
Mechanism of Action of Cyanidin 3-O-Glucoside in Gluconeogenesis and Oxidative Stress-Induced Cancer Cell Senescence


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Mouse Experiments
2.2. Measurement of the Rate of Gluconeogenesis
2.3. Autophagy Analysis
2.4. In Vitro AMPK Activity Assay
2.5. Gene Knockdown with AMPKα and Adiponectin Receptor 1/2 siRNA
2.6. Immunoblotting
2.7. Total RNA Extraction and Quantitative Polymerase Chain Reaction (qPCR)
2.8. Intracellular Calcium Assay
2.9. Reporter Gene (Luciferase) Assay
2.10. Intracellular Reactive Oxygen Species (ROS) Measurement
2.11. Caspase-3 Activity Assay
2.12. Cell Viability Assay
2.13. Senescence-Associated (SA)-β-Galactosidase Assay
2.14. Statistical Analysis
3. Results
3.1. C3G Activates AMPK Phosphorylation and Suppresses the Rate of Gluconeogenesis
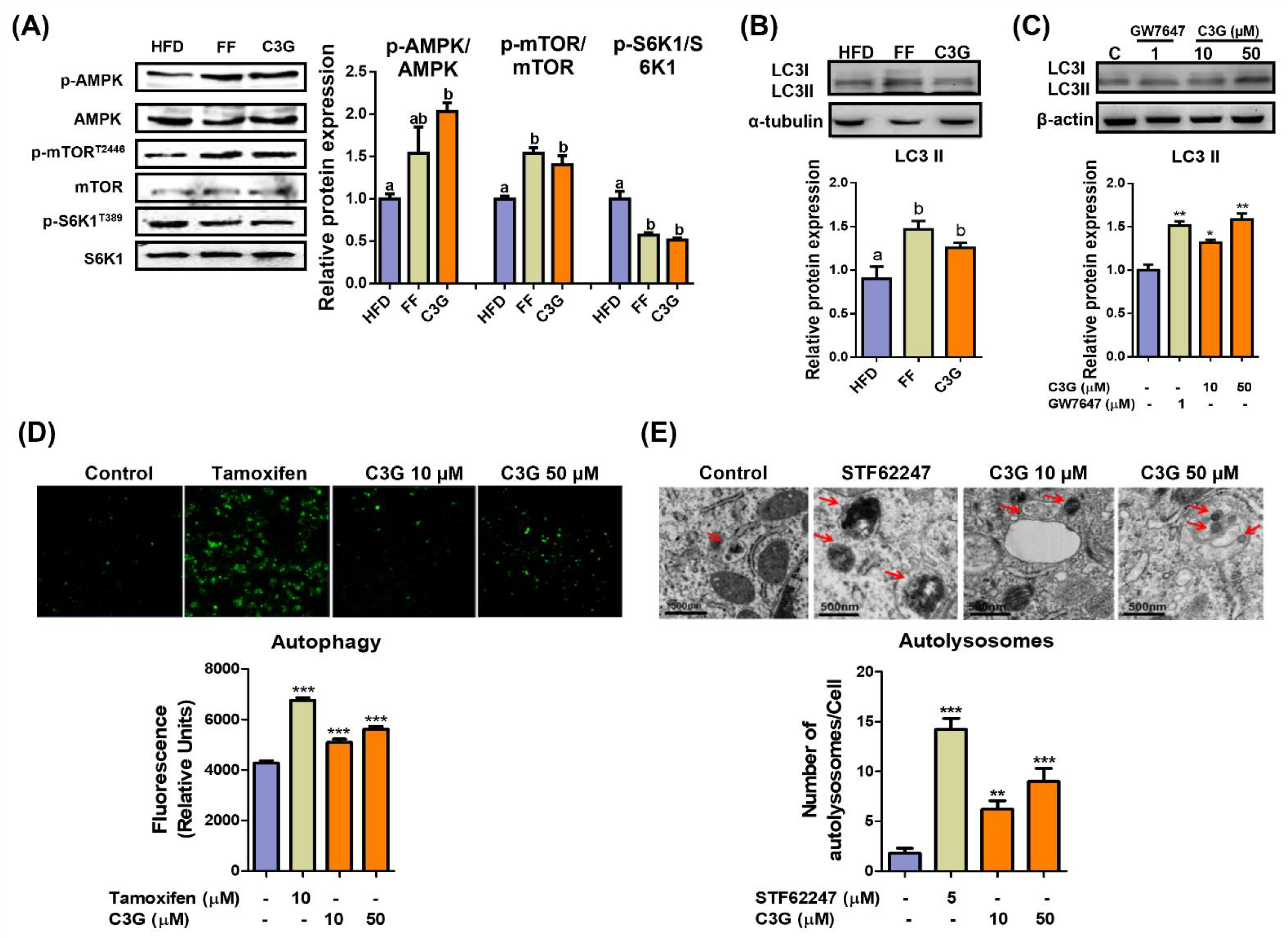
3.2. C3G Induces Hepatic Autophagy through the AMPK-mTORC1 Signaling Axis
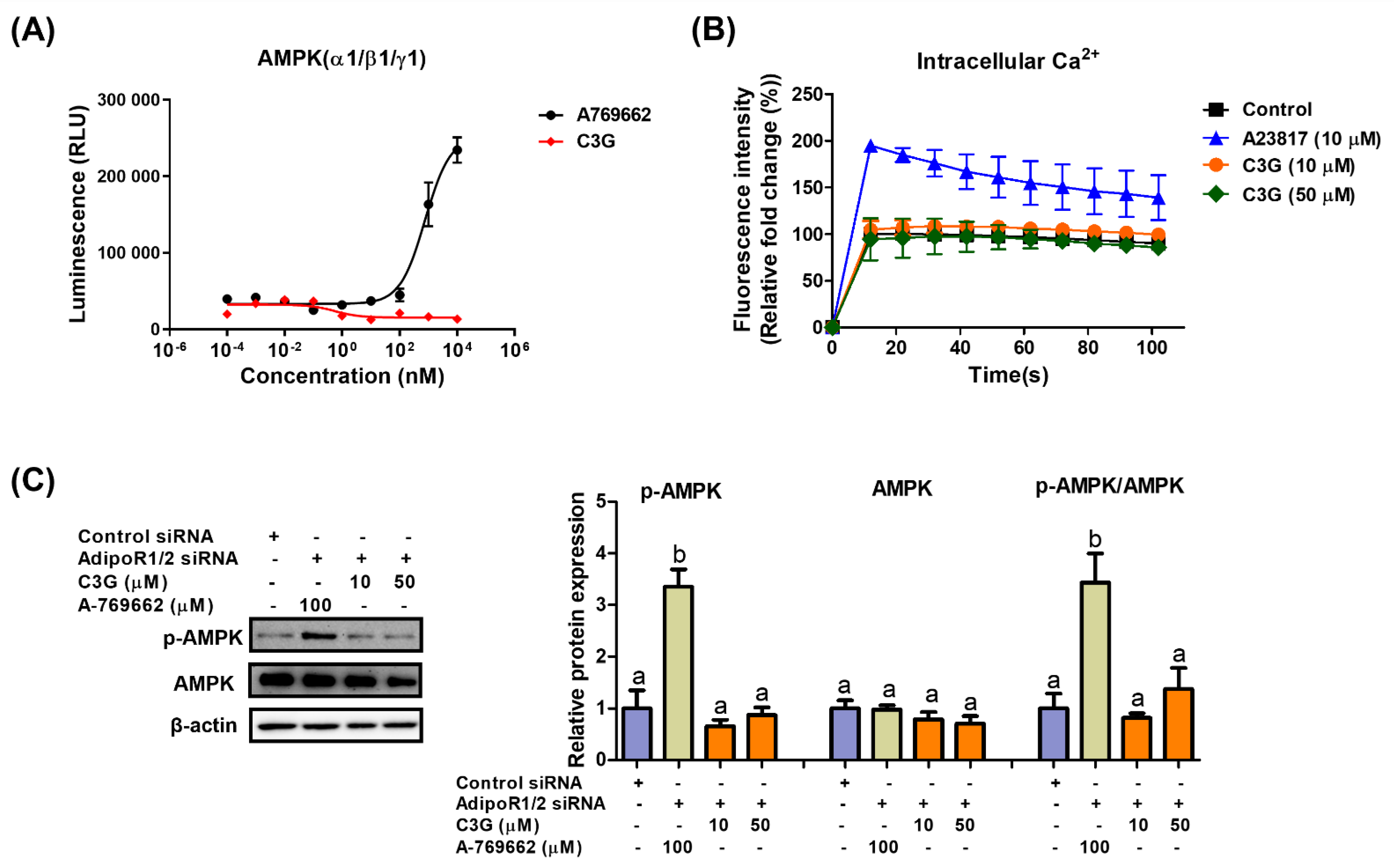
3.3. C3G Activates AMPK via Adiponectin Receptor Signaling
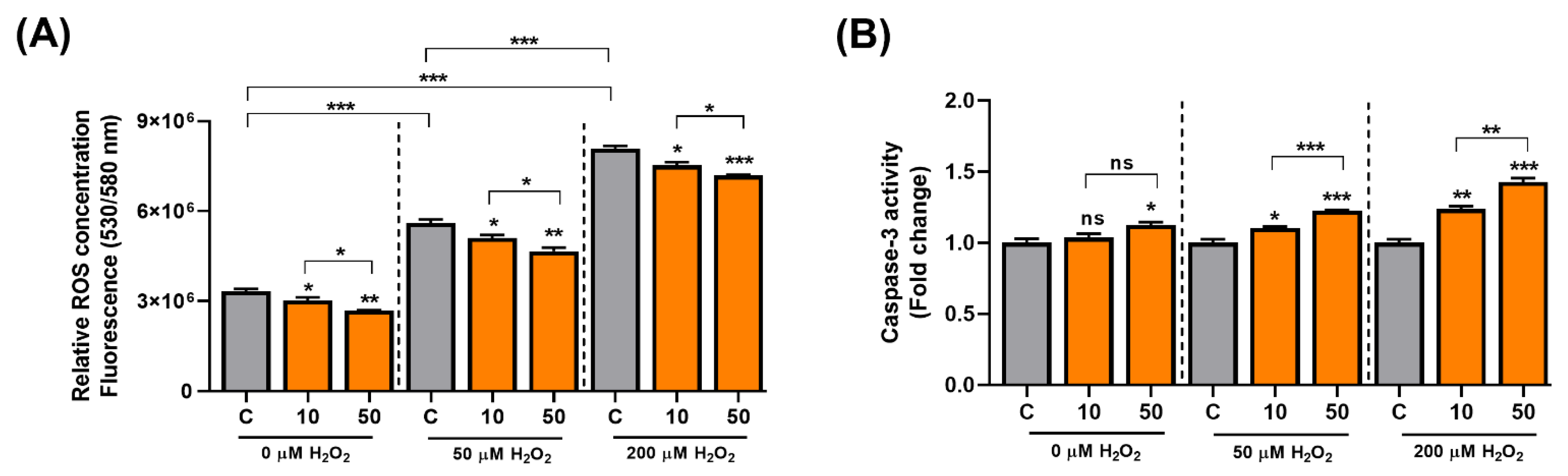
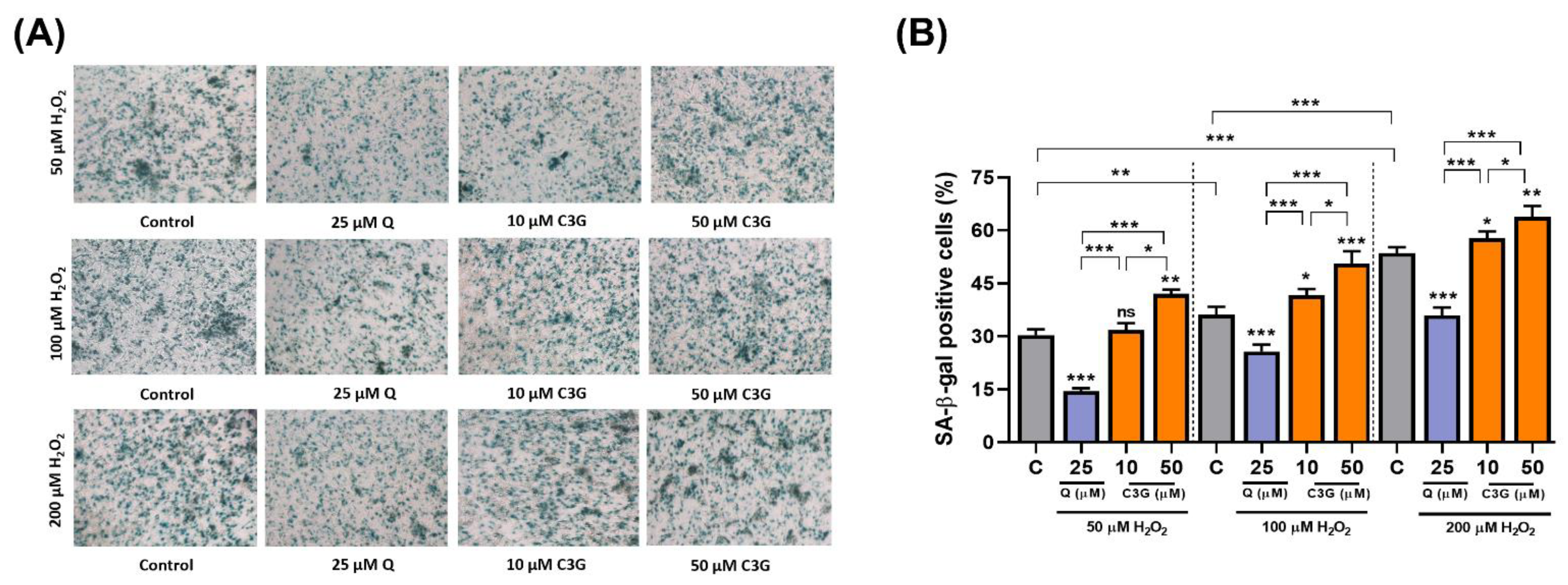
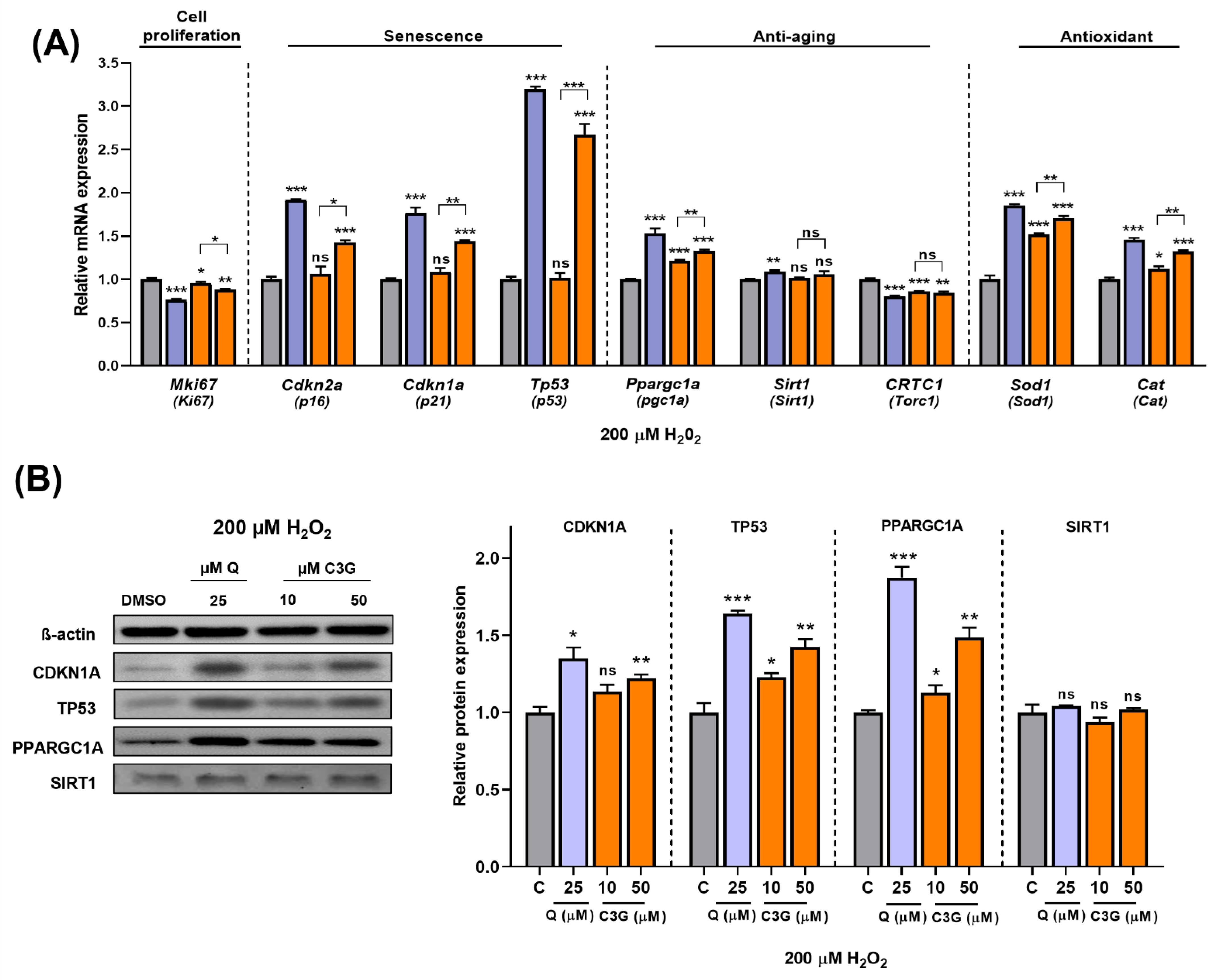
3.4. C3G Is an Antioxidative Molecule That Induces Cellular Senescence in Hepatocarcinoma Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Seo, E.; Kang, H.; Choi, H.; Choi, W.; Jun, H.S. Reactive oxygen species-induced changes in glucose and lipid metabolism contribute to the accumulation of cholesterol in the liver during aging. Aging Cell 2019, 18, e12895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reczek, C.R.; Chandel, N.S. The two faces of reactive oxygen species in cancer. Annu. Rev. Cancer Biol. 2017, 1, 79–98. [Google Scholar] [CrossRef]
