
Coenzyme Q10, Ageing and the Nervous System: An Overview


{kind=link}
Abstract
:1. Introduction
2. Neurological Disorders and Ageing
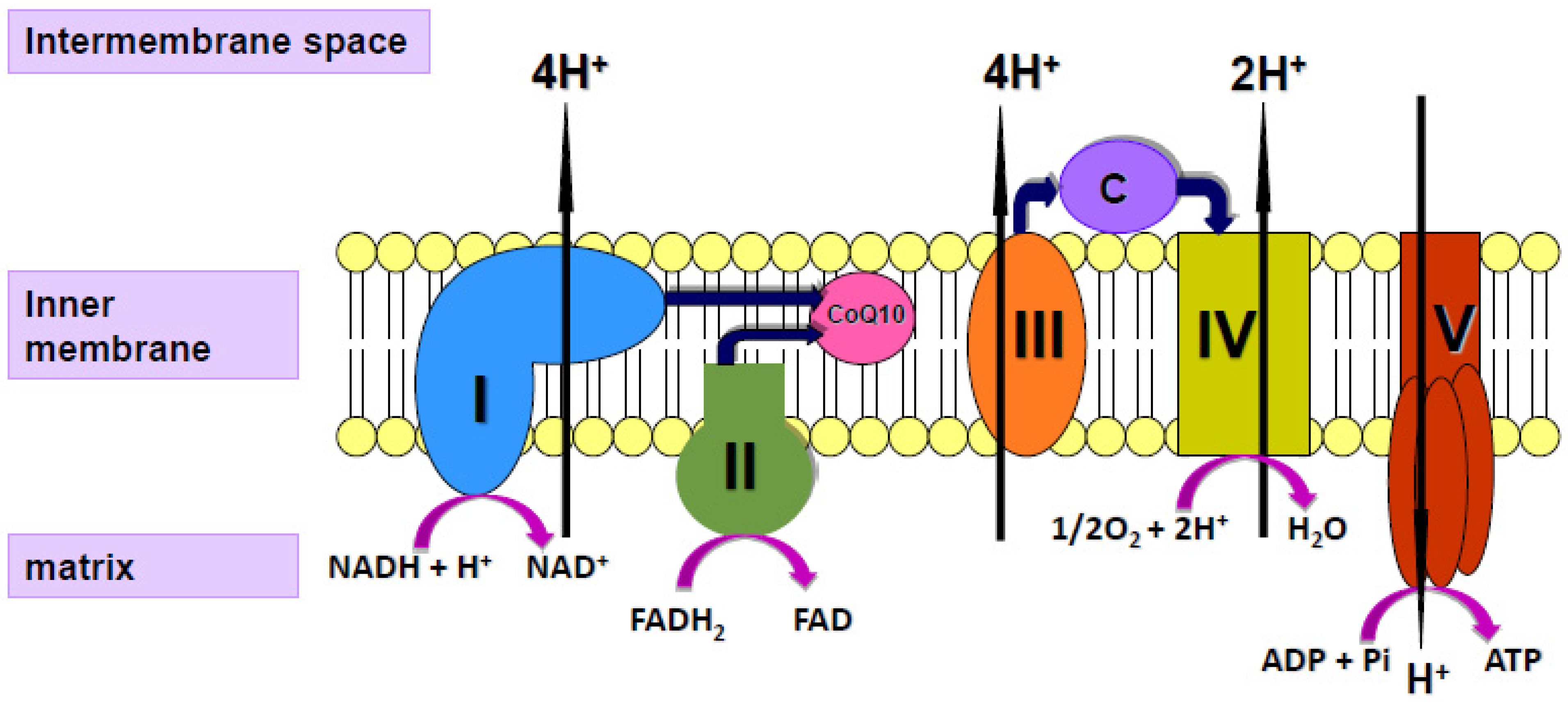
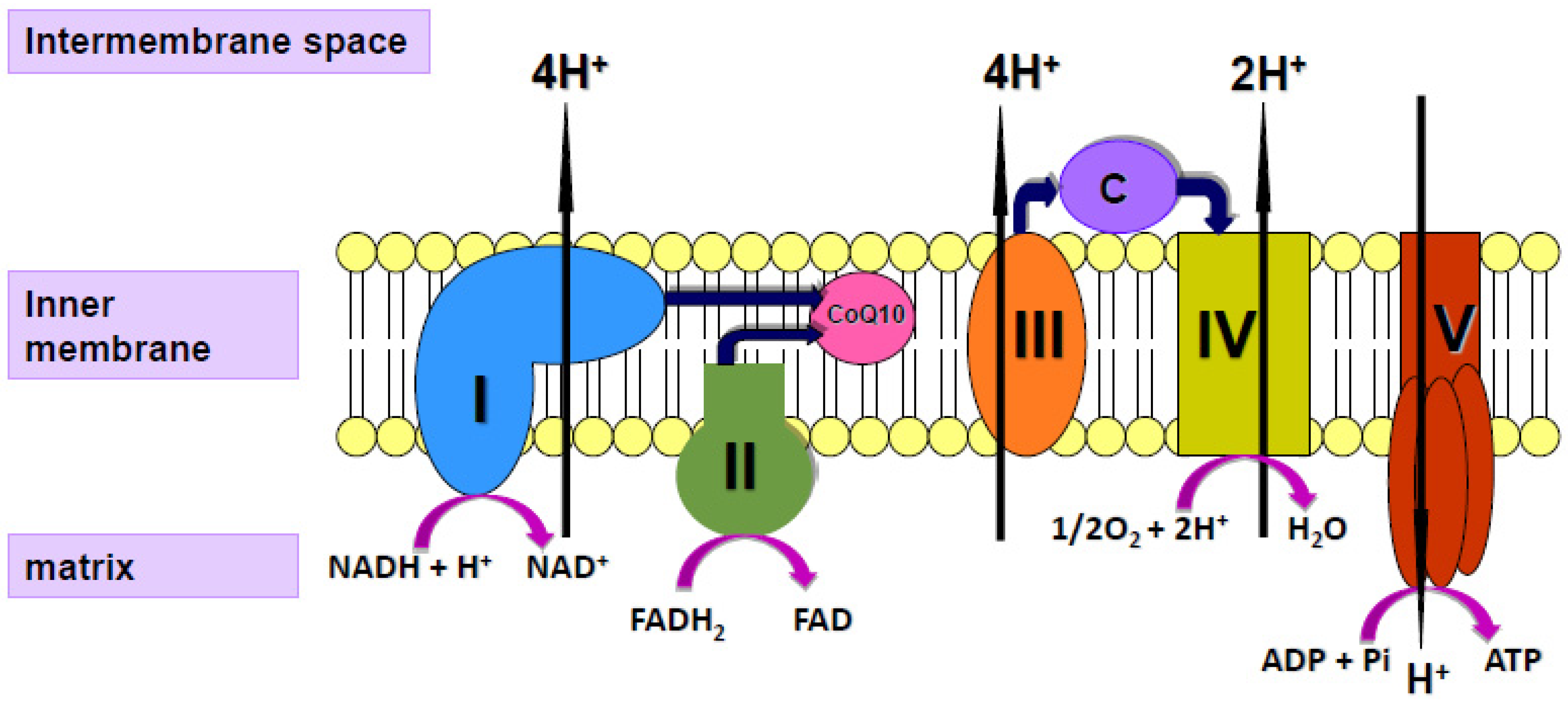
3. Coenzyme Q10 and Ageing
4. Coenzyme Q10 and Neurological Disorders
5. Why Have Clinical Trials of CoQ10 in Neurological Disorders Been Disappointing?
- (i)
- The importance of CoQ10 crystal dispersion since the failure to disperse CoQ10 crystals to single molecules reduces the bioavailability of CoQ10 by approximately 75%;
- (ii)
- That the relative bioavailability of ubiquinone and ubiquinol forms of CoQ10 depends on its CoQ10 crystal dispersion status and carrier oil/excipient composition.
6. Summary
Author Contributions
Funding
Conflicts of Interest
References
- Peters, R. Ageing and the brain. Postgrad. Med. J. 2006, 82, 84–88. [Google Scholar] [CrossRef]
- Maillet, D.; Rajah, M.N. Association between prefrontal activity and volume change in prefrontal and medial temporal lobes in aging and dementia: A review. Ageing Res. Rev. 2013, 12, 479–489. [Google Scholar] [CrossRef]
- Giorgio, A.; Santelli, L.; Tomassini, V.; Bosnell, R.; Smith, S.; de Stefano, N.; Johansen-Berg, H. Age-related changes in grey and white matter structure throughout adulthood. NeuroImage 2010, 51, 943–951. [Google Scholar] [CrossRef] [Green Version]
- Pannese, E. Morphological changes in nerve cells during normal aging. Brain Struct. Funct. 2011, 216, 85–89. [Google Scholar] [CrossRef]
- Valles, S.L.; Iradi, A.; Aldasoro, M.; Vila, J.M.; Aldasoro, C.; De La Torre, J.; Campos-Campos, J.; Jorda, A. Function of Glia in Aging and the Brain Diseases. Int. J. Med Sci. 2019, 16, 1473–1479. [Google Scholar] [CrossRef] [Green Version]
- Carlsson, A. Brain Neurotransmitters in Aging and Dementia: Similar Changes Across Diagnostic Dementia Groups. Gerontology 1987, 33, 159–167. [Google Scholar] [CrossRef]
- Navarro, A.; Boveris, A. Brain mitochondrial dysfunction in aging, neurodegeneration, and Parkinson’s disease. Front. Aging Neurosci. 2010, 2, 34. [Google Scholar] [CrossRef] [Green Version]
- Mariani, E.; Polidori, M.; Cherubini, A.; Mecocci, P. Oxidative stress in brain aging, neurodegenerative and vascular diseases: An overview. J. Chromatogr. B 2005, 827, 65–75. [Google Scholar] [CrossRef]
- Harada, C.N.; Natelson Love, M.C.; Triebel, K.L. Normal Cognitive Aging. Clin. Geriatr. Med. 2013, 29, 737–752. [Google Scholar] [CrossRef] [Green Version]
- Tarumi, T.; Zhang, R. Cerebral blood flow in normal aging adults: Cardiovascular determinants, clinical implications, and aerobic fitness. J. Neurochem. 2018, 144, 595–608. [Google Scholar] [CrossRef] [Green Version]
- Montagne, A.; Barnes, S.R.; Sweeney, M.D.; Halliday, M.R.; Sagare, A.P.; Zhao, Z.; Toga, A.W.; Jacobs, R.E.; Liu, C.Y.; Amezcua, L.; et al. Blood-brain barrier breakdown in the aging human hippocampus. Neuron 2015, 85, 296–302. [Google Scholar] [CrossRef] [Green Version]
