
Shedding a New Light on Skin Aging, Iron- and Redox-Homeostasis and Emerging Natural Antioxidants

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. ‘Intrinsic’ (Chronological) Skin Aging versus ‘Extrinsic’ Skin Photoaging
2.1. Terrestrial Sunlight Radiations
2.2. Photodamaging Effects of Solar UVA and VIS Radiations
2.3. Photodamaging Effects of Terrestrial IRA Radiation
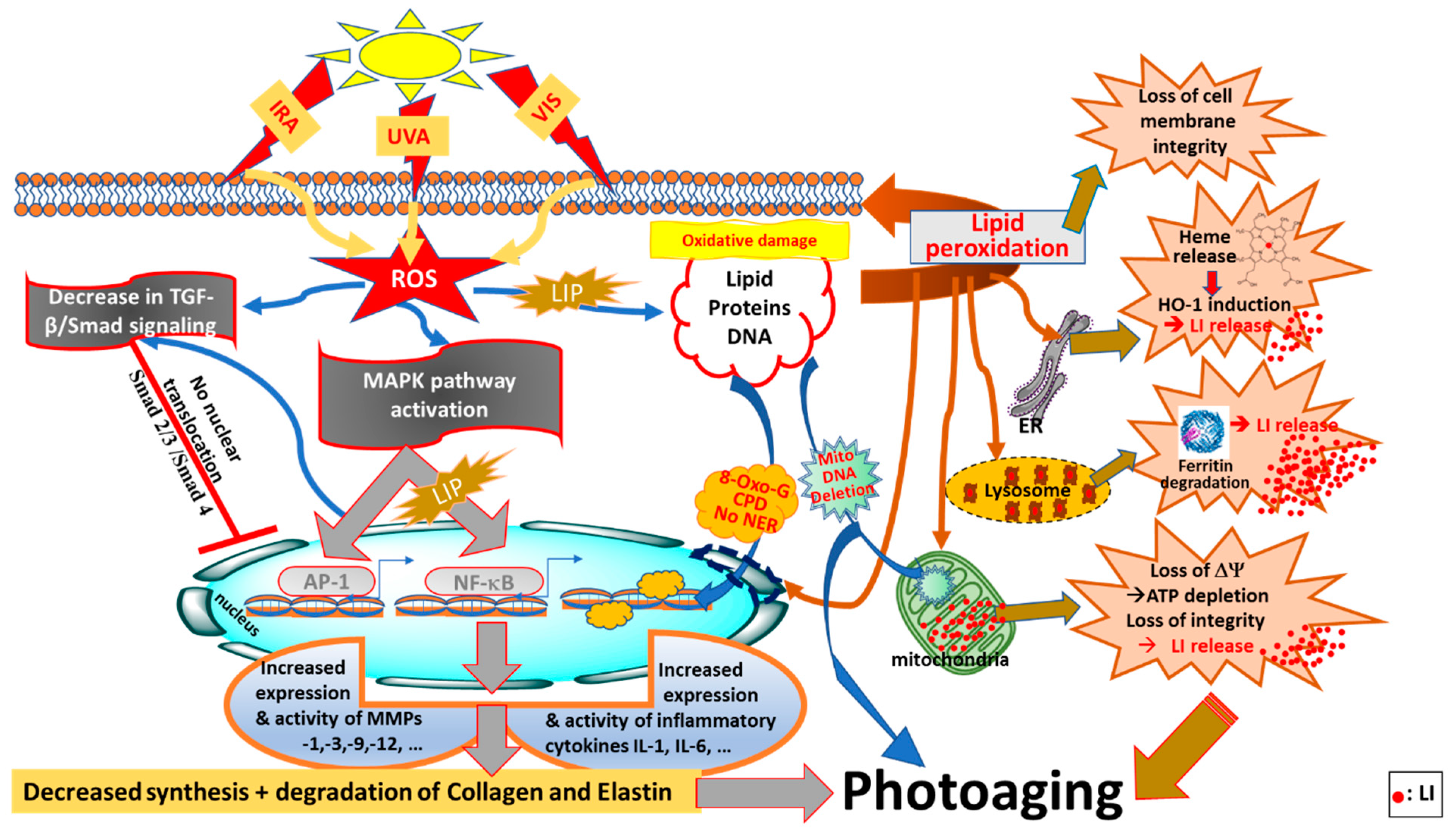
2.4. Photoaging Effects of Solar UVA, VIS and IRA
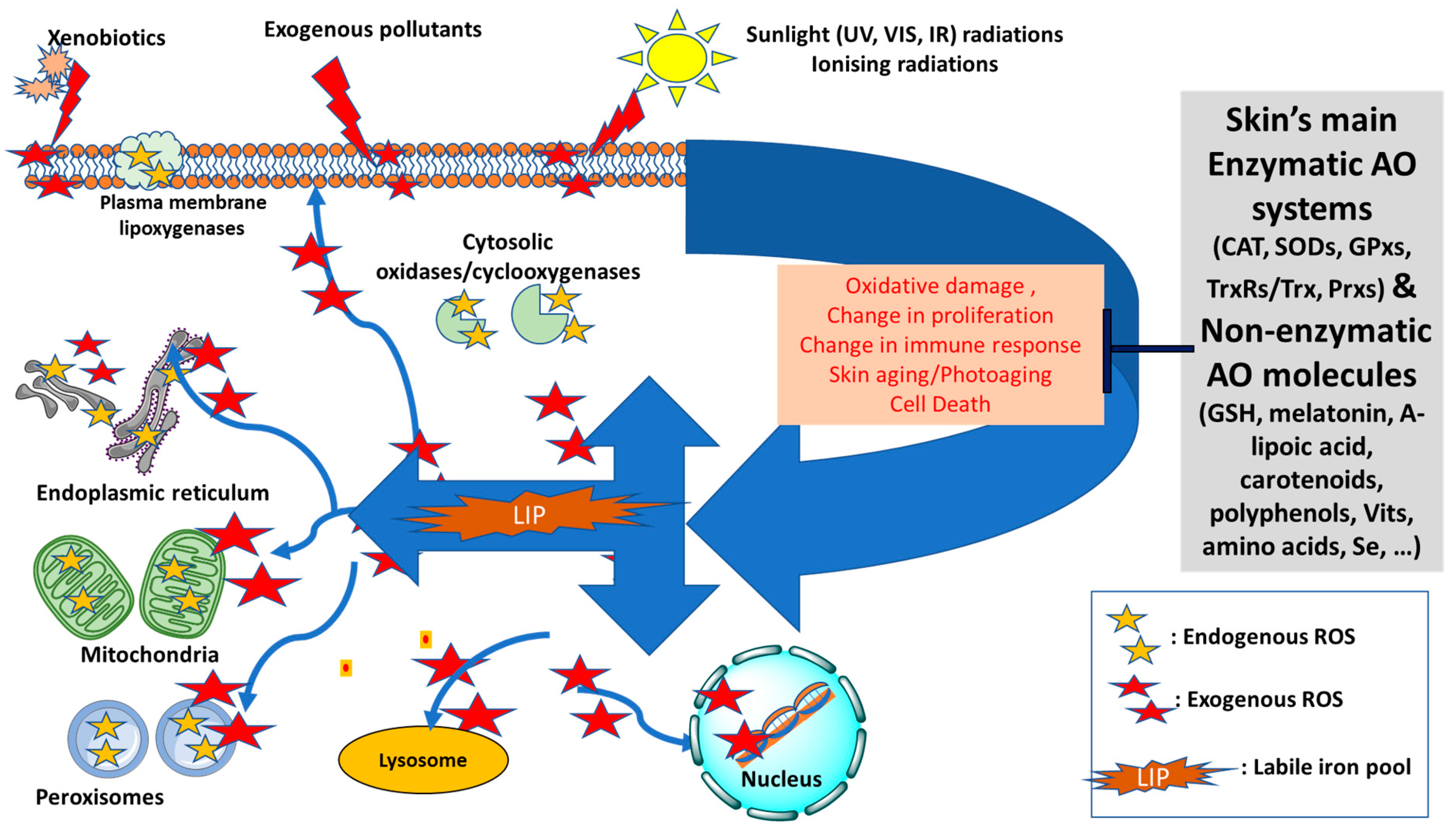
3. Skin, Oxidative Stress and Antioxidant Defense
3.1. Superoxide Dismutase (SOD), a Major Antioxidant Enzyme in the Skin
3.2. Catalase, a Major Antioxidant Enzyme in the Skin
3.3. Glutathione and Thioredoxin Antioxidant Systems in the Skin
3.4. Nuclear Factor E2-Related Factor 2 in Skin Redox Homeostasis
4. Natural-Based Antioxidants for Skin Protection
4.1. Chromanols and Chromenols as Promising Skin Antiaging and Photoprotectants
4.2. Polyphenols as Skin Antiaging and Photoprotecive Agents
4.2.1. Apigenin
4.2.2. Baicalein and Baicalin
4.3. Leontopodium alpinum
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Proksch, E.; Brandner, J.M.; Jensen, J.-M. The skin: An indispensable barrier. Exp. Dermatol. 2008, 17, 1063–1072. [Google Scholar] [CrossRef]
- Madison, K.C. Barrier function of the skin: “La raison d’être” of the epidermis. J. Investig. Dermatol. 2003, 121, 231–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krutmann, J.; Bouloc, A.; Sore, G.; Bernard, B.A.; Passeron, T. The skin aging exposome. J. Dermatol. Sci. 2017, 85, 152–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastore, S.; Korkina, L. Redox imbalance in T cell-mediated skin diseases. Mediat. Inflamm. 2010, 2010, 861949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aroun, A.; Zhong, J.L.; Tyrrell, R.M.; Pourzand, C. Iron, oxidative stress and the example of solar ultraviolet A radiation. Photochem. Photobiol. Sci. 2012, 11, 118–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Addor, F.A.S. Antioxidants in dermatology. An. Bras. Dermatol. 2017, 92, 356–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finkel, T. Redox-dependent signal transduction. FEBS Lett. 2000, 476, 52–54. [Google Scholar] [CrossRef] [Green Version]
- Simon, H.-U.; Haj-Yehia, A.; Levi-Schaffer, F. Role of reactive oxygen species (ROS) in apoptosis induction. Apoptosis 2000, 5, 415–418. [Google Scholar] [CrossRef]
- Fleury, C.; Mignotte, B.; Vayssière, J.-L. Mitochondrial reactive oxygen species in cell death signaling. Biochimie 2002, 84, 131–141. [Google Scholar] [CrossRef]
- Rinnerthaler, M.; Bischof, J.; Streubel, M.K.; Trost, A.; Richter, K. Oxidative stress in aging human skin. Biomolecules 2015, 5, 545–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, H.A.; Mutus, B. Protein disulfide isomerase a multifunctional protein with multiple physiological roles. Front. Chem. 2014, 2, 70. [Google Scholar] [CrossRef] [Green Version]
- Pourzand, C.; Watkin, R.D.; Brown, J.E.; Tyrrell, R.M. Ultraviolet A radiation induces immediate release of iron in human primary skin fibroblasts: The role of ferritin. Proc. Natl. Acad. Sci. USA 1999, 96, 6751–6756. [Google Scholar] [CrossRef] [Green Version]
- Zhong, J.L.; Yiakouvaki, A.; Holley, P.; Tyrrell, R.M.; Pourzand, C. Susceptibility of skin cells to UVA-induced necrotic cell death reflects the intracellular level of labile iron. J. Investig. Dermatol. 2004, 123, 771–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katsarou, A.; Pantopoulos, K. Basics and principles of cellular and systemic iron homeostasis. Mol. Asp. Med. 2020, 75, 100866. [Google Scholar] [CrossRef] [PubMed]
- Che, M.; Wang, R.; Li, X.; Wang, H.-Y.; Zheng, X.S. Expanding roles of superoxide dismutases in cell regulation and cancer. Drug Discov. Today 2016, 21, 143–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reelfs, O.; Eggleston, I.M.; Pourzand, C. Skin protection against UVA-induced iron damage by multiantioxidants and iron chelating drugs/prodrugs. Curr. Drug Metab. 2010, 11, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Pelle, E.; Jian, J.; Zhang, Q.; Muizzuddin, N.; Yang, Q.; Dai, J.; Maes, D.; Pernodet, N.; Yarosh, D.B.; Frenkel, K.; et al. Menopause increases the iron storage protein ferritin in skin. J. Cosmet. Sci. 2013, 64, 175–179. [Google Scholar] [PubMed]
- Jian, J.; Pelle, E.; Huang, X. Iron and menopause: Does increased iron affect the health of postmenopausal women? Antioxid. Redox Signal. 2009, 11, 2939–2943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelle, E.; Jian, J.L.; Declercq, L.; Dong, K.; Yang, Q.; Pourzand, C.; Maes, D.; Pernodet, N.; Yarosh, D.B.; Huang, X. Protection against ultraviolet A-induced oxidative damage in normal human epidermal keratinocytes under post-menopausal conditions by an ultraviolet A-activated caged-iron chelator: A pilot study. Photodermatol. Photoimmunol. Photomed. 2011, 27, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.J.; Fowler, M.; Naftalin, R.J.; Siow, R.C.M. UVA irradiation increases ferrous iron release from human skin fibroblast and endothelial cell ferritin: Consequences for cell senescence and aging. Free Radic. Biol. Med. 2020, 155, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Warraich, U.-E.; Hussain, F.; Kayani, H.U.R. Aging—Oxidative stress, antioxidants and computational modeling. Heliyon 2020, 6, e04107. [Google Scholar] [CrossRef]
- Rudolf, J.; Raad, H.; Taieb, A.; Rezvani, H.R. NADPH oxidases and their roles in skin homeostasis and carcinogenesis. Antioxid. Redox Signal. 2018, 28, 1238–1261. [Google Scholar] [CrossRef]
- Zhang, S.; Duan, E. Fighting against skin aging: The way from bench to bedside. Cell Transplant. 2018, 27, 729–738. [Google Scholar] [CrossRef] [PubMed]
- Reelfs, O.; Abbate, V.; Hider, R.; Pourzand, C. A powerful mitochondria-targeted iron chelator affords high photoprotection against solar ultraviolet A radiation. J. Investig. Dermatol. 2016, 136, 1692–1700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reelfs, O.; Abbate, V.; Cilibrizzi, A.; Pook, M.A.; Hider, R.C.; Pourzand, C. The role of mitochondrial labile iron in Friedreich’s ataxia skin fibroblasts sensitivity to ultraviolet A. Metallomics 2019, 11, 656–665. [Google Scholar] [CrossRef] [Green Version]
- Trachootham, D.; Lu, W.; Ogasawara, M.A.; Valle, N.R.-D.; Huang, P. Redox regulation of cell survival. Antioxid. Redox Signal. 2008, 10, 1343–1374. [Google Scholar] [CrossRef] [Green Version]
- Bickers, D.R.; Athar, M. Oxidative stress in the pathogenesis of skin disease. J. Investig. Dermatol. 2006, 126, 2565–2575. [Google Scholar] [CrossRef] [Green Version]
- Sreedhar, A.; Aguilera-Aguirre, L.; Singh, K.K. Mitochondria in skin health, aging, and disease. Cell Death Dis. 2020, 11, 444. [Google Scholar] [CrossRef]
- Uitto, J. The role of elastin and collagen in cutaneous aging: Intrinsic aging versus photoexposure. J. Drugs Dermatol. 2008, 7, s12–s16. [Google Scholar] [PubMed]
- Fisher, G.J.; Kang, S.; Varani, J.; Bata-Csorgo, Z.; Wan, Y.; Datta, S.; Voorhees, J.J. Mechanisms of photoaging and chronological skin aging. Arch. Dermatol. 2002, 138, 1462–1470. [Google Scholar] [CrossRef]
- Rittié, L.; Fisher, G.J. Natural and sun-induced aging of human skin. Cold Spring Harb. Perspect. Med. 2015, 5, a015370. [Google Scholar] [CrossRef]
- Michniak-Kohn, B.; Leonardi, G.R. An overview about oxidation in clinical practice of skin aging. An. Bras. Dermatol. 2017, 92, 367–374. [Google Scholar] [CrossRef] [Green Version]
- Makrantonaki, E.; Zouboulis, C.C.; William, J. Characteristics and pathomechanisms of endogenously aged skin. Dermatology 2007, 214, 352–360. [Google Scholar] [CrossRef] [PubMed]
- Krutmann, J.; Gilchrest, B.A. Photoaging of skin. In Skin Aging; Gilchrest, B.A., Krutmann, J., Eds.; Springer: New York, NY, USA, 2006; pp. 33–44. [Google Scholar]
- Lavker, R.M. Cutaneous aging: Chronologic versus photoaging. In Photodamage; Gilchrest, B.A., Ed.; Blackwell Science: Cambridge, MA, USA, 1995; pp. 123–135. [Google Scholar]
- Braverman, I.M.; Fonferko, E. Studies in cutaneous aging: I. The elastic fiber network. J. Investig. Dermatol. 1982, 78, 434–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCabe, M.C.; Hill, R.C.; Calderone, K.; Cui, Y.; Yan, Y.; Quan, T.; Fisher, G.J.; Hansen, K.C. Alterations in extracellular matrix composition during aging and photoaging of the skin. Matrix Biol. Plus 2020, 8, 100041. [Google Scholar] [CrossRef] [PubMed]
- Luebberding, S.; Krueger, N.; Kerscher, M. Age-related changes in skin barrier function—Quantitative evaluation of 150 female subjects. Int. J. Cosmet. Sci. 2012, 35, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Fligiel, S.E.; Varani, J.; Datta, S.C.; Kang, S.; Fisher, G.J.; Voorhees, J.J. Collagen degradation in aged/photodamaged skin in vivo and after exposure to matrix metalloproteinase-1 in vitro. J. Investig. Dermatol. 2003, 120, 842–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varani, J.; Dame, M.K.; Rittié, L.; Fligiel, S.E.; Kang, S.; Fisher, G.J.; Voorhees, J.J. Decreased collagen production in chronologically aged skin: Roles of age-dependent alteration in fibroblast function and defective mechanical stimulation. Am. J. Pathol. 2006, 168, 1861–1868. [Google Scholar] [CrossRef] [Green Version]
- Scharffetter–Kochanek, K.; Brenneisen, P.; Wenk, J.; Herrmann, G.; Ma, W.; Kuhr, L.; Meewes, C.; Wlaschek, M. Photoaging of the skin from phenotype to mechanisms. Exp. Gerontol. 2000, 35, 307–316. [Google Scholar] [CrossRef]
- Freitas-Rodríguez, S.; Folgueras, A.R.; López-Otín, C. The role of matrix metalloproteinases in aging: Tissue remodeling and beyond. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2017, 1864, 2015–2025. [Google Scholar] [CrossRef]
- Panich, U.; Slominski, A.T. Editorial: Redox biology of skin aging and carcinogenesis: The role of natural antioxidants as potential protective agents. Front. Pharmacol. 2020, 11, 249. [Google Scholar] [CrossRef] [PubMed]
- Quan, T.; Fisher, G.J. Role of age-associated alterations of the dermal extracellular matrix microenvironment in human skin aging: A mini-review. Gerontology 2015, 61, 427–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Treiber, N.; Maity, P.; Singh, K.; Ferchiu, F.; Wlaschek, M.; Scharffetter-Kochanek, K. The role of manganese superoxide dismutase in skin aging. Dermato-Endocrinology 2012, 4, 232–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sprenger, C.C.; Plymate, S.R.; Reed, M.J. Aging-related alterations in the extracellular matrix modulate the microenvironment and influence tumor progression. Int. J. Cancer 2010, 127, 2739–2748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisher, G.J.; Quan, T.; Purohit, T.; Shao, Y.; Cho, M.K.; He, T.; Varani, J.; Kang, S.; Voorhees, J.J. Collagen fragmentation promotes oxidative stress and elevates matrix metalloproteinase-1 in fibroblasts in aged human skin. Am. J. Pathol. 2009, 174, 101–114. [Google Scholar] [CrossRef] [Green Version]