- Fitzgerald, A.L.; Osman, A.A.; Xie, T.X.; Patel, A.; Skinner, H.; Sandulache, V.; Myers, J.N. Reactive oxygen species and p21(Waf1/Cip1) are both essential for p53-mediated senescence of head and neck cancer cells. Cell Death Dis. 2015, 6, e1678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davalli, P.; Mitic, T.; Caporali, A.; Lauriola, A.; D’Arca, D. ROS, cell senescence, and novel molecular mechanisms in aging and age-related diseases. Oxid. Med. Cell. Longev. 2016, 2016, 3565127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, E.K.; Rogowska-Wrzesinska, A.; Roepstorff, P.; Bulteau, A.L.; Friguet, B. Protein modification and replicative senescence of WI-38 human embryonic fibroblasts. Aging Cell 2010, 9, 252–272. [Google Scholar] [CrossRef]
- Lawless, C.; Jurk, D.; Gillespie, C.S.; Shanley, D.; Saretzki, G.; von Zglinicki, T.; Passos, J.F. A stochastic step model of replicative senescence explains ROS production rate in ageing cell populations. PLoS ONE 2012, 7, e32117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vigneron, A.; Vousden, K.H. p53, ROS and senescence in the control of aging. Aging 2010, 2, 471. [Google Scholar] [CrossRef] [Green Version]
- Passos, J.F.; Nelson, G.; Wang, C.; Richter, T.; Simillion, C.; Proctor, C.J.; Miwa, S.; Olijslagers, S.; Hallinan, J.; Wipat, A. Feedback between p21 and reactive oxygen production is necessary for cell senescence. Mol. Syst. Biol. 2010, 6, 347. [Google Scholar] [CrossRef]
- Malavolta, M.; Costarelli, L.; Giacconi, R.; Piacenza, F.; Basso, A.; Pierpaoli, E.; Marchegiani, F.; Cardelli, M.; Provinciali, M.; Mocchegiani, E. Modulators of cellular senescence: Mechanisms, promises, and challenges from in vitro studies with dietary bioactive compounds. Nutr. Res. 2014, 34, 1017–1035. [Google Scholar] [CrossRef]
- Yan, L.-J. Positive oxidative stress in aging and aging-related disease tolerance. Redox Biol. 2014, 2, 165–169. [Google Scholar] [CrossRef] [Green Version]
