
The Protective Effect of Ubiquinone against the Amyloid Peptide in Endothelial Cells Is Isoprenoid Chain Length-Dependent

 ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Aβ Preparation
2.3. Cell Culture
2.4. Determination of Apoptosis and Necrosis
2.5. NADPH Oxidase Quantification
2.6. Determination of O2−, H2O2, and Free Cytosolic Ca2+
2.7. Mitochondrial Status Characterization
2.8. Determination of Cellular Energy Phenotype
2.9. Determination of Cellular Mitochondrial Respiration
2.10. Aβ25–35 Internalization and Mitochondrial Colocalization in bEnd.3 Cells
2.11. Statistical Analysis
3. Results
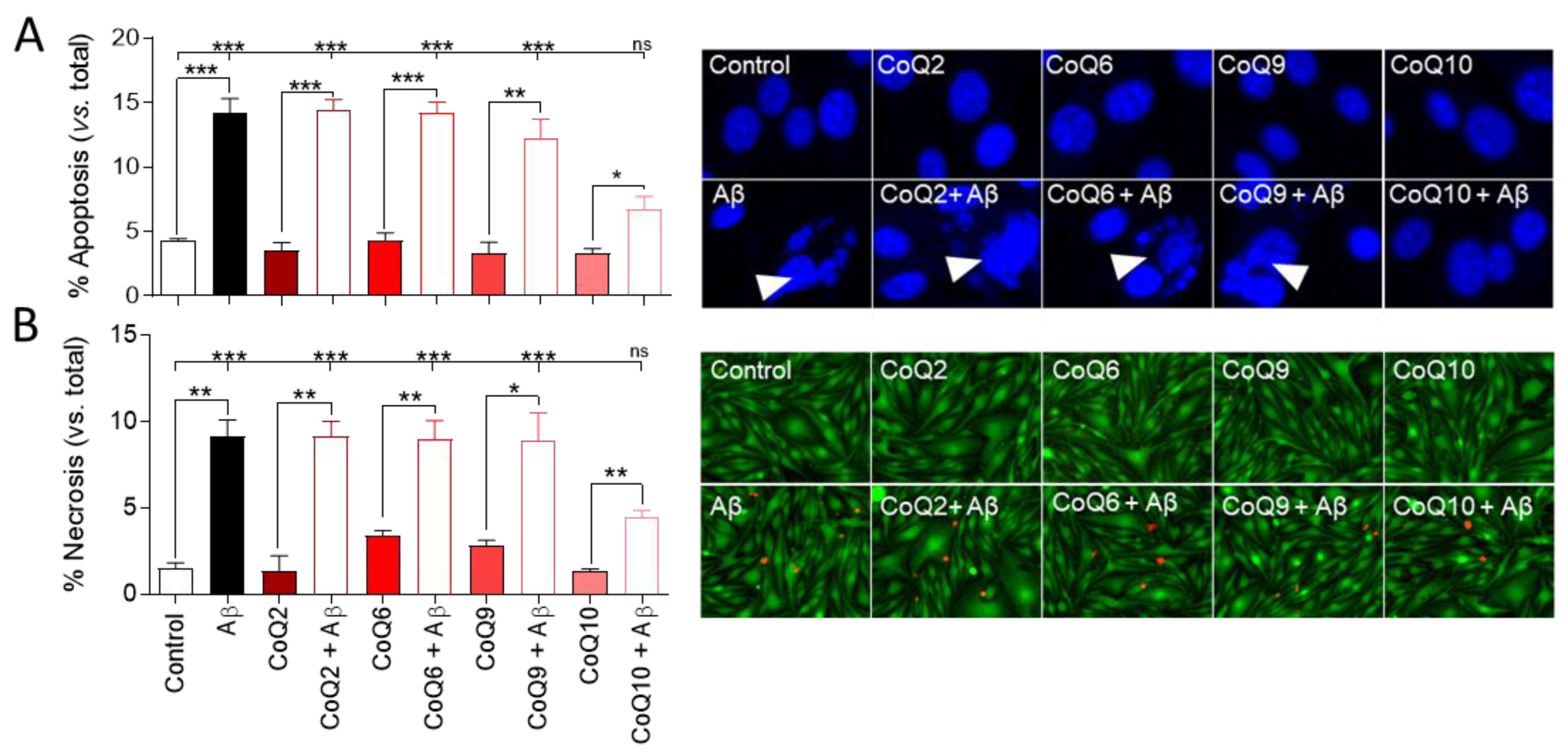
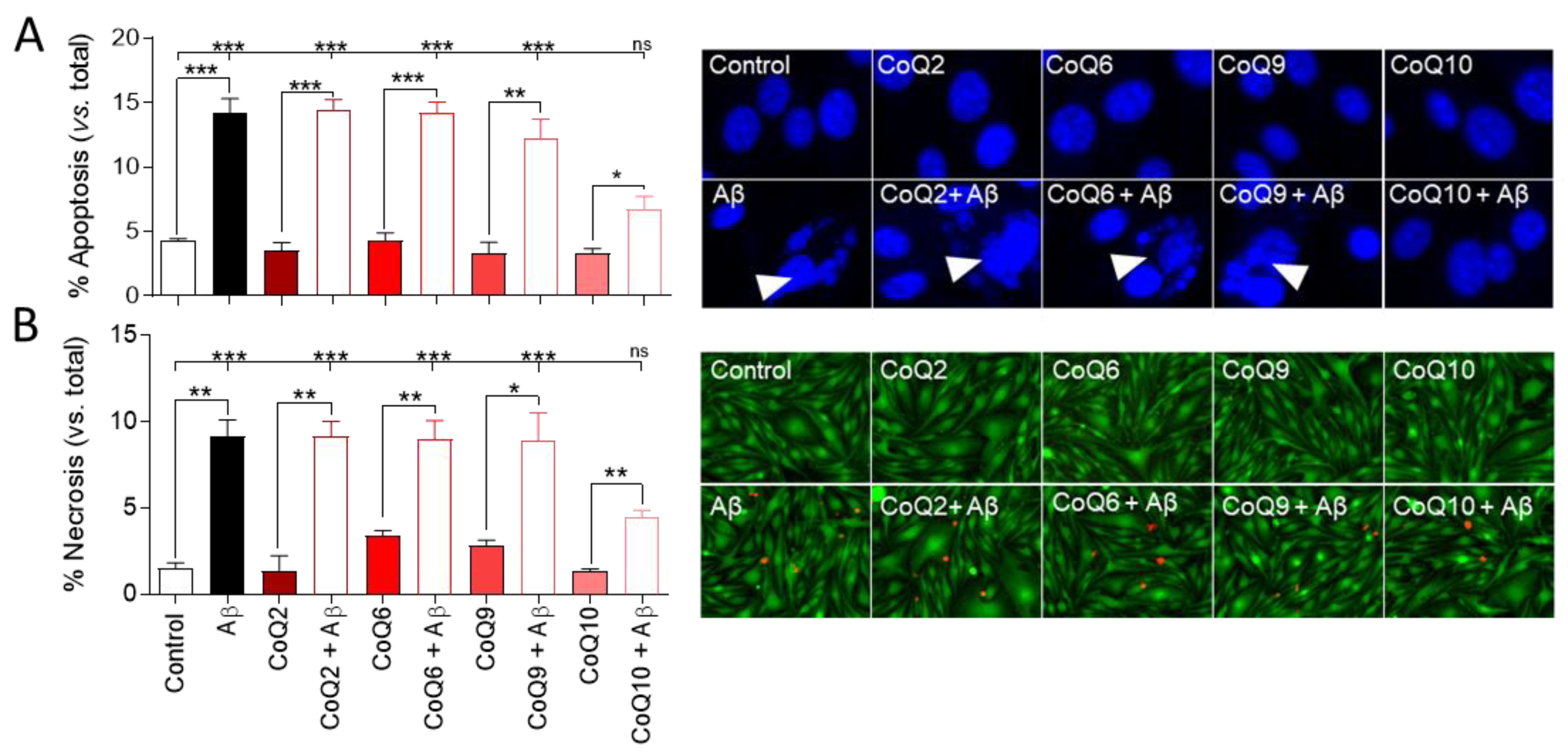
3.1. CoQ10 but Not Shorter Side Chain CoQ Prevents Aβ25–35-Induced Endothelial Cell Death
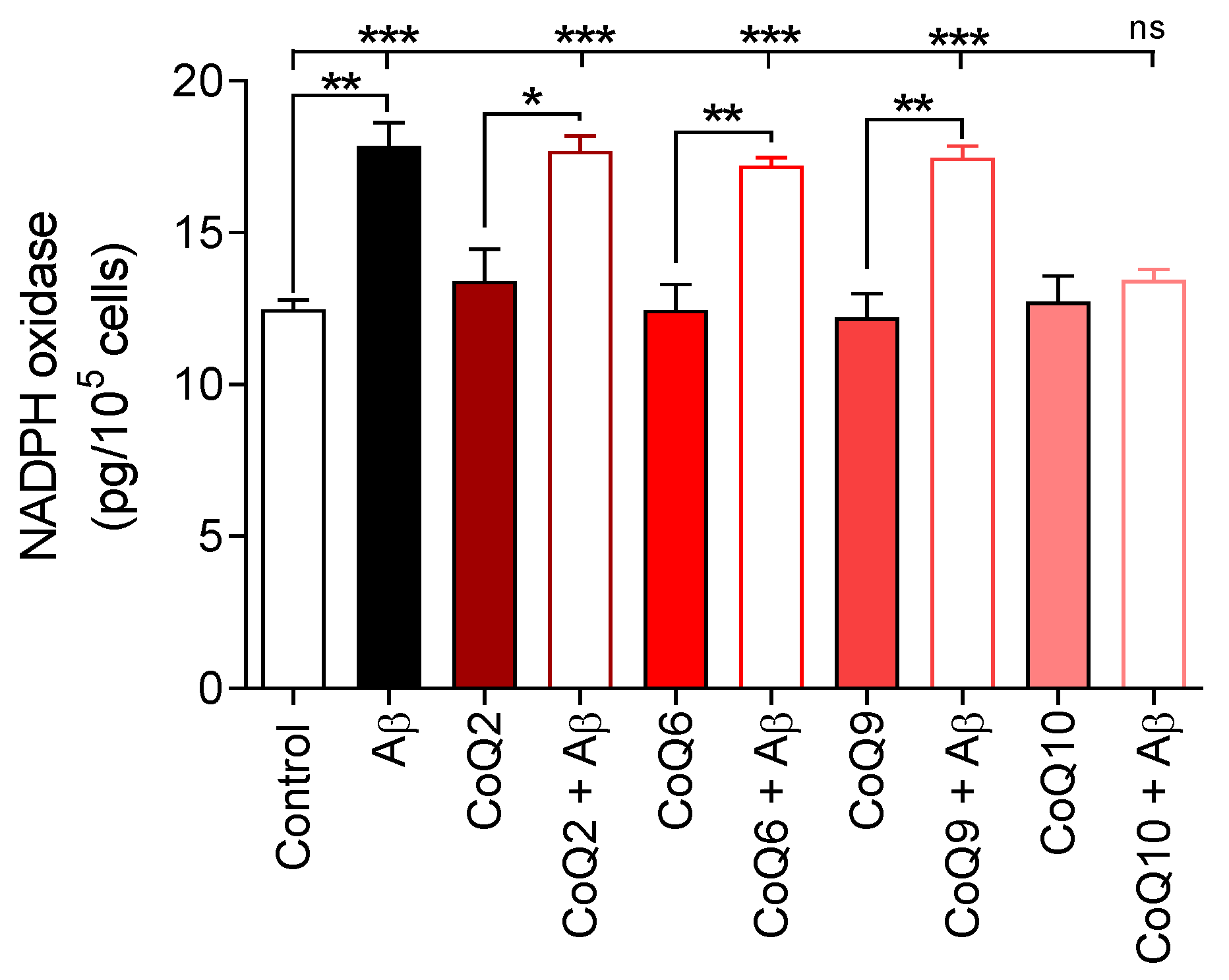
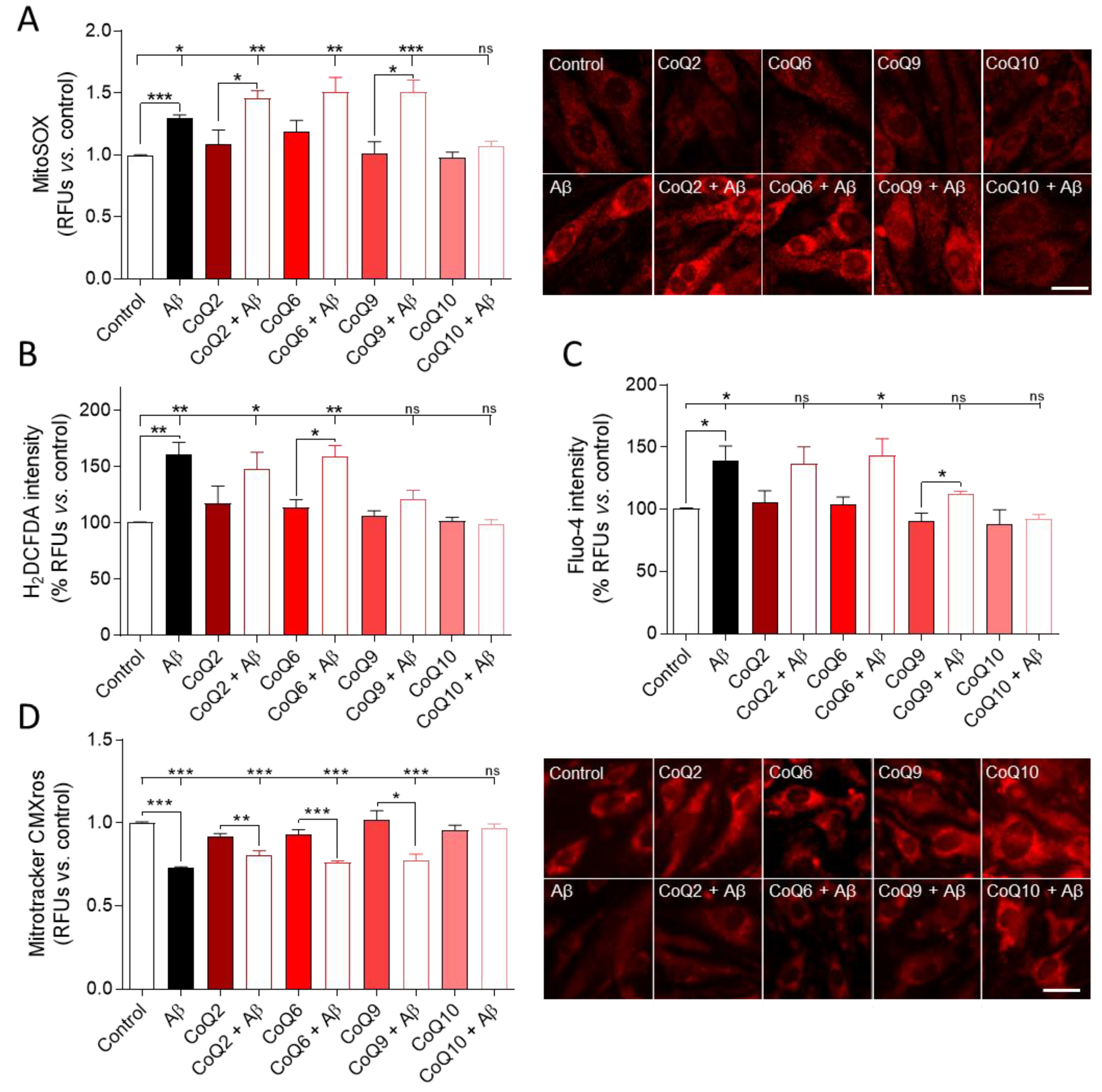
3.2. CoQ10 but Not Shorter Side Chain CoQ Prevents Aβ25–35-Induced Activation of NADPH Oxidase
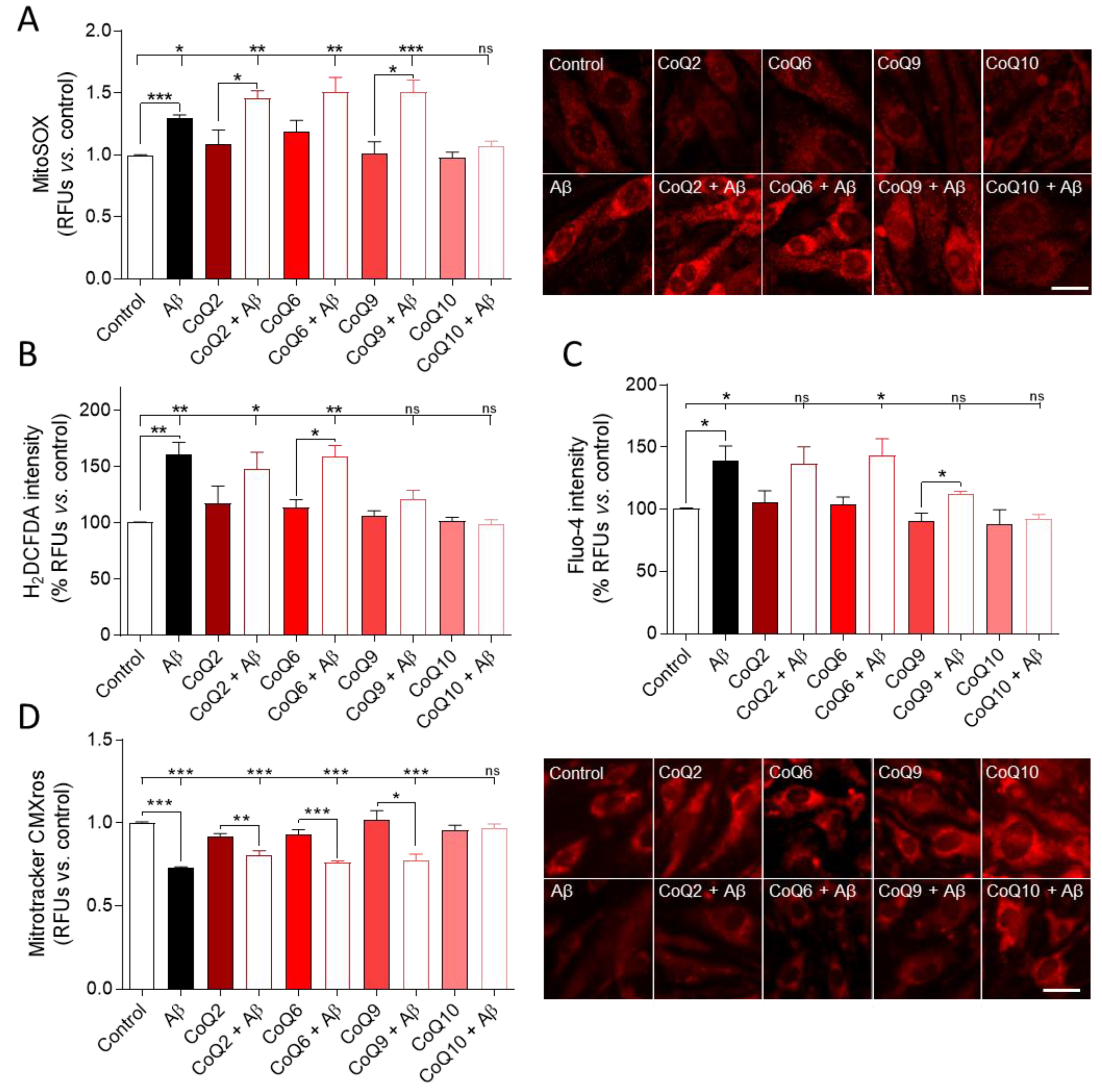
3.3. CoQ10 Prevents the Aβ25–35-Mediated Increase in O2−, H2O2, and Free Cytosolic Ca2+ Levels in Endothelial Cells and Mitochondrial Calcium Depletion in Endothelial Cells
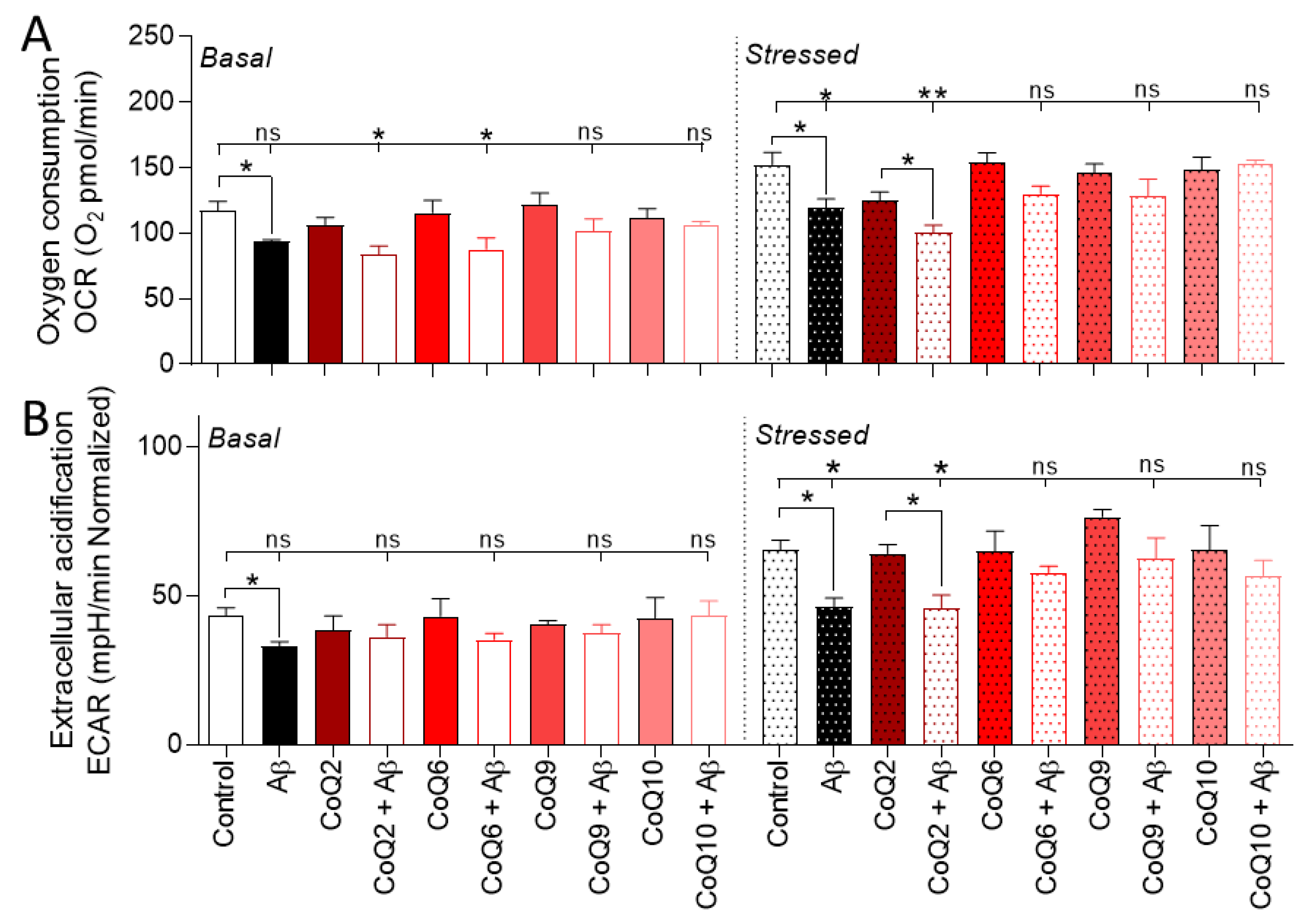
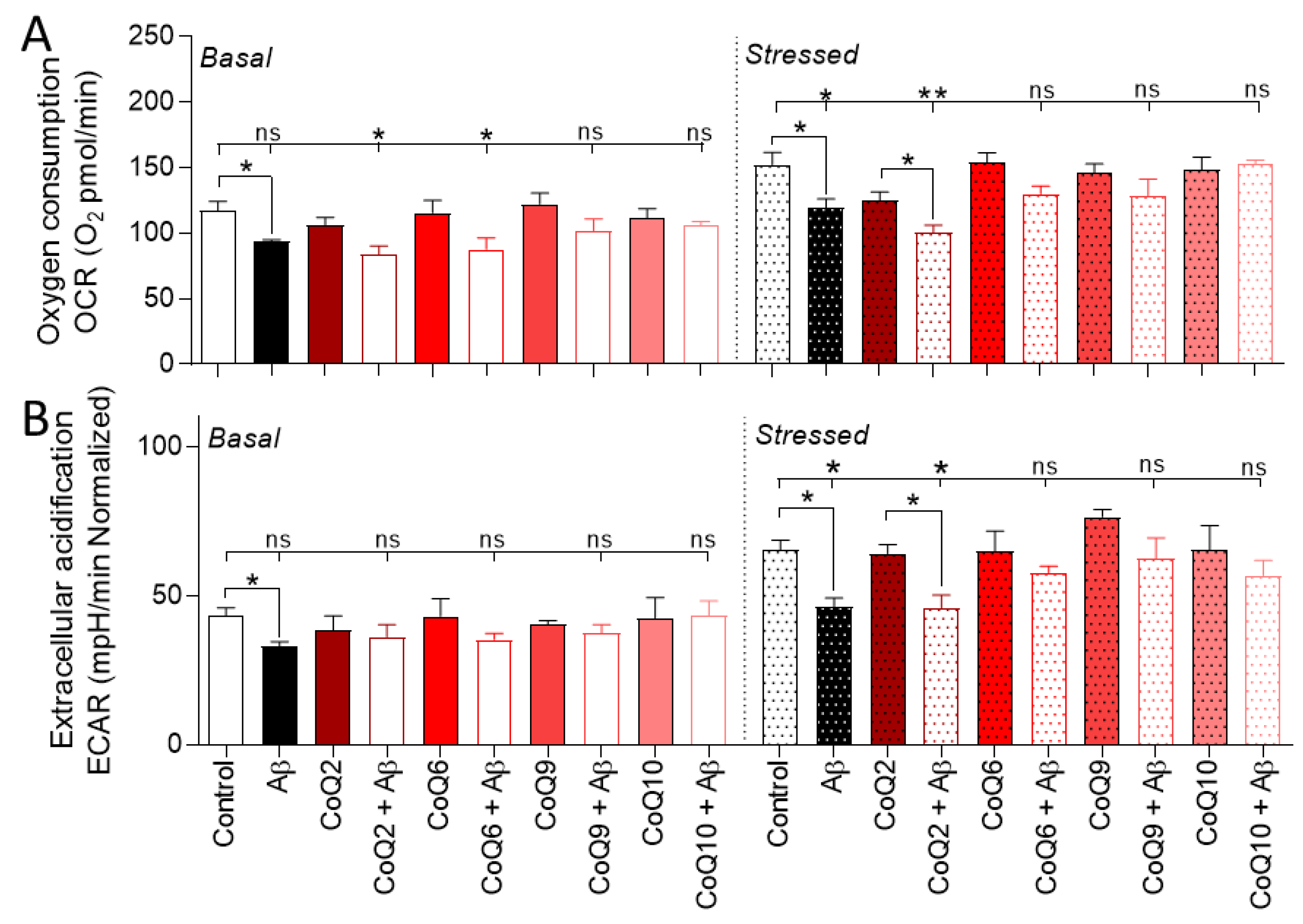
3.4. The CoQ Isoprenoid Chain Length Determines the Protective Effect against Aβ25–35-Mediated Mitochondrial Bioenergetics Alterations
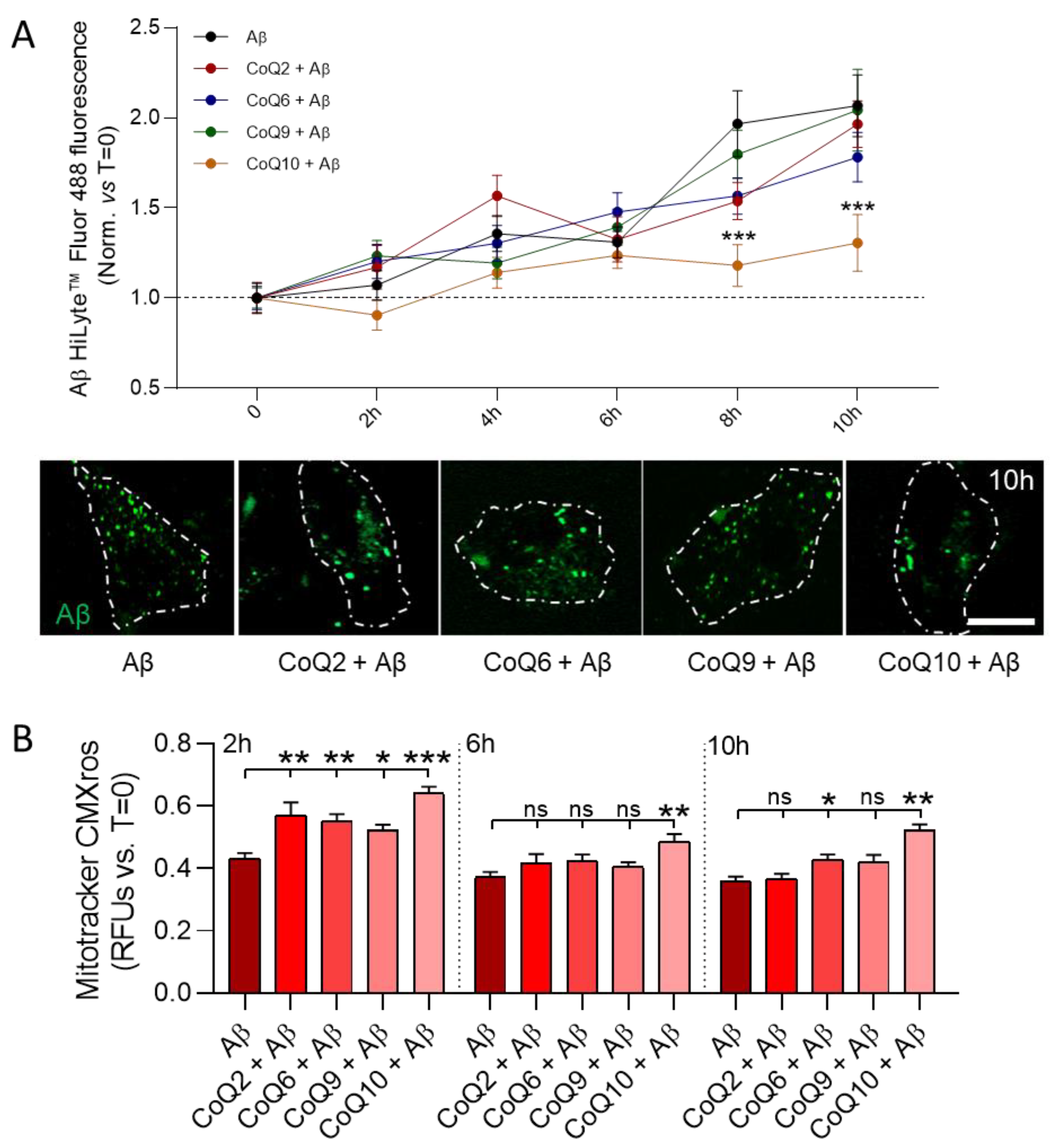
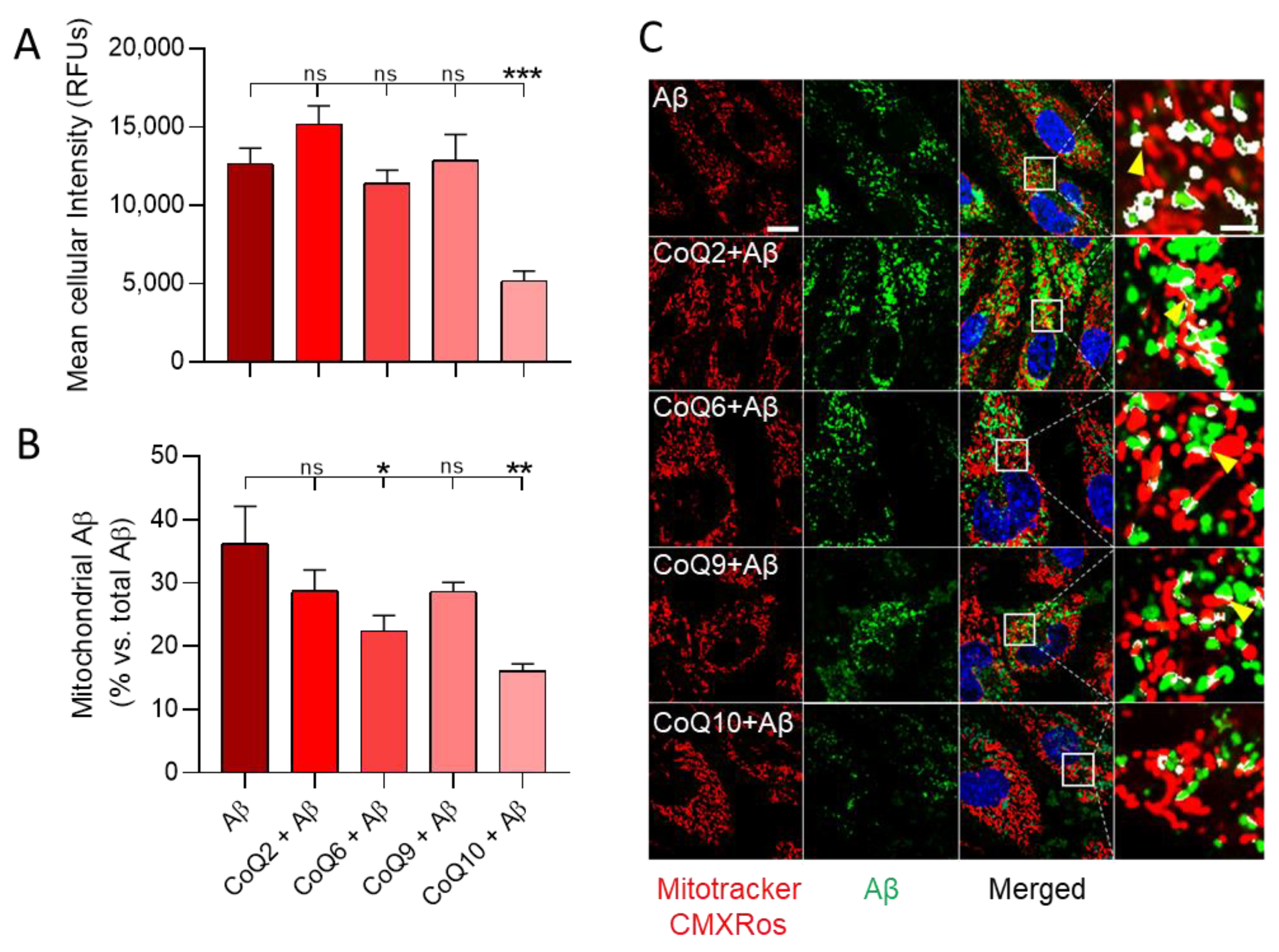
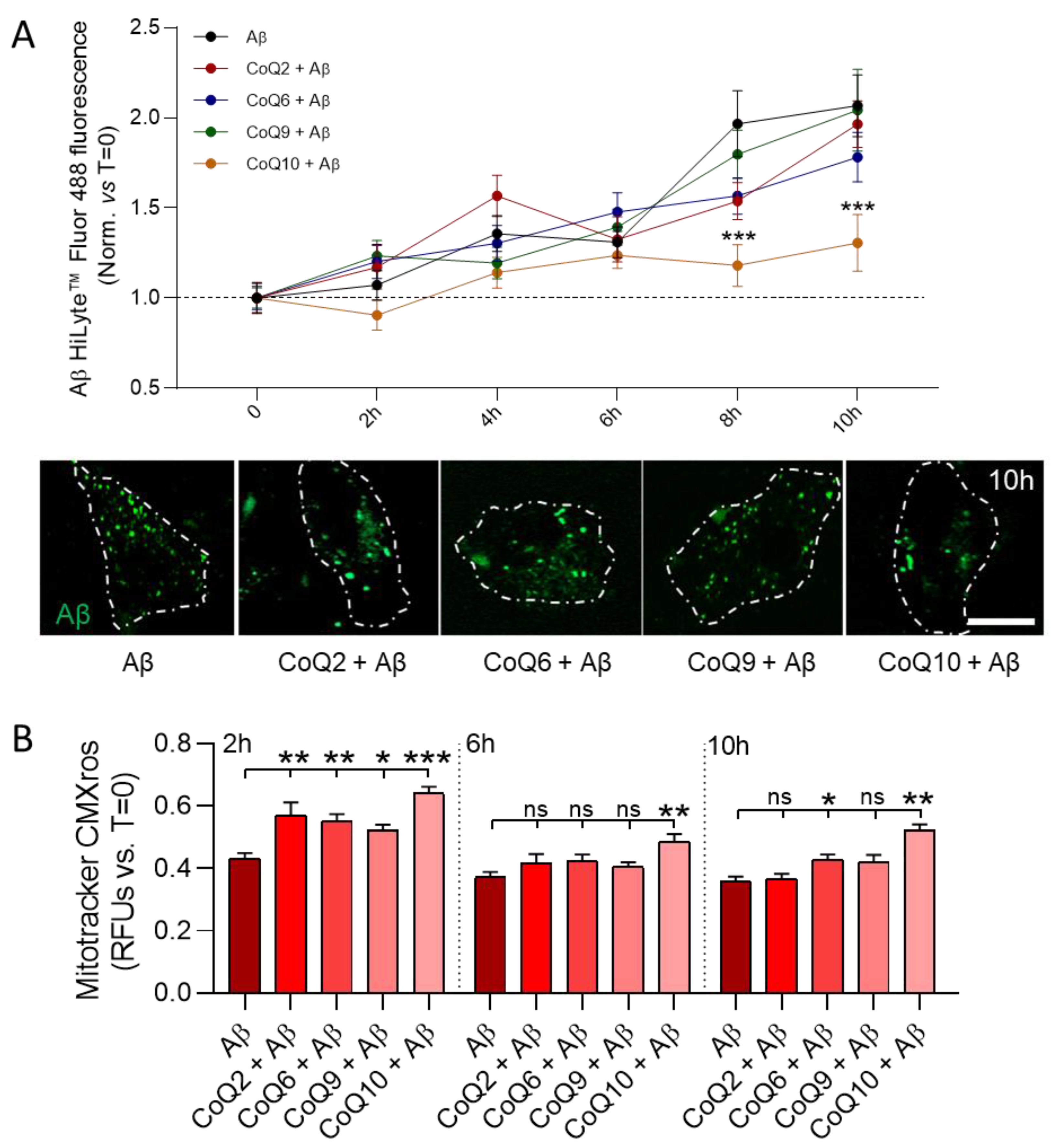
3.5. CoQ10 but Not Other Ubiquinones Delayed and Decreased β-amyloid Incorporation into Mitochondria in Endothelial Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Karran, E.; Mercken, M.; de Strooper, B. The amyloid cascade hypothesis for Alzheimer’s disease: An appraisal for the development of therapeutics. Nat. Rev. Drug. Discov. 2011, 10, 698–712. [Google Scholar] [CrossRef]
- Akinyemi, R.O.; Mukaetova-Ladinska, E.B.; Attems, J.; Ihara, M.; Kalaria, R.N. Vascular risk factors and neurodegeneration in ageing related dementias: Alzheimer’s disease and vascular dementia. Curr. Alzheimer Res. 2013, 10, 642–653. [Google Scholar] [CrossRef]