- Xia, W.; Hammerberg, C.; Li, Y.; He, T.; Quan, T.; Voorhees, J.J.; Fisher, G.J. Expression of catalytically active matrix metalloproteinase-1 in dermal fibroblasts induces collagen fragmentation and functional alterations that resemble aged human skin. Aging Cell 2013, 12, 661–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krutmann, J.; Schroeder, P. Role of mitochondria in photoaging of human skin: The defective powerhouse model. J. Investig. Dermatol. Symp. Proc. 2009, 14, 44–49. [Google Scholar] [CrossRef] [Green Version]
- Stout, R.; Birch-Machin, M. Mitochondria’s role in skin ageing. Biology 2019, 8, 29. [Google Scholar] [CrossRef] [Green Version]
- Kar, S.; Subbaram, S.; Carrico, P.M.; Melendez, J.A. Redox-control of matrix metalloproteinase-1: A critical link between free radicals, matrix remodeling and degenerative disease. Respir. Physiol. Neurobiol. 2010, 174, 299–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawrence, K.P.; Douki, T.; Sarkany, R.P.E.; Acker, S.; Herzog, B.; Young, A.R. The UV/visible radiation boundary region (385–405 nm) damages skin cells and induces “dark” cyclobutane pyrimidine dimers in human skin in vivo. Sci. Rep. 2018, 8, 12722. [Google Scholar] [CrossRef] [PubMed]
- Sondenheimer, K.; Krutmann, J. Novel means for photoprotection. Front. Med. 2018, 5, 162. [Google Scholar] [CrossRef] [Green Version]
- Grether-Beck, S.; Marini, A.; Jaenicke, T.; Krutmann, J. Effective photoprotection of human skin against infrared A radiation by topically applied antioxidants: Results from a vehicle controlled, double-blind, randomized study. Photochem. Photobiol. 2014, 91, 248–250. [Google Scholar] [CrossRef] [PubMed]
- Diffey, B.; Osterwalder, U. Labelled sunscreen SPFs may overestimate protection in natural sunlight. Photochem. Photobiol. Sci. 2017, 16, 1519–1523. [Google Scholar] [CrossRef]
- Young, A.R.; Harrison, G.I.; Chadwick, C.A.; Nikaido, O.; Ramsden, J.; Potten, C.S. The similarity of action spectra for thymine dimers in human epidermis and erythema suggests that DNA is the chromophore for erythema. J. Investig. Dermatol. 1998, 111, 982–988. [Google Scholar] [CrossRef] [PubMed]
- Young, A.R.; Claveau, J.; Rossi, A.B. Ultraviolet radiation and the skin: Photobiology and sunscreen photoprotection. J. Am. Acad. Dermatol. 2017, 76, S100–S109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schieke, S.M.; Schroeder, P.; Krutmann, J. Cutaneous effects of infrared radiation: From clinical observations to molecular response mechanisms. Photodermatol. Photoimmunol. Photomed. 2003, 19, 228–234. [Google Scholar] [CrossRef] [PubMed]
- Tyrrell, R.M. The molecular and cellular pathology of solar ultraviolet radiation. Mol. Asp. Med. 1994, 15, 1–77. [Google Scholar]
- Tyrrell, R.M. Activation of mammalian gene expression by the UV component of sunlight—From models to reality. BioEssays 1996, 18, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, I.V.; Mappes, T.; Schaupp, P.; Lappe, C.; Wahl, S. Ultraviolet radiation oxidative stress affects eye health. J. Biophotonics 2018, 11, e201700377. [Google Scholar] [CrossRef] [Green Version]
- Bruls, W.A.G.; Slaper, H.; Van Der Leun, J.C.; Berrens, L. Transmission of human epidermis and stratum corneum as a function of thickness in the ultraviolet and visible wavelengths. Photochem. Photobiol. 1984, 40, 485–494. [Google Scholar] [CrossRef]
- Vile, G.F.; Tyrrell, R.M. UVA radiation-induced oxidative damage to lipids and proteins in vitro and in human skin fibroblasts is dependent on iron and singlet oxygen. Free Radic. Biol. Med. 1995, 18, 721–730. [Google Scholar] [CrossRef]
- Morlière, P.; Moysan, A.; Santus, R.; Hüppe, G.; Mazière, J.-C.; Dubertret, L. UVA-induced lipid peroxidation in cultured human fibroblasts. Biochim. Biophys. Acta (BBA) Lipids Lipid Metab. 1991, 1084, 261–268. [Google Scholar] [CrossRef]
- Punnonen, K.; Puntala, A.; Ahotupa, M. Effects of ultraviolet A and B irradiation on lipid peroxidation and activity of the antioxidant enzymes in keratinocytes in culture. Photodermatol. Photoimmunol. Photomed. 1991, 8, 3–6. [Google Scholar] [PubMed]
- Kvam, E.; Hejmadi, V.; Ryter, S.; Pourzand, C.; Tyrrell, R.M. Heme oxygenase activity causes transient hypersensitivity to oxidative ultraviolet a radiation that depends on release of iron from heme. Free Radic. Biol. Med. 2000, 28, 1191–1196. [Google Scholar] [CrossRef]
- Aust, S.D.; Morehouse, L.A.; Thomas, C.E. Role of metals in oxygen radical reactions. J. Free Radic. Biol. Med. 1985, 1, 3–25. [Google Scholar] [CrossRef]
- Girotti, A.W. Photosensitized oxidation of membrane lipids: Reaction pathways, cytotoxic effects, and cytoprotective mechanisms. J. Photochem. Photobiol. B Biol. 2001, 63, 103–113. [Google Scholar] [CrossRef]
- Dissemond, J.; Schneider, L.A.; Brenneisen, P.; Briviba, K.; Wenk, J.; Wlaschek, M.; Scharffetter-Kochanek, K. Protective and determining factors for the overall lipid peroxidation in ultraviolet A1-irradiated fibroblasts: In vitro and in vivo investigations. Br. J. Dermatol. 2003, 149, 341–349. [Google Scholar] [CrossRef]
- Wondrak, G.T.; Jacobson, M.K.; Jacobson, E.L. Endogenous UVA-photosensitizers: Mediators of skin photodamage and novel targets for skin photoprotection. Photochem. Photobiol. Sci. 2006, 5, 215–237. [Google Scholar] [CrossRef] [PubMed]
- Grether-Beck, S.; Marini, A.; Jaenicke, T.; Krutmann, J. Photoprotection of human skin beyond ultraviolet radiation. Photodermatol. Photoimmunol. Photomed. 2014, 30, 167–174. [Google Scholar] [CrossRef]
- Karran, P.; Brem, R. Protein oxidation, UVA and human DNA repair. DNA Repair 2016, 44, 178–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajapakse, A.; Suraweera, A.; Boucher, D.; Naqi, A.; O’Byrne, K.; Richard, D.J.; Croft, L.V. Redox regulation in the base excision repair pathway: Old and new players as cancer therapeutic targets. Curr. Med. Chem. 2020, 27, 1901–1921. [Google Scholar] [CrossRef] [PubMed]
- Pinnell, S.R. Cutaneous photodamage, oxidative stress, and topical antioxidant protection. J. Am. Acad. Dermatol. 2003, 48, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Cadet, J.; Douki, T.; Ravanat, J.-L. Oxidatively generated damage to cellular DNA by UVB and UVA radiation. Photochem. Photobiol. 2015, 91, 140–155. [Google Scholar] [CrossRef] [PubMed]
- Courdavault, S.; Baudouin, C.; Charveron, M.; Favier, A.; Cadet, J.; Douki, T. Larger yield of cyclobutane dimers than 8-oxo-7,8-dihydroguanine in the DNA of UVA-irradiated human skin cells. Mutat. Res. Mol. Mech. Mutagen. 2004, 556, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Mouret, S.; Baudouin, C.; Charveron, M.; Favier, A.; Cadet, J.; Douki, T. Cyclobutane pyrimidine dimers are predominant DNA lesions in whole human skin exposed to UVA radiation. Proc. Natl. Acad. Sci. USA 2006, 103, 13765–13770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reelfs, O.; Tyrrell, R.M.; Pourzand, C. Ultraviolet a radiation-induced immediate iron release is a key modulator of the activation of NF-κB in human skin fibroblasts. J. Investig. Dermatol. 2004, 122, 1440–1447. [Google Scholar] [CrossRef] [PubMed]
- Kvam, E.; Noel, A.; Basu-Modak, S.; Tyrrell, R.M. Cyclooxygenase dependent release of heme from microsomal hemeproteins correlates with induction of heme oxygenase 1 transcription in human fibroblasts. Free Radic. Biol. Med. 1999, 26, 511–517. [Google Scholar] [CrossRef]
- Valencia, A.; Kochevar, I.E. Nox1-based NADPH oxidase is the major source of UVA-induced reactive oxygen species in human keratinocytes. J. Investig. Dermatol. 2008, 128, 214–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scharffetter-Kochanek, K.; Wlaschek, M.; Brenneisen, P.; Schauen, M.; Blaudschun, R.; Wenk, J. UV-induced reactive oxygen species in photocarcinogenesis and photoaging. Biol. Chem. 1997, 378, 1247–1258. [Google Scholar]
- Reeve, V.E.; Domanski, D.; Slater, M. Radiation sources providing increased UVA/UVB ratios induce photoprotection dependent on the UVA dose in hairless mice. Photochem. Photobiol. 2006, 82, 406–411. [Google Scholar] [CrossRef]
- Zhong, J.L.; Edwards, G.P.; Raval, C.; Li, H.; Tyrrell, R.M. The role of Nrf2 in ultraviolet A mediated heme oxygenase 1 induction in human skin fibroblasts. Photochem. Photobiol. Sci. 2010, 9, 18–24. [Google Scholar] [CrossRef]
- Soares, M.P.; Marguti, I.; Cunha, A.; Larsen, R. Immunoregulatory effects of HO-1: How does it work? Curr. Opin. Pharmacol. 2009, 9, 482–489. [Google Scholar] [CrossRef]
- Gozzelino, R.; Jeney, V.; Soares, M.P. Mechanisms of cell protection by heme oxygenase-1. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 323–354. [Google Scholar] [CrossRef] [Green Version]
- Nisar, M.F.; Parsons, K.S.G.; Bian, C.X.; Zhong, J.L. UVA irradiation induced heme oxygenase-1: A novel phototherapy for morphea. Photochem. Photobiol. 2014, 91, 210–220. [Google Scholar] [CrossRef] [PubMed]
- Tyrrell, R.M. Modulation of gene expression by the oxidative stress generated in human skin cells by UVA radiation and the restoration of redox homeostasis. Photochem. Photobiol. Sci. 2012, 11, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Zastrow, L.; Groth, N.; Klein, F.; Kockott, D.; Lademann, J.; Renneberg, R.; Ferrero, L. The missing link—Light-induced (280–1600 nm) free radical formation in human skin. Ski. Pharmacol. Physiol. 2009, 22, 31–44. [Google Scholar] [CrossRef]
- Mahmoud, B.H.; Ruvolo, E.; Hexsel, C.L.; Liu, Y.; Owen, M.R.; Kollias, N.; Lim, H.W.; Hamzavi, I.H. Impact of long-wavelength UVA and visible light on melanocompetent skin. J. Investig. Dermatol. 2010, 130, 2092–2097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kvam, E.; Tyrrell, R.M. Induction of oxidative DNA base damage in human skin cells by UV and near visible radiation. Carcinogenesis 1997, 18, 2379–2384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenstein, B.S.; Mitchell, D.L. Action spectra for the induction of pyrimidine(6-4)pyrimidone photoproducts and cyclobutane pyrimidine dimers in normal human skin fibroblasts. Photochem. Photobiol. 1987, 45, 775–780. [Google Scholar] [CrossRef] [PubMed]
- Kielbassa, C.; Roza, L.; Epe, B. Wavelength dependence of oxidative DNA damage induced by UV and visible light. Carcinogenesis 1997, 18, 811–816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiarelli-Neto, O.; Ferreira, A.S.; Martins, W.K.; Pavani, C.; Severino, D.; Faião-Flores, F.; Maria-Engler, S.S.; Aliprandini, E.; Martinez, G.R.; Di Mascio, P.; et al. Melanin photosensitization and the effect of visible light on epithelial cells. PLoS ONE 2014, 9, e113266. [Google Scholar] [CrossRef] [Green Version]
