidermal keratinocyte barrier function. J. Innate. Immun. 2014, 6, 739–753. [Google Scholar] [CrossRef] [PubMed]
- Mori, N.; Yoshikawa, K.; Ohno, M. Psoriasis occuring in young monozygotic twins. J. Dermatol. 1980, 7, 71–73. [Google Scholar] [CrossRef] [PubMed]
- Nair, R.P.; Stuart, P.E.; Nistor, I.; Hiremagalore, R.; Chia, N.V.C.; Jenisch, S.; Weichenthal, M.; Abecasis, G.R.; Lim, H.W.; Christophers, E.; et al. Sequence and Haplotype Analysis Supports HLA-C as the Psoriasis Susceptibility 1 Gene. Am. J. Hum. Genet. 2006, 78, 827–851. [Google Scholar] [CrossRef]
- Lande, R.; Botti, E.; Jandus, C.; Dojcinovic, D.; Fanelli, G.; Conrad, C.; Chamilos, G.; Feldmeyer, L.; Marinari, B.; Chon, S.; et al. The antimicrobial peptide LL37 is a T-cell autoantigen in psoriasis. Nat. Commun. 2014, 5, 5621. [Google Scholar] [CrossRef]
- Menssen, A.; Trommler, P.; Vollmer, S.; Schendel, D.; Albert, E.; Gürtler, L.; Riethmüller, G.; Prinz, J.C. Evidence for an antigen-specific cellular immune response in skin lesions of patients with Psoriasis vulgaris. J. Immunol. 1995, 155, 4078–4083. [Google Scholar]
- Bour, H.; Puisieux, I.; Even, J.; Kourilsky, P.; Favrot, M.; Musette, P.; Nicolas, J.-F. T-cell repertoire analysis in chronic plaque psoriasis suggests an antigen-specific immune response. Hum. Immunol. 1999, 60, 665–676. [Google Scholar] [CrossRef]
- Prinz, J.C.; Vollmer, S.; Boehncke, W.H.; Menssen, A.; Laisney, I.; Trommler, P. Selection of conserved TCR VDJ rearrangements in chronic psoriatic plaques indicates a common antigen in Psoriasis vulgaris. Eur. J. Immunol. 1999, 29, 3360–3368. [Google Scholar] [CrossRef]
- Hawkes, J.E.; Yan, B.Y.; Chan, T.C.; Krueger, J.G. Discovery of the IL-23/IL-17 Signaling Pathway and the Treatment of Psoriasis. J. Immunol. 2018, 201, 1605–1613. [Google Scholar] [CrossRef] [PubMed]
- Mosca, M.; Hong, J.; Hadeler, E.; Hakimi, M.; Liao, W.; Bhutani, T. The Role of IL-17 Cytokines in Psoriasis. Immunotargets. Ther. 2021, 10, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Li, S.; Ying, S.; Tang, S.; Ding, Y.; Li, Y.; Qiao, J.; Fang, H. The IL-23/IL-17 Pathway in Inflammatory Skin Diseases: From Bench to Bedside. Front. Immunol. 2020, 11, 594735. Available online: https://www.frontiersin.org/article/10.3389/fimmu.2020.594735 (accessed on 7 April 2022). [CrossRef]
- Widlansky, M.E.; Gokce, N.; Keaney, J.F.; Vita, J.A. The Clinical Implications of Endothelial Dysfunction. J. Am. Coll. Cardiol. 2003, 42, 1149–1160. [Google Scholar] [CrossRef]
- Arida, A.; Protogerou, A.D.; Kitas, G.D.; Sfikakis, P.P. Systemic inflammatory response and atherosclerosis: The paradigm of chronic inflammatory rheumatic diseases. Int. J. Mol. Sci. 2018, 19, 1890. [Google Scholar] [CrossRef]
- Liao, J.K. Linking endothelial dysfunction with endothelial cell activation. J. Clin. Investig. 2013, 123, 540–541. [Google Scholar] [CrossRef]
- Bacon, P.A. Endothelial cell dysfunction in systemic vasculitis: New developments and therapeutic prospects. Curr. Opin. Rheumatol. 2005, 17, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Tadzic, R.; Mihalj, M.; Vcev, A.; Ennen, J.; Tadzic, A.; Drenjancevic, I. The effects of arterial blood pressure reduction on endocan and soluble endothelial cell adhesion molecules (CAMs) and CAMs ligands expression in hypertensive patients on Ca-channel blocker therapy. Kidney Blood Press. Res. 2013, 37, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Hennekens, C.H.; Roitman-Johnson, B.; Stampfer, M.J.; Allen, J. Plasma concentration of soluble intercellular adhesion molecule 1 and risks of future myocardial infarction in apparently healthy men. Lancet 1998, 351, 88–92. [Google Scholar] [CrossRef]
- Bragulat, E.; De La Sierra, A.; Antonio, T. Endothelial Dysfunction in Salt-Sensitive Essential Hypertension. Hypertension 2001, 37, 444–448. [Google Scholar] [CrossRef]
- Liu, F.Q.; Mu, J.J.; Liu, Z.Q.; Shi, D.C.; Huang, Q.; Yuan, Z.Y.; Lian, Q.F.; Zheng, S.H. Endothelial dysfunction in normotensive salt-sensitive subjects. J. Hum. Hypertens. 2012, 26, 247–252. [Google Scholar] [CrossRef] [PubMed]
- Greaney, J.L.; Dupont, J.J.; Lennon-Edwards, S.L.; Sanders, P.W.; Edwards, D.G.; Farquhar, W.B. Dietary sodium loading impairs microvascular function independent of blood pressure in humans: Role of oxidative stress. J. Physiol. 2012, 590, 5519–5528. [Google Scholar] [CrossRef] [PubMed]
- Barić, L.; Drenjančević, I.; Mihalj, M.; Matić, A.; Stupin, M.; Kolar, L.; Mihaljević, Z.; Mrakovčić-šutić, I.; Šerić, V.; Stupin, A. Enhanced antioxidative defense by vitamins C and E consumption prevents 7-day high-salt diet-induced microvascular endothelial function impairment in young healthy individuals. J. Clin. Med. 2020, 9, 843. [Google Scholar] [CrossRef]
- Halliwell, B. Free radicals, antioxidants, and human disease: Curiosity, cause, or consequence? Lancet 1994, 344, 721–724. [Google Scholar] [CrossRef]
- Betteridge, D.J. What is oxidative stress? Metabolism 2000, 49, 3–8. [Google Scholar] [CrossRef]
- Schieber, M.; Chandel, N.S. ROS Function in Redox Signaling and Oxidative Stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: Oxidative eustress. Redox Biol. 2017, 11, 613–619. [Google Scholar] [CrossRef]
- Li, H.; Horke, S.; Förstermann, U. Vascular oxidative stress, nitric oxide and atherosclerosis. Atherosclerosis 2014, 237, 208–219. [Google Scholar] [CrossRef]
- Förstermann, U.; Xia, N.; Li, H. Roles of Vascular Oxidative Stress and Nitric Oxide in the Pathogenesis of Atherosclerosis. Circ. Res. 2017, 120, 713–735. [Google Scholar] [CrossRef]
- Lenda, D.M.; Sauls, B.A.; Boegehold, M.A. Reactive oxygen species may contribute to reduced endothelium-dependent dilation in rats fed high salt. Am. J. Physiol. Heart Circ. Physiol. 2000, 279, H7–H14. [Google Scholar] [CrossRef]
- Boegehold, M.A. Effect of dietary salt on arteriolar nitric oxide in striated muscle of normotensive rats. Am. J. Physiol. 1993, 264, H1810–H1816. [Google Scholar] [CrossRef] [PubMed]
- Nurkiewicz, T.R.; Boegehold, M.A. High salt intake reduces endothelium-dependent dilation of mouse arterioles via superoxide anion generated from nitric oxide synthase. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 292, R1550–R1556. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Huang, T.; Lombard, J.H. Effect of High-Salt Diet on Vascular Relaxation and Oxidative Stress in Mesenteric Resistance Arteries. J. Vasc. Res. 2007, 44, 382–390. [Google Scholar] [CrossRef] [PubMed]
- Alp, N.J.; Channon, K.M. Regulation of Endothelial Nitric Oxide Synthase by Tetrahydrobiopterin in Vascular Disease. ATVB 2004, 24, 413–420. [Google Scholar] [CrossRef]
- Sena, C.M.; Leandro, A.; Azul, L.; Seiça, R.; Perry, G. Vascular oxidative stress: Impact and therapeutic approaches. Front. Physiol. 2018, 9, 1668. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Drenjancevic-Peric, I.; McEwen, S.; Friesema, J.; Schulta, D.; Yu, M.; Roman, R.J.; Lombard, J.H.; Lombard, J.H. Role of superoxide and angiotensin II suppression in salt-induced changes in endothelial Ca 2 signaling and NO production in rat aorta. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, 929–938. [Google Scholar] [CrossRef]
- Wild, J.; Soehnlein, O.; Dietel, B.; Urschel, K.; Garlichs, C.D.; Cicha, I. Rubbing salt into wounded endothelium: Sodium potentiates proatherogenic effects of TNF-α under non-uniform shear stress. Thromb. Haemost. 2014, 112, 183–195. [Google Scholar] [CrossRef]
- Cosic, A.; Jukic, I.; Stupin, A.; Mihalj, M.; Mihaljevic, Z.; Novak, S.; Vukovic, R.; Drenjancevic, I. Attenuated flow-induced dilatation of middle cerebral arteries is related to increased vascular oxidative stress in rats on a short-term high salt diet. J. Physiol. 2016, 594, 4917–4931. [Google Scholar] [CrossRef]
- Durand, M.J.; Lombard, J.H. Low-dose angiotensin II infusion restores vascular function in cerebral arteries of high salt-fed rats by increasing copper/zinc superoxide dimutase expression. Am. J. Hypertens. 2013, 26, 739–747. [Google Scholar] [CrossRef]
- Lenda, D.M.; Boegehold, M.A. Effect of a High Salt Diet on Microvascular Antioxidant Enzymes. J. Vasc. Res. 2002, 39, 41–50. [Google Scholar] [CrossRef]
- Boveris, A.; Cadenas, E.; Stoppani, A.O.M. Role of ubiquinone in the mitochondrial generation of hydrogen peroxide. Biochem. J. 1976, 156, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Harper, M.-E.; Bevilacqua, L.; Hagopian, K.; Weindruch, R.; Ramsey, J.J. Ageing, oxidative stress, and mitochondrial uncoupling. Acta. Physiol. Scand. 2004, 182, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Ferraris, J.D.; Burg, M.B. Mitochondrial reactive oxygen species contribute to high NaCl-induced activation of the transcription factor TonEBP/OREBP. Am. J. Physiol. Renal. Physiol. 2006, 290, 1169–1176. [Google Scholar] [CrossRef]
- Zhou, X.; Ferraris, J.D.; Cai, Q.; Agarwal, A.; Burg, M.B. Increased reactive oxygen species contribute to high NaCl-induced activation of the osmoregulatory transcription factor TonEBP/OREBP. Am. J. Physiol. Renal. Physiol. 2005, 289, F377–F385. [Google Scholar] [CrossRef]