- Xia, X.; Jiang, Q.; McDermott, J.; Han, J.-D.J. Aging and Alzheimer’s disease: Comparison and associations from molecular to system level. Aging Cell 2018, 17, e12802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reeve, A.; Simcox, E.; Turnbull, D. Ageing and Parkinson’s disease: Why is advancing age the biggest risk factor? Ageing Res. Rev. 2014, 14, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Fang, F.; Ingre, C.; Roos, P.M.; Kamel, F.; Piehl, F. Risk factors for amyotrophic lateral sclerosis. Clin. Epidemiol. 2015, 7, 181–193. [Google Scholar] [CrossRef] [Green Version]
- Kelly-Hayes, M. Influence of Age and Health Behaviors on Stroke Risk: Lessons from Longitudinal Studies. J. Am. Geriatr. Soc. 2010, 58 (Suppl. S2), S325–S328. [Google Scholar] [CrossRef]
- Crane, F.L. Biochemical Functions of Coenzyme Q10. J. Am. Coll. Nutr. 2001, 20, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Heaton, R.A.; Heales, S.; Rahman, K.; Sexton, D.W.; Hargreaves, I. The Effect of Cellular Coenzyme Q10 Deficiency on Lysosomal Acidification. J. Clin. Med. 2020, 9, 1923. [Google Scholar] [CrossRef] [PubMed]
- Schmelzer, C.; Lindner, I.; Rimbach, G.; Niklowitz, P.; Menke, T.; Döring, F. Functions of coenzyme Q10 in inflammation and gene expression. Biofactors 2008, 32, 179–183. [Google Scholar] [CrossRef]
- Weber, C.; Bysted, A.; Hłlmer, G. The coenzyme Q10 content of the average Danish diet. Int. J. Vitam. Nutr. Res. 1997, 67, 123–129. [Google Scholar]
- Yubero, D.; Montero, R.; Artuch, R.; Land, J.M.; Heales, S.J.; Hargreaves, I.P. Biochemical Diagnosis of Coenzyme Q10 Deficiency. Mol. Syndr. 2014, 5, 147–155. [Google Scholar] [CrossRef] [Green Version]
- Duberley, K.E.C.; Hargreaves, I.P.; Chaiwatanasirikul, K.-A.; Heales, S.J.R.; Land, J.M.; Rahman, S.; Mills, K.; Eaton, S. Coenzyme Q10 quantification in muscle, fibroblasts and cerebrospinal fluid by liquid chromatography/tandem mass spectrometry using a novel deuterated internal standard. Rapid Commun. Mass Spectrom. 2013, 27, 924–930. [Google Scholar] [CrossRef]
- Kalén, A.; Appelkvist, E.-L.; Dallner, G. Age-related changes in the lipid compositions of rat and human tissues. Lipids 1989, 24, 579–584. [Google Scholar] [CrossRef]
- Hargreaves, I.P.; Lane, A.; Sleiman, P.M. The coenzyme Q10 status of the brain regions of Parkinson’s disease patients. Neurosci. Lett. 2008, 447, 17–19. [Google Scholar] [CrossRef]
- Duberley, K.; Heales, S.; Abramov, A.; Chalasani, A.; Land, J.; Rahman, S.; Hargreaves, I. Effect of Coenzyme Q10 supplementation on mitochondrial electron transport chain activity and mitochondrial oxidative stress in Coenzyme Q10 deficient human neuronal cells. Int. J. Biochem. Cell Biol. 2014, 50, 60–63. [Google Scholar] [CrossRef]
- Schultz, C.; Del Tredici, K.H.B. Neuropathology of Alzheimer’s Disease. In Alzheimer’s Disease Current Clinical Neurology; Humana Press: Totowa, NJ, USA, 2004. [Google Scholar]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological Alterations in Alzheimer Disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Xu, T.H.; Melcher, K.; Xu, H.E. Amyloid beta: Structure, biology and structure-based therapeutic development. Acta Pharmacol. Sin. 2017, 38, 1205–1235. [Google Scholar]
