
Mechanisms of Non-Alcoholic Fatty Liver Disease in the Metabolic Syndrome. A Narrative Review

 , ,
, ,  , ,
, , 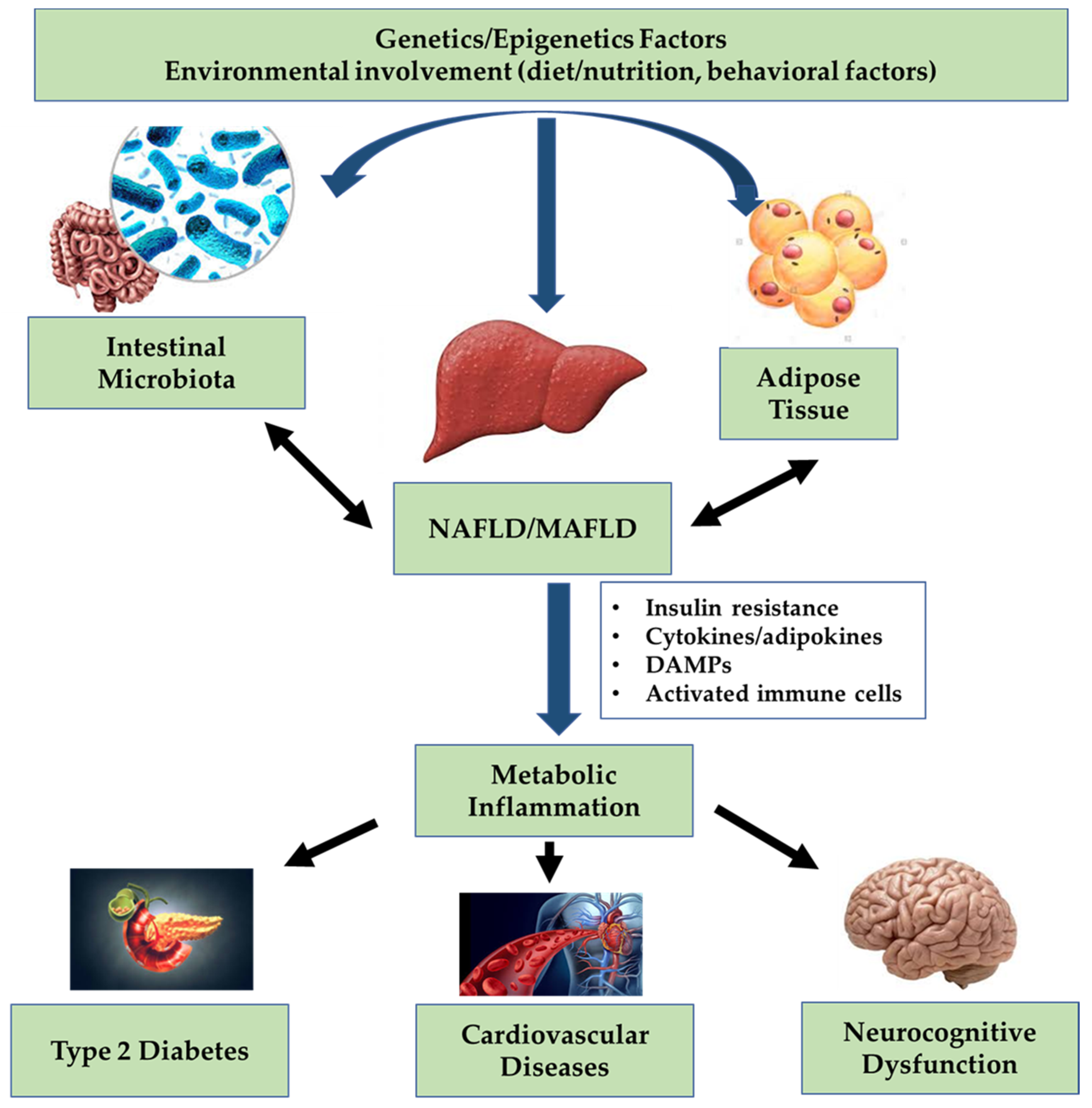{kind=link}
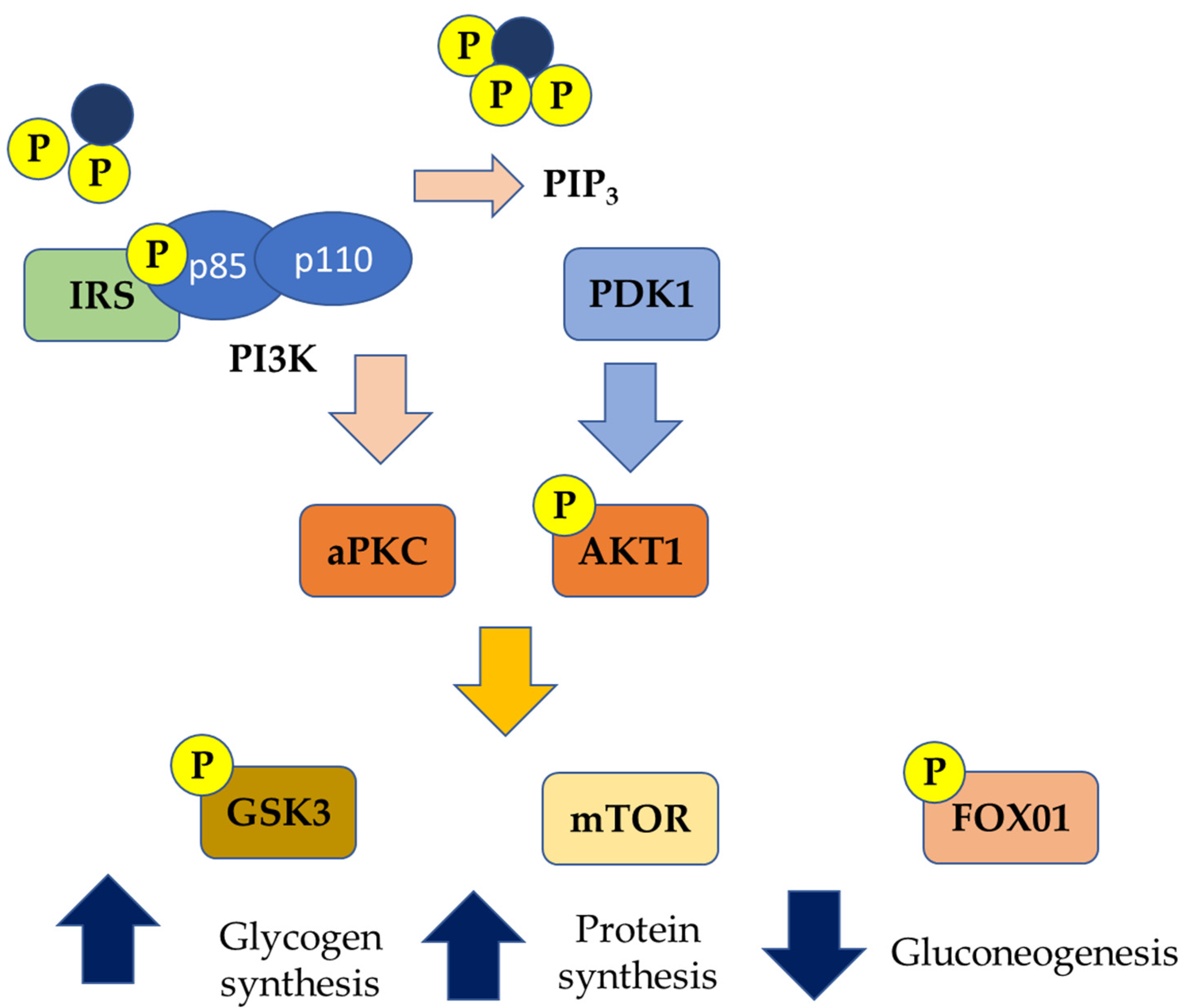{kind=link}
Abstract
1. Introduction
2. Methods
3. NAFLD and MAFLD
4. Metabolic Syndrome and Insulin Resistance
5. The Microbiota
6. Oxidative Stress and Chronic Inflammation
7. Diet and Lifestyle
7.1. Mediterranean Diet
7.2. MD Mechanism: Antioxidant and Lipid Lowering Effects
7.3. Physical Activity
8. Genetics and Epigenetics
9. Endothelial Dysfunction
10. Clinical Impact
11. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| NAFLD | Non-alcoholic fatty liver disease |
| MS | Metabolic syndrome |
| IR | Insulin resistance |
| CV | Cardiovascular |
| HDL | High density lipoproteins |
| FFAs | Free fatty acids |
| ER | Endoplasmic reticulum |
| PDFF | Proton density fat fraction |
| MRI | Magnetic resonance imaging |
| NASH | Non-alcoholic steatohepatitis |
| HCC | Hepatocellular carcinoma |
| T2DM | Type 2 diabetes mellitus |
| BMI | Body Mass Index |
| OGTT | Oral glucose tolerance test |
| HbA1c | Glycated hemoglobin |
| MAFLD | Metabolic associated fatty liver disease |
| IRS | Insulin receptor |
| PI3K | Phosphatidylinositol 3-kinase |
| GLUT-4 | Glucose transporter protein type-4 |
| TNF-α | Tumor necrosis factor-alpha |
| SREBP-1/2 | Sterol regulatory element-binding protein-1/2 |
| ChREBP | Carbohydrate-responsive element-binding protein |
| PPAR-γ | Peroxisome proliferator-activated receptor gamma |
| BA | Bile acids |
| LPS | lipopolysaccharides |
| SCFA | Short-chain fatty acids |
| ROS | Reactive oxygen species |
| O2•− | superoxidation radicals |
| H2O2 | Hydrogen peroxide |
| NADPH | Nicotinamide adenine dinucleotide phosphate hydrogen |
| NF-κB | Nuclear factor kappa B |
| JNK | Jun N-terminal kinase |
| IKK | Inhibitor of nuclear factor kappa-B kinase |
| EASL | European Association for the Study of Liver |
| AASLD | American Association for the Study of Liver Disease |
| PUFA | Polyunsaturated fatty acid |
| MD | Mediterranean diet |
| MUFA | Mono-unsaturated fatty acid |
| IL | Interleukin |
| LDL | Low density lipoprotein |
| VLDL | Very low density lipoprotein |
| SNPs | Single nucleotide polymorphisms |
| PNPLA3 | Patatin like phospholipase domain-containing protein 3 |