- Mori, T.; Cowley, A.W. Renal Oxidative Stress in Medullary Thick Ascending Limbs Produced by Elevated NaCl and Glucose. Hypertension 2004, 43, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Bauer, J.; Ripperger, A.; Frantz, S.; Ergün, S.; Schwedhelm, E.; Benndorf, R.A. Pathophysiology of isoprostanes in the cardiovascular system: Implications of isoprostane-mediated thromboxane A2 receptor activation. Br. J. Pharmacol. 2014, 171, 3115–3131. [Google Scholar] [CrossRef]
- Negri, S.; Faris, P.; Moccia, F. Reactive oxygen species and endothelial ca2+ signaling: Brothers in arms or partners in crime? Int. J. Mol. Sci. 2021, 22, 9821. [Google Scholar] [CrossRef] [PubMed]
- Graier, W.F.; Hoebel, B.G.; Paltauf-Doburzynska, J.; Kostner, G.M. Effects of superoxide anions on endothelial Ca2+ signaling pathways. Arterioscler. Thromb. Vasc. Biol. 1998, 18, 1470–1479. [Google Scholar] [CrossRef]
- Morrow, J.D.; Hill, K.E.; Burk, R.F.; Nammour, T.M.; Badr, K.F.; Roberts, L.J. A series of prostaglandin F2-like compounds are produced in vivo in humans by a non-cyclooxygenase, free radical-catalyzed mechanism. Proc. Natl. Acad. Sci. USA 1990, 87, 9383–9387. [Google Scholar] [CrossRef]
- Kromer, B.M.; Tippins, J.R. Coronary artery constriction by the isoprostane 8-epi prostaglandin F2 alpha. Br. J. Pharmacol. 1996, 119, 1276–1280. [Google Scholar] [CrossRef]
- Kang, K.H.; Morrow, J.D.; Roberts, L.J.; Newman, J.H.; Banerjee, M. Airway and vascular effects of 8-epi-prostaglandin F2 alpha in isolated perfused rat lung. J. Appl. Physiol. 1993, 74, 460–465. [Google Scholar] [CrossRef] [PubMed]
- Lahaie, I.; Hardy, P.; Hou, X.; Hasséssian, H.; Asselin, P.; Lachapelle, P.; Almazan, G.; Varma, D.R.; Morrow, J.D.; Roberts, L.J.; et al. A novel mechanism for vasoconstrictor action of 8-isoprostaglandin F2 alpha on retinal vessels. Am. J. Physiol. 1998, 274, R1406–R1416. [Google Scholar] [PubMed]
- Hou, X.; Gobeil, F.; Peri, K.; Speranza, G.; Marrache, A.M.; Lachapelle, P.; Roberts, J.; Varma, D.R.; Chemtob, S.; Ellis, E.F. Augmented vasoconstriction and thromboxane formation by 15-F(2t)-isoprostane (8-iso-prostaglandin F(2alpha)) in immature pig periventricular brain microvessels. Stroke 2000, 31, 516–524. [Google Scholar] [CrossRef] [PubMed]
- Ochi, H.; Masuda, J.; Gimbrone, M.A. Hyperosmotic stimuli inhibit VCAM-1 expression in cultured endothelial cells via effects on interferon regulatory factor-1 expression and activity. Eur. J. Immunol. 2002, 32, 1821–1831. [Google Scholar] [CrossRef]
- Monteleone, I.; Marafini, I.; Dinallo, V.; Di Fusco, D.; Troncone, E.; Zorzi, F.; Laudisi, F.; Monteleone, G. Sodium chloride-enriched Diet Enhanced Inflammatory Cytokine Production and Exacerbated Experimental Colitis in Mice. J. Crohn’s Colitis 2017, 11, 237–245. [Google Scholar] [CrossRef]
- Winiarska-Mieczan, A.; Mieczan, T.; Wójcik, G. Importance of redox equilibrium in the pathogenesis of psoriasis—impact of antioxidant-rich diet. Nutrients 2020, 12, 1841. [Google Scholar] [CrossRef]
- Zhou, Q.; Mrowietz, U.; Rostami-Yazdi, M. Oxidative stress in the pathogenesis of psoriasis. Free. Radic. Biol. Med. 2009, 47, 891–905. [Google Scholar] [CrossRef]
- Peluso, I.; Morabito, G.; Urban, L.; Ioannone, F.; Serafini, M. Oxidative Stress in Atherosclerosis Development: The Central Role of LDL and Oxidative Burst. Endocr. Metab. Immune Disord.-Drug Targets (Former. Curr. Drug Targets-Immune Endocr. Metab. Disord.) 2012, 12, 351–360. [Google Scholar] [CrossRef]
- Peluso, I.; Cavaliere, A.; Palmery, M. Plasma total antioxidant capacity and peroxidation biomarkers in psoriasis. J. Biomed. Sci. 2016, 23, 52. [Google Scholar] [CrossRef]
- Chimenti, M.S.; Sunzini, F.; Fiorucci, L.; Botti, E.; Fonti, G.L.; Conigliaro, P.; Triggianese, P.; Costa, L.; Caso, F.; Giunta, A.; et al. Potential Role of Cytochrome c and Tryptase in Psoriasis and Psoriatic Arthritis Pathogenesis: Focus on Resistance to Apoptosis and Oxidative Stress. Front. Immunol. 2018, 9, 2363. [Google Scholar] [CrossRef]
- Young, C.N.; Koepke, J.I.; Terlecky, L.J.; Borkin, M.S.; Boyd, S.L.; Terlecky, S.R. Reactive oxygen species in tumor necrosis factor-α-activated primary human keratinocytes: Implications for psoriasis and inflammatory skin disease. J. Investig. Dermatol. 2008, 128, 2606–2614. [Google Scholar] [CrossRef] [PubMed]
- Schulze-Osthoff, K.; Bakker, A.C.; Vanhaesebroeck, B.; Beyaert, R.; Jacob, W.A.; Fiers, W. Cytotoxic activity of tumor necrosis factor is mediated by early damage of mitochondrial functions. Evidence for the involvement of mitochondrial radical generation. J. Biol. Chem. 1992, 267, 5317–5323. [Google Scholar] [CrossRef]
- Goossens, V.; Grooten, J.; De Vos, K.; Fiers, W. Direct evidence for tumor necrosis factor-induced mitochondrial reactive oxygen intermediates and their involvement in cytotoxicity. Proc. Natl. Acad. Sci. USA 1995, 92, 8115–8119. [Google Scholar] [CrossRef] [PubMed]
- Paukkonen, K.; Naukkarinen, A.; Horsmanheimo, M. The development of manifest psoriatic lesions is linked with the appearance of ICAM-1 positivity on keratinocytes. Arch. Dermatol. Res. 1995, 287, 165–170. [Google Scholar] [CrossRef]
- Veale, D.; Rogers, S.; Fitzgerald, O. Immunolocalization of adhesion molecules in psoriatic arthritis, psoriatic and normal skin. Br. J. Dermatol. 1995, 132, 32–38. [Google Scholar] [CrossRef]
- Guérard, S.; Allaeys, I.; Martin, G.; Pouliot, R.; Poubelle, P.E. Psoriatic keratinocytes prime neutrophils for an overproduction of superoxide anions. Arch. Dermatol. Res. 2013, 305, 879–889. [Google Scholar] [CrossRef]
- Ayala, A.; Muñoz, M.F.; Argüelles, S. Lipid peroxidation: Production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxidative Med. Cell. Longev. 2014, 2014, 360438. [Google Scholar] [CrossRef]
- Halliwell, B.; Gutteridge, J.M. Oxygen toxicity, oxygen radicals, transition metals and disease. Biochem. J. 1984, 219, 1–14. [Google Scholar] [CrossRef]
- Esterbauer, H.; Lang, J.; Zadravec, S.; Slater, T.F. Detection of malonaldehyde by high-performance liquid chromatography. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 1984; pp. 319–328. [Google Scholar]
- Esterbauer, H.; Cheeseman, K.H. Determination of aldehydic lipid peroxidation products: Malonaldehyde and 4-hydroxynonenal. Methods Enzym. 1990, 186, 407–421. [Google Scholar]
- Giera, M.; Lingeman, H.; Niessen, W.M.A. Recent Advancements in the LC- and GC-Based Analysis of Malondialdehyde (MDA): A Brief Overview. Chromatographia 2012, 75, 433–440. [Google Scholar] [CrossRef]
- Szepietowski, J.C.; Pietrzak, A.; Michalak-Stoma, A.; Chodorowska, G. Lipid disturbances in psoriasis: An update. Mediat. Inflamm. 2010, 2010, 535612. [Google Scholar]
- Rashmi, R.; Rao, K.S.J.; Basavaraj, K.H. A comprehensive review of biomarkers in psoriasis. Clin. Exp. Dermatol. 2009, 34, 658–663. [Google Scholar] [CrossRef] [PubMed]
- Wiswedel, I. F(2)-isoprostanes: Sensitive biomarkers of oxidative stress in vitro and in vivo: A gas chromatography-mass spectrometric approach. Methods Mol. Biol. 2009, 580, 3–16. [Google Scholar]
- Sikar Aktürk, A.; Özdoğan, H.K.; Bayramgürler, D.; Çekmen, M.B.; Bilen, N.; Kıran, R. Nitric oxide and malondialdehyde levels in plasma and tissue of psoriasis patients. J. Eur. Acad. Dermatol. Venereol. 2012, 26, 833–837. [Google Scholar] [CrossRef] [PubMed]
- Toker, A.; Kadi, M.; Yildirim, A.K.; Aksoy, H.; Akçay, F. Serum lipid profile paraoxonase and arylesterase activities in psoriasis. Cell Biochem. Funct. 2009, 27, 176–180. [Google Scholar] [CrossRef]
- Attwa, E.; Swelam, E. Relationship between smoking-induced oxidative stress and the clinical severity of psoriasis. J. Eur. Acad. Dermatol. Venereol. 2011, 25, 782–787. [Google Scholar] [CrossRef] [PubMed]
- Gabr, S.A.; Al-Ghadir, A.H. Role of cellular oxidative stress and cytochrome c in the pathogenesis of psoriasis. Arch. Dermatol. Res. 2012, 304, 451–457. [Google Scholar] [CrossRef] [PubMed]
- Relhan, V.; Gupta, S.K.; Dayal, S.; Pandey, R.; Lal, H. Blood thiols and malondialdehyde levels in psoriasis. J. Dermatol. 2002, 29, 399–403. [Google Scholar] [CrossRef]
- Asefi, M.; Vaisi-Raygani, A.; Khodarahmi, R.; Nemati, H.; Rahimi, Z.; Vaisi-Raygani, H.; Tavilani, H.; Pourmotabbed, T. Methylentetrahydrofolatereductase (rs1801133) polymorphism and psoriasis: Contribution to oxidative stress, lipid peroxidation and correlation with vascular adhesion protein 1, preliminary report. J. Eur. Acad. Dermatol. Venereol. 2014, 28, 1192–1198. [Google Scholar] [CrossRef]
- Adekunbi, D.A.; Ogunsola, O.A.; Oyelowo, O.T.; Aluko, E.O.; Popoola, A.A.; Akinboboye, O.O. Consumption of high sucrose and/or high salt diet alters sperm function in male Sprague–Dawley rats. Egypt. J. Basic Appl. Sci. 2016, 3, 194–201. [Google Scholar] [CrossRef]
- Solak Tekin, N.; Tekin, I.O.; Barut, F.; Yilmaz Sipahi, E. Accumulation of oxidized low-density lipoprotein in psoriatic skin and changes of plasma lipid levels in psoriatic patients. Mediat. Inflamm. 2007, 2007, 078454. [Google Scholar] [CrossRef] [PubMed]