- Long, J.M.; Holtzman, D.M. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 2019, 179, 312–339. [Google Scholar] [CrossRef] [PubMed]
- Perry, G.; Cash, A.D.; Smith, M.A. Alzheimer Disease and Oxidative Stress. J. Biomed. Biotechnol. 2002, 2, 120–123. [Google Scholar] [CrossRef]
- De Bustos, F.; Molina, J.A.; Jiménez-Jiménez, F.J.; García-Redondo, A.; Gómez-Escalonilla, C.; Porta-Etessam, J.; Berbel, A.; Zurdo, M.; Barcenilla, B.; Parrilla, G.; et al. Serum levels of coenzyme Q10 in patient with Alzheimer’s disease. J. Neural Transm. 2000, 107, 233–239. [Google Scholar] [CrossRef]
- Schippling, S.; Kontush, A.; Arlt, S.; Buhmann, C.; Stürenburg, H.-J.; Mann, U.; Müller-Thomsen, T.; Beisiegel, U. Increased lipoprotein oxidation in Alzheimer’s disease. Free Radic. Biol. Med. 2000, 28, 351–360. [Google Scholar] [CrossRef]
- Yamagishi, K.; Ikeda, A.; Moriyama, Y.; Chei, C.-L.; Noda, H.; Umesawa, M.; Cui, R.; Nagao, M.; Kitamura, A.; Yamamoto, Y.; et al. Serum coenzyme Q10 and risk of disabling dementia: The Circulatory Risk in Communities Study (CIRCS). Atherosclerosis 2014, 237, 400–403. [Google Scholar] [CrossRef] [Green Version]
- Durán-Prado, M.; Frontiñán, J.; Santiago-Mora, R.; Peinado, J.R.; Parrado-Fernandez, C.; Gomez, M.V.; Moreno, M.; Lopez-Dominguez, J.A.; Villalba, J.M.; Alcain, F.J. Coenzyme Q10 Protects Human Endothelial Cells from β-Amyloid Uptake and Oxidative Stress-Induced Injury. PLoS ONE 2014, 9, e109223. [Google Scholar] [CrossRef]
- Komaki, H.; Faraji, N.; Komaki, A.; Shahidi, S.; Etaee, F.; Raoufi, S.; Mirzaei, F. Investigation of protective effects of coenzyme Q10 on impaired synaptic plasticity in a male rat model of Alzheimer’s disease. Brain Res. Bull. 2019, 147, 14–21. [Google Scholar] [CrossRef]
- Galasko, D.R.; Peskind, E.; Clark, C.M.; Quinn, J.F.; Ringman, J.M.; Jicha, G.A.; Cotman, C.; Cottrell, B.; Montine, T.J.; Thomas, R.G.; et al. Antioxidants for Alzheimer disease: A randomized clinical trial with cerebrospinal fluid biomarker measures. Arch. Neurol. 2012, 69, 836–841. [Google Scholar] [CrossRef] [Green Version]
- Gutzmann, H.; Hadler, D. Sustained efficacy and safety of idebenone in the treatment of Alzheimer’s disease: Update on a 2-year double-blind multicentre study. J. Neural Transm. Suppl. 1998, 54, 301–310. [Google Scholar]
- Gutzmann, H.; Kuhl, K.-P.; Hadler, D.; Rapp, M.A. Safety and efficacy of idebenone versus tacrine in patients with Alzheimer’s disease: Results of a randomised, double-blind, parallel-group multicentre study. Pharmacopsychiatry 2002, 35, 12–18. [Google Scholar] [CrossRef]
- Thal, L.J.; Grundman, M.; Berg, J.; Ernstrom, K.; Margolin, R.; Pfeiffer, E.; Weiner, M.F.; Zamrini, E.; Thomas, R.G. Idebenone treatment fails to slow cognitive decline in Alzheimer’s disease. Neurology 2003, 61, 1498–1502. [Google Scholar] [CrossRef] [PubMed]
- Chai, C.; Lim, K.-L. Genetic Insights into Sporadic Parkinson’s Disease Pathogenesis. Curr. Genom. 2014, 14, 486–501. [Google Scholar] [CrossRef] [Green Version]
- Eden, S.; Tanner, M.; Bernstein, A.; Fross, R.; Leimpeter, A.; Bloch, D.; Nelson, L. Incidence of Parkinson’s Disease: Variation by Age, Gender, and Race/Ethnicity. Am. J. Epidemiol. 2003, 157, 1015–1022. [Google Scholar] [CrossRef]
