
Hypothiocyanous Acid Disrupts the Barrier Function of Brain Endothelial Cells



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Methods
2.1. Preparation of HOSCN
2.2. Animals
2.3. Cell Culture and Treatment
2.4. TEER Measurements
2.5. Dextran Permeability Assay
2.6. Viability Assays
2.7. Immunofluorescence Microscopy
2.8. CellProfiler Analysis
2.9. Statistical Analysis
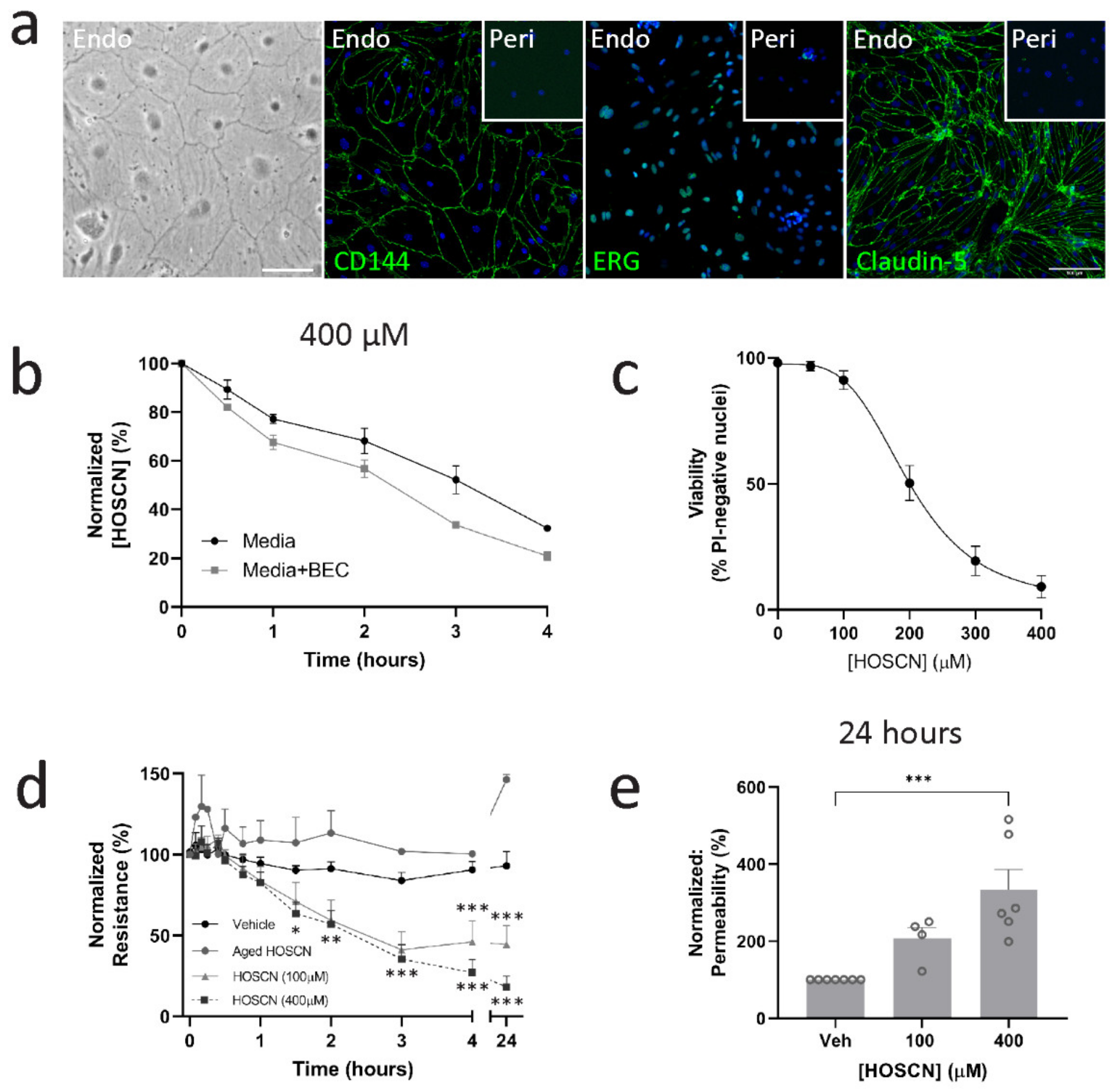
3. Results
3.1. HOSCN Affects Blood–Brain Barrier Permeability Irrespective of Endothelial Cell Death
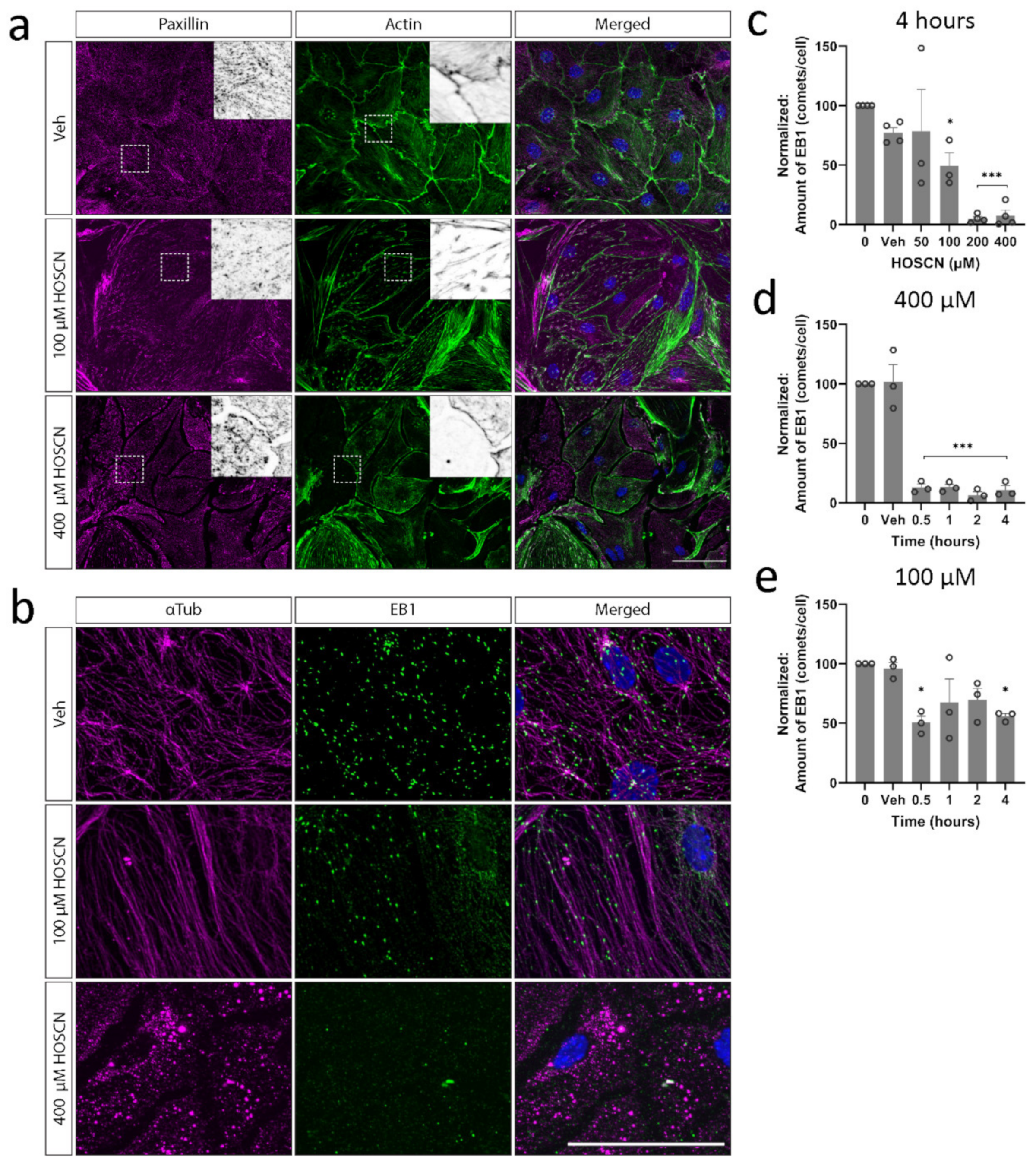
3.2. HOSCN Disrupts Tight and Adherens Junctions in BECs
3.3. HOSCN Disrupts BEC Cytoskeletal Structures
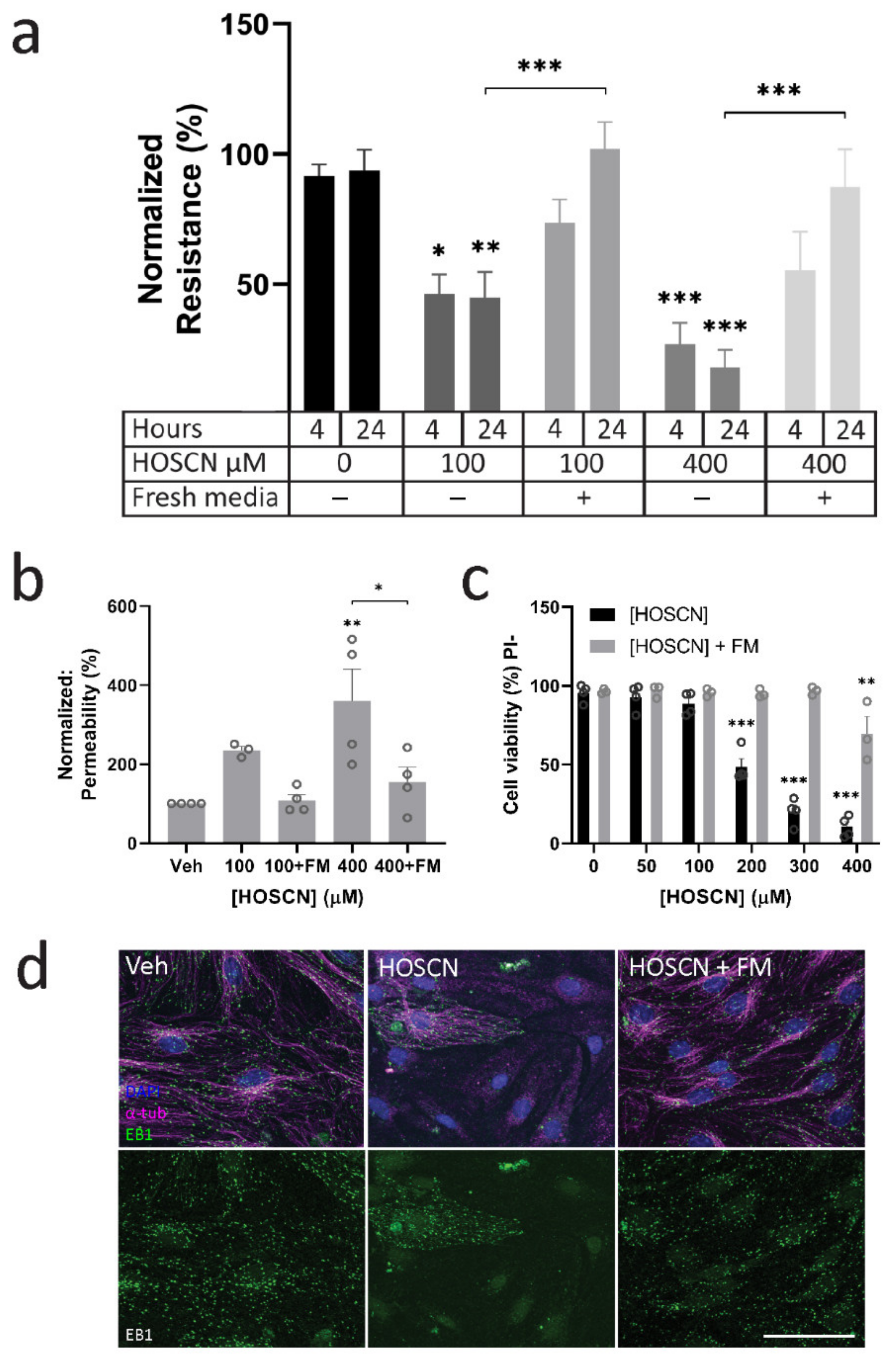
3.4. HOSCN Removal Prevents Barrier Dysfunction
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Armulik, A.; Genové, G.; Mäe, M.; Nisancioglu, M.H.; Wallgard, E.; Niaudet, C.; He, L.; Norlin, J.; Lindblom, P.; Strittmatter, K.; et al. Pericytes regulate the blood-brain barrier. Nature 2010, 468, 557–561. [Google Scholar] [CrossRef] [PubMed]
- Fanning, A.S.; Jameson, B.J.; Jesaitis, L.A.; Anderson, J.M. The tight junction protein ZO-1 establishes a link between the transmembrane protein occludin and the actin cytoskeleton. J. Biol. Chem. 1998, 273, 29745–29753. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.H.; Kuo, K.H.; Leo, J.M. Critical role of actin in modulating BBB permeability. Brain Res. Rev. 2005, 50, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Dejana, E. Endothelial Cell-Cell Junctions: Happy Together. Nat. Rev. 2004, 5, 261–270. [Google Scholar] [CrossRef]
- Nation, D.A.; Sweeney, M.D.; Montagne, A.; Sagare, A.P.; Lina, M.; Pachicano, M.; Sepehrband, F.; Nelson, A.R.; Buennagel, D.P.; Harrington, M.G.; et al. Blood-brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nat. Med. 2019, 25, 270–276. [Google Scholar] [CrossRef]