- Premi, S.; Wallisch, S.; Mano, C.M.; Weiner, A.B.; Bacchiocchi, A.; Wakamatsu, K.; Bechara, E.J.; Halaban, R.; Douki, T.; Brash, D.E. Chemiexcitation of melanin derivatives induces DNA photoproducts long after UV exposure. Science 2015, 347, 842–847. [Google Scholar] [CrossRef] [PubMed]
- Delinasios, G.J.; Karbaschi, M.; Cooke, M.S.; Young, A.R. Vitamin E inhibits the UVAI induction of “light” and “dark” cyclobutane pyrimidine dimers, and oxidatively generated DNA damage, in keratinocytes. Sci. Rep. 2018, 8, 4998, Erratum in: Sci. Rep. 2018, 8, 4998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Opländer, C.; Volkmar, C.M.; Paunel-Görgülü, A.; Van Faassen, E.E.; Heiss, C.; Kelm, M.; Halmer, D.; Mürtz, M.; Pallua, N.; Suschek, C.V. Whole body UVA irradiation lowers systemic blood pressure by release of nitric oxide from intracutaneous photolabile nitric oxide derivates. Circ. Res. 2009, 105, 1031–1040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holliman, G.; Lowe, D.; Cohen, H.; Felton, S.; Raj, K. Ultraviolet radiation-induced production of nitric oxide: A multi-cell and multi-donor analysis. Sci. Rep. 2017, 7, 1105. [Google Scholar] [CrossRef]
- Opländer, C.; Deck, A.; Volkmar, C.M.; Kirsch, M.; Liebmann, J.; Born, M.; van Abeelen, F.; van Faassen, E.E.; Kröncke, K.-D.; Windolf, J.; et al. Mechanism and biological relevance of blue-light (420–453 nm)-induced nonenzymatic nitric oxide generation from photolabile nitric oxide derivates in human skin in vitro and in vivo. Free Radic. Biol. Med. 2013, 65, 1363–1377. [Google Scholar] [CrossRef]
- Randhawa, M.; Seo, I.; Liebel, F.; Southall, M.D.; Kollias, N.; Ruvolo, E. Visible light induces melanogenesis in human skin through a photoadaptive response. PLoS ONE 2015, 10, e0130949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Regazzetti, C.; Sormani, L.; Debayle, D.; Bernerd, F.; Tulic, M.; De Donatis, G.M.; Chignon-Sicard, B.; Rocchi, S.; Passeron, T. Melanocytes sense blue light and regulate pigmentation through opsin-3. J. Investig. Dermatol. 2018, 138, 171–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lan, Y.; Wang, Y.; Lu, H. Opsin 3 is a key regulator of ultraviolet A-induced photoageing in human dermal fibroblast cells. Br. J. Dermatol. 2019, 182, 1228–1244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kochevar, I.E.; Taylor, C.R.; Krutmann, J. Fundamentals of cutaneous photobiology and photoimmunology. In Fitzpatrick’s Dermatology in General Medicine, 7th ed.; Wolff, K., Goldsmith, L.A., Katz, S., Gilchrest, B., Paller, A.S., Lefell, D.J., Eds.; McGraw-Hill: New York, NY, USA, 2008. [Google Scholar]
- Schroeder, P.; Haendeler, J.; Krutmann, J. The role of near infrared radiation in photoaging of the skin. Exp. Gerontol. 2008, 43, 629–632. [Google Scholar] [CrossRef] [PubMed]
- Darvin, M.E.; Haag, S.F.; Meinke, M.C.; Sterry, W.; Lademann, J. Determination of the influence of IR radiation on the antioxidative network of the human skin. J. Biophotonics 2010, 4, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, P.; Calles, C.; Benesova, T.; Macaluso, F.; Krutmann, J. Photoprotection beyond ultraviolet radiation—Effective sun protection has to include protection against infrared A radiation-induced skin damage. Skin Pharmacol. Physiol. 2010, 23, 15–17. [Google Scholar] [CrossRef] [PubMed]
- Jantschitsch, C.; Majewski, S.; Maeda, A.; Schwarz, T.; Schwarz, A. Infrared radiation confers resistance to UV-induced apoptosis via reduction of DNA damage and upregulation of antiapoptotic proteins. J. Investig. Dermatol. 2009, 129, 1271–1279. [Google Scholar] [CrossRef] [Green Version]
- Jantschitsch, C.; Weichenthal, M.; Maeda, A.; Proksch, E.; Schwarz, T.; Schwarz, A. Infrared radiation does not enhance the frequency of ultraviolet radiation-induced skin tumors, but their growth behaviour in mice. Exp. Dermatol. 2011, 20, 346–350. [Google Scholar] [CrossRef] [PubMed]
- Wlaschek, M.; Tantcheva-Poór, I.; Naderi, L.; Ma, W.; Schneider, L.A.; Razi-Wolf, Z.; Schüller, J.; Scharffetter-Kochanek, K. Solar UV irradiation and dermal photoaging. J. Photochem. Photobiol. B Biol. 2001, 63, 41–51. [Google Scholar] [CrossRef]
- Quan, T.; He, T.; Kang, S.; Voorhees, J.J.; Fisher, G.J. Solar ultraviolet irradiation reduces collagen in photoaged human skin by blocking transforming growth factor-β type II receptor/Smad signaling. Am. J. Pathol. 2004, 165, 741–751. [Google Scholar] [CrossRef]
- Meng, X.-M.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-β: The master regulator of fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338. [Google Scholar] [CrossRef] [PubMed]
- Purohit, T.; He, T.; Qin, Z.; Li, T.; Fisher, G.J.; Yan, Y.; Voorhees, J.J.; Quan, T. Smad3-dependent regulation of type I collagen in human dermal fibroblasts: Impact on human skin connective tissue aging. J. Dermatol. Sci. 2016, 83, 80–83. [Google Scholar] [CrossRef] [PubMed]
- Sárdy, M. Role of matrix metalloproteinases in skin ageing. Connect. Tissue Res. 2009, 50, 132–138. [Google Scholar] [CrossRef]
- Kammeyer, A.; Luiten, R.M. Oxidation events and skin aging. Ageing Res. Rev. 2015, 21, 16–29. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Han, J.; Jiang, C.; Zhang, Y. Biomarkers, oxidative stress and autophagy in skin aging. Ageing Res. Rev. 2020, 59, 101036. [Google Scholar] [CrossRef] [PubMed]
- Ågren, M.S.; Schnabel, R.; Christensen, L.H.; Mirastschijski, U. Tumor necrosis factor-α-accelerated degradation of type I collagen in human skin is associated with elevated matrix metalloproteinase (MMP)-1 and MMP-3 ex vivo. Eur. J. Cell Biol. 2015, 94, 12–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell, S.; Degitz, K.; Quirling, M.; Jilg, N.; Page, S.; Brand, K. Involvement of NF-κB signalling in skin physiology and disease. Cell. Signal. 2003, 15, 1–7. [Google Scholar] [CrossRef]
- Hwang, B.-M.; Noh, E.-M.; Kim, J.-S.; Kim, J.-M.; Hwang, J.-K.; Kim, H.-K.; Kang, J.-S.; Kim, D.-S.; Chae, H.-J.; You, Y.-O.; et al. Decursin inhibits UVB-induced MMP expression in human dermal fibroblasts via regulation of nuclear factor-κB. Int. J. Mol. Med. 2012, 31, 477–483. [Google Scholar] [CrossRef] [Green Version]
- Rittié, L.; Fisher, G.J.; Voorhees, J.J. Retinoid therapy for photoaging. In Skin Aging; Gilchrest, B.A., Krutmann, J., Eds.; Springer: Berlin/Heidelberg, Germany, 2006; pp. 143–156. [Google Scholar]
- Jian, J.; Pelle, E.; Yang, Q.; Pernodet, N.; Maes, D.; Huang, X. Iron sensitizes keratinocytes and fibroblasts to UVA-mediated matrix metalloproteinase-1 through TNF-α and ERK activation. Exp. Dermatol. 2011, 20, 249–254. [Google Scholar] [CrossRef]
- Brenneisen, P.; Sies, H.; Scharffetter-Kochanek, K. Ultraviolet-B irradiation and matrix metalloproteinases: From induction via signaling to initial events. Ann. N. Y. Acad. Sci. 2002, 973, 31–43. [Google Scholar] [CrossRef] [PubMed]
- Tewari, A.; Grys, K.; Kollet, J.; Sarkany, R.; Young, A.R. Upregulation of MMP12 and its activity by UVA1 in human skin: Potential implications for photoaging. J. Investig. Dermatol. 2014, 134, 2598–2609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calles, C.; Schneider, M.; Macaluso, F.; Benesova, T.; Krutmann, J.; Schroeder, P. Infrared A radiation influences the skin fibroblast transcriptome: Mechanisms and consequences. J. Investig. Dermatol. 2010, 130, 1524–1536. [Google Scholar] [CrossRef] [Green Version]
- Liebel, F.; Kaur, S.; Ruvolo, E.; Kollias, N.; Southall, M.D. Irradiation of skin with visible light induces reactive oxygen species and matrix-degrading enzymes. J. Investig. Dermatol. 2012, 132, 1901–1907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, S.; Lee, M.J.; Kim, M.S.; Lee, S.; Kim, Y.K.; Lee, D.H.; Lee, C.W.; Cho, K.H.; Chung, J.H. Infrared plus visible light and heat from natural sunlight participate in the expression of MMPs and type I procollagen as well as infiltration of inflammatory cell in human skin in vivo. J. Dermatol. Sci. 2008, 50, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Martin, K.; Sur, R.; Liebel, F.; Tierney, N.; Lyte, P.; Garay, M.; Oddos, T.; Anthonavage, M.; Shapiro, S.; Southall, M. Parthenolide-depleted feverfew (Tanacetum parthenium) protects skin from UV irradiation and external aggression. Arch. Dermatol. Res. 2008, 300, 69–80. [Google Scholar] [CrossRef] [PubMed]
- McAdam, E.; Brem, R.; Karran, P. Oxidative stress–induced protein damage inhibits DNA repair and determines mutation risk and therapeutic efficacy. Mol. Cancer Res. 2016, 14, 612–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pisoschi, A.M.; Pop, A. The role of antioxidants in the chemistry of oxidative stress: A review. Eur. J. Med. Chem. 2015, 97, 55–74. [Google Scholar] [CrossRef] [PubMed]
- Gerschman, R.; Gilbert, D.L.; Nye, S.W.; Dwyer, P.; Fenn, W.O. Oxygen poisoning and x-irradiation: A mechanism in common. Science 1954, 119, 623–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pomatto, L.C.; Davies, K.J. Adaptive homeostasis and the free radical theory of ageing. Free Radic. Biol. Med. 2018, 124, 420–430. [Google Scholar] [CrossRef] [PubMed]
- Farage, M.A.; Miller, K.W.; Elsner, P.; Maibach, H.I. Intrinsic and extrinsic factors in skin ageing: A review. Int. J. Cosmet. Sci. 2008, 30, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Ferri, F.; Olivieri, F.; Cannataro, R.; Caroleo, M.C.; Cione, E. Phytomelatonin regulates keratinocytes homeostasis counteracting aging process. Cosmetics 2019, 6, 27. [Google Scholar] [CrossRef] [Green Version]
- Meng, J.; Lv, Z.; Qiao, X.; Li, X.; Li, Y.; Zhang, Y.; Chen, C. The decay of redox-stress response capacity is a substantive characteristic of aging: Revising the redox theory of aging. Redox Biol. 2016, 11, 365–374. [Google Scholar] [CrossRef]
- Schallreuter, K.U.; Wood, J.M. Thioredoxin reductase—Its role in epidermal redox status. J. Photochem. Photobiol. B Biol. 2001, 64, 179–184. [Google Scholar] [CrossRef]
- Xu, H.; Zheng, Y.-W.; Liu, Q.; Liu, L.-P.; Luo, F.-L.; Zhou, H.-C.; Isoda, H.; Ohkohchi, N.; Li, Y.-M. Reactive oxygen species in skin repair, regeneration, aging, and inflammation. In Reactive Oxygen Species (ROS) in Living Cells; Filip, C., Albu, E., Eds.; IntechOpen: London, UK, 2018. [Google Scholar]
- Masaki, H. Role of antioxidants in the skin: Anti-aging effects. J. Dermatol. Sci. 2010, 58, 85–90. [Google Scholar] [CrossRef]
- Buettner, G.R. Superoxide dismutase in redox biology: The roles of superoxide and hydrogen peroxide. Anti-Cancer Agents Med. Chem. 2011, 11, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Lowenstein, C.J.; Dinerman, J.L.; Snyder, S.H. Nitric-oxide—A physiological messenger. Ann. Intern. Med. 1994, 120, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Bogdan, C.; Röllinghoff, M.; Diefenbach, A. Reactive oxygen and reactive nitrogen intermediates in innate and specific immunity. Curr. Opin. Immunol. 2000, 12, 64–76. [Google Scholar] [CrossRef]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franco, R.; Sánchez-Olea, R.; Reyes-Reyes, E.M.; Panayiotidis, M.I. Environmental toxicity, oxidative stress and apoptosis: Ménage à trois. Mutat. Res. Toxicol. Environ. Mutagen. 2009, 674, 3–22. [Google Scholar] [CrossRef] [PubMed]