- Stefanis, L. α-Synuclein in Parkinson’s Disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009399. [Google Scholar] [CrossRef] [Green Version]
- Al Shahrani, M.; Heales, S.; Hargreaves, I.; Orford, M. Oxidative Stress: Mechanistic Insights into Inherited Mitochondrial Disorders and Parkinson’s Disease. J. Clin. Med. 2017, 6, 100. [Google Scholar] [CrossRef]
- Langston, J.W.; Ballard, P.; Tetrud, J.W.; Irwin, I. Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science 1983, 219, 979–980. [Google Scholar] [CrossRef] [Green Version]
- Schapira, A.H.V.; Cooper, J.M.; Dexter, D.; Clark, J.B.; Jenner, P.; Marsden, C.D. Mitochondrial Complex I Deficiency in Parkinson’s Disease. J. Neurochem. 1990, 54, 823–827. [Google Scholar] [CrossRef] [PubMed]
- Greenamyre, J.T.; Hastings, T.G. Parkinson’s—Divergent Causes, Convergent Mechanisms. Science 2004, 304, 1120–1122. [Google Scholar] [CrossRef]
- Matsubara, T.; Azums, T.; Yoshids, S.; Yamegami, T. Serum coenzyme Q-10 level in Parkinson syndrome. In Biomedical and Clinical Aspects of Coenzyme Q; Folkers, K., Littarru, G.P., Yamagami, T., Eds.; Elsevier Science: Amsterdam, The Netherlands, 1991; Volume 16, pp. 159–166. [Google Scholar]
- Shults, C.W.; Haas, R.D.; Passor, D.; Beal, M.F. Coenzyme Q10 level is reduced in mitochondria from Parkinsonian patients. Ann. Neurol. 1997, 42, 61–65. [Google Scholar]
- Galpern, W.R.; Cudkowicz, M.E. Coenzyme Q treatment of neurodegenerative diseases of ages. Mitochondrion 2007, 7, S146–S153. [Google Scholar] [CrossRef]
- Shults, C.W.; Oakes, D.; Kieburtz, K.; Beal, M.F.; Haas, R.; Plumb, S.; Juncos, J.L.; Nutt, J.; Shoulson, I.; Carter, J.; et al. Effects of coenzyme Q10 in early Parkinson disease: Evidence of slowing of the functional decline. Arch. Neurol. 2002, 59, 1541–1550. [Google Scholar] [CrossRef]
- Neergheen, V.; Chalasani, A.; Wainwright, L.; Yubero, D.; Montero, R.; Artuch, R.; Hargreaves, I. Coenzyme Q10 in the Treatment of Mitochondrial Disease. J. Inborn Errors Metab. Screen. 2017, 5. [Google Scholar] [CrossRef] [Green Version]
- Beal, M.F.; Parkinson Study Group. A randomized clinical trial of high-dosage coenzyme Q10 in early Parkinson disease: No evidence of benefit. JAMA Neurol. 2014, 71, 543–552. [Google Scholar]
- Smith, E.F.; Shaw, P.; De Vos, K.J. The role of mitochondria in amyotrophic lateral sclerosis. Neurosci. Lett. 2019, 710, 132933. [Google Scholar] [CrossRef]
- Cunha-Oliveira, T.; Montezinho, L.; Mendes, C.; Firuzi, O.; Saso, L.; Oliveira, P.J.; Silva, F.S.G. Oxidative Stress in Amyotrophic Lateral Sclerosis: Pathophysiology and Opportunities for Pharmacological Intervention. Oxidative Med. Cell. Longev. 2020, 2020, 5021694. [Google Scholar] [CrossRef]
- Matthews, R.T.; Yang, L.; Browne, S.; Baik, M.; Beal, M.F. Coenzyme Q10 administration increases brain mitochondrial concentrations and exerts neuroprotective effects. Proc. Natl. Acad. Sci. USA 1998, 95, 8892–8897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miquel, E.; Cassina, A.; Martínez-Palma, L.; Souza, J.M.; Bolatto, C.; Rodríguez-Bottero, S.; Logan, A.; Smith, R.A.; Murphy, M.; Barbeito, L.; et al. Neuroprotective effects of the mitochondria-targeted antioxidant MitoQ in a model of inherited amyotrophic lateral sclerosis. Free Radic. Biol. Med. 2014, 70, 204–213. [Google Scholar] [CrossRef]