- Shin, Y.; Choi, S.H.; Kim, E.; Bylykbashi, E.; Kim, J.A.; Chung, S.; Kim, D.Y.; Kamm, R.D.; Tanzi, R.E. Blood–Brain Barrier Dysfunction in a 3D In Vitro Model of Alzheimer’s Disease. Adv. Sci. 2019, 6, 1900962. [Google Scholar] [CrossRef]
- Yang, Y.; Kimura-Ohba, S.; Thompson, J.F.; Salayandia, V.M.; Cosse, M.; Raz, L.; Jalal, F.Y.; Rosenberg, G.A. Vascular tight junction disruption and angiogenesis in spontaneously hypertensive rat with neuroinflammatory white matter injury. Neurobiol. Dis. 2018, 114, 95–110. [Google Scholar] [CrossRef]
- Smyth, L.C.D.; Rustenhoven, J.; Park, T.I.; Schweder, P.; Jansson, D.; Heppner, P.A.; O’Carroll, S.J.; Mee, E.W.; Faull, R.L.M.; Curtis, M.; et al. Unique and shared inflammatory profiles of human brain endothelia and pericytes. J. Neuroinflamm. 2018, 15, 138. [Google Scholar] [CrossRef]
- Kim, S.; Lee, H.; Chung, M.; Jeon, N.L. Engineering of functional, perfusable 3D microvascular networks on a chip. Lab Chip 2013, 13, 1489–1500. [Google Scholar] [CrossRef]
- Winterbourn, C.C.; Kettle, A.J.; Hampton, M.B. Reactive Oxygen Species and Neutrophil Function. Annu. Rev. Biochem. 2016, 85, 765–792. [Google Scholar] [CrossRef]
- Bozonet, S.M.; Scott-Thomas, A.P.; Nagy, P.; Vissers, M.C.M. Hypothiocyanous acid is a potent inhibitor of apoptosis and caspase 3 activation in endothelial cells. Free Radic. Biol. Med. 2010, 49, 1054–1063. [Google Scholar] [CrossRef]
- Wang, J.-G.; Mahmud, S.A.; Nguyen, J.; Slungaard, A. Thiocyanate-Dependent Induction of Endothelial Cell Adhesion Molecule Expression by Phagocyte Peroxidases: A Novel HOSCN-Specific Oxidant Mechanism to Amplify Inflammation. J. Immunol. 2006, 177, 8714–8722. [Google Scholar] [CrossRef] [PubMed]