- Palsamy, P.; Subramanian, S. Ameliorative potential of resveratrol on proinflammatory cytokines, hyperglycemia mediated oxidative stress, and pancreatic beta-cell dysfunction in streptozotocin-nicotinamide-induced diabetic rats. J. Cell. Physiol. 2010, 224, 423–432. [Google Scholar] [CrossRef] [PubMed]
- Gibellini, L.; Pinti, M.; Nasi, M.; De Biasi, S.; Roat, E.; Bertoncelli, L.; Cossarizza, A. Interfering with ROS metabolism in cancer cells: The potential role of quercetin. Cancers 2010, 2, 1288–1311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yılmaz Göler, A.; Biçim, G.; Toprak, K.; Yılmaz, B.; Yalçın, A.; Milisav, I. Hydrogen Peroxide and Quercetin Induced Changes on Cell Viability, Apoptosis and Oxidative Stress in HepG2 Cells. Curr. Nutraceuticals 2021, 2, 47–55. [Google Scholar] [CrossRef]
- Acquaviva, R.; Tomasello, B.; Di Giacomo, C.; Santangelo, R.; La Mantia, A.; Naletova, I.; Sarpietro, M.G.; Castelli, F.; Malfa, G.A. Protocatechuic Acid, a Simple Plant Secondary Metabolite, Induced Apoptosis by Promoting Oxidative Stress through HO-1 Downregulation and p21 Upregulation in Colon Cancer Cells. Biomolecules 2021, 11, 1485. [Google Scholar] [CrossRef]
- Badiola, I.; Santaolalla, F.; Garcia-Gallastegui, P.; Unda, F.; Ibarretxe, G. Biomolecular bases of the senescence process and cancer. A new approach to oncological treatment linked to ageing. Ageing Res. Rev. 2015, 23, 125–138. [Google Scholar] [CrossRef]
- Vurusaner, B.; Poli, G.; Basaga, H. Tumor suppressor genes and ROS: Complex networks of interactions. Free Radic. Biol. Med. 2012, 52, 7–18. [Google Scholar] [CrossRef]
- Alers, S.; Loffler, A.S.; Wesselborg, S.; Stork, B. Role of AMPK-mTOR-Ulk1/2 in the Regulation of Autophagy: Cross Talk, Shortcuts, and Feedbacks. Mol. Cell. Biol. 2012, 32, 2–11. [Google Scholar] [CrossRef] [Green Version]