- Kaufmann, P.; Thompson, J.L.; Levy, G.; Buchsbaum, R.; Shefner, J.; Krivickas, L.S.; Katz, J.; Rollins, Y.; Barohn, R.J.; Jackson, C.E.; et al. Phase II trial of CoQ10 for ALS finds insufficient evidence to justify phase III. Ann. Neurol. 2009, 66, 235–244. [Google Scholar] [CrossRef] [Green Version]
- Rodrigo, R.; Fernandez-Gajardo, R.; Gutierrez, R.; Matamala, J.M.; Carrasco, R.; Miranda-Merchak, A.; Feuerhake, W. Oxidative Stress and Pathophysiology of Ischemic Stroke: Novel Therapeutic Opportunities. CNS Neurol. Disord. Drug Targets 2013, 12, 698–714. [Google Scholar] [CrossRef]
- Liu, F.; Lu, J.; Manaenko, A.; Tang, J.; Hu, Q. Mitochondria in Ischemic Stroke: New Insight and Implications. Aging Dis. 2018, 9, 924–937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simani, L.; Ryan, F.; Hashemifard, S.; Hooshmandi, E.; Madahi, M.; Sahraei, Z.; Rezaei, O.; Heydari, K.; Ramezani, M. Serum Coenzyme Q10 Is Associated with Clinical Neurological Outcomes in Acute Stroke Patients. J. Mol. Neurosci. 2018, 66, 53–58. [Google Scholar] [CrossRef]
- Ramezani, M.; Sahraei, Z.; Simani, L.; Heydari, K.; Shahidi, F. Coenzyme Q10 supplementation in acute ischemic stroke: Is it beneficial in short-term administration? Nutr. Neurosci. 2020, 23, 640–645. [Google Scholar] [CrossRef] [PubMed]
- Mitsui, J.; Matsukawa, T.; Yasuda, T.; Ishiura, H.; Tsuji, S. Plasma Coenzyme Q10 Levels in Patients with Multiple System Atrophy. JAMA Neurol. 2016, 73, 977–980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barca, E.; Kleiner, G.; Tang, G.; Ziosi, M.; Tadesse, S.; Masliah, E.; Louis, E.D.; Faust, P.; Kang, U.J.; Torres, J.; et al. Decreased Coenzyme Q10 Levels in Multiple System Atrophy Cerebellum. J. Neuropathol. Exp. Neurol. 2016, 75, 663–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamoto, F.K.; Okamoto, S.; Mitsui, J.; Sone, T.; Ishikawa, M.; Yamamoto, Y.; Kanegae, Y.; Nakatake, Y.; Imaizumi, K.; Ishiura, H.; et al. The pathogenesis linked to coenzyme Q10 insufficiency in iPSC-derived neurons from patients with multiple-system atrophy. Sci. Rep. 2018, 8, 14215. [Google Scholar] [CrossRef]
- Ahmad, S.S.; Ghani, S.A.; Rajagopal, T.H. Current concepts in the biochemical mechanisms of glaucomatous neurodegeneration. J. Curr. Glau. Pract. 2013, 7, 49–53. [Google Scholar] [CrossRef]
- Qu, J.; Kaufman, Y.; Washington, I. Coenzyme Q10 in the Human Retina. Investig. Opthalmol. Vis. Sci. 2009, 50, 1814–1818. [Google Scholar] [CrossRef] [Green Version]
- Parisi, V.; Centofanti, M.; Gandolfi, S.; Marangoni, D.; Rossetti, L.; Tanga, L.; Tardini, M.; Traina, S.; Ungaro, N.; Vetrugno, M.; et al. Effects of Coenzyme Q10 in Conjunction With Vitamin E on Retinal-evoked and Cortical-evoked Responses in Patients With Open-angle Glaucoma. J. Glaucoma 2014, 23, 391–404. [Google Scholar] [CrossRef]