- Cook, N.L.; Viola, H.M.; Sharov, V.S.; Hool, L.C.; Schoneich, C.; Davies, M.J. Myeloperoxidase-derived oxidants inhibit sarco/endoplasmic reticulum Ca2+ -ATPase activity, and perturb Ca2+ homeostasis in human coronary artery endothelial cells. Free Radic. Biol. Med. 2012, 52, 951–961. [Google Scholar] [CrossRef] [PubMed]
- Van Dalen, C.J.; Whitehouse, M.W.; Winterbourn, C.C.; Kettle, A.J. Thiocyanate and chloride as competing substrates for myeloperoxidase. Biochem. J. 1997, 327, 487–492. [Google Scholar] [CrossRef] [PubMed]
- Pattison, D.I.; Davies, M.J.; Hawkins, C.L. Reactions and reactivity of myeloperoxidase-derived oxidants: Differential biological effects of hypochlorous and hypothiocyanous acids. Free Radic. Res. 2012, 46, 975–995. [Google Scholar] [CrossRef] [PubMed]
- Van Leeuwen, E.; Hampton, M.B.; Smyth, L.C.D. Redox signalling and regulation of the blood-brain barrier. Int. J. Biochem. Cell Biol. 2020, 125, 105794. [Google Scholar] [CrossRef] [PubMed]
- Walter, J.K.; Castro, V.; Voss, M.; Gast, K.; Rueckert, C.; Piontek, J.; Blasig, I.E. Redox-sensitivity of the dimerization of occludin. Cell. Mol. Life Sci. 2009, 66, 3655–3662. [Google Scholar] [CrossRef] [PubMed]
- Walter, J.K.; Rueckert, C.; Voss, M.; Mueller, S.L.; Gast, K.; Blasig, I.E. The Oligomerization of the Coiled Coil-domain of Occluddin Is Redox Sensitive. Ann. N. Y. Acad. Sci. 2009, 27, 19–27. [Google Scholar] [CrossRef]
- Bellmann, C.; Schreivogel, S.; Dabrowski, S.; Schu, M. Highly Conserved Cysteines Are Involved in the Oligomerization of Occludin—Redox Dependency of the Second Extracellular Loop. Antioxid. Redox Signal. 2014, 20, 855–867. [Google Scholar] [CrossRef]
- Wen, H.; Watry, D.D.; Marcondes, M.C.G.; Fox, H.S. Selective Decrease in Paracellular Conductance of Tight Junctions: Role of the First Extracellular Domain of Claudin-5. Mol. Cell. Biol. 2004, 24, 8408–8417. [Google Scholar] [CrossRef]