- Franco, R.; Cidlowski, J.A. Apoptosis and glutathione: Beyond an antioxidant. Cell Death Differ. 2009, 16, 1303–1314. [Google Scholar] [CrossRef] [Green Version]
- Couto, N.; Wood, J.; Barber, J. The role of glutathione reductase and related enzymes on cellular redox homoeostasis network. Free Radic. Biol. Med. 2016, 95, 27–42. [Google Scholar] [CrossRef]
- Fan, Q.; Chen, M.; Fang, X.; Lau, W.B.; Xue, L.; Zhao, L.; Zhang, H.; Liang, Y.-H.; Bai, X.; Niu, H.-Y.; et al. Aging might augment reactive oxygen species (ROS) formation and affect reactive nitrogen species (RNS) level after myocardial ischemia/reperfusion in both humans and rats. AGE 2012, 35, 1017–1026. [Google Scholar] [CrossRef]
- Kaminskyy, V.; Zhivotovsky, B. Free radicals in cross talk between autophagy and apoptosis. Antioxid. Redox Signal. 2014, 21, 86–102. [Google Scholar] [CrossRef] [PubMed]
- Dalby, K.N.; Tekedereli, I.; Lopez-Berestein, G.; Ozpolat, B. Targeting the prodeath and prosurvival functions of autophagy as novel therapeutic strategies in cancer. Autophagy 2010, 6, 322–329. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Charareh, P.; Lei, X.; Zhong, J.L. Autophagy: Multiple mechanisms to protect skin from ultraviolet radiation-driven photoaging. Oxidative Med. Cell. Longev. 2019, 2019, 1–14. [Google Scholar] [CrossRef]
- Sies, H.; Stahl, W.; Sevanian, A. Nutritional, dietary and postprandial oxidative stress. J. Nutr. 2005, 135, 969–972. [Google Scholar] [CrossRef] [PubMed]
- Valko, M.; Rhodes, C.J.; Moncol, J.; Izakovic, M.; Mazur, M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem. Biol. Interact. 2006, 160, 1–40. [Google Scholar] [CrossRef]
- Valacchi, G.; Sticozzi, C.; Pecorelli, A.; Cervellati, F.; Cervellati, C.; Maioli, E. Cutaneous responses to environmental stressors. Ann. N. Y. Acad. Sci. 2012, 1271, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Krutmann, J.; Schalka, S.; Watson, R.E.B.; Wei, L.; Morita, A. Daily photoprotection to prevent photoaging. Photodermatol. Photoimmunol. Photomed. 2021, 37, 482–489. [Google Scholar] [CrossRef]
- Jankovic, A.; Saso, L.; Korac, A.; Korac, B. Relation of redox and structural alterations of rat skin in the function of chronological aging. Oxid. Med. Cell. Longev. 2019, 2019, 2471312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukai, T.; Ushio-Fukai, M. Superoxide dismutases: Role in redox signaling, vascular function, and diseases. Antioxid. Redox Signal. 2011, 15, 1583–1606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altobelli, G.G.; Van Noorden, S.; Balato, A.; Cimini, V. Copper/zinc superoxide dismutase in human skin: Current knowledge. Front. Med. 2020, 7, 183. [Google Scholar] [CrossRef] [PubMed]
- Velarde, M.C.; Flynn, J.; Day, N.U.; Melov, S.; Campisi, J. Mitochondrial oxidative stress caused by Sod2 deficiency promotes cellular senescence and aging phenotypes in the skin. Aging 2012, 4, 3–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhie, G.E.; Seo, J.Y.; Chung, J.H. Modulation of catalase in human skin in vivo by acute and chronic UV radiation. Mol. Cells 2001, 11, 399–404. [Google Scholar]
- Altobelli, G.G.; Van Noorden, S.; Cimini, V. Copper/zinc-superoxide dismutase in human epidermis: An immunochemical study. Front. Med. 2019, 6, 258. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Saito, N.; Takemori, N.; Iizuka, S.; Suzuki, K.; Taniguchi, N.; Iizuka, H. Ultrastructural localization of superoxide dismutase in human skin. Acta Derm.-Venereol. 1993, 73, 41–45. [Google Scholar] [PubMed]
- Alfonso-Prieto, M.; Biarnés, X.; Vidossich, P.; Rovira, C. The molecular mechanism of the catalase reaction. J. Am. Chem. Soc. 2009, 131, 11751–11761. [Google Scholar] [CrossRef] [PubMed]
- Giacomoni, P.U.; Declercq, L.; Hellemans, L.; Maes, D. Aging of human skin: Review of a mechanistic model and first experimental data. IUBMB Life 2000, 49, 259–263. [Google Scholar] [CrossRef]
- Hellemans, L.; Corstjens, H.; Neven, A.; Declercq, L.; Maes, D. Antioxidant enzyme activity in human stratum corneum shows seasonal variation with an age-dependent recovery. J. Investig. Dermatol. 2003, 120, 434–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, M.H.; Rhie, G.-E.; Kim, Y.K.; Park, C.-H.; Cho, K.H.; Kim, K.H.; Eun, H.C.; Chung, J.H. H2O2 accumulation by catalase reduction changes MAP kinase signaling in aged human skin in vivo. J. Investig. Dermatol. 2005, 125, 221–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giordani, A.; Morlière, P.; Aubailly, M.; Santus, R. Photoinactivation of cellular catalase by ultraviolet radiation. Redox Rep. 1997, 3, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Schriner, S.E.; Linford, N.J.; Martin, G.M.; Treuting, P.; Ogburn, C.E.; Emond, M.; Coskun, P.E.; Ladiges, W.; Wolf, N.; Van Remmen, H.; et al. Extension of murine life span by overexpression of catalase targeted to mitochondria. Science 2005, 308, 1909–1911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, D.F.; Chiao, Y.A.; Martin, G.M.; Marcinek, D.J.; Basisty, N.; Quarles, E.K.; Rabinovitch, P.S. Mitochondrial-targeted catalase: Extended longevity and the roles in various disease models. Prog. Mol. Biol. Transl. Sci. 2017, 146, 203–241. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Zou, L.; Zhang, X.; Branco, V.; Wang, J.; Carvalho, C.; Holmgren, A.; Lu, J. Redox signaling mediated by thioredoxin and glutathione systems in the central nervous system. Antioxid. Redox Signal. 2017, 27, 989–1010. [Google Scholar] [CrossRef]
- Lushchak, V.I. Glutathione homeostasis and functions: Potential targets for medical interventions. J. Amino Acids 2012, 2012, 736837. [Google Scholar] [CrossRef] [Green Version]
- Lauterburg, B.H.; Smith, C.V.; Hughes, H.; Mitchell, J.R. Biliary excretion of glutathione and glutathione disulfide in the rat. Regulation and response to oxidative stress. J. Clin. Investig. 1984, 73, 124–133. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.P. Extracellular redox state: Refining the definition of oxidative stress in aging. Rejuvenation Res. 2006, 9, 169–181. [Google Scholar] [CrossRef]
- Bjørklund, G.; Peana, M.; Maes, M.; Dadar, M.; Severin, B. The glutathione system in Parkinson’s disease and its progression. Neurosci. Biobehav. Rev. 2020, 120, 470–478. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Lee, D.G. Naringin-generated ROS promotes mitochondria-mediated apoptosis in Candida albicans. IUBMB Life 2021, 73, 953–967. [Google Scholar] [CrossRef] [PubMed]
- Sreekumar, P.; Ferrington, D.; Kannan, R. Glutathione metabolism and the novel role of mitochondrial GSH in retinal degeneration. Antioxidants 2021, 10, 661. [Google Scholar] [CrossRef] [PubMed]
- Meredith, M.J.; Redd, D.J. Status of the mitochondrial pool of glutathione in the isolated hepatocyte. J. Biol. Chem. 1982, 257, 3747–3753. [Google Scholar] [CrossRef]
- Kumar, C.; Igbaria, A.; D’Autreaux, B.; Planson, A.-G.; Junot, C.; Godat, E.; Bachhawat, A.K.; Delaunay-Moisan, A.; Toledano, M.B. Glutathione revisited: A vital function in iron metabolism and ancillary role in thiol-redox control. EMBO J. 2011, 30, 2044–2056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marí, M.; Morales, A.; Colell, A.; García-Ruiz, C.; Kaplowitz, N.; Fernández-Checa, J.C. Mitochondrial glutathione: Features, regulation and role in disease. Biochim. Biophys. Acta (BBA) Gen. Subj. 2012, 1830, 3317–3328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Görlach, A.; Bertram, K.; Hudecova, S.; Krizanova, O. Calcium and ROS: A mutual interplay. Redox Biol. 2015, 6, 260–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mbemba, F.; Houbion, A.; Raes, M.; Remacle, J. Subcellular localization and modification with ageing of glutathione, gluta-thione peroxidase and glutathione reductase activities in human fibroblasts. Biochim. Biophys. Acta 1985, 838, 211–220. [Google Scholar] [CrossRef]
- Sonthalia, S.; Jha, A.; Lallas, A.; Jain, G.; Jakhar, D. Glutathione for skin lightening: A regnant myth or evidence-based verity? Dermatol. Pract. Concept. 2018, 8, 15–21. [Google Scholar] [CrossRef] [Green Version]
- Exner, R.; Wessner, B.; Manhart, N.; Roth, E. Therapeutic potential of glutathione. Wien. Klin. Wochenschr. 2000, 112, 610–616. [Google Scholar]
- Villarama, C.D.; Maibach, H.I. Glutathione as a depigmenting agent: An overview. Int. J. Cosmet. Sci. 2005, 27, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Dilokthornsakul, W.; Dhippayom, T.; Dilokthornsakul, P. The clinical effect of glutathione on skin color and other related skin conditions: A systematic review. J. Cosmet. Dermatol. 2018, 18, 728–737. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, F.; Hashizume, E.; Chan, G.P.; Kamimura, A. Skin-whitening and skin-condition-improving effects of topical oxidized glutathione: A double-blind and placebo-controlled clinical trial in healthy women. Clin. Cosmet. Investig. Dermatol. 2014, 7, 267–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.; Kumar, S.; Kim, S.H.; Seong, K.Y.; Lee, H.; Kim, C.; Jung, Y.-S.; Yang, S.Y. Odorless glutathione microneedle patches for skin whitening. Pharmaceutics 2020, 12, 100. [Google Scholar] [CrossRef] [Green Version]
- Gaschler, M.M.; Stockwell, B.R. Lipid peroxidation in cell death. Biochem. Biophys. Res. Commun. 2017, 482, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Fundu, T.M.; Kapepula, P.M.; Esimo, J.M.; Remacle, J.; Ngombe, N.K. Subcellular Localization of glutathione peroxidase, change in glutathione system during ageing and effects on cardiometabolic risks and associated diseases. In Glutathione System and Oxidative Stress in Health and Disease; Bagatini, M.D., Ed.; IntechOpen: London, UK, 2020. [Google Scholar]
- Brigelius-Flohé, R.; Maiorino, M. Glutathione peroxidases. Biochim. Biophys. Acta 2013, 1830, 3289–3303. [Google Scholar] [CrossRef]
- Lubos, E.; Loscalzo, J.; Handy, D.E. Glutathione peroxidase-1 in health and disease: From molecular mechanisms to therapeutic opportunities. Antioxid. Redox Signal. 2011, 15, 1957–1997. [Google Scholar] [CrossRef] [Green Version]
- Deponte, M. Glutathione catalysis and the reaction mechanisms of glutathione-dependent enzymes. Biochim. Biophys. Acta (BBA) Gen. Subj. 2013, 1830, 3217–3266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brigelius-Flohé, R.; Flohé, L. Regulatory phenomena in the glutathione peroxidase superfamily. Antioxid. Redox Signal. 2020, 33, 498–516. [Google Scholar] [CrossRef] [PubMed]