| HSC | Hepatic stellate cells |
| LXR | Liver X receptor |
| MBOAT-7 | Membrane Bound O-Acyltransferase Domain Containing 7 |
| GCKR | glucokinase regulatory protein |
| TGF-1 | Transforming growth factor beta 1 |
| PDGF | Platelet-derived growth factor |
| HAT | Histone acetyltransferase |
| HDAC | Histone deacetylases |
| SIRT | Sirtuins |
| SFAs | Saturated fatty acids |
| EDHF | hyperpolarizing factor of endothelial derivation |
| ED | endothelial dysfunction |
| NOS | Nitric oxide synthetase |
| ESM-1 | Endocan |
| HMGB1 | High mobility group box 1 protein |
| CAD | coronary artery disease |
| HCV | hepatitis C virus |
| HBV | hepatitis B virus |
| US | ultrasound |
| CAP | Control attenuation parameter |
| IMT | Intima-media thickness |
| CKD | Chronic kidney disease |
| PIP3 | Phosphatidylinositol (3,4,5)-trisphosphate |
| aPKC | activated protein kinase C |
| AKT1 | Protein kinase B |
| GSK3 | Glycogen synthase kinase 3 |
| mTOR | Mammalian target of rapamycin |
| FOXO1 | Forkhead box protein O1 |
| PDK1 | 3-Phosphoinositide-dependent protein kinase 1 |
| P | phosphorylated |
References
- Alberti, K.G.; Eckel, R.H.; Grundy, S.M.; Zimmet, P.Z.; Cleeman, J.I.; Donato, K.A.; Fruchart, J.C.; James, W.P.; Loria, C.M.; Smith, S.C., Jr. Joint scientific statement. Circulation 2009, 120, 1640–1645. [Google Scholar] [CrossRef]
- Kleiner, D.E.; Makhlouf, H.R. Histology of Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis in Adults and Children. Clin. Liver Dis. 2016, 20, 293–312. [Google Scholar] [CrossRef]
- Grundy, S.M. Metabolic syndrome pandemic. Arter. Thromb. Vasc. Biol. 2008, 28, 629–636. [Google Scholar] [CrossRef]
- Cheung, O.; Sanyal, A.J. Recent advances in nonalcoholic fatty liver disease. Curr. Opin. Gastroenterol. 2010, 26, 202–208. [Google Scholar] [CrossRef]
- Sanyal, A.J. NASH: A global health problem. Hepatol. Res. 2011, 41, 670–674. [Google Scholar] [CrossRef]
- Paschos, P.; Paletas, K. Non alcoholic fatty liver disease and metabolic syndrome. Hippokratia 2009, 13, 9–19. [Google Scholar]
- Kotronen, A.; Yki-Järvinen, H. Fatty liver: A novel component of the metabolic syndrome. Arter. Thromb. Vasc. Biol. 2008, 28, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Lebovitz, H.E. Insulin resistance: Definition and consequences. Exp. Clin. Endocrinol. Diabetes 2001, 109 (Suppl. 2), S135–S148. [Google Scholar] [CrossRef] [PubMed]
