

Copper Depletion Strongly Enhances Ferroptosis via Mitochondrial Perturbation and Reduction in Antioxidative Mechanisms
 , , and
, , and 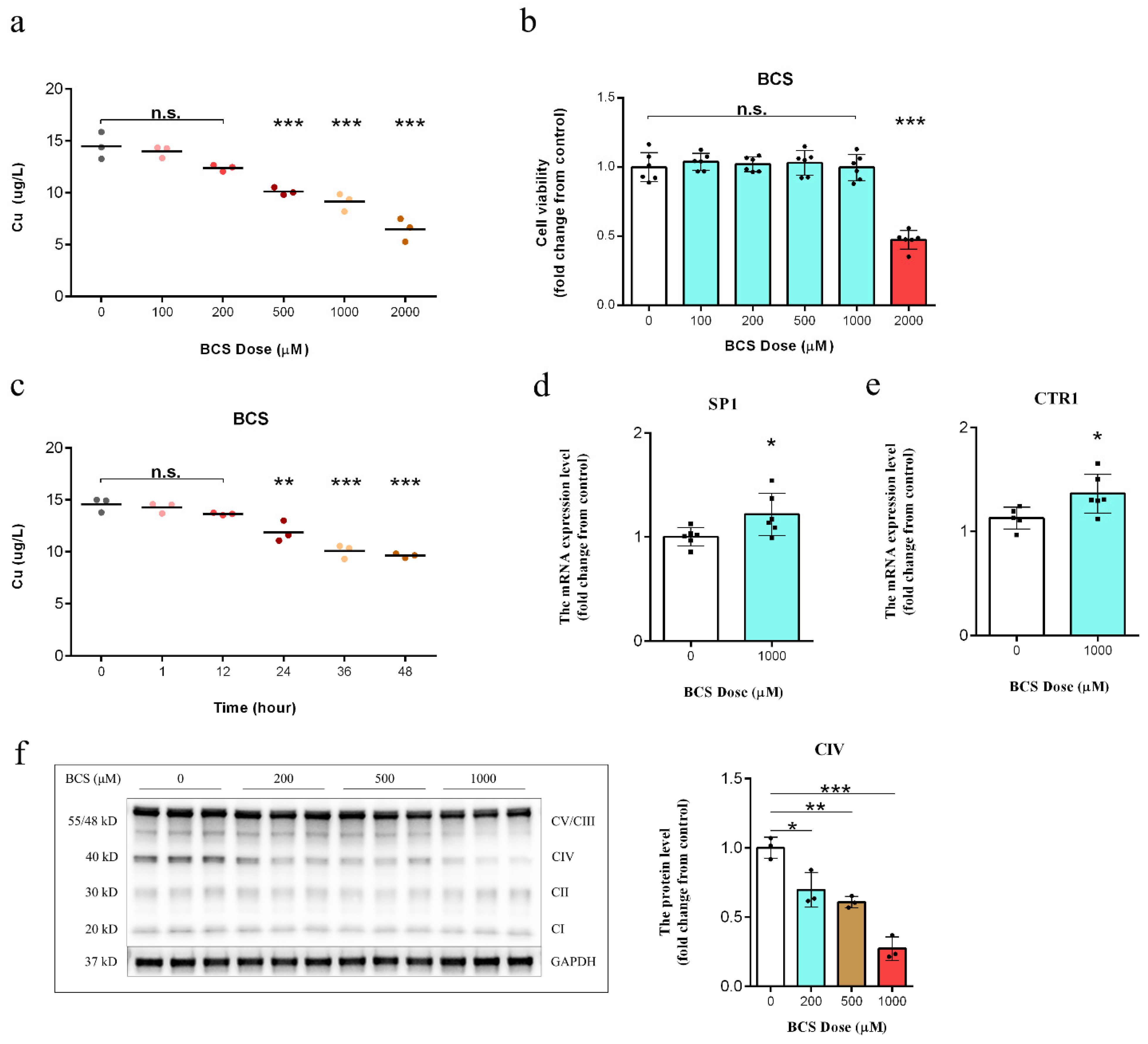{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cells
2.3. Cell Culture
2.4. Measurement of ATP Content
2.5. Cytochrome c Oxidase Activity
2.6. Assay for Mitochondrial Membrane Potential (MMP)
2.7. Determination of Total Cellular ROS Concentrations
2.8. SOD1 Activity
2.9. Measurement of GSH-Px Activity
2.10. Detection of Intracellular GSH and GSSG Levels
2.11. Determination of MDA Concentrations
2.12. Determination of Fe2+ Concentrations
2.13. Determination of Glutamic Concentrations
2.14. Determination of Cysteine Concentrations
2.15. Determination of Lipid Peroxide Concentrations
2.16. Determination of Protein Carbonylation Concentrations
2.17. Measurement of Copper Contents
2.18. RNA Isolation and Analysis
2.19. Western Blot
2.20. Liquid Chromatography–Mass Spectrometry (LC–MS) Analysis
2.21. Statistical Analysis
3. Results
3.1. BCS Treatment Depleted Intracellular Copper Concentrations
3.2. Copper Depletion Impaired Mitochondrial Complex IV
3.3. Copper Depletion Impaired Antioxidant Capacity in DPCs
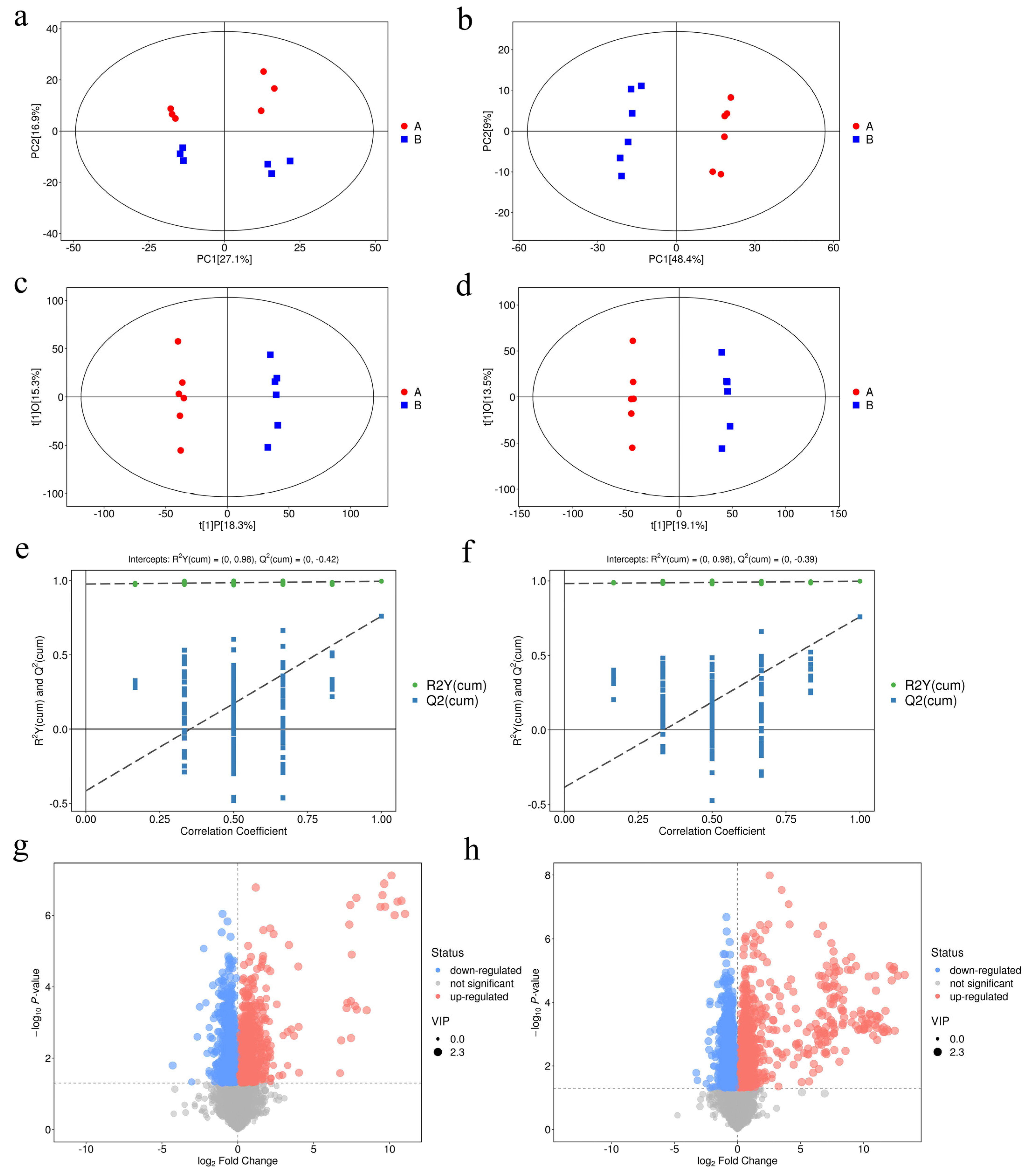
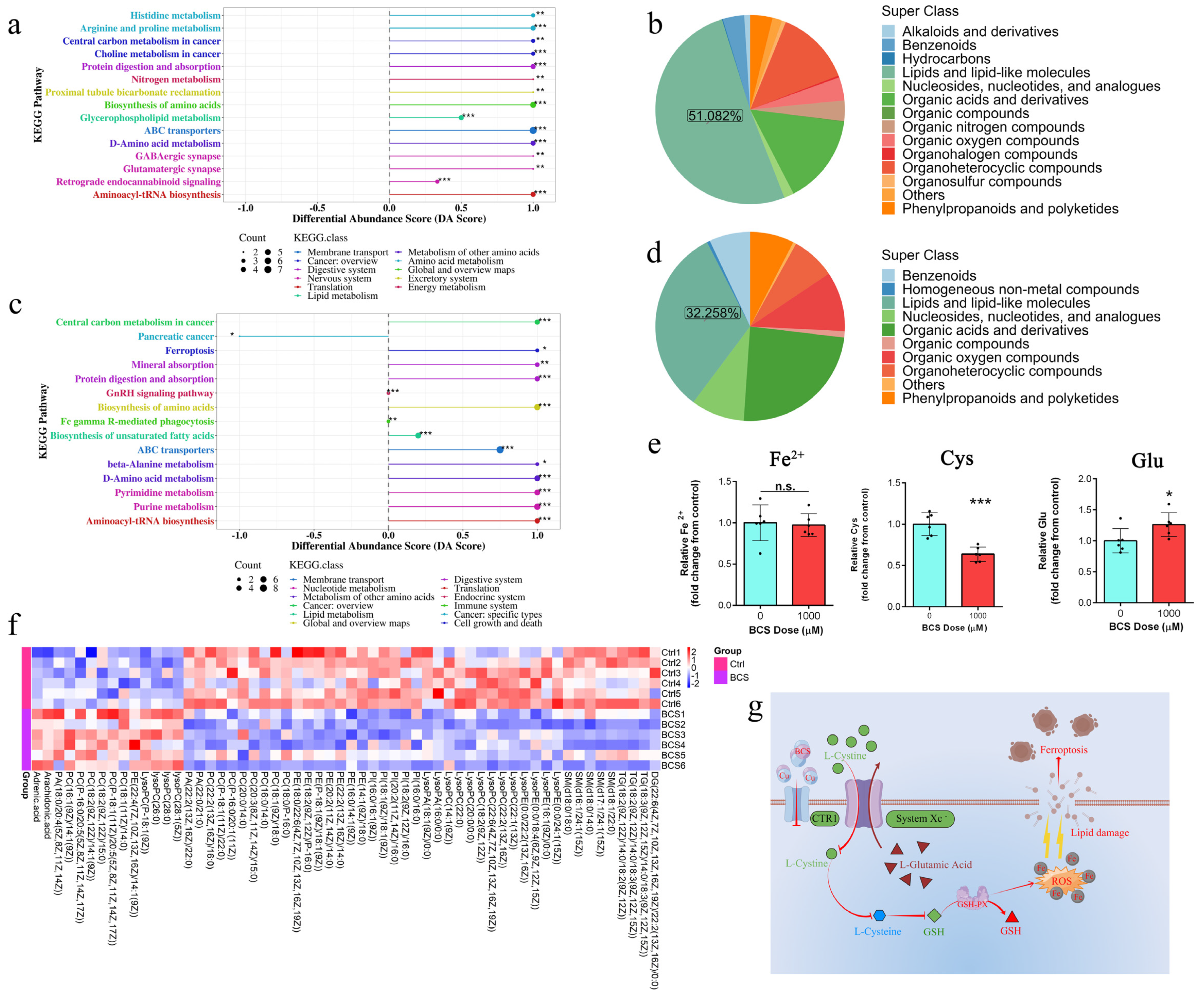
3.4. UHPLC–MS/MS Analysis
3.5. Copper Depletion Induce Ferroptosis
3.6. Copper Depletion Decreased the Sensitivity of DPCs to Erastin
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhou, Q.; Lin, W.; Wang, C.; Sun, F.; Ju, S.; Chen, Q.; Wang, Y.; Chen, Y.; Li, H.; Wang, L.; et al. Neddylation inhibition induces glutamine uptake and metabolism by targeting CRL3SPOP E3 ligase in cancer cells. Nat. Commun. 2022, 13, 3034. [Google Scholar] [CrossRef] [PubMed]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Sancho, P.; Barneda, D.; Heeschen, C. Hallmarks of cancer stem cell metabolism. Br. J. Cancer 2016, 114, 1305–1312. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Ito, K. Metabolism and the Control of Cell Fate Decisions and Stem Cell Renewal. Annu. Rev. Cell Dev. Biol. 2016, 32, 399–409. [Google Scholar] [CrossRef]