- Lamming, D.W.; Ye, L.; Katajisto, P.; Goncalves, M.D.; Saitoh, M.; Stevens, D.M.; Davis, J.G.; Salmon, A.B.; Richardson, A.; Ahima, R.S.; et al. Rapamycin-induced insulin resistance is mediated by mTORC2 loss and uncoupled from longevity. Science 2012, 335, 1638–1643. [Google Scholar] [CrossRef] [Green Version]
- Selman, C.; Tullet, J.M.A.; Wieser, D.; Irvine, E.; Lingard, S.J.; Choudhury, A.I.; Claret, M.; Al-Qassab, H.; Carmignac, D.; Ramadani, F.; et al. Ribosomal protein S6 kinase 1 signaling regulates mammalian life span. Science 2009, 326, 140–144. [Google Scholar] [CrossRef] [Green Version]
- Joshi, T.; Singh, A.K.; Haratipour, P.; Sah, A.N.; Pandey, A.K.; Naseri, R.; Juyal, V.; Farzaei, M.H. Targeting AMPK signaling pathway by natural products for treatment of diabetes mellitus and its complications. J. Cell. Physiol. 2019, 234, 17212–17231. [Google Scholar] [CrossRef]
- Salomone, F.; Godos, J.; Zelber-Sagi, S. Natural antioxidants for non-alcoholic fatty liver disease: Molecular targets and clinical perspectives. Liver Int. 2016, 36, 5–20. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Xu, B.; Sun, J.; Jiang, X.; Bai, W. Anthocyanin supplement as a dietary strategy in cancer prevention and management: A comprehensive review. Crit. Rev. Food Sci. Nutr. 2021, 19, 1–13. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Giusti, M.M. Anthocyanins: Natural colorants with health-promoting properties. Annu. Rev. Food Sci. Technol. 2010, 1, 163–187. [Google Scholar] [CrossRef]
- Mannino, G.; Gentile, C.; Ertani, A.; Serio, G.; Bertea, C.M. Anthocyanins: Biosynthesis, distribution, ecological role, and use of biostimulants to increase their content in plant foods—A review. Agriculture 2021, 11, 212. [Google Scholar] [CrossRef]