- Becatti, M.; Barygina, V.; Mannucci, A.; Emmi, G.; Prisco, D.; Lotti, T.; Fiorillo, C.; Taddei, N. Sirt1 Protects against Oxidative Stress-Induced Apoptosis in Fibroblasts from Psoriatic Patients: A New Insight into the Pathogenetic Mechanisms of Psoriasis. Int. J. Mol. Sci. 2018, 19, 1572. [Google Scholar] [CrossRef] [PubMed]
- Corrocher, R.; Ferrari, S.; De Gironcoli, M.; Bassi, A.; Olivieri, O.; Guarini, P.; Stanzial, A.; Barba, A.L.; Gregolini, L. Effect of fish oil supplementation on erythrocyte lipid pattern, malondialdehyde production and glutathione-peroxidase activity in psoriasis. Clin. Chim. Acta 1989, 179, 121–131. [Google Scholar] [CrossRef]
- Kökçam, I.; Naziroğlu, M. Antioxidants and lipid peroxidation status in the blood of patients with psoriasis. Clin. Chim. Acta. 1999, 289, 23–31. [Google Scholar] [CrossRef]
- Simon, A.R.; Rai, U.; Fanburg, B.L.; Cochran, B.H. Activation of the JAK-STAT pathway by reactive oxygen species. Am. J. Physiol. 1998, 275, C1640–C1652. [Google Scholar] [CrossRef]
- Dhar, A.; Young, M.R.; Colburn, N.H. The role of AP-1, NF-κB and ROS/NOS in skin carcinogenesis: The JB6 model is predictive. Mol. Cell. Biochem. 2002, 234, 185–193. [Google Scholar] [CrossRef]
- Shim, J.-S.; Kwon, Y.-Y.; Han, Y.-S.; Hwang, J.-K. Inhibitory Effect of Panduratin A on UV-induced Activation of Mitogen-Activated Protein Kinases (MAPKs) in Dermal Fibroblast Cells. Planta Med. 2008, 74, 1446–1450. [Google Scholar] [CrossRef]
- Xu, F.; Xu, J.; Xiong, X.; Deng, Y. Salidroside inhibits MAPK, NF-κB, and STAT3 pathways in psoriasis-associated oxidative stress via SIRT1 activation. Redox Rep. 2019, 24, 70–74. [Google Scholar] [CrossRef]
- Baek, J.O.; Byamba, D.; Wu, W.H.; Kim, T.G.; Lee, M.G. Assessment of an imiquimod-induced psoriatic mouse model in relation to oxidative stress. Arch. Dermatol. Res. 2012, 304, 699–706. [Google Scholar] [CrossRef]
- Wu, Z.; Uchi, H.; Morino-Koga, S.; Shi, W.; Furue, M. Resveratrol inhibition of human keratinocyte proliferation via SIRT1/ARNT/ERK dependent downregulation of aquaporin 3. J. Dermatol. Sci. 2014, 75, 16–23. [Google Scholar] [CrossRef]
- Blander, G.; Bhimavarapu, A.; Mammone, T.; Maes, D.; Elliston, K.; Reich, C.; Matsui, M.S.; Guarente, L.; Loureiro, J.J. SIRT1 Promotes Differentiation of Normal Human Keratinocytes. J. Investig. Dermatol. 2009, 129, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Elibol, B.; Kilic, U. High Levels of SIRT1 Expression as a Protective Mechanism Against Disease-Related Conditions. Front. Endocrinol. 2018, 9, 614. [Google Scholar] [CrossRef] [PubMed]
- Becatti, M.; Barygina, V.; Emmi, G.; Silvestri, E.; Taddei, N.; Lotti, T.; Fiorillo, C. SIRT1 activity is decreased in lesional psoriatic skin. Intern. Emerg. Med. 2016, 11, 891–893. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.Y.; Lin, S.L.; Chen, Y.M.; Wu, V.C.; Yang, W.S.; Wu, K.D. A low-salt diet increases the expression of renal sirtuin 1 through activation of the ghrelin receptor in rats. Sci. Rep. 2016, 6, 32787. [Google Scholar] [CrossRef]
- Cargnello, M.; Roux, P.P. Activation and Function of the MAPKs and Their Substrates, the MAPK-Activated Protein Kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83. [Google Scholar] [CrossRef]
- Kyriakis, J.M.; Avruch, J. Mammalian MAPK Signal Transduction Pathways Activated by Stress and Inflammation: A 10-Year Update. Physiol. Rev. 2012, 92, 689–737. [Google Scholar] [CrossRef]
- Huang, J.; Gan, Q.; Han, L.; Li, J.; Zhang, H.; Sun, Y.; Zhang, Z.; Tong, T. SIRT1 overexpression antagonizes cellular senescence with activated ERK/S6k1 signaling in human diploid fibroblasts. PLoS ONE 2008, 3, e1710. [Google Scholar] [CrossRef]
- Johansen, C.; Kragballe, K.; Westergaard, M.; Henningsen, J.; Kristiansen, K.; Iversen, L. The mitogen-activated protein kinases p38 and ERK1/2 are increased in lesional psoriatic skin. Br. J. Dermatol. 2005, 152, 37–42. [Google Scholar] [CrossRef]
- Takahashi, H.; Ibe, M.; Nakamura, S.; Ishida-Yamamoto, A.; Hashimoto, Y.; Iizuka, H. Extracellular regulated kinase and c-Jun N-terminal kinase are activated in psoriatic involved epidermis. J. Dermatol. Sci. 2002, 30, 94–99. [Google Scholar] [CrossRef]
- Conde de la Rosa, L.; Schoemaker, M.H.; Vrenken, T.E.; Buist-Homan, M.; Havinga, R.; Jansen, P.L.M.; Moshage, H. Superoxide anions and hydrogen peroxide induce hepatocyte death by different mechanisms: Involvement of JNK and ERK MAP kinases. J. Hepatol. 2006, 44, 918–929. [Google Scholar] [CrossRef]
- Murakami, T.; Takagi, H.; Suzuma, K.; Suzuma, I.; Ohashi, H.; Watanabe, D.; Ojima, T.; Suganami, E.; Kurimoto, M.; Kaneto, H.; et al. Angiopoietin-1 Attenuates H2O2-induced SEK1/JNK Phosphorylation through the Phosphatidylinositol 3-Kinase/Akt Pathway in Vascular Endothelial Cells. J. Biol. Chem. 2005, 280, 31841–31849. [Google Scholar] [CrossRef] [PubMed]
- Gazel, A.; Banno, T.; Walsh, R.; Blumenberg, M. Inhibition of JNK promotes differentiation of epidermal keratinocytes. J. Biol. Chem. 2006, 281, 20530–20541. [Google Scholar] [CrossRef]
- Yu, X.-J.; Li, C.-Y.; Dai, H.-Y.; Cai, D.-X.; Wang, K.-Y.; Xu, Y.-H.; Chen, L.-M.; Zhou, C.-L. Expression and localization of the activated mitogen-activated protein kinase in lesional psoriatic skin. Exp. Mol. Pathol. 2007, 83, 413–418. [Google Scholar] [CrossRef] [PubMed]
- Klotz, L.O.; Pellieux, C.; Briviba, K.; Pierlot, C.; Aubry, J.M.; Sies, H. Mitogen-activated protein kinase (p38-, JNK-, ERK-) activation pattern induced by extracellular and intracellular singlet oxygen and UVA. Eur. J. Biochem. 1999, 260, 917–922. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, S.; Demirs, J.T.; Kochevar, I.E. p38 mitogen-activated protein kinase mediates bid cleavage, mitochondrial dysfunction, and caspase-3 activation during apoptosis induced by singlet oxygen but not by hydrogen peroxide. J. Biol. Chem. 2000, 275, 25939–25948. [Google Scholar] [CrossRef]
- Guyton, K.Z.; Liu, Y.; Gorospe, M.; Xu, Q.; Holbrook, N.J. Activation of mitogen-activated protein kinase by H2O2. Role in cell survival following oxidant injury. J. Biol. Chem. 1996, 271, 4138–4142. [Google Scholar] [CrossRef] [PubMed]
- Lander, H.M.; Jacovina, A.T.; Davis, R.J.; Tauras, J.M. Differential activation of mitogen-activated protein kinases by nitric oxide-related species. J. Biol. Chem. 1996, 271, 19705–19709. [Google Scholar] [CrossRef]
- Schieke, S.M.; Briviba, K.; Klotz, L.O.; Sies, H. Activation pattern of mitogen-activated protein kinases elicited by peroxynitrite: Attenuation by selenite supplementation. FEBS Lett. 1999, 448, 301–303. [Google Scholar] [CrossRef]
- Soegaard-Madsen, L.; Johansen, C.; Iversen, L.; Kragballe, K. Adalimumab therapy rapidly inhibits p38 mitogen-activated protein kinase activity in lesional psoriatic skin preceding clinical improvement: Adalimumab inhibits p38 MAPK in psoriatic skin. Br. J. Dermatol. 2010, 162, 1216–1223. [Google Scholar] [CrossRef]
- Schottelius, A.J.G.; Moldawer, L.L.; Dinarello, C.A.; Asadullah, K.; Sterry, W.; Edwards, C.K. Biology of tumor necrosis factor-alpha- implications for psoriasis. Exp. Dermatol. 2004, 13, 193–222. [Google Scholar] [CrossRef]
- Fan, H.; Sun, B.; Gu, Q.; Lafond-Walker, A.; Cao, S.; Becker, L.C. Oxygen radicals trigger activation of NF-kappaB and AP-1 and upregulation of ICAM-1 in reperfused canine heart. Am. J. Physiol. Heart Circ. Physiol. 2002, 282, H1778–H1786. [Google Scholar] [CrossRef] [PubMed]
- Bell, S.; Degitz, K.; Quirling, M.; Jilg, N.; Page, S.; Brand, K. Involvement of NF-kappaB signalling in skin physiology and disease. Cell Signal 2003, 15, 1–7. [Google Scholar] [CrossRef]
- Morgan, M.J.; Liu, Z.G. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Lingappan, K. NF-κB in oxidative stress. Curr. Opin. Toxicol. 2018, 7, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Hu, R.; Saw, C.L.-L.; Yu, R.; Kong, A.-N.T. Regulation of NF-E2-related factor 2 signaling for cancer chemoprevention: Antioxidant coupled with antiinflammatory. Antioxid. Redox Signal. 2010, 13, 1679–1698. [Google Scholar] [CrossRef]
- Abdou, A.G.; Hanout, H.M. Evaluation of survivin and NF-kappaB in psoriasis, an immunohistochemical study. J. Cutan. Pathol. 2008, 35, 445–451. [Google Scholar] [CrossRef]
- Anrather, J.; Racchumi, G.; Iadecola, C. NF-kappaB regulates phagocytic NADPH oxidase by inducing the expression of gp91phox. J. Biol. Chem. 2006, 281, 5657–5667. [Google Scholar] [CrossRef]
- Xu, P.; Huecksteadt, T.P.; Hoidal, J.R. Molecular cloning and characterization of the human xanthine dehydrogenase gene (XDH). Genomics 1996, 34, 173–180. [Google Scholar] [CrossRef]
- Kolyada, A.Y.; Savikovsky, N.; Madias, N.E. Transcriptional regulation of the human iNOS gene in vascular-smooth-muscle cells and macrophages: Evidence for tissue specificity. Biochem. Biophys. Res. Commun. 1996, 220, 600–605. [Google Scholar] [CrossRef]