- Al-Qenaei, A.; Yiakouvaki, A.; Reelfs, O.; Santambrogio, P.; Levi, S.; Hall, N.D.; Tyrrell, R.M.; Pourzand, C. Role of intracellular labile iron, ferritin, and antioxidant defence in resistance of chronically adapted Jurkat T cells to hydrogen peroxide. Free Radic. Biol. Med. 2013, 68, 87–100. [Google Scholar] [CrossRef]
- Razygraev, A.V.; Petrosyan, M.A.; Tumasova, Z.N.; Taborskaya, K.I.; Polyanskikh, L.S.; Baziian, E.V.; Balashova, N.N. Changes in the activity of glutathione peroxidase in the blood plasma and serum of rats during postnatal development and aging. Adv. Gerontol. 2019, 9, 283–288. [Google Scholar] [CrossRef]
- Lu, J.; Holmgren, A. The thioredoxin antioxidant system. Free Radic. Biol. Med. 2014, 66, 75–87. [Google Scholar] [CrossRef]
- Yoshida, T.; Oka, S.-I.; Masutani, H.; Nakamura, H.; Yodoi, J. The role of thioredoxin in the aging process: Involvement of oxidative stress. Antioxid. Redox Signal. 2003, 5, 563–570. [Google Scholar] [CrossRef]
- Chae, H.Z.; Kim, H.J.; Kang, S.W.; Rhee, S.G. Characterization of three isoforms of mammalian peroxiredoxin that reduce peroxides in the presence of thioredoxin. Diabetes Res. Clin. Pract. 1999, 45, 101–112. [Google Scholar] [CrossRef]
- Brot, N.; Weissbach, L.; Werth, J. Enzymatic reduction of protein-bound methionine sulfoxide. Proc. Natl. Acad. Sci. USA 1981, 78, 2155–2158. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.-Y.; Kim, J.-R. Thioredoxin as a reducing agent for mammalian methionine sulfoxide reductases B lacking resolving cysteine. Biochem. Biophys. Res. Commun. 2008, 371, 490–494. [Google Scholar] [CrossRef]
- Lillig, C.H.; Holmgren, A. Thioredoxin and related molecules—From biology to health and disease. Antioxid. Redox Signal. 2007, 9, 25–47. [Google Scholar] [CrossRef]
- Holmgren, A.; Lu, J. Thioredoxin and thioredoxin reductase: Current research with special reference to human disease. Biochem. Biophys. Res. Commun. 2010, 396, 120–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, H.; Matsuo, Y.; Fukunaga, A.; Ono, R.; Nishigori, C.; Yodoi, J. Thioredoxin ameliorates cutaneous inflammation by regulating the epithelial production and release of pro-inflammatory cytokines. Front. Immunol. 2013, 4, 269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakurai, K.; Dainichi, T.; Matsumoto, R.; Tian, H.; Yodoi, J.; Miyachi, Y.; Kabashima, K. Topical thioredoxin inhibits IL-6 and IL-1beta production from keratinocytes and is effective for psoriasis-like dermatitis in mice. J. Dermatol. Sci. 2016, 84, e15. [Google Scholar] [CrossRef]
- Jia, J.-J.; Geng, W.-S.; Wang, Z.-Q.; Chen, L.; Zeng, X.-S. The role of thioredoxin system in cancer: Strategy for cancer therapy. Cancer Chemother. Pharmacol. 2019, 84, 453–470. [Google Scholar] [CrossRef]
- Young, J.J.; Patel, A.; Rai, P. Suppression of thioredoxin-1 induces premature senescence in normal human fibroblasts. Biochem. Biophys. Res. Commun. 2010, 392, 363–368. [Google Scholar] [CrossRef]
- Zhang, R.; Al-Lamki, R.; Bai, L.; Streb, J.W.; Miano, J.M.; Bradley, J.; Min, W. Thioredoxin-2 inhibits mitochondria-located ASK1-mediated apoptosis in a JNK-independent manner. Circ. Res. 2004, 94, 1483–1491. [Google Scholar] [CrossRef] [Green Version]
- Miranda-Vizuete, A.; Ljung, J.; Damdimopoulos, A.E.; Gustafsson, J.A.; Oko, R.; Pelto-Huikko, M.; Spyrou, G. Characterization of Sptrx, a novel member of the thioredoxin family specifically expressed in human spermatozoa. J. Biol. Chem. 2001, 276, 31567–31574. [Google Scholar] [CrossRef] [Green Version]
- Conrad, M.; Bornkamm, G.W.; Brielmeier, M. Mitochondrial and cytosolic thioredoxin reductase knockout mice. In Selenium; Hatfield, D.L., Berry, M.J., Gladyshev, V.N., Eds.; Springer: Boston, MA, USA; pp. 195–206.
- Pickering, A.M.; Lehr, M.; Gendron, C.M.; Pletcher, S.D.; Miller, R.A. Mitochondrial thioredoxin reductase 2 is elevated in long-lived primate as well as rodent species and extends fly mean lifespan. Aging Cell 2017, 16, 683–692. [Google Scholar] [CrossRef] [Green Version]
- Argyrou, A.; Blanchard, J.S. Flavoprotein disulfide reductases: Advances in chemistry and function. Prog. Nucleic Acid Res. Mol. Biol. 2004, 78, 89–142. [Google Scholar] [CrossRef]
- Lu, J.; Holmgren, A. Selenoproteins. J. Biol. Chem. 2009, 284, 723–727. [Google Scholar] [CrossRef] [Green Version]
- Schallreuter, K.U.; Moore, J.; Wood, J.M.; Beazley, W.D.; Peters, E.M.; Marles, L.K.; Behrens-Williams, S.C.; Dummer, R.; Blau, N.; Thöny, B. Epidermal H2O2 accumulation alters tetrahydrobiopterin (6BH4) recycling in vitiligo: Identification of a general mechanism in regulation of all 6BH4-dependent processes? J. Investig. Dermatol. 2001, 116, 167–174. [Google Scholar] [CrossRef] [Green Version]
- Schäfer, M.; Werner, S. Nrf2—A regulator of keratinocyte redox signaling. Free Radic. Biol. Med. 2015, 88, 243–252. [Google Scholar] [CrossRef]
- Higgins, L.G.; Kelleher, M.O.; Eggleston, I.M.; Itoh, K.; Yamamoto, M.; Hayes, J.D. Transcription factor Nrf2 mediates an adaptive response to sulforaphane that protects fibroblasts in vitro against the cytotoxic effects of electrophiles, peroxides and redox-cycling agents. Toxicol. Appl. Pharmacol. 2009, 237, 267–280. [Google Scholar] [CrossRef]
- Tian, F.F.; Zhang, F.; Lai, X.D.; Wang, L.J.; Yang, L.; Wang, X.; Singh, G.; Zhong, J.L.L. Nrf2-mediated protection against UVA radiation in human skin keratinocytes. Biosci. Trends 2011, 5, 23–29. [Google Scholar] [CrossRef] [Green Version]
- Tyrrell, R.M.; Pidoux, M. Correlation between endogenous glutathione content and sensitivity of cultured human skin cells to radiation at defined wavelengths in the solar ultraviolet range. Photochem. Photobiol. 1988, 47, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Gruber, F.; Ornelas, C.M.; Karner, S.; Narzt, M.-S.; Nagelreiter, I.M.; Gschwandtner, M.; Bochkov, V.; Tschachler, E. Nrf2 deficiency causes lipid oxidation, inflammation, and matrix-protease expression in DHA-supplemented and UVA-irradiated skin fibroblasts. Free Radic. Biol. Med. 2015, 88, 439–451. [Google Scholar] [CrossRef]
- Canning, P.; Sorrell, F.J.; Bullock, A.N. Structural basis of Keap1 interactions with Nrf2. Free Radic. Biol. Med. 2015, 88, 101–107. [Google Scholar] [CrossRef] [Green Version]
- Da Costa, R.M.; Rodrigues, D.; Pereira, C.A.; Silva, J.; Alves, J.V.; Lobato, N.S.; Tostes, R.C. Nrf2 as a potential mediator of cardiovascular risk in metabolic diseases. Front. Pharmacol. 2019, 10, 382. [Google Scholar] [CrossRef] [Green Version]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999, 13, 76–86. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, A.; Kang, M.-I.; Okawa, H.; Ohtsuji, M.; Zenke, Y.; Chiba, T.; Igarashi, K.; Yamamoto, M. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol. Cell. Biol. 2004, 24, 7130–7139. [Google Scholar] [CrossRef] [Green Version]
- Kaspar, J.W.; Jaiswal, A.K. An autoregulatory loop between Nrf2 and Cul3-Rbx1 controls their cellular abundance. J. Biol. Chem. 2010, 285, 21349–21358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baird, L.; Yamamoto, M. The molecular mechanisms regulating the KEAP1-NRF2 pathway. Mol. Cell. Biol. 2020, 40, e00099-20. [Google Scholar] [CrossRef] [PubMed]
- Rushmore, T.; Morton, M.; Pickett, C. The antioxidant responsive element. Activation by oxidative stress and identification of the DNA consensus sequence required for functional activity. J. Biol. Chem. 1991, 266, 11632–11639. [Google Scholar] [CrossRef]
- Robledinos-Antón, N.; Fernández-Ginés, R.; Manda, G.; Cuadrado, A. Activators and inhibitors of NRF2: A review of their potential for clinical development. Oxid. Med. Cell. Longev. 2019, 2019, 9372182. [Google Scholar] [CrossRef]
- Venugopal, R.; Jaiswal, A.K. Nrf1 and Nrf2 positively and c-Fos and Fra1 negatively regulate the human antioxidant response element-mediated expression of NAD(P)H:quinone oxidoreductase1 gene. Proc. Natl. Acad. Sci. USA 1996, 93, 14960–14965. [Google Scholar] [CrossRef] [Green Version]
- Hayes, J.D.; Chanas, S.A.; Henderson, C.J.; McMahon, M.; Sun, C.; Moffat, G.J.; Wolf, C.R.; Yamamoto, M. The Nrf2 transcription factor contributes both to the basal expression of glutathione S-transferases in mouse liver and to their induction by the chemopreventive synthetic antioxidants, butylated hydroxyanisole and ethoxyquin. Biochem. Soc. Trans. 2000, 28, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Harvey, C.J.; Thimmulappa, R.K.; Singh, A.; Blake, D.J.; Ling, G.; Wakabayashi, N.; Fujii, J.; Myers, A.; Biswal, S. Nrf2-regulated glutathione recycling independent of biosynthesis is critical for cell survival during oxidative stress. Free Radic. Biol. Med. 2009, 46, 443–453. [Google Scholar] [CrossRef] [Green Version]
- Brigelius-Flohé, R.; Müller, M.; Lippmann, D.; Kipp, A.P. The Yin and Yang of Nrf2-regulated selenoproteins in carcinogenesis. Int. J. Cell Biol. 2012, 2012, 486147. [Google Scholar] [CrossRef] [Green Version]
- McMahon, M.; Itoh, K.; Yamamoto, M.; Chanas, S.A.; Henderson, C.; McLellan, L.I.; Wolf, C.R.; Cavin, C.; Hayes, J.D. The cap’n’collar basic leucine zipper transcription factor Nrf2 (NF-E2 p45-related factor 2) controls both constitutive and inducible expression of intestinal detoxification and glutathione biosynthetic enzymes. Cancer Res. 2001, 61, 3299–3307. [Google Scholar]
- Reisman, S.A.; Lee, C.-Y.I.; Meyer, C.J.; Proksch, J.W.; Ward, K.W. Topical application of the synthetic triterpenoid RTA 408 activates Nrf2 and induces cytoprotective genes in rat skin. Radiat. Res. 2013, 306, 447–454. [Google Scholar] [CrossRef]
- Zhu, H.; Jia, Z.; Zhang, L.; Yamamoto, M.; Misra, H.P.; Trush, M.A.; Li, Y. Antioxidants and phase 2 enzymes in macrophages: Regulation by Nrf2 signaling and protection against oxidative and electrophilic stress. Exp. Biol. Med. 2008, 233, 463–474. [Google Scholar] [CrossRef] [PubMed]
- Chun, K.-S.; Kundu, J.; Kundu, J.K.; Surh, Y.-J. Targeting Nrf2-Keap1 signaling for chemoprevention of skin carcinogenesis with bioactive phytochemicals. Toxicol. Lett. 2014, 229, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Kawachi, Y.; Xu, X.; Taguchi, S.; Sakurai, H.; Nakamura, Y.; Ishii, Y.; Fujisawa, Y.; Furuta, J.; Takahashi, T.; Itoh, K.; et al. Attenuation of UVB-induced sunburn reaction and oxidative DNA damage with no alterations in UVB-induced skin carcinogenesis in Nrf2 gene-deficient mice. J. Investig. Dermatol. 2008, 128, 1773–1779. [Google Scholar] [CrossRef] [Green Version]
- Hirota, A.; Kawachi, Y.; Itoh, K.; Nakamura, Y.; Xu, X.; Banno, T.; Takahashi, T.; Yamamoto, M.; Otsuka, F. Ultraviolet A irradiation induces NF-E2-related factor 2 activation in dermal fibroblasts: Protective role in UVA-induced apoptosis. J. Investig. Dermatol. 2005, 124, 825–832. [Google Scholar] [CrossRef] [Green Version]