- Feher, J.; Kovacs, B.; Kovács, I.; Schveoller, M.; Papale, A.; Gabrieli, C.B. Improvement of Visual Functions and Fundus Alterations in Early Age-Related Macular Degeneration Treated with a Combination of Acetyl-L-Carnitine, n-3 Fatty Acids, and Coenzyme Q10. Ophthalmologica 2005, 219, 154–166. [Google Scholar] [CrossRef] [PubMed]
- Kernt, M.; Hirneiss, C.; Neubauer, A.S.; Ulbig, M.W.; Kampik, A. Coenzyme Q10 prevents human lens epithelial cells from light-induced apoptotic cell death by reducing oxidative stress and stabilizing BAX / Bcl-2 ratio. Acta Ophthalmol. 2010, 88, e78–e86. [Google Scholar] [CrossRef]
- Fogagnolo, P.; Sacchi, M.; Ceresara, G.; Paderni, R.; Lapadula, P.; Orzalesi, N.; Rossetti, L. The effects of topical coenzyme Q10 and vitamin E D-α-tocopheryl polyethylene glycol 1000 succinate after cataract surgery. Ophthalmologica 2013, 229, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Kayiklik, A.; Guvenmez, O. Application of Vitamin E + Coenzyme Q Therapy during FAKO + IOL Implantation. Med Arch. 2019, 73, 109–112. [Google Scholar] [CrossRef] [PubMed]
- Sriram, K.; Pai, K.S.; Boyd, M.R.; Ravindranath, V. Evidence for generation of oxidative stress in brain by MPTP: In vitro and in vivo studies in mice. Brain Res. 1997, 749, 44–52. [Google Scholar] [CrossRef]
- Beal, M.; Matthews, R.T.; Tieleman, A.; Shults, C.W. Coenzyme Q10 attenuates the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) induced loss of striatal dopamine and dopaminergic axons in aged mice. Brain Res. 1998, 783, 109–114. [Google Scholar] [CrossRef]
- Cleren, C.; Yang, L.; Lorenzo, B.; Calingasan, N.Y.; Schomer, A.; Sireci, A.; Wille, E.J.; Beal, M.F. Therapeutic effects of coenzyme Q10 (CoQ10) and reduced CoQ10 in the MPTP model of Parkinsonism. J. Neurochem. 2008, 104, 1613–1621. [Google Scholar] [CrossRef] [PubMed]
- Dumont, M.; Kipiani, K.; Yu, F.; Wille, E.; Katz, M.; Calingasan, N.Y.; Gouras, G.K.; Lin, M.T.; Beal, M.F. Coenzyme Q10 decreases amyloid pathology and improves behavior in a transgenic mouse model of Alzheimer’s disease. J. Alzheimers Dis. 2011, 27, 211–223. [Google Scholar] [CrossRef]
- Takahashi, M.; Takahashi, K. Water-soluble CoQ10 as A Promising Anti-aging Agent for Neurological Dysfunction in Brain Mitochondria. Antioxidants 2019, 8, 61. [Google Scholar] [CrossRef] [Green Version]
- Subramiam, S.R.; Chesselet, M.F. Mitochondrial dysfunction and oxidative stress in Parkinson’s disease. Prog. Neurobiol. 2013, 106–107, 17–32. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Wang, W.; Li, L.; Perry, G.; Lee, H.G.; Zhu, X. Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim. Biophys. Acta 2014, 1842, 1240–1247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obrador, E.; Salvador, R.; López-Blanch, R.; Jihad-Jebbar, A.; Vallés, S.L.; Estrela, J.M. Oxidative Stress, Neuroinflammation and Mitochondria in the Pathophysiology of Amyotrophic Lateral Sclerosis. Antioxidants 2020, 9, 901. [Google Scholar] [CrossRef]
- Narne, P.; Pandey, V.; Phanithi, P.B. Interplay between mitochondrial metabolism and oxidative stress in ischemic stroke: An epigenetic connection. Mol. Cell. Neurosci. 2017, 82, 176–194. [Google Scholar] [CrossRef] [PubMed]