- Hughes, J.E.; Srinivasan, S.; Lynch, K.R.; Proia, R.L.; Ferdek, P.; Hedrick, C.C. Sphingosine-1-phosphate induces an antiinflammatory phenotype in macrophages. Circ. Res. 2008, 102, 950–958. [Google Scholar] [CrossRef]
- Guo, Z.; Shao, L.; Du, Q.; Park, K.S.; Geller, D.A. Identification of a classic cytokine-induced enhancer upstream in the human iNOS promoter. FASEB J. 2007, 21, 535–542. [Google Scholar] [CrossRef] [PubMed]
- Morris, K.R.; Lutz, R.D.; Choi, H.-S.; Kamitani, T.; Chmura, K.; Chan, E.D. Role of the NF-kappaB signaling pathway and kappaB cis-regulatory elements on the IRF-1 and iNOS promoter regions in mycobacterial lipoarabinomannan induction of nitric oxide. Infect. Immun. 2003, 71, 1442–1452. [Google Scholar] [CrossRef] [PubMed]
- Mazière, C.; Conte, M.A.; Mazière, J.C. Activation of JAK2 by the oxidative stress generated with oxidized low-density lipoprotein. Free Radic. Biol. Med. 2001, 31, 1334–1340. [Google Scholar] [CrossRef]
- Sirsjö, A.; Karlsson, M.; Gidlöf, A.; Rollman, O.; Törmä, H. Increased expression of inducible nitric oxide synthase in psoriatic skin and cytokine-stimulated cultured keratinocytes. Br. J. Dermatol. 1996, 134, 643–648. [Google Scholar] [CrossRef] [PubMed]
- Kadam, D.P.; Suryakar, A.N.; Ankush, R.D.; Kadam, C.Y.; Deshpande, K.H. Role of oxidative stress in various stages of psoriasis. Indian J. Clin. Biochem. 2010, 25, 388–392. [Google Scholar] [CrossRef]
- Kute, P.K.; Muddeshwar, M.G.; Sonare, R. Pro-Oxidant and Anti-Oxidant Status in Patients of Psoriasis with Relation to Smoking and Alcoholism. Jemds 2019, 8, 2677–2680. [Google Scholar]
- Wójcik, P.; Biernacki, M.; Wroński, A.; Łuczaj, W.; Waeg, G.; Žarković, N.; Skrzydlewska, E. Altered Lipid Metabolism in Blood Mononuclear Cells of Psoriatic Patients Indicates Differential Changes in Psoriasis Vulgaris and Psoriatic Arthritis. Int. J. Mol. Sci. 2019, 20, 4249. [Google Scholar] [CrossRef]
- Vanizor Kural, B.; Orem, A.; Cimşit, G.; Yandi, Y.E.; Calapoglu, M. Evaluation of the atherogenic tendency of lipids and lipoprotein content and their relationships with oxidant-antioxidant system in patients with psoriasis. Clin. Chim. Acta 2003, 328, 71–82. [Google Scholar] [CrossRef]
- Matoshvili, M.; Katsitadze, A.; Sanikidze, T.; Tophuria, D.; D’Epiro, S.; Richetta, A. Evaluation of blood redox-balance, nitric oxide content and CCR6 rs3093024 in the genetic susceptibility during psoriasis. Georgian Med. News 2015, 240, 37–43. [Google Scholar]
- Jantsch, J.; Schatz, V.; Friedrich, D.; Schröder, A.; Kopp, C.; Siegert, I.; Maronna, A.; Wendelborn, D.; Linz, P.; Binger, K.J.; et al. Cutaneous Na+ storage strengthens the antimicrobial barrier function of the skin and boosts macrophage-driven host defense. Cell Metab. 2015, 21, 493–501. [Google Scholar] [CrossRef]
- Szabó, G.; Magyar, Z. Electrolyte concentrations in subcutaneous tissue fluid and lymph. Lymphology 1982, 15, 174–177. [Google Scholar] [PubMed]
- Wiig, H.; Schröder, A.; Neuhofer, W.; Jantsch, J.; Kopp, C.; Karlsen, T.V.; Boschmann, M.; Goss, J.; Bry, M.; Rakova, N.; et al. Immune cells control skin lymphatic electrolyte homeostasis and blood pressure. J. Clin. Investig. 2013, 123, 2803–2815. [Google Scholar] [CrossRef] [PubMed]
- Suckling, R.J.; He, F.J.; Markandu, N.D.; MacGregor, G.A. Dietary salt influences postprandial plasma sodium concentration and systolic blood pressure. Kidney Int. 2012, 81, 407–411. [Google Scholar] [CrossRef]
- Selvarajah, V.; Connolly, K.; McEniery, C.; Wilkinson, I. Skin Sodium and Hypertension: A Paradigm Shift? Curr. Hypertens. Rep. 2018, 20, 94. [Google Scholar] [CrossRef]
- Nikpey, E.; Karlsen, T.V.; Rakova, N.; Titze, J.M.; Tenstad, O.; Wiig, H. High-Salt Diet Causes Osmotic Gradients and Hyperosmolality in Skin without Affecting Interstitial Fluid and Lymph. Hypertension 2017, 69, 660–668. [Google Scholar] [CrossRef] [PubMed]
- Mauro, T.M. Ode to Salt: Commentary on “Skin Sodium Accumulates in Psoriasis and Reflects Disease Severity”. J. Investig. Dermatol. 2022, 142, 16–17. [Google Scholar] [CrossRef] [PubMed]
- Raphael, I.; Nalawade, S.; Eagar, T.N.; Forsthuber, T.G. T cell subsets and their signature cytokines in autoimmune and inflammatory diseases. Cytokine 2015, 74, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Rainard, P.; Cunha, P.; Martins, R.P.; Gilbert, F.B.; Germon, P.; Foucras, G. Type 3 immunity: A perspective for the defense of the mammary gland against infections. Vet. Res. 2020, 51, 129. [Google Scholar] [CrossRef] [PubMed]
- Kamata, M.; Tada, Y. Efficacy and Safety of Biologics for Psoriasis and Psoriatic Arthritis and Their Impact on Comorbidities: A Literature Review. Int. J. Mol. Sci. 2020, 21, 1690. [Google Scholar] [CrossRef]
- Omenetti, S.; Pizarro, T.T. The Treg/Th17 Axis: A Dynamic Balance Regulated by the Gut Microbiome. Front. Immunol. 2015, 6, 639. [Google Scholar] [CrossRef]
- Skrobot, A.; Demkow, U.; Wachowska, M. Immunomodulatory Role of Vitamin D: A Review. Adv. Exp. Med. Biol. 2018, 1108, 13–23. [Google Scholar] [PubMed]
- Szymczak, I.; Pawliczak, R. The Active Metabolite of Vitamin D3 as a Potential Immunomodulator. Scand. J. Immunol. 2016, 83, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Sitkovsky, M.; Lukashev, D. Regulation of immune cells by local-tissue oxygen tension: HIF1 alpha and adenosine receptors. Nat. Rev. Immunol. 2005, 5, 712–721. [Google Scholar] [CrossRef] [PubMed]
- Mehling, R.; Schwenck, J.; Lemberg, C.; Trautwein, C.; Zizmare, L.; Kramer, D.; Müller, A.; Fehrenbacher, B.; Gonzalez-Menendez, I.; Quintanilla-Martinez, L.; et al. Immunomodulatory role of reactive oxygen species and nitrogen species during T cell-driven neutrophil-enriched acute and chronic cutaneous delayed-type hypersensitivity reactions. Theranostics 2021, 11, 470–490. [Google Scholar] [CrossRef]
- Castro, C.N.; Freitag, J.; Berod, L.; Lochner, M.; Sparwasser, T. Microbe-associated immunomodulatory metabolites: Influence on T cell fate and function. Mol. Immunol. 2015, 68, 575–584. [Google Scholar] [CrossRef]
- Zielinski, C.E. Regulation of T cell responses by ionic salt signals. Cells 2021, 10, 2365. [Google Scholar] [CrossRef]
- Toney, G.M.; Vallon, V.; Stockand, J.D. Intrinsic control of sodium excretion in the distal nephron by inhibitory purinergic regulation of the epithelial Na(+) channel. Curr. Opin. Nephrol. Hypertens. 2012, 21, 52–60. [Google Scholar] [CrossRef]
- Knepper, M.A.; Kwon, T.-H.; Nielsen, S. Molecular physiology of water balance. N. Engl. J. Med. 2015, 372, 1349–1358. [Google Scholar] [CrossRef]
- Müller, D.N.; Wilck, N.; Haase, S.; Kleinewietfeld, M.; Linker, R.A. Sodium in the microenvironment regulates immune responses and tissue homeostasis. Nat. Rev. Immunol. 2019, 19, 243–254. [Google Scholar] [CrossRef]
- Wang, P.; Deger, M.S.; Kang, H.; Ikizler, T.A.; Titze, J.; Gore, J.C. Sex differences in sodium deposition in human muscle and skin. Magn. Reson. Imaging 2017, 36, 93–97. [Google Scholar] [CrossRef]
- Titze, J.; Rakova, N.; Kopp, C.; Dahlmann, A.; Jantsch, J.; Luft, F.C. Balancing wobbles in the body sodium. Nephrol. Dial. Transplant 2016, 31, 1078–1081. [Google Scholar] [CrossRef] [PubMed]
- Matthias, J.; Maul, J.; Noster, R.; Meinl, H.; Chao, Y.-Y.; Gerstenberg, H.; Jeschke, F.; Gasparoni, G.; Welle, A.; Walter, J.; et al. Sodium chloride is an ionic checkpoint for human TH2 cells and shapes the atopic skin microenvironment. Sci. Transl. Med. 2019, 11, eaau0683. [Google Scholar] [CrossRef] [PubMed]
- Fischereder, M.; Michalke, B.; Schmöckel, E.; Habicht, A.; Kunisch, R.; Pavelic, I.; Szabados, B.; Schönermarck, U.; Nelson, P.J.; Stangl, M. Sodium storage in human tissues is mediated by glycosaminoglycan expression. Am. J. Physiol. Renal. Physiol. 2017, 313, F319–F325. [Google Scholar] [CrossRef] [PubMed]
- Machnik, A.; Neuhofer, W.; Jantsch, J.; Dahlmann, A.; Tammela, T.; Machura, K.; Park, J.-K.; Beck, F.-X.; Müller, D.N.; Derer, W.; et al. Macrophages regulate salt-dependent volume and blood pressure by a vascular endothelial growth factor-C-dependent buffering mechanism. Nat. Med. 2009, 15, 545–552. [Google Scholar] [CrossRef] [PubMed]
- Kopp, C.; Linz, P.; Wachsmuth, L.; Dahlmann, A.; Horbach, T.; Schöfl, C.; Renz, W.; Santoro, D.; Niendorf, T.; Müller, D.N.; et al. (23)Na magnetic resonance imaging of tissue sodium. Hypertension 2012, 59, 167–172. [Google Scholar] [CrossRef]
- Wei, Y.; Lu, C.; Chen, J.; Cui, G.; Wang, L.; Yu, T.; Yang, Y.; Wu, W.; Ding, Y.; Li, L.; et al. High salt diet stimulates gut Th17 response and exacerbates TNBS-induced colitis in mice. Oncotarget 2016, 8, 70–82. [Google Scholar] [CrossRef]
- Matthias, J.; Heink, S.; Picard, F.; Zeiträg, J.; Kolz, A.; Chao, Y.-Y.; Soll, D.; de Almeida, G.P.; Glasmacher, E.; Jacobsen, I.D.; et al. Salt generates antiinflammatory Th17 cells but amplifies pathogenicity in proinflammatory cytokine microenvironments. J. Clin. Investig. 2020, 130, 4587–4600. [Google Scholar] [CrossRef]
- Lou, Y.; Zhang, F.; Luo, Y.; Wang, L.; Huang, S.; Jin, F. Serum and Glucocorticoid Regulated Kinase 1 in Sodium Homeostasis. Int. J. Mol. Sci. 2016, 17, 1307. [Google Scholar] [CrossRef]