- Gowd, V.; Jia, Z.; Chen, W. Anthocyanins as promising molecules and dietary bioactive components against diabetes–A review of recent advances. Trends Food Sci. Technol. 2017, 68, 1–13. [Google Scholar] [CrossRef]
- Zhu, W.; Jia, Q.; Wang, Y.; Zhang, Y.; Xia, M. The anthocyanin cyanidin-3-O-β-glucoside, a flavonoid, increases hepatic glutathione synthesis and protects hepatocytes against reactive oxygen species during hyperglycemia: Involvement of a cAMP–PKA-dependent signaling pathway. Free Radic. Biol. Med. 2012, 52, 314–327. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Wu, C.; Kim, Y.-S.; Yang, S.O.; Kim, Y.; Kim, J.-S.; Jeong, M.-Y.; Lee, J.H.; Kim, B.; Lee, S. A dietary anthocyanin cyanidin-3-O-glucoside binds to PPARs to regulate glucose metabolism and insulin sensitivity in mice. Commun. Biol. 2020, 3, 514. [Google Scholar] [CrossRef]
- Wu, C.; Hwang, S.H.; Jia, Y.; Choi, J.; Kim, Y.J.; Choi, D.; Pathiraja, D.; Choi, I.G.; Koo, S.H.; Lee, S.J. Olfactory receptor 544 reduces adiposity by steering fuel preference toward fats. J. Clin. Investig. 2017, 127, 4118–4123. [Google Scholar] [CrossRef] [Green Version]
- Steinberg, G.R.; Carling, D. AMP-activated protein kinase: The current landscape for drug development. Nat. Rev. Drug Discov. 2019, 18, 527–551. [Google Scholar] [CrossRef]
- Bechmann, L.P.; Hannivoort, R.A.; Gerken, G.; Hotamisligil, G.S.; Trauner, M.; Canbay, A. The interaction of hepatic lipid and glucose metabolism in liver diseases. J. Hepatol. 2012, 56, 952–964. [Google Scholar] [CrossRef] [Green Version]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell. Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayr, B.; Montminy, M. Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat. Rev. Mol. Cell. Biol. 2001, 2, 599–609. [Google Scholar] [CrossRef] [PubMed]