- Sohmiya, M.; Tanaka, M.; Suzuki, Y.; Tanino, Y.; Okamoto, K.; Yamamoto, Y. An increase of oxidized coenzyme Q-10 occurs in the plasma of sporadic ALS patients. J. Neurol. Sci. 2005, 228, 49–53. [Google Scholar] [CrossRef]
- Sahoo, S.; Aurich, M.K.; Jonsson, J.J.; Thiele, I. Membrane transporters in a human genome scale metabolic knowledge base and their implications in disease. Front. Physiol. 2014, 5, 1–24. [Google Scholar] [CrossRef] [Green Version]
- Takekawa, Y.; Sato, Y.; Yamaki, Y.; Imai, M.; Noto, K.; Sumi, M.; Takekuma, Y.; Iseki, K.; Sugawara, M. An Approach to Improve Intestinal Absorption of Poorly Absorbed Water-Insoluble Components via Niemann–Pick C1-Like 1. Biol. Pharm. Bull. 2016, 39, 301–307. [Google Scholar] [CrossRef] [Green Version]
- Mantle, D.; Dybring, A. Bioavailability of Coenzyme Q10: An Overview of the Absorption Process and Subsequent Metabolism. Antioxidants 2020, 9, 386. [Google Scholar] [CrossRef] [PubMed]
- López-Lluch, G.; del Pozo-Cruz, J.; Sánchez-Cuesta, A.; Cortés-Rodríguez, A.B.; Navas, P. Bioavailability of coenzyme Q10 supplements depends on carrier lipids and solubilization. Nutrition 2019, 57, 133–140. [Google Scholar] [CrossRef]
- Duncan, A.J.; Heales, S.J.; Mills, K.; Eaton, S.; Land, J.M.; Hargreaves, I.P. Determination of Coenzyme Q10 Status in Blood Mononuclear Cells, Skeletal Muscle, and Plasma by HPLC with Di-Propoxy-Coenzyme Q10 as an Internal Standard. Clin. Chem. 2005, 51, 2380–2382. [Google Scholar] [CrossRef] [Green Version]
- Hargreaves, I.; Heaton, R.A.; Mantle, D. Disorders of Human Coenzyme Q10 Metabolism: An Overview. Int. J. Mol. Sci. 2020, 21, 6695. [Google Scholar] [CrossRef] [PubMed]
- Wainwright, L. Mechanisms of Coenzyme Q10 Blood-Brain Barrier Transport. Ph.D. Thesis, University College London, London, UK, 2018. [Google Scholar]
- Suárez-Rivero, J.M.; Pastor-Maldonado, C.J.; Povea-Cabello, S.; Álvarez-Córdoba, M.; Villalón-García, I.; Munuera-Cabeza, M.; Suárez-Carrillo, A.; Talaverón-Rey, M.; Sánchez-Alcázar, J.A. Coenzyme Q10 analogues: Benefits and challenges for therapeutics. Antioxidants 2021, 10, 236. [Google Scholar] [CrossRef] [PubMed]
- Wainwright, L.; Hargreaves, I.P.; Georgian, A.R.; Turner, C.; Dalton, R.N.; Abbott, N.J.; Heales, S.J.R.; Preston, J.E. CoQ10 Deficient Endothelial Cell Culture Model for the Investigation of CoQ10 Blood–Brain Barrier Transport. J. Clin. Med. 2020, 9, 3236. [Google Scholar] [CrossRef]
- Park, H.W.; Park, C.G.; Park, M.; Lee, S.H.; Park, H.R.; Lim, J.; Paek, S.H.; Bin Choy, Y. Intrastriatal administration of coenzyme Q10 enhances neuroprotection in a Parkinson’s disease rat model. Sci. Rep. 2020, 10, 9572. [Google Scholar] [CrossRef]
- Gross, R.E.; Watts, R.L.; Hauser, R.A.; Bakay, R.A.; Reichmann, H.; von Kummer, R.; Ondo, W.G.; Reissig, E.; Eisner, W.; Steiner-Schulze, H.; et al. Intrastriatal transplantation of microcarrier-bound human retinal pigment epithelial cells versus sham surgery in patients with advanced Parkinson’s disease: A double-blind, randomised, controlled trial. Lancet Neurol. 2011, 10, 509–519. [Google Scholar] [CrossRef]