- Stebut, E.V.; Boehncke, W.; Ghoreschi, K.; Gori, T. IL-17A in Psoriasis and Beyond: Cardiovascular and Metabolic Implications. Front. Immunol. 2020, 10, 3096. [Google Scholar] [CrossRef]
- Mehta, N.N.; Azfar, R.S.; Shin, D.B.; Neimann, A.L.; Troxel, A.B.; Gelfand, J.M. Patients with severe psoriasis are at increased risk of cardiovascular mortality: Cohort study using the General Practice Research Database. Eur. Heart J. 2010, 31, 1000–1006. [Google Scholar] [CrossRef]
- Cybulsky, M.I.; Gimbrone, M.A. Endothelial expression of a mononuclear leukocyte adhesion molecule during atherogenesis. Science 1991, 251, 788–791. [Google Scholar] [CrossRef] [PubMed]
- Ewenstein, M.G. Vascular biology of von Willebrand factor. In Vascular Endothelium: Physiology, Pathology and Therapeutic Opportunities; Schattauer: Sttutgart, Germany, 1997; pp. 107–122. [Google Scholar]
- Verma, S.; Anderson, T.J. Fundamentals of endothelial function for the clinical cardiologist. Circulation 2002, 105, 546–549. [Google Scholar] [CrossRef] [PubMed]
- Bonetti, P.O.; Lerman, L.O.; Lerman, A. Endothelial dysfunction: A marker of atherosclerotic risk. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Gimbrone, M.A.; Topper, J.N.; Nagel, T.; Anderson, K.R.; Garcia-Cardeña, G. Endothelial dysfunction, hemodynamic forces, and atherogenesis. Ann. N. Y. Acad. Sci. 2000, 902, 230–239. [Google Scholar] [CrossRef]
- Lerman, A.; Burnett, J.C. Intact and altered endothelium in regulation of vasomotion. Circulation 1992, 86, III12-19. [Google Scholar] [PubMed]
- Aboyans, V.; Criqui, M.H.; Denenberg, J.O.; Knoke, J.D.; Ridker, P.M.; Fronek, A. Risk factors for progression of peripheral arterial disease in large and small vessels. Circulation 2006, 113, 2623–2629. [Google Scholar] [CrossRef]
- Libby, P. Inflammation in atherosclerosis. Nature 2002, 420, 868–874. [Google Scholar] [CrossRef]
- Ross, R.; Glomset, J.A. The pathogenesis of atherosclerosis (first of two parts). N. Engl. J. Med. 1976, 295, 369–377. [Google Scholar] [CrossRef]
- De Simone, C.; Di Giorgio, A.; Sisto, T.; Carbone, A.; Ghitti, F.; Tondi, P.; Santoliquido, A. Endothelial dysfunction in psoriasis patients: Cross-sectional case-control study. Eur. J. Dermatol. 2011, 21, 510–514. [Google Scholar] [CrossRef]
- Ulusoy, R.E.; Karabudak, O.; Yokusoglu, M.; Kilicaslan, F.; Kirilmaz, A.; Cebeci, B.S. Noninvasive assessment of impaired endothelial function in psoriasis. Rheumatol. Int. 2010, 30, 479–483. [Google Scholar] [CrossRef]
- Usta, M.; Yurdakul, S.; Aral, H.; Turan, E.; Oner, E.; Bercik, B.; Alibaz, F.; Salih, M.; Guvenen, G. Vascular endothelial function assessed by a noninvasive ultrasound method and serum asymmetric dimethylarginine concentrations in mild-to-moderate plaque-type psoriatic patients. Clin. Biochem. 2011, 44, 1080–1084. [Google Scholar] [CrossRef] [PubMed]
- Karadag, A.S.; Yavuz, B.; Ertugrul, D.T.; Akin, K.O.; Yalcin, A.A.; Deveci, O.S.; Ata, N.; Kucukazman, M.; Dal, K. Is psoriasis a pre-atherosclerotic disease? Increased insulin resistance and impaired endothelial function in patients with psoriasis: Psoriasis and atherosclerosis. Int. J. Dermatol. 2010, 49, 642–646. [Google Scholar] [CrossRef] [PubMed]
- Martyn-Simmons, C.L.; Ranawaka, R.R.; Chowienczyk, P.; Crook, M.A.; Marber, M.S.; Smith, C.H.; Barker, J.N.W.N. A prospective case-controlled cohort study of endothelial function in patients with moderate to severe psoriasis. Br. J. Dermatol. 2011, 164, 26–32. [Google Scholar] [CrossRef]
- Yiu, K.-H.; Yeung, C.-K.; Chan, H.-T.; Wong, R.M.Y.; Tam, S.; Lam, K.-F.; Yan, G.H.; Yue, W.S.; Chan, H.H.; Tse, H.-F. Increased arterial stiffness in patients with psoriasis is associated with active systemic inflammation: Arterial stiffness in psoriasis patients. Br. J. Dermatol. 2011, 614, 514–520. [Google Scholar] [CrossRef] [PubMed]
- Balci, D.D.; Balci, A.; Karazincir, S.; Ucar, E.; Iyigun, U.; Yalcin, F.; Seyfeli, E.; Inandi, T.; Egilmez, E. Increased carotid artery intima-media thickness and impaired endothelial function in psoriasis. J. Eur. Acad. Dermatol. Venereol. 2009, 23, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Jensen, P.; Zachariae, C.; Hansen, P.; Skov, L. Normal Endothelial Function in Patients with Mild-to-Moderate Psoriasis: A Case-control Study. Acta. Derm. Venerol. 2011, 91, 516–520. [Google Scholar] [CrossRef]
- Cracowski, J.; Roustit, M. Human Skin Microcirculation. In Comprehensive Physiology; Terjung, R., Ed.; Wiley: Hoboken, NJ, USA, 2020; pp. 1105–1154. [Google Scholar]
- Alba, B.K.; Greaney, J.L.; Ferguson, S.B.; Alexander, L.M. Endothelial function is impaired in the cutaneous microcirculation of adults with psoriasis through reductions in nitric oxide-dependent vasodilation. Am. J. Physiol.-Heart Circ. Physiol. 2018, 314, H343–H349. [Google Scholar] [CrossRef]
- Abdou, A.G.; Hammam, M.; Saad, E.; Hassan, R.A.A. The significance of endocan immunohistochemical expression in chronic plaque psoriasis. J. Cosmet. Dermatol. 2022, 21, 380–386. [Google Scholar] [CrossRef]
- Balta, I.; Balta, S.; Demirkol, S.; Mikhailidis, D.P.; Celik, T.; Akhan, M.; Kurt, O.; Kurt, Y.G.; Aydin, I.; Kilic, S. Elevated serum levels of endocan in patients with Psoriasis vulgaris: Correlations with cardiovascular risk and activity of disease. Br. J. Dermatol. 2013, 169, 1066–1070. [Google Scholar] [CrossRef]
- Chen, J.; Zhu, Z.; Li, Q.; Lin, Y.; Dang, E.; Meng, H.; Sha, N.; Bai, H.; Wang, G.; An, S.; et al. Neutrophils Enhance Cutaneous Vascular Dilation and Permeability to Aggravate Psoriasis by Releasing Matrix Metallopeptidase 9. J. Investig. Dermatol. 2021, 141, 787–799. [Google Scholar] [CrossRef]
- Garshick, M.S.; Barrett, T.J.; Wechter, T.; Azarchi, S.; Scher, J.U.; Neimann, A.; Katz, S.; Fuentes-Duculan, J.; Cannizzaro, M.V.; Jelic, S.; et al. Inflammasome Signaling and Impaired Vascular Health in Psoriasis. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 787–798. [Google Scholar] [CrossRef]
- Magenta, A.; D’Agostino, M.; Sileno, S.; Di Vito, L.; Uras, C.; Abeni, D.; Martino, F.; Barillà, F.; Madonna, S.; Albanesi, C.; et al. The Oxidative Stress-Induced miR-200c Is Upregulated in Psoriasis and Correlates with Disease Severity and Determinants of Cardiovascular Risk. Oxid. Med. Cell Longev. 2019, 2019, 8061901. [Google Scholar] [CrossRef] [PubMed]
- Angel, K.; Provan, S.A.; Gulseth, H.L.; Mowinckel, P.; Kvien, T.K.; Atar, D. Tumor Necrosis Factor-α Antagonists Improve Aortic Stiffness in Patients with Inflammatory Arthropathies: A Controlled Study. Hypertension 2010, 55, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Avgerinou, G.; Tousoulis, D.; Siasos, G.; Oikonomou, E.; Maniatis, K.; Papageorgiou, N.; Paraskevopoulos, T.; Miliou, A.; Koumaki, D.; Latsios, G.; et al. Anti-tumor necrosis factor alpha treatment with adalimumab improves significantly endothelial function and decreases inflammatory process in patients with chronic psoriasis. Int. J. Cardiol. 2011, 151, 382–383. [Google Scholar] [CrossRef] [PubMed]
- Molina-Leyva, A.; Garrido-Pareja, F.; Ruiz-Carrascosa, J.C.; Ruiz-Villaverde, R. La inhibición del TNF-α puede disminuir los biomarcadores de disfunción endotelial en pacientes con psoriasis de moderada-grave: Un estudio cuasiexperimental eco doppler a 52 semanas. Med. Clínica 2018, 150, 465–468. [Google Scholar] [CrossRef] [PubMed]
- Pina, T.; Corrales, A.; Lopez-Mejias, R.; Armesto, S.; Gonzalez-Lopez, M.A.; Gómez-Acebo, I.; Ubilla, B.; Remuzgo-Martínez, S.; Gonzalez-Vela, M.C.; Blanco, R.; et al. Anti-Tumor Necrosis Factor-Alpha Therapy Improves Endothelial Function and Arterial Stiffness in Patients Anti-tumor necrosis factor-alpha therapy improves endothelial function and arterial stiffness in patients with moderate to severe psoriasis: A 6-month prospective study. J. Dermatol. 2016, 43, 1267–1272. [Google Scholar]
- von Stebut, E.; Reich, K.; Thaçi, D.; Koenig, W.; Pinter, A.; Körber, A.; Rassaf, T.; Waisman, A.; Mani, V.; Yates, D.; et al. Impact of Secukinumab on Endothelial Dysfunction and Other Cardiovascular Disease Parameters in Psoriasis Patients over 52 Weeks. J. Investig. Dermatol. 2019, 139, 1054–1062. [Google Scholar] [CrossRef]
- Wegner, J.; Karbach, S.; Drosos, I.; Schnorbus, B.; Muxel, S.; Schmidt, F.; Wenzel, P.; Waisman, A.; Münzel, T.; Gori, T.; et al. TNF-α blockade may lead to improvement of vascular function in psoriasis patients. Exp. Dermatol. 2022, 31, 237–241. [Google Scholar] [CrossRef]
- Nakao, M.; Nakamura, K.; Fukasawa, T.; Shida, R.; Ito, A.; Ichimura, Y.; Takahashi, T.; Mitsui, A.; Yoshizaki, A.; Shibata, S.; et al. Assessment of endothelial function during the loading phase of infliximab in psoriasis: A potential predictor of its drug survival. Int. J. Dermatol. 2019, 58, 54–59. [Google Scholar] [CrossRef]
- Mallbris, L.; Pernow, J.; Stahle, M. Endothelial Function and Inflammatory Activity in Patients with Recent Onset of Severe Plaque Psoriasis. TODJ Open Dermatol. J. 2008, 2, 64–68. [Google Scholar] [CrossRef]