- Dentin, R.; Liu, Y.; Koo, S.-H.; Hedrick, S.; Vargas, T.; Heredia, J.; Yates, J.; Montminy, M. Insulin modulates gluconeogenesis by inhibition of the coactivator TORC2. Nature 2007, 449, 366–369. [Google Scholar] [CrossRef] [PubMed]
- Quinn, P.; Yeagley, D. Insulin regulation of PEPCK gene expression: A model for rapid and reversible modulation. Endocr Metab Immune Disord. Drug Targets 2005, 5, 423–437. [Google Scholar] [CrossRef]
- Housley, M.P.; Udeshi, N.D.; Rodgers, J.T.; Shabanowitz, J.; Puigserver, P.; Hunt, D.F.; Hart, G.W. A PGC-1α-O-GlcNAc transferase complex regulates FoxO transcription factor activity in response to glucose. J. Biol. Chem. 2009, 284, 5148–5157. [Google Scholar] [CrossRef] [Green Version]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [Green Version]
- Ueno, T.; Komatsu, M. Autophagy in the liver: Functions in health and disease. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 170–184. [Google Scholar] [CrossRef]
- Alessi, D.R.; Sakamoto, K.; Bayascas, J.R. LKB1-dependent signaling pathways. Annu. Rev. Biochem. 2006, 75, 137–163. [Google Scholar] [CrossRef]
- Hemminki, A. The molecular basis and clinical aspects of Peutz-Jeghers syndrome. Cell. Mol. Life Sci. 1999, 55, 735–750. [Google Scholar] [CrossRef]
- Kim, J.; Kim, Y.C.; Fang, C.; Russell, R.C.; Kim, J.H.; Fan, W.; Liu, R.; Zhong, Q.; Guan, K.L. Differential regulation of distinct Vps34 complexes by AMPK in nutrient stress and autophagy. Cell 2013, 152, 290–303. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gowans, G.J.; Hawley, S.A.; Ross, F.A.; Hardie, D.G. AMP is a true physiological regulator of AMP-activated protein kinase by both allosteric activation and enhancing net phosphorylation. Cell Metab. 