- Snow, B.; Mulroy, E.; Bok, A.; Simpson, M.; Smith, A.; Taylor, K.; Lockhart, M.; Lam, B.J.; Frampton, C.; Schweder, P.; et al. A phase IIb, randomised, double-blind, placebo-controlled, dose-ranging investigation of the safety and efficacy of NTCELL ® [immunoprotected (alginate-encapsulated) porcine choroid plexus cells for xenotransplantation] in patients with Parkinson’s disease. Parkinsonism Relat. Disord. 2019, 61, 88–93. [Google Scholar] [CrossRef]
- Lee, P.; Ulatowski, L.M. Vitamin E: Mechanism of transport and regulation in the CNS. IUBMB Life 2019, 71, 424–429. [Google Scholar] [CrossRef]
- Wanke, M.; Dallner, G.; Swiezewska, E. Subcellular localization of plastoquinone and ubiquinone synthesis in spinach cells. Biochim. Biophys. Acta (BBA)-Biomembr. 2000, 1463, 188–194. [Google Scholar] [CrossRef] [Green Version]
- Jin, G.; Kubo, H.; Kashiba, M.; Horinouchi, R.; Hasegawa, M.; Suzuki, M.; Sagawa, T.; Oizumi, M.; Fujisawa, A.; Tsukamoto, H.; et al. Saposin B Is a Human Coenzyme Q10-Binding/Transfer Protein. J. Clin. Biochem. Nutr. 2008, 42, 167–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prasuhn, J.; Brüggemann, N.; Hessler, N.; Berg, D.; Gasser, T.; Brockmann, K.; Olbrich, D.; Ziegler, A.; König, I.R.; Klein, C.; et al. An omics-based strategy using coenzyme Q10 in patients with Parkinson’s disease: Concept evaluation in a double-blind randomized placebo-controlled parallel group trial. Neurol. Res. Pract. 2019, 1, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mantle, D.; Hargreaves, I.P. Ataxia and coenzyme Q10: An overview. Br. J. Neurosci. Nurs. 2018, 14, 108–114. [Google Scholar] [CrossRef]
- Musumeci, O.; Naini, A.; Slonim, A.E.; Skavin, N.; Hadjigeorgiou, G.L.; Krawiecki, N.; Weissman, B.M.; Tsao, C.-Y.; Mendell, J.R.; Shanske, S.; et al. Familial cerebellar ataxia with muscle coenzyme Q10 deficiency. Neurology 2001, 56, 849–855. [Google Scholar] [CrossRef] [Green Version]
- Lamperti, C.; Naini, A.; Hirano, M.; De Vivo, D.; Bertini, E.; Servidei, S.; Valeriani, M.; Lynch, D.; Banwell, B.; Berg, M.; et al. Cerebellar ataxia and coenzyme Q10 deficiency. Neurology 2003, 60, 1206–1208. [Google Scholar] [CrossRef] [PubMed]
- Hidaka, T.; Fujii, K.; Funahashi, I.; Fukutomi, N.; Hosoe, K. Safety assessment of CoQ10. Biofactors 2008, 32, 199–208. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mantle, D.; Heaton, R.A.; Hargreaves, I.P. Coenzyme Q10, Ageing and the Nervous System: An Overview. Antioxidants 2022, 11, 2. https://doi.org/10.3390/antiox11010002
Mantle D, Heaton RA, Hargreaves IP. Coenzyme Q10, Ageing and the Nervous System: An Overview. Antioxidants. 2022; 11(1):2. https://doi.org/10.3390/antiox11010002
Chicago/Turabian StyleMantle, David, Robert A. Heaton, and Iain P. Hargreaves. 2022. "Coenzyme Q10, Ageing and the Nervous System: An Overview" Antioxidants 11, no. 1: 2. https://doi.org/10.3390/antiox11010002
APA StyleMantle, D., Heaton, R. A., & Hargreaves, I. P. (2022). Coenzyme Q10, Ageing and the Nervous System: An Overview. Antioxidants, 11(1), 2. https://doi.org/10.3390/antiox11010002