- Gisondi, P.; Fantin, F.; Del Giglio, M.; Valbusa, F.; Marino, F.; Zamboni, M.; Girolomoni, G. Chronic plaque psoriasis is associated with increased arterial stiffness. Dermatology 2009, 218, 110–113. [Google Scholar] [CrossRef] [PubMed]
- Erfan, G.; Guzel, S.; Alpsoy, S.; Rifaioglu, E.N.; Kaya, S.; Kucukyalcın, V.; Topcu, B.; Kulac, M. Serum YKL-40, a potential biomarker for psoriasis or endothelial dysfunction in psoriasis? Mol. Cell. Biochem. 2014, 400, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Haberka, M.; Bańska-Kisiel, K.; Bergler-Czop, B.; Biedroń, M.; Brzezińska-Wcisło, L.; Okopień, B.; Gąsior, Z. Mild to moderate psoriasis is associated with oxidative stress, subclinical atherosclerosis, and endothelial dysfunction. Pol. Arch. Intern. Med. 2018, 128, 434–439. [Google Scholar] [PubMed]
- Holzer, G.; Hoke, M.; Sabeti-Sandor, S.; Perkmann, T.; Rauscher, A.; Strassegger, B.; Radakovic, S.; Tanew, A. Disparate effects of adalimumab and fumaric acid esters on cardiovascular risk factors in psoriasis patients: Results from a prospective, randomized, observer-blinded head-to-head trial. J. Eur. Acad. Dermatol. Venereol. 2021, 35, 441–449. [Google Scholar] [CrossRef] [PubMed]
- Erturan, I.; Köroǧlu, B.K.; Adiloǧlu, A.; Ceyhan, A.M.; Akkaya, V.B.; Tamer, N.; Başak, P.Y.; Korkmaz, S.; Ersoy, I.H.; Kilinç, O. Evaluation of serum sCD40L and homocysteine levels with subclinical atherosclerosis indicators in patients with psoriasis: A pilot study. Int. J. Dermatol. 2014, 53, 503–509. [Google Scholar] [CrossRef]
- Białecka, A.; Białecki, M.; Serafin, Z.; Czajkowski, R. Atherosclerosis attacks in patients with Psoriasis vulgaris but without a relationship with the severity and course of the disease. Postepy Derm. Alergol. 2021, 38, 673–681. [Google Scholar] [CrossRef]
- Bańska-Kisiel, K.; Haberka, M.; Bergler-Czop, B.; Brzezińska-Wcisło, L.; Okopień, B.; Gąsior, Z. Carotid intima-media thickness in patients with mild or moderate psoriasis. Postepy Dermatol. Alergol. 2016, 33, 286–289. [Google Scholar] [CrossRef]
- Troitzsch, P.; Paulista Markus, M.R.; Dörr, M.; Felix, S.B.; Jünger, M.; Schminke, U.; Schmidt, C.-O.; Völzke, H.; Baumeister, S.E.; Arnold, A. Psoriasis is associated with increased intima-media thickness--the Study of Health in Pomerania (SHIP). Atherosclerosis 2012, 225, 486–490. [Google Scholar] [CrossRef]
- Oliveira AN de Simões, M.M.; Simões, R.; Malachias, M.V.B.; Rezende, B.A. Cardiovascular Risk in Psoriasis Patients: Clinical, Functional and Morphological Parameters. Arq. Bras. Cardiol. 2019, 113, 242–249. [Google Scholar]
- Fabi, M.; Chessa, M.A.; Panizza, D.; Dormi, A.; Gazzano, A.; Patrizi, A.; Bardazzi, F.; Rocca, A.; Filice, E.; Neri, I.; et al. Psoriasis and Cardiovascular Risk in Children: The Usefulness of Carotid Intima-Media Thickness. Pediatr. Cardiol. 2022, 1–9. [Google Scholar] [CrossRef]
- Awad, S.M.; Attallah, D.A.; Salama, R.H.; Mahran, A.M.; Abu El-Hamed, E. Serum levels of psoriasin (S100A7) and koebnerisin (S100A15) as potential markers of atherosclerosis in patients with psoriasis. Clin. Exp. Dermatol. 2018, 43, 262–267. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.-H.; Chen, Y.; Zhen, Z.; Yeung, C.-K.; Chan, J.; Chan, H.H.; Tse, H.-F.; Yiu, K.-H. Relation between endothelial progenitor cells and arterial stiffness in patients with psoriasis. J. Dermatol. 2016, 43, 888–893. [Google Scholar] [CrossRef]
- El-Mongy, S.; Fathy, H.; Abdelaziz, A.; Omran, E.; George, S.; Neseem, N.; El-Nour, N. Subclinical atherosclerosis in patients with chronic psoriasis: A potential association. J. Eur. Acad. Dermatol. Venereol. 2010, 24, 661–666. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Lopez, A.; Blasco-Morente, G.; Perez-Lopez, I.; Tercedor-Sanchez, J.; Arias-Santiago, S. Studying the effect of systemic and biological drugs on intima-media thickness in patients suffering from moderate and severe psoriasis. J. Eur. Acad. Dermatol. Venereol. 2018, 32, 1492–1498. [Google Scholar] [CrossRef] [PubMed]
- Piros, É.A.; Szabó, Á.; Rencz, F.; Brodszky, V.; Szalai, K.; Galajda, N.; Szilveszter, B.; Dósa, E.; Merkely, B.; Holló, P. Impact of Interleukin-17 Inhibitor Therapy on Arterial Intima-media Thickness among Severe Psoriatic Patients. Life 2021, 11, 919. [Google Scholar] [CrossRef]
- Jókai, H.; Szakonyi, J.; Kontár, O.; Marschalkó, M.; Szalai, K.; Kárpáti, S.; Holló, P. Impact of effective tumor necrosis factor-alfa inhibitor treatment on arterial intima-media thickness in psoriasis: Results of a pilot study. J. Am. Acad. Dermatol. 2013, 69, 523–529. [Google Scholar] [CrossRef]
- Ikonomidis, I.; Makavos, G.; Papadavid, E.; Varoudi, M.; Andreadou, I.; Gravanis, K.; Theodoropoulos, K.; Pavlidis, G.; Triantafyllidi, H.; Parissis, J.; et al. Similarities in Coronary Function and Myocardial Deformation Between Psoriasis and Coronary Artery Disease: The Role of Oxidative Stress and Inflammation. Can. J. Cardiol. 2015, 31, 287–295. [Google Scholar] [CrossRef]
- Robati, R.M.; Partovi-Kia, M.; Haghighatkhah, H.R.; Younespour, S.; Abdollahimajd, F. Increased serum leptin and resistin levels and increased carotid intima-media wall thickness in patients with psoriasis: Is psoriasis associated with atherosclerosis? J. Am. Acad. Dermatol. 2014, 71, 642–648. [Google Scholar] [CrossRef]
- Antonucci, V.A.; Tengattini, V.; Balestri, R.; Patrizi, A.; Filippini, M.; Bardazzi, F. Intima-media thickness in an Italian psoriatic population: Correlation with lipidic serum levels, PASI and BMI. J. Eur. Acad. Dermatol. Venereol. 2014, 28, 512–515. [Google Scholar] [CrossRef]
- Marovt, M.; Marko, P.B.; Pirnat, M.; Ekart, R. Effect of biologics targeting interleukin-23/-17 axis on subclinical atherosclerosis: Results of a pilot study. Clin. Exp. Dermatol. 2020, 45, 560–564. [Google Scholar] [CrossRef]
- Elsheikh, R.G.; Amin, T.E.-S.; El-Ashmawy, A.A.; Abdalla, S.I.A.E.-F. Evaluation of subclinical atherosclerosis in Egyptian psoriatic patients. J. Saudi Heart Assoc. 2014, 26, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Yang, H.S.; Lee, Y.W.; Choe, Y.B.; Ahn, K.J. Evaluation of the Beta Stiffness Index and Carotid Intima-Media Thickness in Asian Patients with Psoriasis. Angiology 2015, 66, 889–895. [Google Scholar] [CrossRef] [PubMed]
- Patschan, D.; Sugiarto, N.; Henze, E.; Mößner, R.; Mohr, J.; Müller, G.A.; Patschan, S. Early endothelial progenitor cells and vascular stiffness in psoriasis and psoriatic arthritis. Eur. J. Med. Res. 2018, 23, 56. [Google Scholar] [CrossRef] [PubMed]
- Balta, I.; Balta, S.; Demirkol, S.; Celik, T.; Ekiz, O.; Cakar, M.; Sarlak, H.; Ozoguz, P.; Iyisoy, A. Aortic arterial stiffness is a moderate predictor of cardiovascular disease in patients with Psoriasis vulgaris. Angiology 2014, 65, 74–78. [Google Scholar] [CrossRef]
- Dregan, A. Arterial stiffness association with chronic inflammatory disorders in the UK Biobank study. Heart 2018, 104, 1257–1262. [Google Scholar] [CrossRef]
- Choi, B.G.; Kim, M.J.; Yang, H.S.; Lee, Y.W.; Choe, Y.B.; Ahn, K.J. Assessment of Arterial Stiffness in Korean Patients with Psoriasis by Cardio-Ankle Vascular Index. Angiology 2017, 68, 608–613. [Google Scholar] [CrossRef]
- Hansen, P.R.; Isaksen, J.L.; Jemec, G.B.; Ellervik, C.; Kanters, J.K. Arterial stiffness in subjects with psoriasis: A cross-sectional population study. Eur. J. Dermatol. 2018, 28, 683–685. [Google Scholar]
- Jensen, P.; Zachariae, C.; Christensen, R.; Geiker, N.R.W.; Schaadt, B.K.; Stender, S.; Astrup, A.; Hansen, P.R.; Skov, L. Effect of weight loss on the cardiovascular risk profile of obese patients with psoriasis. Acta. Derm. Venereol. 2014, 94, 691–694. [Google Scholar] [CrossRef]
- Sunbul, M.; Seckin, D.; Durmus, E.; Ozgen, Z.; Bozbay, M.; Bozbay, A.; Kivrak, T.; Oguz, M.; Sari, I.; Ergun, T.; et al. Assessment of arterial stiffness and cardiovascular hemodynamics by oscillometric method in psoriasis patients with normal cardiac functions. Heart Vessel. 2015, 30, 347–354. [Google Scholar] [CrossRef]
- Altekin, E.R. Determination of subclinical atherosclerosis in plaque type psoriasis patients without traditional risk factors for atherosclerosis. Arch. Turk. Soc. Cardiol. 2012, 40, 574–580. [Google Scholar] [CrossRef]
- Enany, B.; El Zohiery, A.K.; Elhilaly, R.; Badr, T. Carotid intima-media thickness and serum leptin in psoriasis. Herz 2012, 37, 527–533. [Google Scholar] [CrossRef] [PubMed]
- Divito, L.; Abeni, D.; Uras, C.; Magenta, A.; Cirapica, R.; Capogrossi, M.; Melillo, G. Role of psoriasis on subclinical cardiovascular disease. Minerva. Med. 2018, 109, 255–258. [Google Scholar]
- Asha, K.; Sharma, S.B.; Singal, A.; Aggarwal, A. Association of carotid intima-media thickness with leptin and apoliprotein b/apoliprotein a-I ratio reveals imminent predictors of subclinical atherosclerosis in psoriasis patients. Acta Med. 2014, 57, 21–27. [Google Scholar]