- Clark, H.M.; Hagedorn, T.D.; Landino, L.M. Hypothiocyanous acid oxidation of tubulin cysteines inhibits microtubule polymerization. Arch. Biochem. Biophys. 2014, 541, 67–73. [Google Scholar] [CrossRef]
- Summers, F.A.; Forsman Quigley, A.; Hawkins, C.L. Identification of proteins susceptible to thiol oxidation in endothelial cells exposed to hypochlorous acid and N-chloramines. Biochem. Biophys. Res. Commun. 2012, 425, 157–161. [Google Scholar] [CrossRef]
- Nagy, P.; Jameson, G.N.L.; Winterbourn, C.C. Kinetics and mechanisms of the reaction of hypothiocyanous acid with 5-thio-2-nitrobenzoic acid and reduced glutathione. Chem. Res. Toxicol. 2009, 22, 1833–1840. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, B.; Kolli, A.R. Transepithelial/Transendothelial Electrical Resistance (TEER) to Measure the Integrity of Blood-Brain Barrier. Blood-Brain Barrier 2019, 142, 99–114. [Google Scholar]
- Taddei, A.; Giampietro, C.; Conti, A.; Orsenigo, F.; Breviario, F.; Pirazzoli, V.; Potente, M.; Daly, C.; Dimmeler, S.; Dejana, E. Endothelial adherens junctions control tight junctions by VE-cadherin-mediated upregulation of claudin-5. Nat. Cell Biol. 2008, 10, 923–934. [Google Scholar] [CrossRef]
- Shi, Y.; Zhang, L.; Pu, H.; Mao, L.; Hu, X.; Jiang, X.; Xunming, J.; Stetler, R.A.; Zhang, F.; Liu, X.; et al. Rapid endothelial cytoskeletal reorganization enables early blood-brain barrier disruption and long-term ischaemic reperfusion brain injury. Nat. Commun. 2016, 7, 10523. [Google Scholar] [CrossRef]
- Dugina, V.; Alieva, I.; Khromova, N.; Kireev, I.; Gunning, P.W.; Kopnin, P. Interaction of microtubules with the actin cytoskeleton via cross-talk of EB1-containing +TIPs and y-actin in epithelial cells. Oncotarget 2016, 7, 18–20. [Google Scholar] [CrossRef]