- Gęgotek, A.; Skrzydlewska, E. The role of transcription factor Nrf2 in skin cells metabolism. Arch. Dermatol. Res. 2015, 307, 385–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schäfer, M.; Dütsch, S.; auf dem Keller, U.; Werner, S. Nrf2: A central regulator of UV protection in the epidermis. Cell Cycle 2010, 9, 2917–2918. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.; Shin, J.-M.; Jang, S.; Choi, D.-K.; Seo, M.-S.; Kim, H.-R.; Sohn, K.-C.; Im, M.; Seo, Y.-J.; Lee, J.-H.; et al. Role of nuclear factor E2-related factor 2 (Nrf2) in epidermal differentiation. Arch. Dermatol. Res. 2014, 306, 677–682. [Google Scholar] [CrossRef] [PubMed]
- Piao, M.S.; Choi, J.-Y.; Lee, D.-H.; Yun, S.J.; Lee, J.-B.; Lee, S.-C. Differentiation-dependent expression of NADP(H):quinone oxidoreductase-1 via NF-E2 related factor-2 activation in human epidermal keratinocytes. J. Dermatol. Sci. 2011, 62, 147–153. [Google Scholar] [CrossRef]
- Marrot, L.; Jones, C.; Perez, P.; Meunier, J.R. The significance of Nrf2 pathway in (photo)-oxidative stress response in mela-nocytes and keratinocytes of the human epidermis. Pigment. Cell Melanoma Res. 2008, 21, 79–88. [Google Scholar] [CrossRef]
- Kokot, A.; Metze, D.; Mouchet, N.; Galibert, M.-D.; Schiller, M.; Luger, T.A.; Böhm, M. α-melanocyte-stimulating hormone counteracts the suppressive effect of UVB on Nrf2 and Nrf-dependent gene expression in human skin. Endocrinology 2009, 150, 3197–3206. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.Y.; Kim, J.; Ahn, Y.; Lee, E.J.; Hwang, S.; Almurayshid, A.; Park, K.; Chung, H.-J.; Kim, H.J.; Lee, S.-H.; et al. Autophagy induction can regulate skin pigmentation by causing melanosome degradation in keratinocytes and melanocytes. Pigment. Cell Melanoma Res. 2019, 33, 403–415. [Google Scholar] [CrossRef] [PubMed]
- Park, H.J.; Jo, D.S.; Choi, H.; Bae, J.-E.; Park, N.Y.; Kim, J.B.; Choi, J.Y.; Kim, Y.H.; Oh, G.S.; Chang, J.H.; et al. Melasolv induces melanosome autophagy to inhibit pigmentation in B16F1 cells. PLoS ONE 2020, 15, e0239019. [Google Scholar] [CrossRef] [PubMed]
- Hseu, Y.-C.; Gowrisankar, Y.V.; Wang, L.-W.; Zhang, Y.-Z.; Chen, X.-Z.; Huang, P.-J.; Yen, H.-R.; Yang, H.-L. The in vitro and in vivo depigmenting activity of pterostilbene through induction of autophagy in melanocytes and inhibition of UVA-irradiated α-MSH in keratinocytes via Nrf2-mediated antioxidant pathways. Redox Biol. 2021, 44, 102007. [Google Scholar] [CrossRef]
- Komatsu, M.; Kurokawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.-S.; Ueno, I.; Sakamoto, A.; Tong, K.I.; et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 2010, 12, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Sample, A.; Zhao, B.; Wu, C.; Qian, S.; Shi, X.; Aplin, A.; He, Y.-Y. The autophagy receptor adaptor p62 is up-regulated by UVA radiation in melanocytes and in melanoma cells. Photochem. Photobiol. 2017, 94, 432–437. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhang, C.-F.; Rossiter, H.; Eckhart, L.; Koenig, U.; Karner, S.; Mildner, M.; Bochkov, V.N.; Tschachler, E.; Gruber, F. Autophagy is induced by UVA and promotes removal of oxidized phospholipids and protein aggregates in epidermal keratinocytes. J. Investig. Dermatol. 2013, 133, 1629–1637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.-L.; Lin, C.-P.; Gowrisankar, Y.V.; Huang, P.-J.; Chang, W.-L.; Shrestha, S.; Hseu, Y.-C. The anti-melanogenic effects of ellagic acid through induction of autophagy in melanocytes and suppression of UVA-activated α-MSH pathways via Nrf2 activation in keratinocytes. Biochem. Pharmacol. 2021, 185, 114454. [Google Scholar] [CrossRef]
- Kim, Y.R.; Oh, J.E.; Kim, M.S.; Kang, M.R.; Park, S.W.; Han, J.Y.; Eom, H.S.; Yoo, N.J.; Lee, S.H. Oncogenic NRF2 mutations in squamous cell carcinomas of oesophagus and skin. J. Pathol. 2010, 220, 446–451. [Google Scholar] [CrossRef]
- Surh, Y.-J.; Kundu, J.K.; Na, H.-K. Nrf2 as a master redox switch in turning on the cellular signaling involved in the induction of cytoprotective genes by some chemopreventive phytochemicals. Planta Med. 2008, 74, 1526–1539. [Google Scholar] [CrossRef] [Green Version]
- Tebay, L.E.; Robertson, H.; Durant, S.T.; Vitale, S.R.; Penning, T.M.; Dinkova-Kostova, A.T.; Hayes, J.D. Mechanisms of activation of the transcription factor Nrf2 by redox stressors, nutrient cues, and energy status and the pathways through which it attenuates degenerative disease. Free Radic. Biol. Med. 2015, 88, 108–146. [Google Scholar] [CrossRef] [Green Version]
- Tong, Y.-H.; Zhang, B.; Fan, Y.; Lin, N.-M. Keap1–Nrf2 pathway: A promising target towards lung cancer prevention and therapeutics. Chronic Dis. Transl. Med. 2015, 1, 175–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dohi, Y.; Ikura, T.; Hoshikawa, Y.; Katoh, Y.; Ota, K.; Nakanome, A.; Muto, A.; Omura, S.; Ohta, T.; Ito, A.; et al. Bach1 inhibits oxidative stress–induced cellular senescence by impeding p53 function on chromatin. Nat. Struct. Mol. Biol. 2008, 15, 1246–1254. [Google Scholar] [CrossRef]
- Wang, M.; Lei, M.; Chang, L.; Xing, Y.; Guo, Y.; Pourzand, C.; Bartsch, J.W.; Chen, J.; Luo, J.; Karisma, V.W.; et al. Bach2 regulates autophagy to modulate UVA-induced photoaging in skin fibroblasts. Free Radic. Biol. Med. 2021, 169, 304–316. [Google Scholar] [CrossRef]
- Ma, Q.; He, X. Molecular basis of electrophilic and oxidative defense: Promises and perils of Nrf2. Pharmacol. Rev. 2012, 64, 1055–1081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Q. Role of Nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinkova-Kostova, A.T.; Fahey, J.W.; Talalay, P. Chemical structures of inducers of nicotinamide quinone oxidoreductase 1 (NQO1). Methods Enzymol. 2004, 382, 423–448. [Google Scholar] [CrossRef]
- Dunaway, S.; Odin, R.; Zhou, L.; Ji, L.; Zhang, Y.; Kadekaro, A.L. Natural antioxidants: Multiple mechanisms to protect skin from solar radiation. Front. Pharmacol. 2018, 9, 392. [Google Scholar] [CrossRef] [Green Version]
- Saewan, N.; Jimtaisong, A. Natural products as photoprotection. J. Cosmet. Dermatol. 2015, 14, 47–63. [Google Scholar] [CrossRef]
- He, H.; Li, A.; Li, S.; Tang, J.; Li, L.; Xiong, L. Natural components in sunscreens: Topical formulations with sun protection factor (SPF). Biomed. Pharmacother. 2020, 134, 111161. [Google Scholar] [CrossRef]
- Petruk, G.; Del Giudice, R.; Rigano, M.M.; Monti, D.M. Antioxidants from plants protect against skin photoaging. Oxid. Med. Cell. Longev. 2018, 2018, 1454936. [Google Scholar] [CrossRef] [Green Version]
- Mejía-Giraldo, J.C.; Gallardo, C.; Puertas-Mejía, M.A. Selected extracts from high mountain plants as potential sunscreens with antioxidant capacity. Photochem. Photobiol. 2021, 98, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Birringer, M.; Siems, K.; Maxones, A.; Frank, J.; Lorkowski, S. Natural 6-hydroxy-chromanols and -chromenols: Structural diversity, biosynthetic pathways and health implications. RSC Adv. 2018, 8, 4803–4841. [Google Scholar] [CrossRef] [Green Version]
- Yiakouvaki, A.; Savović, J.; Al-Qenaei, A.; Dowden, J.; Pourzand, C. Caged-iron chelators a novel approach towards protecting skin cells against UVA-induced necrotic cell death. J. Investig. Dermatol. 2006, 126, 2287–2295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franks, A.T.; Wang, Q.; Franz, K.J. A multifunctional, light-activated prochelator inhibits UVA-induced oxidative stress. Bioorgan. Med. Chem. Lett. 2015, 25, 4843–4847. [Google Scholar] [CrossRef] [Green Version]
- Nowicka, B.; Trela-Makowej, A.; Latowski, D.; Strzalka, K.; Szymańska, R. Antioxidant and signaling role of plastid-derived isoprenoid quinones and chromanols. Int. J. Mol. Sci. 2021, 22, 2950. [Google Scholar] [CrossRef]
- Bissett, D.L. Common cosmeceuticals. Clin. Dermatol. 2009, 27, 435–445. [Google Scholar] [CrossRef]
- McDaniel, D.; Farris, P.; Valacchi, G. Atmospheric skin aging—Contributors and inhibitors. J. Cosmet. Dermatol. 2018, 17, 124–137. [Google Scholar] [CrossRef]
- Kim, S.; Kim, J.; Lee, Y.I.; Jang, S.; Song, S.Y.; Lee, W.J.; Lee, J.H. Particulate matter-induced atmospheric skin aging is aggravated by UVA and inhibited by a topical L-ascorbic acid compound. Photodermatol. Photoimmunol. Photomed. 2021. [Google Scholar] [CrossRef]
- Wallert, M.; Kluge, S.; Schubert, M.; Koeberle, A.; Werz, O.; Birringer, M.; Lorkowski, S. Diversity of chromanol and chromenol structures and functions: An emerging class of anti-inflammatory and anti-carcinogenic agents. Front. Pharmacol. 2020, 11, 362. [Google Scholar] [CrossRef]
- Hur, S.; Lee, H.; Kim, Y.; Lee, B.-H.; Shin, J.; Kim, T.-Y. Sargaquinoic acid and sargachromenol, extracts of Sargassum sagamianum, induce apoptosis in HaCaT cells and mice skin: Its potentiation of UVB-induced apoptosis. Eur. J. Pharmacol. 2008, 582, 1–11. [Google Scholar] [CrossRef]
- Kim, J.-A.; Ahn, B.-N.; Kong, C.-S.; Kim, S.-K. Protective effect of chromene isolated from Sargassum horneri against UV-A-induced damage in skin dermal fibroblasts. Exp. Dermatol. 2012, 21, 630–631. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.I.; Seo, Y. Chromanols from Sargassum siliquastrum and their antioxidant activity in HT 1080 cells. Chem. Pharm. Bull. 2011, 59, 757–761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Orazio, N.; Gemello, E.; Gammone, M.A.; De Girolamo, M.; Ficoneri, C.; Riccioni, G. Fucoxantin: A treasure from the sea. Mar. Drugs 2012, 10, 604–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urikura, I.; Sugawara, T.; Hirata, T. Protective effect of fucoxanthin against UVB-induced skin photoaging in hairless mice. Biosci. Biotechnol. Biochem. 2011, 75, 757–760. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Luna, A.; Ávila-Román, J.; González-Rodríguez, M.L.; Cózar, M.J.; Rabasco, A.M.; Motilva, V.; Talero, E. Fucoxanthin-containing cream prevents epidermal hyperplasia and UVB-induced skin erythema in mice. Mar. Drugs 2018, 16, 378. [Google Scholar] [CrossRef] [Green Version]
- Bernstein, E.F.; Sarkas, H.W.; Ba, P.B.; Bouche, D. Beyond sun protection factor: An approach to environmental protection with novel mineral coatings in a vehicle containing a blend of skincare ingredients. J. Cosmet. Dermatol. 2019, 19, 407–415. [Google Scholar] [CrossRef] [Green Version]
- Souza, C.; Campos, P.M.M. Development and photoprotective effect of a sunscreen containing the antioxidants spirulina and dimethylmethoxy chromanol on sun-induced skin damage. Eur. J. Pharm. Sci. 2017, 104, 52–64. [Google Scholar] [CrossRef]
- Nonell, S.; García-Díaz, M.; Viladot, J.L.; Delgado, R. Singlet molecular oxygen quenching by the antioxidant dimethylmethoxy chromanol in solution and inex vivoporcine skin. Int. J. Cosmet. Sci. 2013, 35, 272–280. [Google Scholar] [CrossRef]
- Gimenez, A.; Laporta, O.; Ollagnier, M.; Vincendet, M.; Delgado, R. Protection and Detox of the Skin by Bioinspired Molecules. Available online: https://www.teknoscienze.com/tks_issue/vol-132/skin-care-vol-132/ (accessed on 22 October 2021).