- Arias-Santiago, S.; Orgaz-Molina, J.; Castellote-Caballero, L.; Arrabal-Polo, M.Á.; García-Rodriguez, S.; Perandrés-López, R.; Ruiz, J.C.; Naranjo-Sintes, R.; Zubiaur, M.; Sancho, J.; et al. Atheroma plaque, metabolic syndrome and inflammation in patients with psoriasis. Eur. J. Dermatol. 2012, 22, 337–344. [Google Scholar] [CrossRef]
- Feldman, S.R.; Tian, H.; Gilloteau, I.; Mollon, P.; Shu, M. Economic burden of comorbidities in psoriasis patients in the United States: Results from a retrospective U.S. database. BMC Health Serv. Res. 2017, 17, 337. [Google Scholar] [CrossRef]
- Aurangabadkar, S. Comorbidities in psoriasis. Indian J. Dermatol. Venereol. Leprol. 2013, 79, 529–534. [Google Scholar] [CrossRef]
- Abuabara, K.; Azfar, R.S.; Shin, D.B.; Neimann, A.L.; Troxel, A.B.; Gelfand, J.M. Cause-specific mortality in patients with severe psoriasis: A population-based cohort study in the U.K. Br. J. Dermatol. 2010, 163, 586–592. [Google Scholar] [CrossRef]
- Masson, W.; Rossi, E.; Galimberti, M.L.; Krauss, J.; Navarro Estrada, J.; Galimberti, R. Mortality in patients with psoriasis. A retrospective cohort study. Med. Clin. 2017, 148, 483–488. [Google Scholar] [CrossRef]
- Gelfand, J.M.; Troxel, A.B.; Lewis, J.D.; Kurd, S.K.; Shin, D.B.; Wang, X.; Margolis, D.J.; Strom, B.L. The risk of mortality in patients with psoriasis: Results from a population-based study. Arch. Dermatol. 2007, 143, 1493–1499. [Google Scholar] [CrossRef]
- Langan, S.M.; Seminara, N.M.; Shin, D.B.; Troxel, A.B.; Kimmel, S.E.; Mehta, N.N.; Margolis, D.J.; Gelfand, J.M. Prevalence of metabolic syndrome in patients with psoriasis: A population-based study in the United Kingdom. J. Investig. Dermatol. 2012, 132, 556–562. [Google Scholar] [CrossRef]
- Takeshita, J.; Wang, S.; Shin, D.B.; Mehta, N.N.; Kimmel, S.E.; Margolis, D.J.; Troxel, A.B.; Gelfand, J.M. Effect of psoriasis severity on hypertension control a population-based study in the United Kingdom. JAMA Dermatol. 2015, 151, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Gelfand, J.M.; Neimann, A.L.; Shin, D.B.; Wang, X.; Margolis, D.J.; Troxel, A.B. Risk of Myocardial Infarction in Patients with Psoriasis. JAMA 2006, 296, 1735–1741. [Google Scholar] [CrossRef] [PubMed]
- Gelfand, J.M.; Dommasch, E.D.; Shin, D.B.; Azfar, R.S.; Kurd, S.K.; Wang, X.; Troxel, A.B. The risk of stroke in patients with psoriasis. J. Investig. Dermatol. 2009, 129, 2411–2418. [Google Scholar] [CrossRef] [PubMed]
- Egeberg, A.; Skov, L.; Joshi, A.A.; Mallbris, L.; Gislason, G.H.; Wu, J.J.; Rodante, J.; Lerman, J.B.; Ahlman, M.A.; Gelfand, J.M.; et al. The relationship between duration of psoriasis, vascular inflammation, and cardiovascular events. J. Am. Acad. Dermatol. 2017, 77, 650–656.e3. [Google Scholar] [CrossRef] [PubMed]
- Herédi, E.; Végh, J.; Pogácsás, L.; Gáspár, K.; Varga, J.; Kincse, G.; Zeher, M.; Szegedi, A.; Gaál, J. Subclinical cardiovascular disease and it’s improvement after long-term TNF-α inhibitor therapy in severe psoriatic patients. J. Eur. Acad. Dermatol. Venereol. 2016, 30, 1531–1536. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.C.S.; Lan, C.C.E. Psoriasis and cardiovascular comorbidities: Focusing on severe vascular events, cardiovascular risk factors and implications for treatment. Int. J. Mol. Sci. 2017, 18, 2211. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, A.W.; Harskamp, C.T.; Ledo, L.; Rogers, J.H.; Armstrong, E.J. Coronary Artery Disease in Patients with Psoriasis Referred for Coronary Angiography. Am. J. Cardiol. 2012, 109, 976–980. [Google Scholar] [CrossRef]
- Li, W.-Q.; Han, J.-L.; Manson, J.E.; Rimm, E.B.; Rexrode, K.M.; Curhan, G.C.; Qureshi, A.A. Psoriasis and risk of nonfatal cardiovascular disease in U.S. women: A cohort study: Nonfatal CVD in women with psoriasis. Br. J. Dermatol. 2012, 166, 811–818. [Google Scholar] [CrossRef]
- Kaplan, M.J. Cardiometabolic risk in psoriasis: Differential effects of biologic agents. Vasc. Health Risk Manag. 2008, 4, 1229–1235. [Google Scholar] [CrossRef]
- Azfar, R.S.; Gelfand, J.M. Psoriasis and metabolic disease: Epidemiology and pathophysiology. Curr. Opin. Rheumatol. 2008, 20, 416–422. [Google Scholar] [CrossRef]
- Kremers, H.M.; McEvoy, M.T.; Dann, F.J.; Gabriel, S.E. Heart disease in psoriasis. J. Am. Acad. Dermatol. 2007, 57, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Naik, H.B.; Natarajan, B.; Stansky, E.; Ahlman, M.A.; Teague, H.; Salahuddin, T.; Ng, Q.; Joshi, A.A.; Krishnamoorthy, P.; Dave, J.; et al. Severity of Psoriasis Associates with Aortic Vascular Inflammation Detected by FDG PET/CT and Neutrophil Activation in a Prospective Observational Study. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 2667–2676. [Google Scholar] [CrossRef] [PubMed]
- Mehta, N.N.; Yu, Y.; Saboury, B.; Foroughi, N.; Krishnamoorthy, P.; Raper, A.; Baer, A.; Antigua, J.; Van Voorhees, A.S.; Torigian, D.A.; et al. Systemic and vascular inflammation in patients with moderate to severe psoriasis as measured by [18F]-fluorodeoxyglucose positron emission tomography-computed tomography (FDG-PET/CT): A pilot study. Arch. Dermatol. 2011, 147, 1031–1039. [Google Scholar] [CrossRef] [PubMed]
- Mehta, N.N.; Torigian, D.A.; Gelfand, J.M.; Saboury, B.; Alavi, A. Quantification of atherosclerotic plaque activity and vascular inflammation using [18-F] fluorodeoxyglucose positron emission tomography/computed tomography (FDG-PET/CT). J. Vis. Exp. 2012, e3777. [Google Scholar] [CrossRef]
- Amin, M.; Lee, E.B.; Tsai, T.-F.; Wu, J.J. Psoriasis and Co-morbidity. Acta. Derm. Venereol. 2020, 100, adv00033. [Google Scholar] [CrossRef]
- Schmitt, J.; Wozel, G. The psoriasis area and severity index is the adequate criterion to define severity in chronic plaque-type psoriasis. Dermatology 2005, 210, 194–199. [Google Scholar] [CrossRef]
- Feldman, S.R. Psoriasis assessment tools in clinical trials. Ann. Rheum. Dis. 2005, 64, ii65–ii68. [Google Scholar] [CrossRef]
- Lew-Kaya, D.; Lue, J.; Sefton, J.; Walker, P. Evaluating psoriasis severity: Limitations of the PASI and advantages of the overall lesional assessment. J. Am. Acad. Dermatol. 2004, 50, P153. [Google Scholar] [CrossRef]
- Samarasekera, E.J.; Neilson, J.M.; Warren, R.B.; Parnham, J.; Smith, C.H. Incidence of Cardiovascular Disease in Individuals with Psoriasis: A Systematic Review and Meta-Analysis. J. Investig. Dermatol. 2013, 133, 2340–2346. [Google Scholar] [CrossRef]
- Armstrong, E.J.; Harskamp, C.T.; Armstrong, A.W. Psoriasis and Major Adverse Cardiovascular Events: A Systematic Review and Meta-Analysis of Observational Studies. JAHA J. Am. Heart Assoc. 2013, 2, e000062. [Google Scholar] [CrossRef]
- Brezinski, E.A.; Follansbee, M.R.; Armstrong, E.J.; Armstrong, A.W. Endothelial dysfunction and the effects of TNF inhibitors on the endothelium in psoriasis and psoriatic arthritis: A systematic review. Curr. Pharm. Des. 2014, 20, 513–528. [Google Scholar] [CrossRef] [PubMed]
- Boehncke, W.H. Systemic inflammation and cardiovascular comorbidity in psoriasis patients: Causes and consequences. Front. Immunol. 2018, 9, 579. [Google Scholar] [CrossRef] [PubMed]
- Poredos, P.; Poredos, A.V.; Gregoric, I. Endothelial Dysfunction and Its Clinical Implications. Angiology 2021, 72, 604–615. [Google Scholar] [CrossRef] [PubMed]
- Holowatz, L.A.; Kenney, W.L. Acute localized administration of tetrahydrobiopterin and chronic systemic atorvastatin treatment restore cutaneous microvascular function in hypercholesterolaemic humans. J. Physiol. 2011, 589, 4787–4797. [Google Scholar] [CrossRef]
- Anderson, T.J. Assessment and treatment of endothelial dysfunction in humans. J. Am. Coll. Cardiol. 1999, 34, 631–638. [Google Scholar] [CrossRef]
- Ross, R. The pathogenesis of atherosclerosis--an update. N. Engl. J. Med. 1986, 314, 488–500. [Google Scholar] [CrossRef]
- Ross, R. Atherosclerosis--an inflammatory disease. N. Engl. J. Med. 1999, 340, 115–126. [Google Scholar] [CrossRef]
- Yeboah, J.; Folsom, A.R.; Burke, G.L.; Johnson, C.; Polak, J.F.; Post, W.; Lima, J.A.; Crouse, J.R.; Herrington, D.M. Predictive Value of Brachial Flow-Mediated Dilation for Incident Cardiovascular Events in a Population-Based Study: The Multi-Ethnic Study of Atherosclerosis. Circulation 2009, 120, 502–509. [Google Scholar] [CrossRef]
- Brown, I.J.; Tzoulaki, I.; Candeias, V.; Elliott, P. Salt intakes around the world: Implications for public health. Int. J. Epidemiol. 2009, 38, 791–813. [Google Scholar] [CrossRef]
- World Health Organization. Prevention of Cardiovascular Disease: Guidelines for Assessment and Management of Total Cardiovascular Risk. 2007. Available online: https://apps.who.int/iris/handle/10665/43685 (accessed on 1 April 2022).
- World Health Organization. WHO Global Sodium Benchmarks for Different Food Categories; World Health Organization: Geneva, Switzerland, 2021; Available online: https://apps.who.int/iris/handle/10665/341081 (accessed on 1 April 2022).
- World Health Organization. Guideline: Sodium Intake for Adults and Children; World Health Organization: Geneva, Switzerland, 2012; Available online: https://apps.who.int/iris/handle/10665/77985 (accessed on 1 April 2022).