2013, 18, 556–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zha, D.; Wu, X.; Gao, P. Adiponectin and its receptors in diabetic kidney disease: Molecular mechanisms and clinical potential. Endocrinology 2017, 158, 2022–2034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadowaki, T.; Yamauchi, T. Adiponectin and adiponectin receptors. Endocr. Rev. 2005, 26, 439–451. [Google Scholar] [CrossRef] [Green Version]
- Kadowaki, T.; Yamauchi, T.; Kubota, N.; Hara, K.; Ueki, K.; Tobe, K. Adiponectin and adiponectin receptors in insulin resistance, diabetes, and the metabolic syndrome. J. Clin. Investig. 2006, 116, 1784–1792. [Google Scholar] [CrossRef] [Green Version]
- Belfort, R.; Harrison, S.A.; Brown, K.; Darland, C.; Finch, J.; Hardies, J.; Balas, B.; Gastaldelli, A.; Tio, F.; Pulcini, J. A placebo-controlled trial of pioglitazone in subjects with nonalcoholic steatohepatitis. N. Engl. J. Med. 2006, 355, 2297–2307. [Google Scholar] [CrossRef] [Green Version]
- Yamauchi, T.; Kamon, J.; Ito, Y.; Tsuchida, A.; Yokomizo, T.; Kita, S.; Sugiyama, T.; Miyagishi, M.; Hara, K.; Tsunoda, M. Cloning of adiponectin receptors that mediate antidiabetic metabolic effects. Nature 2003, 423, 762–769. [Google Scholar] [CrossRef]
- Combs, T.P.; Pajvani, U.B.; Berg, A.H.; Lin, Y.; Jelicks, L.A.; Laplante, M.; Nawrocki, A.R.; Rajala, M.W.; Parlow, A.F.; Cheeseboro, L. A transgenic mouse with a deletion in the collagenous domain of adiponectin displays elevated circulating adiponectin and improved insulin sensitivity. Endocrinology 2004, 145, 367–383. [Google Scholar] [CrossRef] [Green Version]
- Miller, R.A.; Chu, Q.; Le Lay, J.; Scherer, P.E.; Ahima, R.S.; Kaestner, K.H.; Foretz, M.; Viollet, B.; Birnbaum, M.J. Adiponectin suppresses gluconeogenic gene expression in mouse hepatocytes independent of LKB1-AMPK signaling. J. Clin. Investig. 2011, 121, 2518–2528. [Google Scholar] [CrossRef] [Green Version]