- Liu, H.; Guo, H.; Jian, Z.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; Zhao, L. Copper Induces Oxidative Stress and Apoptosis in the Mouse Liver. Oxidative Med. Cell. Longev. 2020, 2020, 1359164. [Google Scholar] [CrossRef]
- Lee, H.; Zandkarimi, F.; Zhang, Y.; Meena, J.K.; Kim, J.; Zhuang, L.; Tyagi, S.; Ma, L.; Westbrook, T.F.; Steinberg, G.R.; et al. Energy-stress-mediated AMPK activation inhibits ferroptosis. Nat. Cell Biol. 2020, 22, 225–234. [Google Scholar] [CrossRef]
- Toyama, E.Q.; Herzig, S.; Courchet, J.; Lewis, T.L., Jr.; Losón, O.C.; Hellberg, K.; Young, N.P.; Chen, H.; Polleux, F.; Chan, D.C.; et al. Metabolism. AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science 2016, 351, 275–281. [Google Scholar] [CrossRef]
- Ibrahim, A.; Yucel, N.; Kim, B.; Arany, Z. Local Mitochondrial ATP Production Regulates Endothelial Fatty Acid Uptake and Transport. Cell Metab. 2020, 32, 309–319.e7. [Google Scholar] [CrossRef]
- Ramchandani, D.; Berisa, M.; Tavarez, D.A.; Li, Z.; Miele, M.; Bai, Y.; Lee, S.B.; Ban, Y.; Dephoure, N.; Hendrickson, R.C.; et al. Copper depletion modulates mitochondrial oxidative phosphorylation to impair triple negative breast cancer metastasis. Nat. Commun. 2021, 12, 7311. [Google Scholar] [CrossRef]
- Kim, B.E.; Nevitt, T.; Thiele, D.J. Mechanisms for copper acquisition, distribution, and regulation. Nat. Chem. Biol. 2008, 4, 176–185. [Google Scholar] [CrossRef]
- Barrientos, A.; Barros, M.H.; Valnot, I.; Rötig, A.; Rustin, P.; Tzagoloff, A. Cytochrome oxidase in health and disease. Gene 2002, 286, 53–63. [Google Scholar] [CrossRef]