- Ezzati, A.; Wang, C.; Lipton, R.B.; Altschul, D.; Katz, M.J.; Dickson, D.W.; Derby, C.A. Association Between Vascular Pathology and Rate of Cognitive Decline Independent of Alzheimer’s Disease Pathology. J. Am. Geriatr. Soc. 2017, 65, 1836–1841. [Google Scholar] [CrossRef] [PubMed]
- Kennelly, S.P.; Lawlor, B.A.; Kenny, R.A. Blood pressure and the risk for dementia: A double edged sword. Ageing Res. Rev. 2009, 8, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Guglielmotto, M.; Aragno, M.; Autelli, R.; Giliberto, L.; Novo, E.; Colombatto, S.; Danni, O.; Parola, M.; Smith, M.A.; Perry, G.; et al. The up-regulation of BACE1 mediated by hypoxia and ischemic injury: Role of oxidative stress and HIF1alpha. J. Neurochem. 2009, 108, 1045–1056. [Google Scholar] [CrossRef]
- Zhang, X.; Zhou, K.; Wang, R.; Cui, J.; Lipton, S.A.; Liao, F.F.; Xu, H.; Zhang, Y.W. Hypoxia-inducible factor 1alpha (HIF-1alpha)-mediated hypoxia increases BACE1 expression and beta-amyloid generation. J. Biol. Chem. 2007, 282, 10873–10880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Small, H.Y.; Migliarino, S.; Czesnikiewicz-Guzik, M.; Guzik, T.J. Hypertension: Focus on autoimmunity and oxidative stress. Free Radic. Biol. Med. 2018, 125, 104–115. [Google Scholar] [CrossRef]
- Tsai, K.L.; Chen, L.H.; Chiou, S.H.; Chiou, G.Y.; Chen, Y.C.; Chou, H.Y.; Chen, L.K.; Chen, H.Y.; Chiu, T.H.; Tsai, C.S.; et al. Coenzyme Q10 suppresses oxLDL-induced endothelial oxidative injuries by the modulation of LOX-1-mediated ROS generation via the AMPK/PKC/NADPH oxidase signaling pathway. Mol. Nutr. Food Res. 2011, 55 (Suppl. 2), S227–S240. [Google Scholar] [CrossRef]
- Tsai, K.L.; Huang, Y.H.; Kao, C.L.; Yang, D.M.; Lee, H.C.; Chou, H.Y.; Chen, Y.C.; Chiou, G.Y.; Chen, L.H.; Yang, Y.P.; et al. A novel mechanism of coenzyme Q10 protects against human endothelial cells from oxidative stress-induced injury by modulating NO-related pathways. J. Nutr. Biochem. 2012, 23, 458–468. [Google Scholar] [CrossRef] [PubMed]