- Smyth, L.C.D.; Murray, H.C.; Hill, M.; van Leeuwen, E.; Highet, B.; Magon, N.J.; Osanlouy, M.; Mathiesen, S.N.; Mockett, B.; Singh-Bains, M.K.; et al. Neutrophil-vascular interactions drive myeloperoxidase accumulation in the brain in Alzheimer’s disease. Acta Neuropathol. Commun. 2022. accepted. [Google Scholar]
- Yu, G.; Liang, Y.; Huang, Z.; Jones, D.W.; Pritchard, K.A., Jr.; Zhang, H. Inhibition of myeloperoxidase oxidant production by N-acetyl lysyltyrosylcysteine amide reduces brain damage in a murine model of stroke. J. Neuroinflamm. 2016, 13, 119. [Google Scholar] [CrossRef]
- Jucaite, A.; Svenningsson, P.; Rinne, J.O.; Cselenyi, Z.; Varnäs, K.; Johnström, P.; Amini, N.; Kirjavainen, A.; Helin, S.; Minkwitz, M.; et al. Effect of the myeloperoxidase inhibitor AZD3241 on microglia: A PET study in Parkinson’s disease. Brain 2015, 138, 2687–2700. [Google Scholar]
- Volkman, R.; Ben-zur, T.; Kahana, A.; Garty, B.Z.; Offen, D. Myeloperoxidase Deficiency Inhibits Cognitive Decline in the 5XFAD Mouse Model of Alzheimer’s Disease. Front. Neurosci. 2019, 13, 990. [Google Scholar] [CrossRef] [PubMed]
- Cheng, D.; Talib, J.; Stanley, C.P.; Rashid, I.; Michaëlsson, E.; Lindstedt, E.L.; Croft, K.; Kettle, T.; Maghzal, G.J.; Stocker, R. Inhibition of MPO (myeloperoxidase) attenuates endothelial dysfunction in mouse models of vascular inflammation and atherosclerosis. Arter. Thromb. Vasc. Biol. 2019, 39, 1448–1457. [Google Scholar] [CrossRef] [PubMed]
- Brennan, M.; Gaur, A.; Pahuja, A.; Lusis, A.J.; Reynolds, W.F. Mice lacking myeloperoxidase are more susceptible to experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2001, 112, 97–105. [Google Scholar] [CrossRef]