- Ahn, J.; Chae, H.-S.; Pel, P.; Kim, Y.-M.; Choi, Y.; Kim, J.; Chin, Y.-W. Dilignans with a chromanol motif discovered by molecular networking from the stem barks of Magnolia obovata and their proprotein convertase subtilisin/kexin type 9 expression inhibitory activity. Biomolecules 2021, 11, 463. [Google Scholar] [CrossRef]
- Nichols, J.A.; Katiyar, S.K. Skin photoprotection by natural polyphenols: Anti-inflammatory, antioxidant and DNA repair mechanisms. Arch. Dermatol. Res. 2009, 302, 71–83. [Google Scholar] [CrossRef] [Green Version]
- Cavinato, M.; Waltenberger, B.; Baraldo, G.; Grade, C.V.C.; Stuppner, H.; Jansen-Dürr, P. Plant extracts and natural compounds used against UVB-induced photoaging. Biogerontology 2017, 18, 499–516. [Google Scholar] [CrossRef] [PubMed]
- Bosch, R.; Philips, N.; Suárez-Pérez, J.A.; Juarranz, A.; Devmurari, A.; Chalensouk-Khaosaat, J.; González, S. Mechanisms of photoaging and cutaneous photocarcinogenesis, and photoprotective strategies with phytochemicals. Antioxidants 2015, 4, 248–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabinovich, L.; Kazlouskaya, V. Herbal sun protection agents: Human studies. Clin. Dermatol. 2018, 36, 369–375. [Google Scholar] [CrossRef] [PubMed]
- F’Guyer, S.; Afaq, F.; Mukhtar, H. Photochemoprevention of skin cancer by botanical agents. Photodermatol. Photoimmunol. Photomed. 2003, 19, 56–72. [Google Scholar] [CrossRef]
- Kostyuk, V.; Potapovich, A.; Albuhaydar, A.R.; Mayer, W.; De Luca, C.; Korkina, L. Natural substances for prevention of skin photoaging: Screening systems in the development of sunscreen and rejuvenation cosmetics. Rejuvenation Res. 2018, 21, 91–101. [Google Scholar] [CrossRef]
- Halliwell, B. Dietary polyphenols: Good, bad, or indifferent for your health? Cardiovasc. Res. 2007, 73, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Mira, L.; Fernandez, M.T.; Santos, M.; Rocha, R.; Florêncio, M.H.; Jennings, K.R. Interactions of flavonoids with iron and copper ions: A mechanism for their antioxidant activity. Free Radic. Res. 2002, 36, 1199–1208. [Google Scholar] [CrossRef]
- Perron, N.R.; Brumaghim, J.L. A Review of the antioxidant mechanisms of polyphenol compounds related to iron binding. Cell Biochem. Biophys. 2009, 53, 75–100. [Google Scholar] [CrossRef]
- Basu-Modak, S.; Ali, D.; Gordon, M.; Polte, T.; Yiakouvaki, A.; Pourzand, C.; Rice-Evans, C.; Tyrrell, R.M. Suppression of UVA-mediated release of labile iron by epicatechin—A link to lysosomal protection. Free Radic. Biol. Med. 2006, 41, 1197–1204. [Google Scholar] [CrossRef]
- Imam, M.U.; Zhang, S.; Ma, J.; Wang, H.; Wang, F. Antioxidants mediate both iron homeostasis and oxidative stress. Nutrients 2017, 9, 671. [Google Scholar] [CrossRef]
- Domaszewska-Szostek, A.; Puzianowska-Kuźnicka, M.; Kuryłowicz, A. Flavonoids in skin senescence prevention and treatment. Int. J. Mol. Sci. 2021, 22, 6814. [Google Scholar] [CrossRef]
- Shankar, E.; Goel, A.; Gupta, K.; Gupta, S. Plant flavone apigenin: An emerging anticancer agent. Curr. Pharmacol. Rep. 2017, 3, 423–446. [Google Scholar] [CrossRef]
- Zakaria, N.N.A.; Okello, E.J.; Howes, M.-J. Antioxidant, anti-collagenase, anti-elastase and anti-tyrosinase activities of an aqueous Cosmos caudatus Kunth (Asteraceae) leaf extract. Trop. J. Nat. Prod. Res. 2020, 4, 1124–1130. [Google Scholar] [CrossRef]
- Brimson, J.M.; Onlamoon, N.; Tencomnao, T.; Thitilertdecha, P. Clerodendrum petasites S. Moore: The therapeutic potential of phytochemicals, hispidulin, vanillic acid, verbascoside, and apigenin. Biomed. Pharmacother. 2019, 118, 109319. [Google Scholar] [CrossRef]
- Ma, X.; Lin, Y.; Liu, Y.; Li, W.; He, J.; Fang, M.; Lin, D. Effects of apigenin treatment on random skin flap survival in rats. Front. Pharmacol. 2021, 12, 625733. [Google Scholar] [CrossRef]
- Zhu, D.; Chen, B.; Xiang, Z.; Lin, J.; Miao, Z.; Wang, Y.; Wu, Y.; Zhou, Y. Apigenin enhances viability of random skin flaps by activating autophagy. Phytother. Res. 2021, 35, 3848–3860. [Google Scholar] [CrossRef]
- Zhang, B.; Wang, J.; Zhao, G.; Lin, M.; Lang, Y.; Zhang, D.; Feng, D.; Tu, C. Apigenin protects human melanocytes against oxidative damage by activation of the Nrf2 pathway. Cell Stress Chaperon. 2020, 25, 277–285. [Google Scholar] [CrossRef]
- Park, C.-H.; Min, S.-Y.; Yu, H.-W.; Kim, K.; Kim, S.; Lee, H.-J.; Kim, J.-H.; Park, Y.-J. Effects of apigenin on RBL-2H3, RAW264.7, and HaCaT cells: Anti-allergic, anti-inflammatory, and skin-protective activities. Int. J. Mol. Sci. 2020, 21, 4620. [Google Scholar] [CrossRef]
- Nam, E.J.; Yoo, G.; Lee, J.Y.; Kim, M.; Jhin, C.; Son, Y.J.; Kim, S.Y.; Jung, S.H.; Nho, C.W. Glycosyl flavones from Humulus japonicus suppress MMP-1 production via decreasing oxidative stress in UVB irradiated human dermal fibroblasts. BMB Rep. 2020, 53, 379–384. [Google Scholar] [CrossRef]
- Choi, S.; Youn, J.; Kim, K.; Joo, D.H.; Shin, S.; Lee, J.; Lee, H.K.; An, I.-S.; Kwon, S.; Youn, H.J.; et al. Apigenin inhibits UVA-induced cytotoxicity in vitro and prevents signs of skin aging in vivo. Int. J. Mol. Med. 2016, 38, 627–634. [Google Scholar] [CrossRef] [Green Version]
- Britto, S.M.; Shanthakumari, D.; Agilan, B.; Radhiga, T.; Kanimozhi, G.; Prasad, N.R. Apigenin prevents ultraviolet-B radiation induced cyclobutane pyrimidine dimers formation in human dermal fibroblasts. Mutat. Res. Toxicol. Environ. Mutagen. 2017, 821, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Salehi, B.; Sharifi-Rad, J.; Cappellini, F.; Reiner, Ž.; Zorzan, D.; Imran, M.; Sener, B.; Kilic, M.; El-Shazly, M.; Fahmy, N.M.; et al. The therapeutic potential of anthocyanins: Current approaches based on their molecular mechanism of action. Front. Pharmacol. 2020, 11, 1300. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.; Park, H.; Kim, H.P. Effects of flavonoids on senescence-associated secretory phenotype formation from bleomycin-induced senescence in BJ fibroblasts. Biochem. Pharmacol. 2015, 96, 337–348. [Google Scholar] [CrossRef] [PubMed]
- Yoshihisa, Y.; Andoh, T.; Rehman, M.U.; Shimizu, T. The regulation of protein kinase casein kinase II by apigenin is involved in the inhibition of ultraviolet B-induced macrophage migration inhibitory factor-mediated hyperpigmentation. Phytother. Res. 2020, 34, 1320–1328. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, J.; Cheng, X.; Yi, B.; Zhang, X.; Li, Q. Apigenin induces dermal collagen synthesis via Smad2/3 signaling pathway. Eur. J. Histochem. 2015, 59, 2467. [Google Scholar] [CrossRef]
- Arterbery, V.E.; Gupta, S. Apigenin as an anti-aging skin treatment. J. Clin. Cosmet. Dermatol. 2018, 2, 1–8. [Google Scholar] [CrossRef]
- Pavel, T.I.; Chircov, C.; Rădulescu, M.; Grumezescu, A.M. Regenerative wound dressings for skin cancer. Cancers 2020, 12, 2954. [Google Scholar] [CrossRef]
- Chou, T.-H.; Nugroho, D.; Chang, J.-Y.; Cheng, Y.-S.; Liang, C.-H.; Deng, M.-J. Encapsulation and characterization of nanoemulsions based on an anti-oxidative polymeric amphiphile for topical apigenin delivery. Polymers 2021, 13, 1016. [Google Scholar] [CrossRef]
- Perez, C.A.; Wei, Y.; Guo, M. Iron-binding and anti-Fenton properties of baicalein and baicalin. J. Inorg. Biochem. 2009, 103, 326–332. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Li, H.; Zhao, Y.; Gao, Z. Dietary supplementation of baicalin and quercetin attenuates iron overload induced mouse liver injury. Eur. J. Pharmacol. 2006, 535, 263–269. [Google Scholar] [CrossRef]
- Zhao, Y.; Li, H.; Gao, Z.; Xu, H. Effects of dietary baicalin supplementation on iron overload-induced mouse liver oxidative injury. Eur. J. Pharmacol. 2005, 509, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Li, H.; Gao, Z.; Gong, Y.; Xu, H. Effects of flavonoids extracted from Scutellaria baicalensis Georgi on hemin–nitrite–H2O2 induced liver injury. Eur. J. Pharmacol. 2006, 536, 192–199. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Perez, C.; Wei, Y.; Rapoza, E.; Su, G.; Bou-Abdallah, F.; Chasteen, N.D. Iron-binding properties of plant phenolics and cranberry’s bio-effects. Dalton Trans. 2007, 4951–4961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatcher, H.C.; Singh, R.N.; Torti, F.M.; Torti, S.V. Synthetic and natural iron chelators: Therapeutic potential and clinical use. Futur. Med. Chem. 2009, 1, 1643–1670. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.Z.; Leung, H.W.; Lai, M.Y.; Wu, C.H. Baicalein induced cell cycle arrest and apoptosis in human lung squamous car-cinoma CH27 cells. Anticancer Res. 2005, 25, 959–964. [Google Scholar]
- Lee, J.-H.; Li, Y.-C.; Ip, S.-W.; Hsu, S.-C.; Chang, N.-W.; Tang, N.-Y.; Yu, C.-S.; Chou, S.-T.; Lin, S.-S.; Lino, C.-C.; et al. The role of Ca2+ in baicalein-induced apoptosis in human breast MDA-MB-231 cancer cells through mitochondria- and caspase-3-dependent pathway. Anticancer Res. 2008, 28, 1701–1711. [Google Scholar]
- Wang, S.-C.; Chen, S.-F.; Lee, Y.-M.; Chuang, C.-L.; Bau, D.-T.; Lin, S.-S. Baicalin scavenges reactive oxygen species and protects human keratinocytes against UVC-induced cytotoxicity. In Vivo 2013, 27, 707–714. [Google Scholar]
- Liu, H.; Dong, Y.; Gao, Y.; Du, Z.; Wang, Y.; Cheng, P.; Chen, A.; Huang, H. The fascinating effects of baicalein on cancer: A review. Int. J. Mol. Sci. 2016, 17, 1681. [Google Scholar] [CrossRef] [Green Version]
- Yin, B.; Li, W.; Qin, H.; Yun, J.; Sun, X. The use of Chinese skullcap (Scutellaria baicalensis) and its extracts for sustainable animal production. Animals 2021, 11, 1039. [Google Scholar] [CrossRef]
- Xie, Y.; Song, X.; Sun, X.; Huang, J.; Zhong, M.; Lotze, M.T.; Zeh, H.J., 3rd; Kang, R.; Tang, D. Identification of baicalein as a ferroptosis inhibitor by natural product library screening. Biochem. Biophys. Res. Commun. 2016, 473, 775–780. [Google Scholar] [CrossRef]
- Yang, M.; Li, X.; Li, H.; Zhang, X.; Liu, X.; Song, Y. Baicalein inhibits RLS3-induced ferroptosis in melanocytes. Biochem. Biophys. Res. Commun. 2021, 561, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Ai, Y.; Yan, B.; Wang, X. The oxidoreductases POR and CYB5R1 catalyze lipid peroxidation to execute ferroptosis. Mol. Cell. Oncol. 2021, 8, 1881393. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Cao, F.; Yin, H.L.; Huang, Z.J.; Lin, Z.T.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, present and future. Cell Death Dis. 2020, 11, 88. [Google Scholar] [CrossRef] [PubMed]
- Min, W.; Liu, X.; Qian, Q.; Lin, B.; Wu, D.; Wang, M.; Ahmad, I.; Yusuf, N.; Luo, D. The effects of baicalin against UVA-induced photoaging in skin fibroblasts. Am. J. Chin. Med. 2014, 42, 709–727. [Google Scholar] [CrossRef]
- Sun, X.; Ou, Z.; Chen, R.; Niu, X.; Chen, D.; Kang, R.; Tang, D. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology 2016, 63, 173–184. [Google Scholar] [CrossRef]
- La Rosa, P.; Petrillo, S.; Turchi, R.; Berardinelli, F.; Schirinzi, T.; Vasco, G.; Lettieri-Barbato, D.; Fiorenza, M.T.; Bertini, E.S.; Aquilano, K.; et al. The Nrf2 induction prevents ferroptosis in Friedreich’s ataxia. Redox Biol. 2020, 38, 101791. [Google Scholar] [CrossRef]
- Shi, L.; Hao, Z.; Zhang, S.; Wei, M.; Lu, B.; Wang, Z.; Ji, L. Baicalein and baicalin alleviate acetaminophen-induced liver injury by activating Nrf2 antioxidative pathway: The involvement of ERK1/2 and PKC. Biochem. Pharmacol. 2018, 150, 9–23. [Google Scholar] [CrossRef]
- Ma, J.; Li, S.; Zhu, L.; Guo, S.; Yi, X.; Cui, T.; He, Y.; Chang, Y.; Liu, B.; Li, C.; et al. Baicalein protects human vitiligo melanocytes from oxidative stress through activation of NF-E2-related factor2 (Nrf2) signaling pathway. Free Radic. Biol. Med. 2018, 129, 492–503. [Google Scholar] [CrossRef]
- Zhou, Y.; Yang, Z.-Y.; Tang, R.-C. Bioactive and UV protective silk materials containing baicalin—The multifunctional plant extract from Scutellaria baicalensis Georgi. Mater. Sci. Eng. C Mater. Biol. Appl. 2016, 67, 336–344. [Google Scholar] [CrossRef]
- Zhou, B.-R.; Luo, D.; Wei, F.-D.; Chen, X.-E.; Gao, J. Baicalin protects human fibroblasts against ultraviolet B-induced cyclobutane pyrimidine dimers formation. Arch. Dermatol. Res. 2008, 300, 331–334. [Google Scholar] [CrossRef]
- Choi, H.-J.; Yun, M.-Y.; Won, E.-Y.; Lee, J.-H.; Jung, J.-I. Bioconversion from Scutellaria baicalensis (baicalin) feremted with Leatiporus sulphureus into enriched-baicalein and anti-wrinkle effects. Pharmacogn. Mag. 2018, 14, 453. [Google Scholar] [CrossRef]
- Zhou, B.-R.; Jin, S.-L.; Chen, X.-E.; Lin, X.-F.; Cai, B.-X.; Gao, J.; Luo, D. Protective effect of the Baicalin against DNA damage induced by ultraviolet B irradiation to mouse epidermis. Photodermatol. Photoimmunol. Photomed. 2008, 24, 175–182. [Google Scholar] [CrossRef]
- Kim, D.H.; Kim, H.K.; Park, S.; Kim, J.Y.; Zou, Y.; Cho, K.H.; Kim, Y.S.; Kim, D.H.; Yu, B.P.; Choi, J.S.; et al. Short-term feeding of baicalin inhibits age-associated NF-κB activation. Mech. Ageing Dev. 2006, 127, 719–725. [Google Scholar] [CrossRef]
- Kimura, Y.; Sumiyoshi, M. Effects of baicalein and wogonin isolated from Scutellaria baicalensis roots on skin damage in acute UVB-irradiated hairless mice. Eur. J. Pharmacol. 2011, 661, 124–132. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.-A.; Ma, L.-W.; Yin, Z.-Q.; Hu, Y.-Y.; Xu, Y.; Wu, D.; Permatasari, F.; Luo, D.; Zhou, B.-R. The protective effect of Baicalin against UVB irradiation induced photoaging: An in vitro and in vivo study. PLoS ONE 2014, 9, e99703. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.F.; Ma, K.H.; Chang, Y.J.; Lo, L.C.; Jhap, T.Y.; Su, Y.H.; Liu, P.S.; Chueh, S.H. Baicalein inhibits matrix metalloproteinase 1 expression via activation of TRPV 1-Ca-ERK pathway in ultraviolet B–irradiated human dermal fibroblasts. Exp. Dermatol. 2019, 28, 568–575. [Google Scholar] [CrossRef]
- Kim, B.; Kim, J.E.; Lee, S.M.; Kim, H.S. Effect of Scutellaria baicalensis extract on skin barrier function via peroxisome prolif-eratoractivated receptor-α. Korean J. Chem. Eng. 2014, 31, 104–108. [Google Scholar] [CrossRef]
- Kao, T.-T.; Wang, M.-C.; Chen, Y.-H.; Chung, Y.-T.; Hwang, P.-A. Propylene glycol improves stability of the anti-inflammatory compounds in Scutellaria baicalensis extract. Processes 2021, 9, 894. [Google Scholar] [CrossRef]
- Plamed. Scutellaria baicalensis Root Extract Application in Cosmetics. 2019. Available online: http://www.plamed.cn/scutellaria-baicalensis-root-extract-application-in-cosmetics/ (accessed on 8 September 2021).
- Tauchen, J.; Kokoska, L. The chemistry and pharmacology of Edelweiss: A review. Phytochem. Rev. 2016, 16, 295–308. [Google Scholar] [CrossRef]
- Cho, W.K.; Kim, H.-I.; Kim, S.-Y.; Seo, H.H.; Song, J.; Kim, J.; Shin, D.S.; Jo, Y.; Choi, H.; Lee, J.H.; et al. Anti-aging effects of Leontopodium alpinum (Edelweiss) Callus culture extract through transcriptome profiling. Genes 2020, 11, 230. [Google Scholar] [CrossRef] [Green Version]
- Daniela, L.; Alla, P.; Maurelli, R.; Elena, D.; Giovanna, P.; Vladimir, K.; Roberto, D.T.; Chiara, D.L.; Saveria, P.; Liudmila, K. Anti-inflammatory effects of concentrated ethanol extracts of edelweiss (Leontopodium alpinum Cass.) Callus cultures towards human keratinocytes and endothelial cells. Mediat. Inflamm. 2012, 2012, 498373. [Google Scholar] [CrossRef] [PubMed]
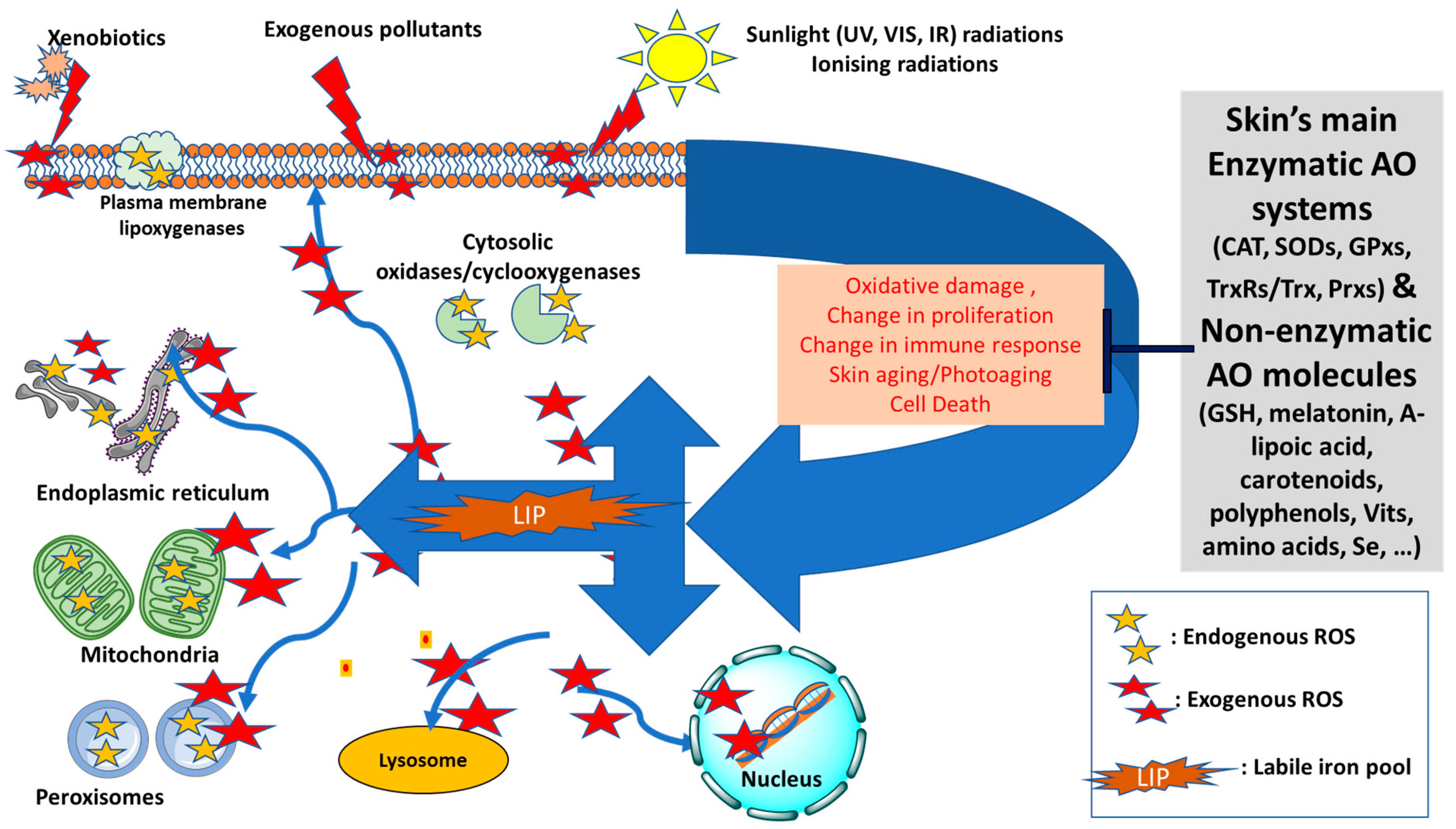
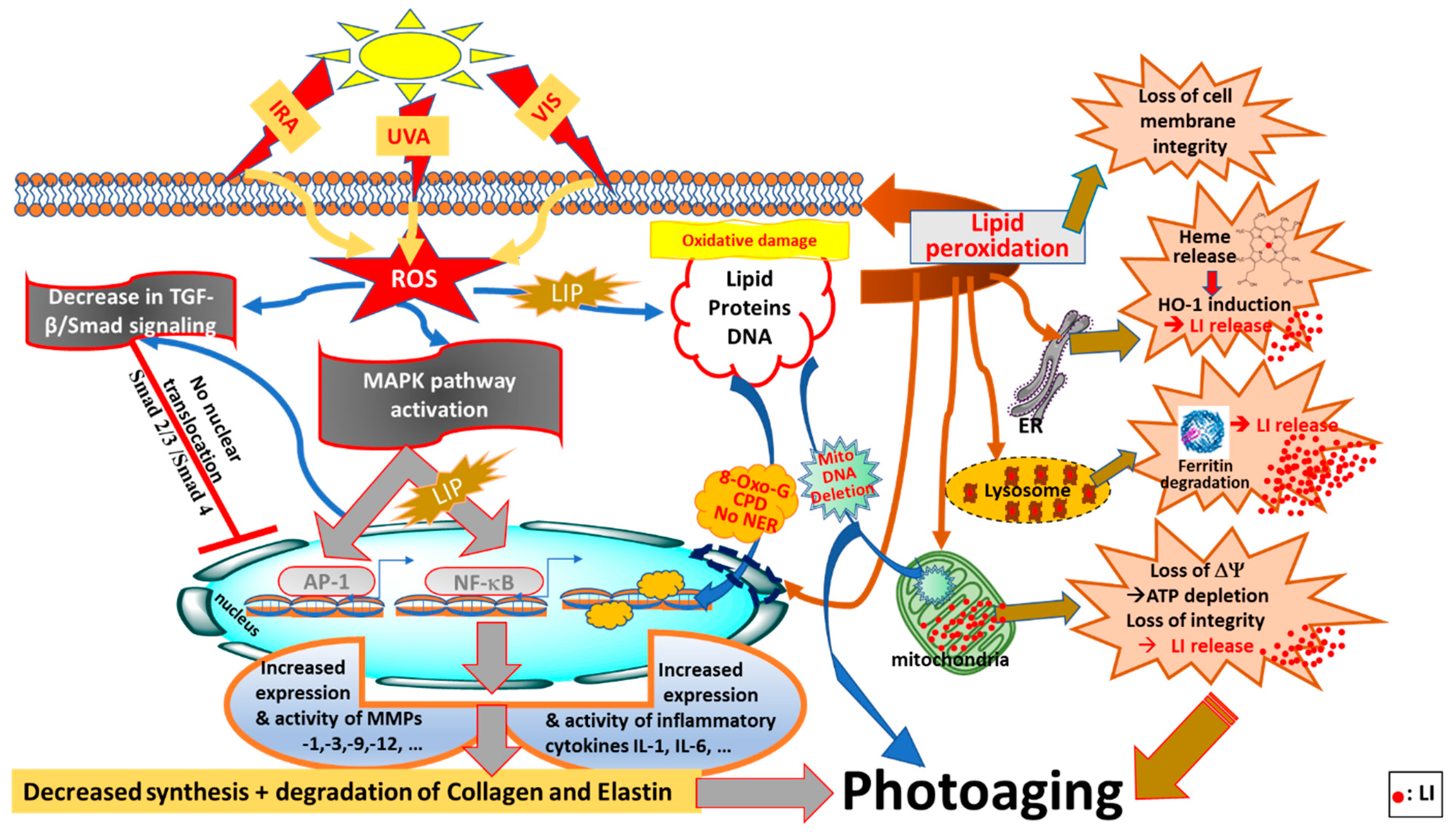
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pourzand, C.; Albieri-Borges, A.; Raczek, N.N. Shedding a New Light on Skin Aging, Iron- and Redox-Homeostasis and Emerging Natural Antioxidants. Antioxidants 2022, 11, 471. https://doi.org/10.3390/antiox11030471
Pourzand C, Albieri-Borges A, Raczek NN. Shedding a New Light on Skin Aging, Iron- and Redox-Homeostasis and Emerging Natural Antioxidants. Antioxidants. 2022; 11(3):471. https://doi.org/10.3390/antiox11030471
Chicago/Turabian StylePourzand, Charareh, Andrea Albieri-Borges, and Nico N. Raczek. 2022. "Shedding a New Light on Skin Aging, Iron- and Redox-Homeostasis and Emerging Natural Antioxidants" Antioxidants 11, no. 3: 471. https://doi.org/10.3390/antiox11030471
APA StylePourzand, C., Albieri-Borges, A., & Raczek, N. N. (2022). Shedding a New Light on Skin Aging, Iron- and Redox-Homeostasis and Emerging Natural Antioxidants. Antioxidants, 11(3), 471. https://doi.org/10.3390/antiox11030471