- Tsuneki, H.; Sekizaki, N.; Suzuki, T.; Kobayashi, S.; Wada, T.; Okamoto, T.; Kimura, I.; Sasaoka, T. Coenzyme Q10 prevents high glucose-induced oxidative stress in human umbilical vein endothelial cells. Eur. J. Pharmacol. 2007, 566, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Tsuneki, H.; Tokai, E.; Suzuki, T.; Seki, T.; Okubo, K.; Wada, T.; Okamoto, T.; Koya, S.; Kimura, I.; Sasaoka, T. Protective effects of coenzyme Q10 against angiotensin II-induced oxidative stress in human umbilical vein endothelial cells. Eur. J. Pharmacol. 2013, 701, 218–227. [Google Scholar] [CrossRef]
- Blanc, E.M.; Toborek, M.; Mark, R.J.; Hennig, B.; Mattson, M.P. Amyloid beta-peptide induces cell monolayer albumin permeability, impairs glucose transport, and induces apoptosis in vascular endothelial cells. J. Neurochem. 1997, 68, 1870–1881. [Google Scholar] [CrossRef] [PubMed]
- Hsu, M.-J.; Hsu, C.Y.; Chen, B.C.; Chen, M.C.; Ou, G.; Lin, C.H. Apoptosis signal-regulating kinase 1 in amyloid beta peptide-induced cerebral endothelial cell apoptosis. J. Neurosci. Off. J. Soc. Neurosci. 2007, 27, 5719–5729. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, T.M.; Leaver, H.A. Brain endothelial cell death: Modes, signaling pathways, and relevance to neural development, homeostasis, and disease. Mol. Neurobiol. 2010, 42, 52–63. [Google Scholar] [CrossRef]
- Suo, Z.; Fang, C.; Crawford, F.; Mullan, M. Superoxide free radical and intracellular calcium mediate A beta(1-42) induced endothelial toxicity. Brain Res. 1997, 762, 144–152. [Google Scholar] [CrossRef]
- Xu, J.; Chen, S.; Ku, G.; Ahmed, S.H.; Xu, J.; Chen, H.; Hsu, C.Y. Amyloid beta peptide-induced cerebral endothelial cell death involves mitochondrial dysfunction and caspase activation. J. Cereb. Blood Flow Metab. 2001, 21, 702–710. [Google Scholar] [CrossRef] [Green Version]
- Duran-Prado, M.; Frontinan, J.; Santiago-Mora, R.; Peinado, J.R.; Parrado-Fernandez, C.; Gomez-Almagro, M.V.; Moreno, M.; Lopez-Dominguez, J.A.; Villalba, J.M.; Alcain, F.J. Coenzyme Q10 protects human endothelial cells from beta-amyloid uptake and oxidative stress-induced injury. PLoS ONE 2014, 9, e109223. [Google Scholar]
- Frontinan-Rubio, J.; Sancho-Bielsa, F.J.; Peinado, J.R.; LaFerla, F.M.; Gimenez-Llort, L.; Duran-Prado, M.; Alcain, F.J. Sex-dependent co-occurrence of hypoxia and beta-amyloid plaques in hippocampus and entorhinal cortex is reversed by long-term treatment with ubiquinol and ascorbic acid in the 3xTg-AD mouse model of Alzheimer’s disease. Mol. Cell Neurosci. 2018, 92, 67–81. [Google Scholar] [CrossRef]
- Garcia-Carpintero, S.; Dominguez-Bertalo, J.; Pedrero-Prieto, C.; Frontinan-Rubio, J.; Amo-Salas, M.; Duran-Prado, M.; Garcia-Perez, E.; Vaamonde, J.; Alcain, F.J. Ubiquinol Supplementation Improves Gender-Dependent Cerebral Vasoreactivity and Ameliorates Chronic Inflammation and Endothelial Dysfunction in Patients with Mild Cognitive Impairment. Antioxidants 2021, 10, 143. [Google Scholar] [CrossRef] [PubMed]
- Crane, F.L. Biochemical functions of coenzyme Q10. J. Am. Coll. Nutr. 2001, 20, 591–598. [Google Scholar] [CrossRef]
- Meganathan, R. Ubiquinone biosynthesis in microorganisms. FEMS Microbiol. Lett. 2001, 203, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Turunen, M.; Olsson, J.; Dallner, G. Metabolism and function of coenzyme Q. Biochim. Biophys. Acta 2004, 1660, 171–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Divakaruni, A.S.; Paradyse, A.; Ferrick, D.A.; Murphy, A.N.; Jastroch, M. Analysis and interpretation of microplate-based oxygen consumption and pH data. Methods Enzym. 2014, 547, 309–354. [Google Scholar]
- Frontiñan-Rubio, J.; Gomez, M.V.; González, V.J.; Durán-Prado, M.; Vázquez, E. Sublethal exposure of small few-layer graphene promotes metabolic alterations in human skin cells. Sci. Rep. 2020, 1, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 2018, 14, 450–464. [Google Scholar] [CrossRef]
- Abramov, Y.A.; Canevari, L.; Duchen, M.R. Beta-amyloid peptides induce mitochondrial dysfunction and oxidative stress in astrocytes and death of neurons through activation of NADPH oxidase. J. Neurosci. 2004, 24, 565–575. [Google Scholar] [CrossRef] [Green Version]
- Bianca, V.D.; Dusi, S.; Bianchini, E.; Dal Pra, I.; Rossi, F. beta-amyloid activates the O-2 forming NADPH oxidase in microglia, monocytes, and neutrophils. A possible inflammatory mechanism of neuronal damage in Alzheimer’s disease. J. Biol. Chem. 1999, 274, 15493–15499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, L.; Anrather, J.; Zhou, P.; Frys, K.; Pitstick, R.; Younkin, S.; Carlson, G.A.; Iadecola, C. NADPH-oxidase-derived reactive oxygen species mediate the cerebrovascular dysfunction induced by the amyloid beta peptide. J. Neurosci. 2005, 25, 1769–1777. [Google Scholar] [CrossRef] [PubMed]
- Park, L.; Zhou, P.; Pitstick, R.; Capone, C.; Anrather, J.; Norris, E.H.; Younkin, L.; Younkin, S.; Carlson, G.; McEwen, B.S.; et al. Nox2-derived radicals contribute to neurovascular and behavioral dysfunction in mice overexpressing the amyloid precursor protein. Proc. Natl. Acad. Sci. USA 2008, 105, 1347–1352. [Google Scholar] [CrossRef] [Green Version]
- Cahill-Smith, S.; Li, J.M. Oxidative stress, redox signalling and endothelial dysfunction in ageing-related neurodegenerative diseases: A role of NADPH oxidase 2. Br. J. Clin. Pharmacol. 2014, 78, 441–453. [Google Scholar] [CrossRef] [Green Version]
- Zalba, G.; Beaumont, F.J.; San Jose, G.; Fortuno, A.; Fortuno, M.A.; Etayo, J.C.; Diez, J. Vascular NADH/NADPH oxidase is involved in enhanced superoxide production in spontaneously hypertensive rats. Hypertension 2000, 35, 1055–1061. [Google Scholar] [CrossRef] [Green Version]
- Frey, R.S.; Ushio-Fukai, M.; Malik, A.B. NADPH oxidase-dependent signaling in endothelial cells: Role in physiology and pathophysiology. Antioxid. Redox Signal. 2009, 11, 791–810. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Ayala, D.J.; Brea-Calvo, G.; Lopez-Lluch, G.; Navas, P. Coenzyme Q distribution in HL-60 human cells depends on the endomembrane system. Biochim. Biophys. Acta 2005, 1713, 129–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lass, A.; Agarwal, S.; Sohal, R.S. Mitochondrial ubiquinone homologues, superoxide radical generation, and longevity in different mammalian species. J. Biol. Chem. 1997, 272, 19199–19204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez-Ayala, D.J.; Lopez-Lluch, G.; Garcia-Valdes, M.; Arroyo, A.; Navas, P. Specificity of coenzyme Q10 for a balanced function of respiratory chain and endogenous ubiquinone biosynthesis in human cells. Biochim. Biophys. Acta. 2005, 1706, 174–183. [Google Scholar] [CrossRef] [Green Version]
- Gutierrez-Casado, E. Mitochondrial Phisyology and Autophagic Signaling Regulation through Nutritional Interventions with Different Lipid Sources and Antioxidants. Ph.D. Thesis, University of Córdoba, Córdoba, Spain, 2018; p. 264. [Google Scholar]
- Du, H.; Yan, S.S. Mitochondrial permeability transition pore in Alzheimer’s disease: Cyclophilin D and amyloid beta. Biochim. Biophys. Acta 2010, 1802, 198–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fontaine, E.; Ichas, F.; Bernardi, P. A ubiquinone-binding site regulates the mitochondrial permeability transition pore. J. Biol. Chem. 1998, 273, 25734–25740. [Google Scholar] [CrossRef] [Green Version]
- Papucci, L.; Schiavone, N.; Witort, E.; Donnini, M.; Lapucci, A.; Tempestini, A.; Formigli, L.; Zecchi-Orlandini, S.; Orlandini, G.; Carella, G.; et al. Coenzyme q10 prevents apoptosis by inhibiting mitochondrial depolarization independently of its free radical scavenging property. J. Biol. Chem. 2003, 278, 28220–28228. [Google Scholar] [CrossRef] [Green Version]
- Bello, R.I.; Gomez-Diaz, C.; Buron, M.I.; Alcain, F.J.; Navas, P.; Villalba, J.M. Enhanced anti-oxidant protection of liver membranes in long-lived rats fed on a coenzyme Q10-supplemented diet. Exp. Gerontol. 2005, 40, 694–706. [Google Scholar] [CrossRef]
- Quintana, D.D.; Garcia, J.A.; Anantula, Y.; Rellick, S.L.; Engler-Chiurazzi, E.B.; Sarkar, S.N.; Brown, C.M.; Simpkins, J.W.; Huisa, B. Amyloid-β Causes Mitochondrial Dysfunction via a Ca2+-Driven Upregulation of Oxidative Phosphorylation and Superoxide Production in Cerebrovascular Endothelial Cells. J. Alzheimer’s Dis. 2020, 75, 119–138. [Google Scholar] [CrossRef]
- Li, Y.; Lu, J.; Cao, X.; Zhao, H.; Gao, L.; Xia, P.; Pei, G. A Newly Synthesized Rhamnoside Derivative Alleviates Alzheimer’s Amyloid-beta-Induced Oxidative Stress, Mitochondrial Dysfunction, and Cell Senescence through Upregulating SIRT3. Oxid. Med. Cell Longev. 2020, 2020, 7698560. [Google Scholar]
- Long, J.; He, P.; Shen, Y.; Li, R. New evidence of mitochondria dysfunction in the female Alzheimer’s disease brain: Deficiency of estrogen receptor-beta. J. Alzheimers Dis. 2012, 30, 545–558. [Google Scholar] [CrossRef]
- Park, Y.H.; Shin, S.J.; Kim, H.S.; Hong, S.B.; Kim, S.; Nam, Y.; Kim, J.J.; Lim, K.; Kim, J.S.; Kim, J.I.; et al. Omega-3 Fatty Acid-Type Docosahexaenoic Acid Protects against Abeta-Mediated Mitochondrial Deficits and Pathomechanisms in Alzheimer’s Disease-Related Animal Model. Int. J. Mol. Sci. 2020, 21, 3879. [Google Scholar] [CrossRef] [PubMed]
- Sadli, N.; Barrow, C.J.; McGee, s.; Suphioglu, C. Effect of DHA and coenzymeQ10 against Abeta- and zinc-induced mitochondrial dysfunction in human neuronal cells. Cell Physiol. Biochem. 2013, 32, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Sotolongo, K.; Ghiso, J.; Rostagno, A. Nrf2 activation through the PI3K/GSK-3 axis protects neuronal cells from Abeta-mediated oxidative and metabolic damage. Alzheimers Res. Ther. 2020, 12, 13. [Google Scholar] [CrossRef] [Green Version]
- Readnower, R.D.; Sauerbeck, A.D.; Sullivan, P.G. Mitochondria, Amyloid β, and Alzheimer’s Disease. Int. J. Alzheimer’s Dis. 2011, 2011, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Rhein, V.; Baysang, G.; Rao, S.; Meier, F.; Bonert, A.; Müller-Spahn, F.; Eckert, A. Amyloid-beta Leads to Impaired Cellular Respiration, Energy Production and Mitochondrial Electron Chain Complex Activities in Human Neuroblastoma Cells. Cell. Mol. Neurobiol. 2009, 29, 1063–1071. [Google Scholar] [CrossRef] [Green Version]
- Kagan, B.L.; Thundimadathil, J. Amyloid peptide pores and the beta sheet conformation. Adv. Exp. Med. Biol. 2010, 677, 150–167. [Google Scholar]
- Ono, K.; Hasegawa, K.; Naiki, H.; Yamada, M. Preformed beta-amyloid fibrils are destabilized by coenzyme Q10 in vitro. Biochem. Biophys. Res. Commun. 2005, 330, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Agmo Hernandez, V.; Eriksson, E.K.; Edwards, K. Ubiquinone-10 alters mechanical properties and increases stability of phospholipid membranes. Biochim. Biophys. Acta 2015, 1848, 2233–2243. [Google Scholar] [CrossRef]
- Edlund, C.; Holmberg, K.; Dallner, G.; Norrby, E.; Kristensson, K. Ubiquinone-10 protects neurons from virus-induced degeneration. J. Neurochem. 1994, 63, 634–639. [Google Scholar] [CrossRef]
- De Cabo, R.; Cabello, R.; Rios, M.; Lopez-Lluch, G.; Ingram, D.K.; Lane, M.A.; Navas, P. Calorie restriction attenuates age-related alterations in the plasma membrane antioxidant system in rat liver. Exp. Gerontol. 2004, 39, 297–304. [Google Scholar] [CrossRef]
- Halagappa, V.K.; Guo, Z.; Pearson, M.; Matsuoka, Y.; Cutler, R.G.; Laferla, F.M.; Mattson, M.P. Intermittent fasting and caloric restriction ameliorate age-related behavioral deficits in the triple-transgenic mouse model of Alzheimer’s disease. Neurobiol. Dis. 2007, 26, 212–220. [Google Scholar] [CrossRef]
- Qin, W.; Chachich, M.; Lane, M.; Roth, G.; Bryant, M.; de Cabo, R.; Ottinger, M.A.; Mattison, J.; Ingram, D.; Gandy, S.; et al. Calorie restriction attenuates Alzheimer’s disease type brain amyloidosis in Squirrel monkeys (Saimiri sciureus). J. Alzheimers Dis. 2006, 10, 417–422. [Google Scholar] [CrossRef]
- Fernandez-Del-Rio, L.; Rodriguez-Lopez, S.; Gutierrez-Casado, E.; Gonzalez-Reyes, J.A.; Clarke, C.F.; Buron, M.I.; Villalba, J.M. Regulation of hepatic coenzyme Q biosynthesis by dietary omega-3 polyunsaturated fatty acids. Redox. Biol. 2021, 46, 102061. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frontiñán-Rubio, J.; Rabanal-Ruiz, Y.; Durán-Prado, M.; Alcain, F.J. The Protective Effect of Ubiquinone against the Amyloid Peptide in Endothelial Cells Is Isoprenoid Chain Length-Dependent. Antioxidants 2021, 10, 1806. https://doi.org/10.3390/antiox10111806
Frontiñán-Rubio J, Rabanal-Ruiz Y, Durán-Prado M, Alcain FJ. The Protective Effect of Ubiquinone against the Amyloid Peptide in Endothelial Cells Is Isoprenoid Chain Length-Dependent. Antioxidants. 2021; 10(11):1806. https://doi.org/10.3390/antiox10111806
Chicago/Turabian StyleFrontiñán-Rubio, Javier, Yoana Rabanal-Ruiz, Mario Durán-Prado, and Francisco Javier Alcain. 2021. "The Protective Effect of Ubiquinone against the Amyloid Peptide in Endothelial Cells Is Isoprenoid Chain Length-Dependent" Antioxidants 10, no. 11: 1806. https://doi.org/10.3390/antiox10111806
APA StyleFrontiñán-Rubio, J., Rabanal-Ruiz, Y., Durán-Prado, M., & Alcain, F. J. (2021). The Protective Effect of Ubiquinone against the Amyloid Peptide in Endothelial Cells Is Isoprenoid Chain Length-Dependent. Antioxidants, 10(11), 1806. https://doi.org/10.3390/antiox10111806