- Morgan, P.E.; Pattison, D.I.; Talib, J.; Summers, F.A.; Harmer, J.A.; Celermajer, D.S.; Hawkins, C.; Davies, M. High plasma thiocyanate levels in smokers are a key determinant of thiol oxidation induced by myeloperoxidase. Free Radic. Biol. Med. 2011, 51, 1815–1822. [Google Scholar] [CrossRef]
- Kevil, C.G.; Ohno, N.; Gute, D.C.; Okayama, N.; Robinson, S.A.; Chaney, E.; Alexander, J. Role of cadherin internalization in hydrogen peroxide-mediated endothelial permeability. Free Radic. Biol. Med. 1998, 24, 1015–1022. [Google Scholar] [CrossRef]
- Tornavaca, O.; Chia, M.; Dufton, N.; Almagro, L.O.; Conway, D.E.; Randi, A.M.; Schwartz, M.A.; Matter, K.; Balda, M.S. ZO-1 controls endothelial adherens junctions, cell–cell tension, angiogenesis, and barrier formation. J. Cell Biol. 2015, 208, 821–838. [Google Scholar] [CrossRef]
- Lee, M.J.; Thangada, S.; Claffey, K.P.; Ancellin, N.; Liu, C.H.; Kluk, M.; Volpi, M.; Sha’Afi, R.I.; Hla, T. Vascular endothelial cell adherens junction assembly and morphogenesis induced by sphingosine-1-phosphate. Cell 1999, 99, 301–312. [Google Scholar] [CrossRef]
- Blanchoin, L.; Boujemaa-Paterski, R.; Sykes, C.; Plastino, J. Actin dynamics, architecture, and mechanics in cell motility. Physiol. Rev. 2014, 94, 235–263. [Google Scholar] [CrossRef]
- Üllen, A.; Fauler, G.; Bernhart, E.; Nusshold, C.; Reicher, H.; Leis, H.J.; Malle, E.; Sattler, W. Phloretin ameliorates 2-chlorohexadecanal-mediated brain microvascular endothelial cell dysfunction in vitro. Free Radic. Biol. Med. 2012, 53, 1770–1781. [Google Scholar] [CrossRef]
- Nusshold, C.; Üllen, A.; Kogelnik, N.; Bernhart, E.; Reicher, H. Assessment of electrophile damage in a human brain endothelial cell line utilizing a clickable alkyne analogue of 2- chlorohexadecanal. Free Radic. Biol. Med. 2016, 90, 59–74. [Google Scholar] [CrossRef]
- Dudek, S.M.; Garcia, J.G.N. Cytoskeletal regulation of pulmonary vascular permeability. J. Appl. Physiol. 2001, 91, 1487–1500. [Google Scholar] [CrossRef]
- Shaji, C.A.; Robinson, B.D.; Yeager, A.; Beeram, M.R.; Davis, M.L.; Isbell, C.L.; Huang, J.H.; Tharakan, B. The Tri-phasic Role of Hydrogen Peroxide in Blood-Brain Barrier Endothelial cells. Sci. Rep. 2019, 9, 133. [Google Scholar] [CrossRef]
- Lee, H.; Namkoong, K.; Kim, D.; Kim, K.; Cheong, Y.; Kim, S.; Lee, W.-B.; Kim, K.-Y. Hydrogen peroxide-induced alterations of tight junction proteins in bovine brain microvascular endothelial cells. Microvasc. Res. 2004, 68, 231–238. [Google Scholar] [CrossRef]
- Ochoa, L.; Waypa, G.; Mahoney, J.R.; Rodriguez, L.; Minnear, F.L. Contrasting effects of hypochlorous acid and hydrogen peroxide on endothelial permeability: Prevention with cAMP drugs. Am. J. Respir. Crit. Care Med. 1997, 156 Pt I, 1247–1255. [Google Scholar] [CrossRef][Green Version]
- Schreibelt, G.; Kooij, G.; Reijerkerk, A.; van Doorn, R.; Gringhuis, S.I.; van der Pol, S.; Weksler, B.B.; Romero, I.A.; Couraud, P.; Piontek, J.; et al. Reactive oxygen species alter brain endothelial tight junction dynamics via RhoA, PI3 kinase, and PKB signaling. FASEB J. 2007, 21, 3666–3676. [Google Scholar] [CrossRef]
- Birukova, A.A.; Arce, F.T.; Moldobaeva, N.; Dudek, S.M.; Garcia, J.G.N.; Lal, R.; Birukov, K.G. Endothelial permeability is controlled by spatially defined cytoskeletal mechanics: Atomic force microscopy force mapping of pulmonary endothelial monolayer. Nanomed. Nanotechnol. Biol. Med. 2009, 5, 30–41. [Google Scholar] [CrossRef]
- Jiao, H.; Wang, Z.; Liu, Y. Specific Role of Tight Junction Proteins Claudin-5, Occludin, and ZO-1 of the Blood–Brain Barrier in a Focal Cerebral Ischemic Insult. J. Mol. Neurosci. 2011, 44, 130–139. [Google Scholar] [CrossRef]