- Fryer, L.G.; Parbu-Patel, A.; Carling, D. The anti-diabetic drugs rosiglitazone and metformin stimulate AMP-activated protein kinase through distinct signaling pathways. J. Biol. Chem. 2002, 277, 25226–25232. [Google Scholar] [CrossRef] [Green Version]
- Miller, R.A.; Birnbaum, M.J. An energetic tale of AMPK-independent effects of metformin. J. Clin. Investig. 2010, 120, 2267–2270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Huang, T.; Li, P.; Ai, J.; Liu, J.; Bai, W.; Tian, L. Dietary fiber modulates the fermentation patterns of cyanidin-3-O-glucoside in a fiber-type dependent manner. Foods 2021, 10, 1386. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jia, Y.; Wu, C.; Rivera-Piza, A.; Kim, Y.-J.; Lee, J.H.; Lee, S.-J. Mechanism of Action of Cyanidin 3-O-Glucoside in Gluconeogenesis and Oxidative Stress-Induced Cancer Cell Senescence. Antioxidants 2022, 11, 749. https://doi.org/10.3390/antiox11040749
Jia Y, Wu C, Rivera-Piza A, Kim Y-J, Lee JH, Lee S-J. Mechanism of Action of Cyanidin 3-O-Glucoside in Gluconeogenesis and Oxidative Stress-Induced Cancer Cell Senescence. Antioxidants. 2022; 11(4):749. https://doi.org/10.3390/antiox11040749
Chicago/Turabian StyleJia, Yaoyao, Chunyan Wu, Adriana Rivera-Piza, Yeon-Ji Kim, Ji Hae Lee, and Sung-Joon Lee. 2022. "Mechanism of Action of Cyanidin 3-O-Glucoside in Gluconeogenesis and Oxidative Stress-Induced Cancer Cell Senescence" Antioxidants 11, no. 4: 749. https://doi.org/10.3390/antiox11040749
APA StyleJia, Y., Wu, C., Rivera-Piza, A., Kim, Y.-J., Lee, J. H., & Lee, S.-J. (2022). Mechanism of Action of Cyanidin 3-O-Glucoside in Gluconeogenesis and Oxidative Stress-Induced Cancer Cell Senescence. Antioxidants, 11(4), 749. https://doi.org/10.3390/antiox11040749