- Banerji, M.A.; Lebowitz, J.; Chaiken, R.L.; Gordon, D.; Kral, J.G.; Lebovitz, H.E. Relationship of visceral adipose tissue and glucose disposal is independent of sex in black NIDDM subjects. Am. J. Physiol. 1997, 273, E425–E432. [Google Scholar] [CrossRef]
- Park, H.K.; Kwak, M.K.; Kim, H.J.; Ahima, R.S. Linking resistin, inflammation, and cardiometabolic diseases. Korean J. Intern. Med. 2017, 32, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Sasso, F.C.; Pafundi, P.C.; Marfella, R.; Calabrò, P.; Piscione, F.; Furbatto, F.; Esposito, G.; Galiero, R.; Gragnano, F.; Rinaldi, L.; et al. Adiponectin and insulin resistance are related to restenosis and overall new PCI in subjects with normal glucose tolerance: The prospective AIRE Study. Cardiovasc. Diabetol. 2019, 18, 24. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Hotamisligil, G.S. Nonalcoholic fatty liver disease: Cytokine-adipokine interplay and regulation of insulin resistance. Gastroenterology 2006, 131, 934–945. [Google Scholar] [CrossRef] [PubMed]
- Lewis, G.F.; Carpentier, A.; Adeli, K.; Giacca, A. Disordered fat storage and mobilization in the pathogenesis of insulin resistance and type 2 diabetes. Endocr. Rev. 2002, 23, 201–229. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Role of endoplasmic reticulum stress and c-Jun NH2-terminal kinase pathways in inflammation and origin of obesity and diabetes. Diabetes 2005, 54, S73–S78. [Google Scholar] [CrossRef] [PubMed]
- Acierno, C.; Caturano, A.; Pafundi, P.C.; Nevola, R.; Adinolfi, L.E.; Sasso, F.C. Nonalcoholic fatty liver disease and type 2 diabetes: Pathophysiological mechanisms shared between the two faces of the same coin. Explor. Med. 2020, 1, 287–306. [Google Scholar] [CrossRef]
- Lonardo, A.; Nascimbeni, F.; Maurantonio, M.; Marrazzo, A.; Rinaldi, L.; Adinolfi, L.E. Nonalcoholic fatty liver disease: Evolving paradigms. World J. Gastroenterol. 2017, 23, 6571–6592. [Google Scholar] [CrossRef]
- Targher, G.; Bertolini, L.; Padovani, R.; Rodella, S.; Tessari, R.; Zenari, L.; Day, C.; Arcaro, G. Prevalence of nonalcoholic fatty liver disease and its association with cardiovascular disease among type 2 diabetic patients. Diabetes Care. 2007, 30, 1212–1218. [Google Scholar] [CrossRef] [PubMed]
- Neuschwander-Tetri, B.A.; Caldwell, S.H. Nonalcoholic steatohepatitis: Summary of an AASLD Single Topic Conference. Hepatology 2003, 37, 1202–1219. [Google Scholar] [CrossRef] [PubMed]
- European Association for the Study of the Liver (EASL); European Association for the Study of Diabetes (EASD); European Association for the Study of Obesity (EASO). EASL-EASD-EASO Clinical Practice Guidelines for the management of non-alcoholic fatty liver disease. J. Hepatol. 2016, 64, 1388–1402. [Google Scholar] [CrossRef]
- Masarone, M.; Rosato, V.; Aglitti, A.; Bucci, T.; Caruso, R.; Salvatore, T.; Sasso, F.C.; Tripodi, M.F.; Persico, M. Liver biopsy in type 2 diabetes mellitus: Steatohepatitis represents the sole feature of liver damage. PLoS ONE 2017, 12, e0178473. [Google Scholar] [CrossRef]
- Sasso, F.C.; Pafundi, P.C.; Marfella, R.; Calabrò, P.; Piscione, F.; Furbatto, F.; Esposito, G.; Galiero, R.; Gragnano, F.; Rinaldi, L.; et al. Nonalcoholic Steatohepatitis Council. Nonalcoholic Steatohepatitis Is the Fastest Growing Cause of Hepatocellular Carcinoma in Liver Transplant Candidates. Clin. Gastroenterol. Hepatol. 2019, 17, 748–755.e3. [Google Scholar] [CrossRef]
- Younossi, Z.; Tacke, F.; Arrese, M.; Chander Sharma, B.; Mostafa, I.; Bugianesi, E.; Wai-Sun Wong, V.; Yilmaz, Y.; George, J.; Fan, J.; et al. Global Perspectives on Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis. Hepatology 2019, 69, 2672–2682. [Google Scholar] [CrossRef] [PubMed]
- Eslam, M.; Newsome, P.N.; Sarin, S.K.; Anstee, Q.M.; Targher, G.; Romero-Gomez, M.; Zelber-Sagi, S.; Wai-Sun Wong, V.; Dufour, J.F.; Schattenberg, J.M.; et al. A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement. J. Hepatol. 2020, 73, 202–209. [Google Scholar] [CrossRef]
- Lee, H.W.; Wong, G.L.; Kwok, R.; Choi, K.C.; Chan, C.K.; Shu, S.S.; Leung, J.K.; Chim, A.M.; Luk, A.O.; Ma, R.C.; et al. Serial Transient Elastography Examinations to Monitor Patients With Type 2 Diabetes: A Prospective Cohort Study. Hepatology 2020, 72, 1230–1241. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Golabi, P.; de Avila, L.; Paik, J.M.; Srishord, M.; Fukui, N.; Qiu, Y.; Burns, L.; Afendy, A.; Nader, F. The global epidemiology of NAFLD and NASH in patients with type 2 diabetes: A systematic review and meta-analysis. J. Hepatol. 2019, 71, 793–801. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Chen, C.; Cui, J.; Lu, J.; Yan, C.; Wei, X.; Zhao, X.; Li, N.; Li, S.; Xue, G.; et al. Fatty Liver Disease Caused by High-Alcohol-Producing Klebsiella pneumoniae. Cell Metab. 2019, 30, 1172. [Google Scholar] [CrossRef] [PubMed]