- Piehl, C.; Piontek, J.; Cording, J.; Wolburg, H.; Blasig, I.E. Participation of the second extracellular loop of claudin-5 in paracellular tightening against ions, small and large molecules. Cell. Mol. Life Sci. 2010, 67, 2131–2140. [Google Scholar] [CrossRef]
- Piontek, J.; Winkler, L.; Wolburg, H.; Mu, S.L.; Zuleger, N.; Piehl, C.; Wiesner, B.; Krause, G.; Blasig, I.E. Formation of tight junction: Determinants of homophilic interaction between classic claudins. FASEB J. 2008, 22, 146–158. [Google Scholar] [CrossRef]
- Nitta, T.; Hata, M.; Gotoh, S.; Seo, Y.; Sasaki, H.; Hashimoto, N.; Furuse, M.; Tsukita, S. Size-selective loosening of the blood-brain barrier in claudin-5–deficient mice. J. Cell Biol. 2003, 161, 653–660. [Google Scholar] [CrossRef]
- Chang, F.; Flavahan, S.; Flavahan, N.A. Impaired activity of adherens junctions contributes to endothelial dilator dysfunction in ageing rat arteries. J. Physiol. 2017, 595, 5143–5158. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
van Leeuwen, E.; Hampton, M.B.; Smyth, L.C.D. Hypothiocyanous Acid Disrupts the Barrier Function of Brain Endothelial Cells. Antioxidants 2022, 11, 608. https://doi.org/10.3390/antiox11040608
van Leeuwen E, Hampton MB, Smyth LCD. Hypothiocyanous Acid Disrupts the Barrier Function of Brain Endothelial Cells. Antioxidants. 2022; 11(4):608. https://doi.org/10.3390/antiox11040608
Chicago/Turabian Stylevan Leeuwen, Eveline, Mark B. Hampton, and Leon C. D. Smyth. 2022. "Hypothiocyanous Acid Disrupts the Barrier Function of Brain Endothelial Cells" Antioxidants 11, no. 4: 608. https://doi.org/10.3390/antiox11040608
APA Stylevan Leeuwen, E., Hampton, M. B., & Smyth, L. C. D. (2022). Hypothiocyanous Acid Disrupts the Barrier Function of Brain Endothelial Cells. Antioxidants, 11(4), 608. https://doi.org/10.3390/antiox11040608