- Wilck, N.; Balogh, A.; Markó, L.; Bartolomaeus, H.; Müller, D.N. The role of sodium in modulating immune cell function. Nat. Rev. Nephrol. 2019, 15, 546–558. [Google Scholar] [CrossRef]
- Haase, S.; Wilck, N.; Kleinewietfeld, M.; Müller, D.N.; Linker, R.A. Sodium chloride triggers Th17 mediated autoimmunity. J. Neuroimmunol. 2019, 329, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Sharif, K.; Amital, H.; Shoenfeld, Y. The role of dietary sodium in autoimmune diseases: The salty truth. Autoimmun. Rev. 2018, 17, 1069–1073. [Google Scholar] [CrossRef] [PubMed]
- Toussirot, E.; Béreau, M.; Vauchy, C.; Saas, P. Could sodium chloride be an environmental trigger for immune-mediated diseases? An overview of the experimental and clinical evidence. Front. Physiol. 2018, 9, 440. [Google Scholar] [CrossRef] [PubMed]
- Pona, A.; Haidari, W.; Kolli, S.S.; Feldman, S.R. Diet and psoriasis. Dermatol. Online J. 2019, 25. [Google Scholar] [CrossRef]
- Garbicz, J.; Całyniuk, B.; Górski, M.; Buczkowska, M.; Piecuch, M.; Kulik, A.; Rozentryt, P. Nutritional Therapy in Persons Suffering from Psoriasis. Nutrients 2021, 14, 119. [Google Scholar] [CrossRef]
- Rucević, I.; Perl, A.; Barisić-Drusko, V.; Adam-Perl, M. The role of the low energy diet in Psoriasis vulgaris treatment. Coll. Antropol. 2003, 27 (Suppl. 1), 41–48. [Google Scholar]
- Schena, D.; Chieregato, G.C.; de Gironcoli, M.; Girelli, D.; Olivieri, O.; Stanzial, A.M.; Corrocher, R.; Bassi, A.; Ferrari, S.; Perazzoli, P. Increased erythrocyte membrane arachidonate and platelet malondialdehyde (MDA) production in psoriasis: Normalization after fish-oil. Acta. Derm. Venereol. Suppl. 1989, 146, 42–44. [Google Scholar]
- Lassus, A.; Dahlgren, A.L.; Halpern, M.J.; Santalahti, J.; Happonen, H.P. Effects of dietary supplementation with polyunsaturated ethyl ester lipids (Angiosan) in patients with psoriasis and psoriatic arthritis. J. Int. Med. Res. 1990, 18, 68–73. [Google Scholar] [CrossRef]
- Kragballe, K.; Fogh, K. A low-fat diet supplemented with dietary fish oil (Max-EPA) results in improvement of psoriasis and in formation of leukotriene B5. Acta. Derm. Venereol. 1989, 69, 23–28. [Google Scholar]
- Thongprayoon, C.; Cheungpasitporn, W.; Petnak, T.; Ghamrawi, R.; Thirunavukkarasu, S.; Chewcharat, A.; Bathini, T.; Vallabhajosyula, S.; Kashani, K.B. The prognostic importance of serum sodium levels at hospital discharge and one-year mortality among hospitalized patients. Int. J. Clin. Pract. 2020, 74, e13581. [Google Scholar] [CrossRef]
- Knezović, A.; Kolobarić, N.; Drenjančević, I.; Mihaljević, Z.; Šušnjara, P.; Jukić, I.; Stupin, M.; Kibel, A.; Marczi, S.; Mihalj, M.; et al. Role of Oxidative Stress in Vascular Low-Grade Inflammation Initiation Due to Acute Salt Loading in Young Healthy Individuals. Antioxidants 2022, 11, 444. [Google Scholar] [CrossRef] [PubMed]
- Jablonski, K.L.; Racine, M.L.; Geolfos, C.J.; Gates, P.E.; Chonchol, M.; McQueen, M.B.; Seals, D.R. Dietary Sodium Restriction Reverses Vascular Endothelial Dysfunction in Middle-Aged/Older Adults with Moderately Elevated Systolic Blood Pressure. J. Am. Coll. Cardiol. 2013, 61, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Dickinson, K.M.; Keogh, J.B.; Clifton, P.M. Effects of a low-salt diet on flow-mediated dilatation in humans. Am. J. Clin. Nutr. 2009, 89, 485–490. [Google Scholar] [CrossRef] [PubMed]
- Kyriakou, A.; Patsatsi, A.; Vyzantiadis, T.-A.; Sotiriadis, D. Serum levels of TNF-α, IL-12/23p40, and IL-17 in plaque psoriasis and their correlation with disease severity. J. Immunol. Res. 2014, 2014, 467541. [Google Scholar] [CrossRef]
- Barrea, L.; Balato, N.; Di Somma, C.; Macchia, P.E.; Napolitano, M.; Savanelli, M.C.; Esposito, K.; Colao, A.; Savastano, S. Nutrition and psoriasis: Is there any association between the severity of the disease and adherence to the Mediterranean diet? J. Transl. Med. 2015, 13, 18. [Google Scholar] [CrossRef]
- Galland, L. Diet and inflammation. Nutr. Clin. Pract. 2010, 25, 634–640. [Google Scholar] [CrossRef]
- Solis, M.Y.; Melo NS de Macedo, M.E.M.; Carneiro, F.P.; Sabbag, C.Y.; Lancha Junior, A.H.; Frangella, V.S. Nutritional status and food intake of patients with systemic psoriasis and psoriatic arthritis associated. Einstein 2012, 10, 44–52. [Google Scholar] [CrossRef]
- Naldi, L.; Parazzini, F.; Peli, L.; Chatenoud, L.; Cainelli, T. Dietary factors and the risk of psoriasis. Results of an Italian case-control study. Br. J. Dermatol. 1996, 134, 101–106. [Google Scholar]
- Duarte, G.; Barbosa, L.O.; Rosa, M.E.A. The management of psoriasis through diet. PTT Psoriasis Targets Ther. 2012, 2, 45–53. [Google Scholar] [CrossRef]
- Wong, A.P.; Kalinovsky, T.; Niedzwiecki, A.; Rath, M. Efficacy of nutritional treatment in patients with psoriasis: A case report. Exp. Ther. Med. 2015, 10, 1071–1073. [Google Scholar] [CrossRef][Green Version]
- Amin, S.; Adil, M.; Alam, M. Role of dietary intervention in psoriasis: A review. Indian J. Clin. Dermatol. 2018, 1, 13. [Google Scholar]
- Engin, K.N. Alpha-tocopherol: Looking beyond an antioxidant. Mol. Vis. 2009, 15, 855–860. [Google Scholar] [PubMed]
- Rietjens, I.M.C.M.; Boersma, M.G.; de Haan, L.; Spenkelink, B.; Awad, H.M.; Cnubben, N.H.P.; van Zanden, J.J.; van der Woude, H.; Alink, G.M.; Koeman, J.H. The pro-oxidant chemistry of the natural antioxidants vitamin C, vitamin E, carotenoids and flavonoids. Environ. Toxicol. Pharmacol. 2002, 11, 321–333. [Google Scholar] [CrossRef]
- Thiele, J.J.; Schroeter, C.; Hsieh, S.N.; Podda, M.; Packer, L. The antioxidant network of the stratum corneum. Curr. Probl. Dermatol. 2001, 29, 26–42. [Google Scholar] [PubMed]
- Liu, X.; Yang, G.; Luo, M.; Lan, Q.; Shi, X.; Deng, H.; Wang, N.; Xu, X.; Zhang, C. Serum vitamin E levels and chronic inflammatory skin diseases: A systematic review and meta-analysis. PLoS ONE 2021, 16, e0261259. [Google Scholar] [CrossRef]
- Pujari, V.M.; Ireddy, S.; Itagi, I.; Kumar, H.S. The serum levels of malondialdehyde, vitamin e and erythrocyte catalase activity in psoriasis patients. J. Clin. Diagn. Res. 2014, 8, CC14–CC16. [Google Scholar] [CrossRef]
- Kharaeva, Z.; Gostova, E.; De Luca, C.; Raskovic, D.; Korkina, L. Clinical and biochemical effects of coenzyme Q10, vitamin E, and selenium supplementation to psoriasis patients. Nutrition 2009, 25, 295–302. [Google Scholar] [CrossRef]
- Kanda, N.; Hoashi, T.; Saeki, H. Nutrition and psoriasis. Int. J. Mol. Sci. 2020, 21, 5405. [Google Scholar] [CrossRef]
- Harvima, R.J.; Jägerroos, H.; Kajander, E.O.; Harvima, I.T.; Aalto, M.L.; Neittaanmäki, H.; Naukkarinen, A.; Kantola, M.; Miettinen, U.K.; Horsmanheimo, M. Screening of effects of selenomethionine-enriched yeast supplementation on various immunological and chemical parameters of skin and blood in psoriatic patients. Acta. Derm. Venereol. 1993, 73, 88–91. [Google Scholar]
- Fairris, G.M.; Lloyd, B.; Hinks, L.; Perkins, P.J.; Clayton, B.E. The effect of supplementation with selenium and vitamin E in psoriasis. Ann. Clin. Biochem. 1989, 26 Pt 1, 83–88. [Google Scholar] [CrossRef]
- Oppedisano, F.; Macrì, R.; Gliozzi, M.; Musolino, V.; Carresi, C.; Maiuolo, J.; Bosco, F.; Nucera, S.; Caterina Zito, M.; Guarnieri, L.; et al. The Anti-Inflammatory and Antioxidant Properties of n-3 PUFAs: Their Role in Cardiovascular Protection. Biomedicines 2020, 8, 306. [Google Scholar] [CrossRef] [PubMed]
- Myśliwiec, H.; Baran, A.; Harasim-Symbor, E.; Myśliwiec, P.; Milewska, A.J.; Chabowski, A.; Flisiak, I. Serum fatty acid profile in psoriasis and its comorbidity. Arch. Dermatol. Res. 2017, 309, 371–380. [Google Scholar] [CrossRef] [PubMed]
- Guida, B.; Napoleone, A.; Trio, R.; Nastasi, A.; Balato, N.; Laccetti, R.; Cataldi, M. Energy-restricted, n-3 polyunsaturated fatty acids-rich diet improves the clinical response to immuno-modulating drugs in obese patients with plaque-type psoriasis: A randomized control clinical trial. Clin. Nutr. 2014, 33, 399–405. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.K.; Ellis, C.N.; Tellner, D.C.; Anderson, T.F.; Voorhees, J.J. Double-blind, placebo-controlled study to evaluate the efficacy of fish oil and low-dose UVB in the treatment of psoriasis. Br. J. Dermatol. 1989, 120, 801–807. [Google Scholar] [CrossRef] [PubMed]
- Danno, K.; Sugie, N. Combination therapy with low-dose etretinate and eicosapentaenoic acid for Psoriasis vulgaris. J. Dermatol. 1998, 25, 703–705. [Google Scholar] [CrossRef] [PubMed]
- Stupin, A.; Mihalj, M.; Kolobarić, N.; Šušnjara, P.; Kolar, L.; Mihaljević, Z.; Matić, A.; Stupin, M.; Jukić, I.; Kralik, Z.; et al. Anti-Inflammatory Potential of n-3 Polyunsaturated Fatty Acids Enriched Hen Eggs Consumption in Improving Microvascular Endothelial Function of Healthy Individuals-Clinical Trial. Int. J. Mol. Sci. 2020, 21, 4149. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krajina, I.; Stupin, A.; Šola, M.; Mihalj, M. Oxidative Stress Induced by High Salt Diet—Possible Implications for Development and Clinical Manifestation of Cutaneous Inflammation and Endothelial Dysfunction in Psoriasis vulgaris. Antioxidants 2022, 11, 1269. https://doi.org/10.3390/antiox11071269
Krajina I, Stupin A, Šola M, Mihalj M. Oxidative Stress Induced by High Salt Diet—Possible Implications for Development and Clinical Manifestation of Cutaneous Inflammation and Endothelial Dysfunction in Psoriasis vulgaris. Antioxidants. 2022; 11(7):1269. https://doi.org/10.3390/antiox11071269
Chicago/Turabian StyleKrajina, Ivana, Ana Stupin, Marija Šola, and Martina Mihalj. 2022. "Oxidative Stress Induced by High Salt Diet—Possible Implications for Development and Clinical Manifestation of Cutaneous Inflammation and Endothelial Dysfunction in Psoriasis vulgaris" Antioxidants 11, no. 7: 1269. https://doi.org/10.3390/antiox11071269
APA StyleKrajina, I., Stupin, A., Šola, M., & Mihalj, M. (2022). Oxidative Stress Induced by High Salt Diet—Possible Implications for Development and Clinical Manifestation of Cutaneous Inflammation and Endothelial Dysfunction in Psoriasis vulgaris. Antioxidants, 11(7), 1269. https://doi.org/10.3390/antiox11071269