- Romeo, S.; Sanyal, A.; Valenti, L. Leveraging Human Genetics to Identify Potential New Treatments for Fatty Liver Disease. Cell Metab. 2020, 31, 35–45. [Google Scholar] [CrossRef]
- Trepo, E.; Valenti, L. Update on NAFLD genetics: From new variants to the clinic. J. Hepatol. 2020, 72, 1196–1209. [Google Scholar] [CrossRef]
- Pelusi, S.; Valenti, L. Hepatic fat as clinical outcome and therapeutic target for nonalcoholic fatty liver disease. Liver Int. 2019, 39, 250–256. [Google Scholar] [CrossRef]
- Dongiovanni, P.; Stender, S.; Pietrelli, A.; Mancina, R.M.; Cespiati, A.; Petta, S.; Pelusi, S.; Pingitore, P.; Badiali, S.; Maggioni, M.; et al. Causal relationship of hepatic fat with liver damage and insulin resistance in nonalcoholic fatty liver. J. Intern. Med. 2018, 283, 356–370. [Google Scholar] [CrossRef]
- Eslam, M.; George, J. Genetic Insights for Drug Development in NAFLD. Trends Pharmacol. Sci. 2019, 40, 506–516. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Huang, J.; Wang, M.; Kumar, R.; Liu, Y.; Liu, S.; Wu, Y.; Wang, X.; Zhu, Y. Comparison of MAFLD and NAFLD diagnostic criteria in real world. Liver Int. 2020, 40, 2082–2089. [Google Scholar] [CrossRef] [PubMed]
- Shiha, G.; Korenjak, M.; Eskridge, W.; Casanovas, T.; Velez-Moller, P.; Högström, S.; Richardson, B.; Munoz, C.; Sigurðardóttir, S.; Coulibaly, A.; et al. Redefining fatty liver disease: An international patient perspective. Lancet Gastroenterol. Hepatol. 2021, 6, 73–79. [Google Scholar] [CrossRef]
- Wai-Sun Wong, V.; Lai-Hung Wong, G.; Woo, J.; Abrigo, J.M.; Ka-Man Chan, C.; She-Ting Shu, S.; Ka-Yu Leung, J.; Mei-Ling Chim, A.; Pik-Shan Kong, A.; Chung-Yan Lui, G.; et al. Impact of the new definition of metabolic associated fatty liver disease on the epidemiology of the disease. Clin. Gastroenterol. Hepatol. 2020, 30. [Google Scholar] [CrossRef] [PubMed]
- Thevis, M.; Thomas, A.; Schänzer, W. Insulin. Handb. Exp. Pharmacol. 2010, 15, 209–226. [Google Scholar] [CrossRef]
- Sonksen, P.; Sonksen, J. Insulin: Understanding its action in health and disease. Br. J. Anaesth. 2000, 85, 69–79. [Google Scholar] [CrossRef]
- