
Sanghuangporus sanghuang Mycelium Prevents Paracetamol-Induced Hepatotoxicity through Regulating the MAPK/NF-κB, Keap1/Nrf2/HO-1, TLR4/PI3K/Akt, and CaMKKβ/LKB1/AMPK Pathways and Suppressing Oxidative Stress and Inflammation


Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Source of Material
2.3. Sample Preparation
2.4. Animals
2.5. Experimental Protocol
2.6. Analysis of Biochemical Markers
2.7. Histopathological Examination
2.8. Analysis of MDA
2.9. Analysis of GSH
2.10. Analysis of Serum NO, TNF-α, IL-1β, and IL-6
2.11. Western Blot Analysis
2.12. Statistical Analysis
3. Results
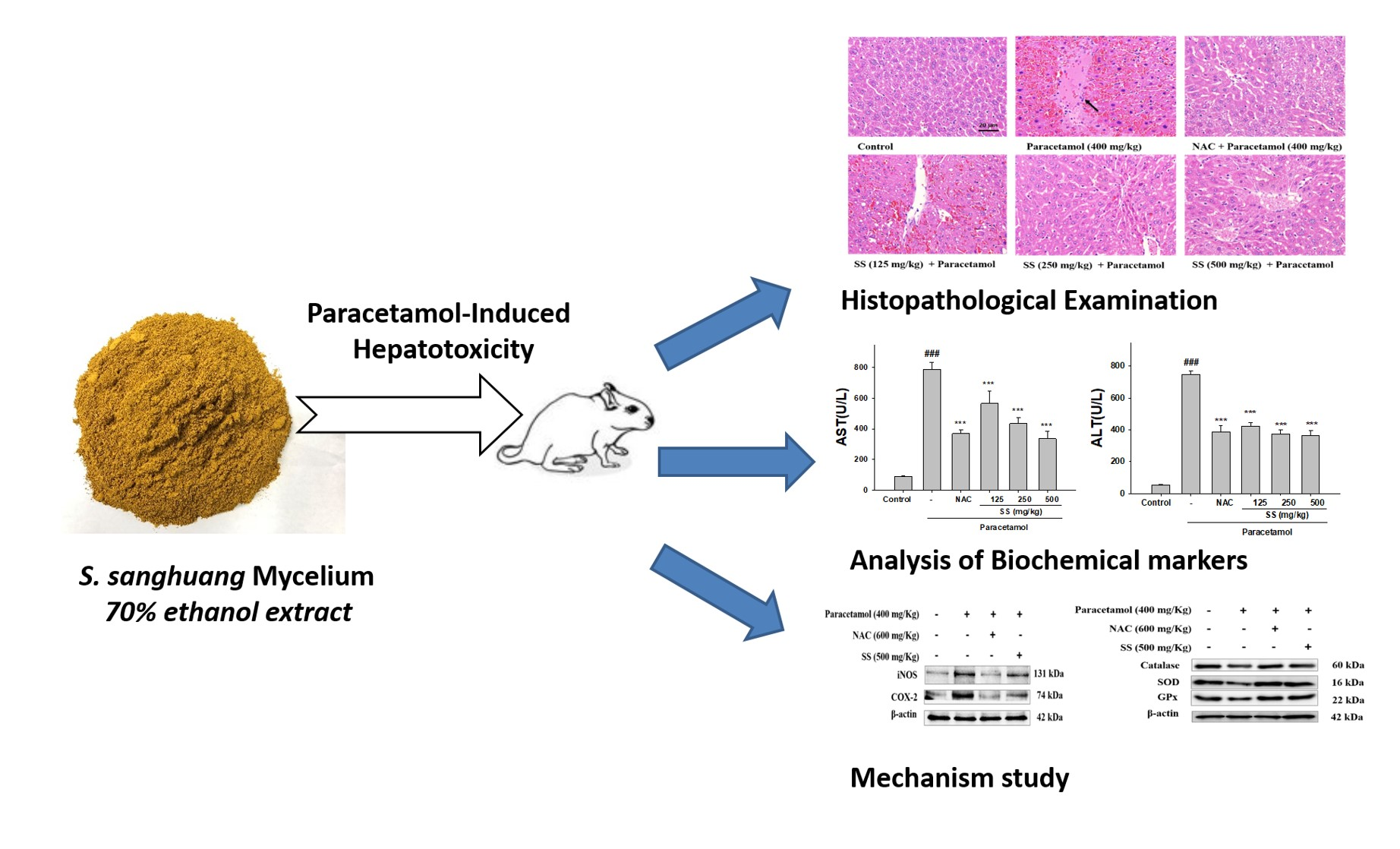
3.1. The Preventive Effect of SS on Hepatocellular Damage
3.2. SS Alleviates Paracetamol Hepatotoxicity
3.3. Inhibition of Paracetamol-Induced Lipid Peroxidation and Preservation of the Levels of GSH by SS
3.4. Inhibition of Paracetamol-Induced Liver Inflammation
3.5. SS Inhibited Paracetamol-Induced iNOS, COX-2, and NF-κB Pathway Protein Expression
3.6. SS Inhibited Paracetamol’s Induction of MAPK Signaling Pathway
3.7. SS Relieved Oxidative Stress and Activated Protective Antioxidant Mechanisms via Keap1/Nrf2/HO-1 Signaling after Paracetamol Challenge
3.8. SS Relieved CYP2E1 Expression after Paracetamol Challenge
3.9. SS Regulated TLR4/PI3K/Akt Signaling Pathway after Paracetamol Challenge
3.10. SS Regulated CaMKKβ/LKB1/AMPK Signaling Pathway after Paracetamol Challenge
3.11. Blocking AMPK Synergistically with Compound C to Increase Anti-Inflammatory Capacity of SS
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dai, G.; He, L.; Chou, N.; Wan, Y. Acetaminophen metabolism does not contribute to gender difference in its hepatotoxicity in mouse. Toxicol. Sci. 2006, 92, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Du, K.; Ramachandran, A.; Jaeschke, H. Oxidative stress during acetaminophen hepatotoxicity: Sources, pathophysiological role and therapeutic potential. Redox Biol. 2016, 10, 148–156. [Google Scholar] [CrossRef]
- Graham, G.G.; Scott, K.F. Mechanism of action of paracetamol. Am. J. Ther. 2005, 12, 46–55. [Google Scholar] [CrossRef]
- Singh, D.; Cho, W.C.; Upadhyay, G. Drug-induced liver toxicity and prevention by herbal antioxidants: An overview. Front. Physiol. 2015, 16. [Google Scholar] [CrossRef] [PubMed]
- Hinson, J.A.; Roberts, D.W.; James, L.P. Mechanisms of acetaminophen-induced liver necrosis. Handb. Exp. Pharmacol. 2010, 196, 369–405. [Google Scholar]
- James, L.P.; Mayeux, P.R.; Hinson, J.A. Acetaminophen-induced hepatotoxicity. Drug Metab. Dispos. 2003, 31, 1499–1506. [Google Scholar] [CrossRef] [PubMed]
- Woolbright, B.L.; Jaeschke, H. Role of the inflammasome in acetaminophen-induced liver injury and acute liver failure. J. Hepatol. 2017, 66, 836–848. [Google Scholar] [CrossRef] [PubMed]
- Eugenio-Pérez, D.; Montes de Oca-Solano, H.A.; Pedraza-Chaverri, J. Role of food-derived antioxidant agents against acetaminophen-induced hepatotoxicity. Pharm. Biol. 2016, 54, 2340–2352. [Google Scholar] [CrossRef] [PubMed]
- Cichoż-Lach, H.; Michalak, A. Oxidative stress as a crucial factor in liver diseases. World J. Gastroenterol. 2014, 20, 8082–8091. [Google Scholar] [CrossRef]
- Jadeja, R.N.; Upadhyay, K.K.; Devkar, R.V.; Khurana, S. Naturally occurring Nrf2 activators: Potential in treatment of liver injury. Oxid. Med. Cell Longev. 2016, 2016. [Google Scholar] [CrossRef]
- Nguyen, N.U.; Stamper, B.D. Polyphenols reported to shift paracetamol-induced changes in MAPK signaling and toxicity outcomes. Chem. Biol. Interact. 2017, 277, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Zhang, G.; Huang, L.; Pang, H.; Zhang, N.; Chen, Y.; Wang, G. Hepatoprotective effect of polyphenol-enriched fraction from folium microcos on oxidative stress and apoptosis in acetaminophen-induced liver injury in mice. Oxid. Med. Cell Longev. 2017, 2017. [Google Scholar] [CrossRef] [PubMed]
- Ning, C.; Gao, X.; Wang, C.; Kong, Y.; Liu, Z.; Sun, H.; Sun, P.; Huo, X.; Ma, X.; Meng, Q.; et al. Ginsenoside Rg1 protects against acetaminophen-induced liver injury via activating Nrf2 signaling pathway in vivo and in vitro. Regul. Toxicol. Pharmacol. 2018, 98, 58–68. [Google Scholar] [CrossRef]
- Lv, H.; Hong, L.; Tian, Y.; Yin, C.; Zhu, C.; Feng, H. Corilagin alleviates acetaminophen-induced hepatotoxicity via enhancing the AMPK/GSK3beta-Nrf2 signaling pathway. Cell Commun. Signal. 2019, 17. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Wei, S.; Zhou, H.; Shen, G.; Gan, X.; Zhou, S.; Qiu, J.; Shi, C.; Lu, L. Hyperglycemia exacerbates acetaminophen-induced acute liver injury by promoting liver-resident macrophage proinflammatory response via AMPK/PI3K/AKT-mediated oxidative stress. Cell Death Discov. 2019, 5. [Google Scholar] [CrossRef] [PubMed]
- Shu, G.; Qiu, Y.; Hao, J.; Fu, Q.; Deng, X. γ-Oryzanol alleviates acetaminophen-induced liver injury: Roles of modulating AMPK/GSK3beta/Nrf2 and NF-κB signaling pathways. Food Funct. 2019, 10, 6858–6872. [Google Scholar] [CrossRef]
- Leng, J.; Wang, Z.; Fu, C.L.; Zhang, J.; Ren, S.; Hu, J.N.; Jiang, S.; Wang, Y.P.; Chen, C.; Li, W. NF-κB and AMPK/PI3K/Akt signaling pathways are involved in the protective effects of Platycodon grandiflorum saponins against acetaminophen-induced acute hepatotoxicity in mice. Phytother. Res. 2018, 32, 2235–2246. [Google Scholar] [CrossRef]
- Lin, W.C.; Deng, J.S.; Huang, S.S.; Wu, S.H.; Lin, H.Y.; Huang, G.J. Evaluation of antioxidant, anti-inflammatory and anti-proliferative activities of ethanol extracts from different varieties of sanghuang species. RSC Adv. 2017, 7, 7780–7788. [Google Scholar] [CrossRef]
- Lin, W.C.; Deng, J.S.; Huang, S.S.; Wu, S.H.; Chen, C.C.; Lin, W.R.; Lin, H.Y.; Huang, G.J. Anti-inflammatory activity of Sanghuangporus sanghuang mycelium. Int. J. Mol. Sci. 2017, 18, 347. [Google Scholar] [CrossRef]
- Lin, W.C.; Deng, J.S.; Huang, S.S.; Lin, W.R.; Wu, S.H.; Lin, H.Y.; Huang, G.J. Anti-inflammatory activity of Sanghuangporus sanghuang by suppressing TLR4-mediated PI3K/AKT/mTOR/IKKß signaling pathway. RSC Adv. 2017, 7, 21234–21251. [Google Scholar] [CrossRef]
- Cai, C.; Ma, J.; Han, C.; Jin, Y.; Zhao, G.; He, X. Extraction and antioxidant activity of total triterpenoids in the mycelium of a medicinal fungus, Sanghuangporus sanghuang. Sci. Rep. 2019, 9. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.Y.; Deng, J.S.; Huang, W.C.; Jiang, W.P.; Huang, G.J. Attenuation of lipopolysaccharide-induced acute lung injury by hispolon in mice, through regulating the TLR4/PI3K/Akt/mTOR and Keap1/Nrf2/HO-1 pathways, and suppressing oxidative stress-mediated er stress-induced apoptosis and autophagy. Nutrients 2020, 12, 1742. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Song, J.; Wei, H.; Wang, Y.; Huang, X.; Liu, Y.; Lu, N.; He, L.; Lv, G.; Ding, H.; et al. Structural characterization and hypoglycemic activity of an intracellular polysaccharide from Sanghuangporus sanghuang mycelia. Int. J. Biol. Macromol. 2020, 164, 3305–3314. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.T.; Deng, J.S.; Huang, W.C.; Shieh, P.C.; Chung, M.I.; Huang, G.J. Salvianolic acid C against acetaminophen-induced acute liver injury by attenuating inflammation, oxidative stress, and apoptosis through inhibition of the Keap1/Nrf2/HO-1 signaling. Oxid. Med. Cell Longev. 2019, 2019. [Google Scholar] [CrossRef] [PubMed]
- Fazil, M.A.; Saulol Hamid, N.F.; Raslan, A.F.; Mohamed, M.N.; Nurhusien, Y. Histopathological changes of acetaminophen-induced liver injury and subsequent liver regeneration in BALB/C and ICR mice. Vet. World 2019, 12, 1682–1688. [Google Scholar]
- Jiang, W.P.; Huang, S.S.; Matsuda, Y.; Saito, H.; Uramaru, N.; Ho, H.Y.; Wu, J.B.; Huang, G.J. Protective effects of tormentic acid, a major component of suspension cultures of Eriobotrya japonica cells, on acetaminophen-induced hepatotoxicity in mice. Molecules 2017, 22, 830. [Google Scholar] [CrossRef]
- Fakurazi, S.; Hairuszah, I.; Nanthini, U. Moringa oleifera Lam prevents acetaminophen induced liver injury through restoration of glutathione level. Food Chem. Toxicol. 2008, 46, 2611–2615. [Google Scholar] [CrossRef]
- Deng, J.S.; Jiang, W.P.; Chen, C.C.; Lee, L.Y.; Li, P.Y.; Huang, W.C.; Liao, J.C.; Chen, H.Y.; Huang, S.S.; Huang, G.J. Cordyceps cicadae mycelia ameliorate cisplatin induced acute kidney injury by suppressing the TLR4/NFκB/MAPK and activating the HO 1/Nrf2 and Sirt 1/AMPK pathways in mice. Oxid. Med. Cell Longev. 2020, 2020. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Zhou, M.; Wang, Z.; Fu, Y.; Jia, M.; Wang, X.; Liu, M.; Zhang, Y.; Sun, Y.; Zhou, Y.; et al. Progranulin alleviates podocyte injury via regulating CAMKK/AMPK-mediated autophagy under diabetic conditions. J. Mol. Med. 2019, 97, 1507–1520. [Google Scholar] [CrossRef]
- Chao, X.; Wang, H.; Jaeschke, H.; Ding, W.X. Role and mechanisms of autophagy in acetaminophen-induced liver injury. Liver Int. 2018, 38, 1363–1374. [Google Scholar] [CrossRef] [PubMed]
- Jaeschke, H.; Ramachandran, A. Mechanisms and pathophysiological significance of sterile inflammation during acetaminophen hepatotoxicity. Food Chem. Toxicol. 2020, 138. [Google Scholar] [CrossRef]
- Han, D.; Shinohara, M.; Ybanez, M.D.; Saberi, B.; Kaplowitz, N. Signal transduction pathways involved in drug-induced liver injury. Handb. Exp. Pharmacol. 2010, 196, 267–310. [Google Scholar]
- Chen, Y.; Dong, H.; Thompson, D.C.; Shertzer, H.G.; Nebert, D.W.; Vasiliou, V. Glutathione defense mechanism in liver injury: Insights from animal models. Food Chem. Toxicol. 2013, 60, 38–44. [Google Scholar] [CrossRef]
- Papackova, Z.; Heczkova, M.; Dankova, H.; Sticova, E.; Lodererova, A.; Bartonova, L.; Poruba, M.; Cahova, M. Silymarin prevents acetaminophen-induced hepatotoxicity in mice. PLoS ONE. 2018, 13, e0191353. [Google Scholar] [CrossRef]
- Yang, R.; Song, C.; Chen, J.; Zhou, L.; Jiang, X.; Cao, X.; Sun, Y.; Zhang, Q. Limonin ameliorates acetaminophen-induced hepatotoxicity by activating Nrf2 antioxidative pathway and inhibiting NF-κB inflammatory response via upregulating Sirt1. Phytomedicine 2020, 69. [Google Scholar] [CrossRef] [PubMed]
- Vuong, L.D.; Nguyen, Q.N.; Truong, V.L. Anti-inflammatory and anti-oxidant effects of combination between sulforaphane and acetaminophen in LPS-stimulated RAW 264.7 macrophage cells. Immunopharmacol. Immunotoxicol. 2019, 41, 413–419. [Google Scholar] [CrossRef] [PubMed]
- Ren, S.; Leng, J.; Xu, X.Y.; Jiang, S.; Wang, Y.P.; Yan, X.T.; Liu, Z.; Chen, C.; Wang, Z.; Li, W. Ginsenoside Rb1, a major saponin from Panax ginseng, exerts protective effects against acetaminophen-induced hepatotoxicity in mice. Am. J. Chin. Med. 2019, 47, 1815–1831. [Google Scholar] [CrossRef]
- Yi, R.K.; Song, J.L.; Lim, Y.I.; Kim, Y.K.; Park, K.Y. Preventive effect of the Korean traditional health drink (taemyeongcheong) on acetaminophen-induced hepatic damage in ICR mice. Prev. Nutr. Food Sci. 2015, 20, 52–59. [Google Scholar] [CrossRef] [PubMed]
- El-Shafey, M.M.; Abd-Allah, G.M.; Mohamadin, A.M.; Harisa, G.I.; Mariee, A.D. Quercetin protects against acetaminophen-induced hepatorenal toxicity by reducing reactive oxygen and nitrogen species. Pathophysiology 2015, 22, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.M.; Ball, J.G.; Wright, M.S.; Meter, S.V.; Valentovic, M.A. Novel protective mechanisms for S-adenosyl-L-methionine against acetaminophen hepatotoxicity: Improvement of key antioxidant enzymatic function. Toxicol. Lett. 2012, 212, 320–328. [Google Scholar] [CrossRef] [PubMed]
- Bourdi, M.; Korrapati, M.C.; Chakraborty, M.; Yee, S.B.; Pohl, L.R. Protective role of c-Jun N-terminal kinase 2 in acetaminophen-induced liver injury. Biochem. Biophys. Res. Commun. 2008, 374, 6–10. [Google Scholar] [CrossRef]
- Noh, J.R.; Kim, Y.H.; Hwang, J.H.; Gang, G.T.; Kim, K.S.; Lee, I.K.; Yun, B.S.; Lee, C.H. Davallialactone protects against acetaminophen overdose-induced liver injuries in mice. Food Chem. Toxicol. 2013, 58, 14–21. [Google Scholar] [CrossRef]
- Huang, G.J.; Deng, J.S.; Huang, S.S.; Lee, C.Y.; Hou, W.C.; Wang, S.Y.; Sung, P.J.; Kuo, Y.H. Hepatoprotective effects of eburicoic acid and dehydroeburicoic acid from Antrodia camphorata in a mouse model of acute hepatic injury. Food Chem. 2013, 141, 3020–3027. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.J.; Deng, J.S.; Huang, S.S.; Shao, Y.Y.; Chen, C.C.; Kuo, Y.H. Protective effect of antrosterol from Antrodia camphorata submerged whole broth against carbon tetrachloride-induced acute liver injury in mice. Food Chem. 2012, 132, 709–716. [Google Scholar] [CrossRef]
- Yayeh, T.; Hong, M.; Jia, Q.; Lee, Y.C.; Kim, H.J.; Hyun, E.; Kim, T.W.; Rhee, M.H. Pistaciachinensis inhibits NO production and upregulates HO-1 induction via PI3K/Akt pathway in paracetamol stimulated macrophage cells. Am. J. Chin. Med. 2012, 40, 1085–1097. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Hao, W.; Hu, J.; Mi, X.; Han, Y.; Ren, S.; Jiang, S.; Wang, Y.; Li, X.; Li, W. Maltol improves paracetamol-induced hepatotoxicity by inhibiting oxidative stress and inflammation response via NF-κB and PI3K/Akt signal pathways. Antioxidants 2019, 8, 395. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.H.; Kim, H.W.; Kim, M.Y.; Kim, T.W.; Kim, E.N.; Kim, Y.; Chung, S.; Kim, Y.S.; Choi, B.S.; Kim, Y.S.; et al. Cinacalcet-mediated activation of the CaMKKbeta-LKB1-AMPK pathway attenuates diabetic nephropathy in db/db mice by modulation of apoptosis and autophagy. Cell Death Dis. 2018, 9. [Google Scholar] [CrossRef]
- Fogarty, S.; Ross, F.A.; Ciruelos, D.V.; Gray, A.; Gowans, G.J.; Hardie, D.G. AMPK causes cell cycle arrest in LKB1-deficient cells via activation of CAMKK2. Mol. Cancer Res. 2016, 14, 683–695. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.C.; Qiao, J.Y.; Wang, B.Y.; Bai, M.; Shen, J.D.; Cheng, Y.X. Paeoniflorin ameliorates fructose-induced insulin resistance and hepatic steatosis by activating LKB1/AMPK and AKT pathways. Nutrients 2018, 10, 1024. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, A.F.; Bettaieb, A.; Donohoe, D.R.; Alani, D.S.; Han, A.; Zhao, Y.; Whelan, J. Concurrent regulation of LKB1 and CaMKK2 in the activation of AMPK in castrate-resistant prostate cancer by a well-defined polyherbal mixture with anticancer properties. BMC Complement. Altern. Med. 2018, 18. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.; Bissa, B.; Brecht, L.; Allers, L.; Choi, S.W.; Gu, Y.; Zbinden, M.; Burge, M.R.; Timmins, G.; Hallows, K.; et al. AMPK is activated during lysosomal damage via a galectin-ubiquitin signal transduction system. Autophagy 2020, 16, 1550–1552. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Yang, D.; Gong, X.; Ge, P.; Dai, J.; Lin, L.; Zhang, L. Protective benefits of AMP-activated protein kinase in hepatic ischemia-reperfusion injury. Am. J. Transl. Res. 2017, 9, 823–829. [Google Scholar] [PubMed]
- Zhou, X.; Cao, Y.; Ao, G.; Hu, L.; Liu, H.; Wu, J.; Wang, X.; Jin, M.; Zheng, S.; Zhen, X.; et al. CaMKKβ-dependent activation of AMP-activated protein kinase is critical to suppressive effects of hydrogen sulfide on neuroinflammation. Antioxid. Redox Signal. 2014, 21, 1741–1758. [Google Scholar] [CrossRef] [PubMed]
- Saberi, B.; Ybanez, M.D.; Johnson, H.S.; Gaarde, W.A.; Han, D.; Kaplowitz, N. Protein kinase C (PKC) participates in acetaminophen hepatotoxicity through c-jun-N-terminal kinase (JNK)-dependent and -independent signaling pathways. Hepatology 2014, 59, 1543–1554. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, W.-P.; Deng, J.-S.; Huang, S.-S.; Wu, S.-H.; Chen, C.-C.; Liao, J.-C.; Chen, H.-Y.; Lin, H.-Y.; Huang, G.-J. Sanghuangporus sanghuang Mycelium Prevents Paracetamol-Induced Hepatotoxicity through Regulating the MAPK/NF-κB, Keap1/Nrf2/HO-1, TLR4/PI3K/Akt, and CaMKKβ/LKB1/AMPK Pathways and Suppressing Oxidative Stress and Inflammation. Antioxidants 2021, 10, 897. https://doi.org/10.3390/antiox10060897
Jiang W-P, Deng J-S, Huang S-S, Wu S-H, Chen C-C, Liao J-C, Chen H-Y, Lin H-Y, Huang G-J. Sanghuangporus sanghuang Mycelium Prevents Paracetamol-Induced Hepatotoxicity through Regulating the MAPK/NF-κB, Keap1/Nrf2/HO-1, TLR4/PI3K/Akt, and CaMKKβ/LKB1/AMPK Pathways and Suppressing Oxidative Stress and Inflammation. Antioxidants. 2021; 10(6):897. https://doi.org/10.3390/antiox10060897
Chicago/Turabian StyleJiang, Wen-Ping, Jeng-Shyan Deng, Shyh-Shyun Huang, Sheng-Hua Wu, Chin-Chu Chen, Jung-Chun Liao, Hung-Yi Chen, Hui-Yi Lin, and Guan-Jhong Huang. 2021. "Sanghuangporus sanghuang Mycelium Prevents Paracetamol-Induced Hepatotoxicity through Regulating the MAPK/NF-κB, Keap1/Nrf2/HO-1, TLR4/PI3K/Akt, and CaMKKβ/LKB1/AMPK Pathways and Suppressing Oxidative Stress and Inflammation" Antioxidants 10, no. 6: 897. https://doi.org/10.3390/antiox10060897
APA StyleJiang, W.-P., Deng, J.-S., Huang, S.-S., Wu, S.-H., Chen, C.-C., Liao, J.-C., Chen, H.-Y., Lin, H.-Y., & Huang, G.-J. (2021). Sanghuangporus sanghuang Mycelium Prevents Paracetamol-Induced Hepatotoxicity through Regulating the MAPK/NF-κB, Keap1/Nrf2/HO-1, TLR4/PI3K/Akt, and CaMKKβ/LKB1/AMPK Pathways and Suppressing Oxidative Stress and Inflammation. Antioxidants, 10(6), 897. https://doi.org/10.3390/antiox10060897