- Partridge, R.S.; Monroe, S.M.; Parks, J.K.; Johnson, K.; Parker, W.D., Jr.; Eaton, G.R.; Eaton, S.S. Spin trapping of azidyl and hydroxyl radicals in azide-inhibited rat brain submitochondrial particles. Arch. Biochem. Biophys. 1994, 310, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Park, E.; Chung, S.W. ROS-mediated autophagy increases intracellular iron levels and ferroptosis by ferritin and transferrin receptor regulation. Cell Death Dis. 2019, 10, 822. [Google Scholar] [CrossRef] [PubMed]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascón, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef]
- Gao, M.; Yi, J.; Zhu, J.; Minikes, A.M.; Monian, P.; Thompson, C.B.; Jiang, X. Role of Mitochondria in Ferroptosis. Mol. Cell 2019, 73, 354–363.e3. [Google Scholar] [CrossRef]
- Chen, X.; Kang, R.; Kroemer, G.; Tang, D. Broadening horizons: The role of ferroptosis in cancer. Nat. Rev. Clin. Oncol. 2021, 18, 280–296. [Google Scholar] [CrossRef]
- Pisoschi, A.M.; Pop, A. The role of antioxidants in the chemistry of oxidative stress: A review. Eur. J. Med. Chem. 2015, 97, 55–74. [Google Scholar] [CrossRef]
- Fukai, T.; Ushio-Fukai, M. Superoxide dismutases: Role in redox signaling, vascular function, and diseases. Antioxid. Redox Signal. 2011, 15, 1583–1606. [Google Scholar] [CrossRef]
- Hu, C.L.; Nydes, M.; Shanley, K.L.; Morales Pantoja, I.E.; Howard, T.A.; Bizzozero, O.A. Reduced expression of the ferroptosis inhibitor glutathione peroxidase-4 in multiple sclerosis and experimental autoimmune encephalomyelitis. J. Neurochem. 2019, 148, 426–439. [Google Scholar] [CrossRef]
- Pitman, K.E.; Alluri, S.R.; Kristian, A.; Aarnes, E.K.; Lyng, H.; Riss, P.J.; Malinen, E. Influx rate of 18F-fluoroaminosuberic acid reflects cystine/glutamate antiporter expression in tumour xenografts. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 2190–2198. [Google Scholar] [CrossRef]
- Dixon, S.J.; Patel, D.N.; Welsch, M.; Skouta, R.; Lee, E.D.; Hayano, M.; Thomas, A.G.; Gleason, C.E.; Tatonetti, N.P.; Slusher, B.S.; et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. eLife 2014, 3, e02523. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [PubMed]
- Li, J.L.; Liu, M.; Zhou, J.; Chen, Q.R.; Chen, Y.; Chen, S.; Shen, J.Y.; Wu, X. Isolation, Culture and Identification of Rabbit Dermal Papilla Cells. Chin. J. Rabbit Farming 2020, 3, 4–7. [Google Scholar]
- Li, F.; Liu, H.; Wu, X.; Liu, M.; Yue, Z.; Liu, L.; Li, F. Copper Modulates Mitochondrial Oxidative Phosphorylation to Enhance Dermal Papilla Cells Proliferation in Rex Rabbits. Int. J. Mol. Sci. 2022, 23, 6209. [Google Scholar] [CrossRef]
- Li, F.; Liu, L.; Chen, X.; Zhang, B.; Li, F. Dietary Copper Supplementation Increases Growth Performance by Increasing Feed Intake, Digestibility, and Antioxidant Activity in Rex Rabbits. Biol. Trace Elem. Res. 2021, 199, 4614–4623. [Google Scholar] [CrossRef]
- Yuan, S.; Chen, S.; Xi, Z.; Liu, Y. Copper-finger protein of Sp1: The molecular basis of copper sensing. Met. Integr. Biometal Sci. 2017, 9, 1169–1175. [Google Scholar] [CrossRef]
- Liang, Z.D.; Tsai, W.B.; Lee, M.Y.; Savaraj, N.; Kuo, M.T. Specificity protein 1 (sp1) oscillation is involved in copper homeostasis maintenance by regulating human high-affinity copper transporter 1 expression. Mol. Pharmacol. 2012, 81, 455–464. [Google Scholar] [CrossRef]
- Horn, D.; Barrientos, A. Mitochondrial copper metabolism and delivery to cytochrome c oxidase. IUBMB Life 2008, 60, 421–429. [Google Scholar] [CrossRef]
- Tsvetkov, P.; Coy, S.; Petrova, B.; Dreishpoon, M.; Verma, A.; Abdusamad, M.; Rossen, J.; Joesch-Cohen, L.; Humeidi, R.; Spangler, R.D.; et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science 2022, 375, 1254–1261. [Google Scholar] [CrossRef]
- Krishnamoorthy, L.; Cotruvo, J.A., Jr.; Chan, J.; Kaluarachchi, H.; Muchenditsi, A.; Pendyala, V.S.; Jia, S.; Aron, A.T.; Ackerman, C.M.; Wal, M.N.; et al. Copper regulates cyclic-AMP-dependent lipolysis. Nat. Chem. Biol. 2016, 12, 586–592. [Google Scholar] [CrossRef]
- Song, I.S.; Chen, H.H.; Aiba, I.; Hossain, A.; Liang, Z.D.; Klomp, L.W.; Kuo, M.T. Transcription factor Sp1 plays an important role in the regulation of copper homeostasis in mammalian cells. Mol. Pharmacol. 2008, 74, 705–713. [Google Scholar] [CrossRef] [PubMed]
- Spinelli, J.B.; Haigis, M.C. The multifaceted contributions of mitochondria to cellular metabolism. Nat. Cell Biol. 2018, 20, 745–754. [Google Scholar] [CrossRef] [PubMed]
- Moloney, J.N.; Cotter, T.G. ROS signalling in the biology of cancer. Semin. Cell Dev. Biol. 2018, 80, 50–64. [Google Scholar] [CrossRef] [PubMed]
- Giorgio, M.; Trinei, M.; Migliaccio, E.; Pelicci, P.G. Hydrogen peroxide: A metabolic by-product or a common mediator of ageing signals? Nat. Rev. Mol. Cell Biol. 2007, 8, 722–728. [Google Scholar] [CrossRef]
- Caihong, L.; Ping, C. Cytochrome c oxidase and chronic obstructive pulmonary disease. Int. J. Respir 2008, 28, 1436–1439. [Google Scholar]
- Hauck, A.K.; Huang, Y.; Hertzel, A.V.; Bernlohr, D.A. Adipose oxidative stress and protein carbonylation. J. Biol. Chem. 2019, 294, 1083–1088. [Google Scholar] [CrossRef]
- Gaschler, M.M.; Stockwell, B.R. Lipid peroxidation in cell death. Biochem. Biophys. Res. Commun. 2017, 482, 419–425. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Fetherolf, M.; Boyd, S.D.; Winkler, D.D.; Winge, D.R. Oxygen-dependent activation of Cu,Zn-superoxide dismutase-1. Metallomics 2017, 9, 1047–1059. [Google Scholar] [CrossRef]
- Xie, Y.; Hou, W.; Song, X.; Yu, Y.; Huang, J.; Sun, X.; Kang, R.; Tang, D. Ferroptosis: Process and function. Cell Death Differ. 2016, 23, 369–379. [Google Scholar] [CrossRef]
- Yang, Y.; Luo, M.; Zhang, K.; Zhang, J.; Gao, T.; Connell, D.O.; Yao, F.; Mu, C.; Cai, B.; Shang, Y.; et al. Nedd4 ubiquitylates VDAC2/3 to suppress erastin-induced ferroptosis in melanoma. Nat. Commun. 2020, 11, 433. [Google Scholar] [CrossRef] [PubMed]
- Shimada, K.; Skouta, R.; Kaplan, A.; Yang, W.S.; Hayano, M.; Dixon, S.J.; Brown, L.M.; Valenzuela, C.A.; Wolpaw, A.J.; Stockwell, B.R. Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat. Chem. Biol. 2016, 12, 497–503. [Google Scholar] [CrossRef] [PubMed]
- Koppula, P.; Zhang, Y.; Zhuang, L.; Gan, B. Amino acid transporter SLC7A11/xCT at the crossroads of regulating redox homeostasis and nutrient dependency of cancer. Cancer Commun. 2018, 38, 12. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Li, D.; Alesi, G.N.; Fan, J.; Kang, H.B.; Lu, Z.; Boggon, T.J.; Jin, P.; Yi, H.; Wright, E.R.; et al. Glutamate dehydrogenase 1 signals through antioxidant glutathione peroxidase 1 to regulate redox homeostasis and tumor growth. Cancer Cell 2015, 27, 257–270. [Google Scholar] [CrossRef]
- Hirayama, T.; Nagasawa, H. Chemical tools for detecting Fe ions. J. Clin. Biochem. Nutr. 2017, 60, 39–48. [Google Scholar] [CrossRef]
- Liu, J.; Hu, Z.; Liu, D.; Zheng, A.; Ma, Q. Glutathione metabolism-mediated ferroptosis reduces water-holding capacity in beef during cold storage. Food Chem. 2023, 398, 133903. [Google Scholar] [CrossRef]
- Kim, C.S.; Ding, X.; Allmeroth, K.; Biggs, L.C.; Kolenc, O.I.; L’Hoest, N.; Chacón-Martínez, C.A.; Edlich-Muth, C.; Giavalisco, P.; Quinn, K.P.; et al. Glutamine Metabolism Controls Stem Cell Fate Reversibility and Long-Term Maintenance in the Hair Follicle. Cell Metab. 2020, 32, 629–642.e8. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Liu, Y.; Du, T.; Yang, H.; Lei, L.; Guo, M.; Ding, H.F.; Zhang, J.; Wang, H.; Chen, X.; et al. ATF3 promotes erastin-induced ferroptosis by suppressing system Xc. Cell Death Differ. 2020, 27, 662–675. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Feng, Y.; Li, H.; Chen, X.; Wang, G.; Xu, S.; Li, Y.; Zhao, L. Ferrostatin-1 alleviates lipopolysaccharide-induced acute lung injury via inhibiting ferroptosis. Cell. Mol. Biol. Lett. 2020, 25, 10. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Z.; Kong, B.; Fang, J.; Qin, T.; Dai, C.; Shuai, W.; Huang, H. Ferrostatin-1 alleviates lipopolysaccharide-induced cardiac dysfunction. Bioengineered 2021, 12, 9367–9376. [Google Scholar] [CrossRef] [PubMed]
- Basit, F.; van Oppen, L.M.; Schöckel, L.; Bossenbroek, H.M.; van Emst-de Vries, S.E.; Hermeling, J.C.; Grefte, S.; Kopitz, C.; Heroult, M.; Hgm Willems, P.; et al. Mitochondrial complex I inhibition triggers a mitophagy-dependent ROS increase leading to necroptosis and ferroptosis in melanoma cells. Cell Death Dis. 2017, 8, e2716. [Google Scholar] [CrossRef] [PubMed]
- Degterev, A.; Linkermann, A. Generation of small molecules to interfere with regulated necrosis. Cell. Mol. Life Sci. CMLS 2016, 73, 2251–2267. [Google Scholar] [CrossRef] [PubMed]
- Bogacz, M.; Krauth-Siegel, R.L. Tryparedoxin peroxidase-deficiency commits trypanosomes to ferroptosis-type cell death. eLife 2018, 7, e37503. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, F.; Wu, X.; Liu, H.; Liu, M.; Yue, Z.; Wu, Z.; Liu, L.; Li, F. Copper Depletion Strongly Enhances Ferroptosis via Mitochondrial Perturbation and Reduction in Antioxidative Mechanisms. Antioxidants 2022, 11, 2084. https://doi.org/10.3390/antiox11112084
Li F, Wu X, Liu H, Liu M, Yue Z, Wu Z, Liu L, Li F. Copper Depletion Strongly Enhances Ferroptosis via Mitochondrial Perturbation and Reduction in Antioxidative Mechanisms. Antioxidants. 2022; 11(11):2084. https://doi.org/10.3390/antiox11112084
Chicago/Turabian StyleLi, Fan, Xiaojing Wu, Hongli Liu, Mengqi Liu, Zhengkai Yue, Zhenyu Wu, Lei Liu, and Fuchang Li. 2022. "Copper Depletion Strongly Enhances Ferroptosis via Mitochondrial Perturbation and Reduction in Antioxidative Mechanisms" Antioxidants 11, no. 11: 2084. https://doi.org/10.3390/antiox11112084
APA StyleLi, F., Wu, X., Liu, H., Liu, M., Yue, Z., Wu, Z., Liu, L., & Li, F. (2022). Copper Depletion Strongly Enhances Ferroptosis via Mitochondrial Perturbation and Reduction in Antioxidative Mechanisms. Antioxidants, 11(11), 2084. https://doi.org/10.3390/antiox11112084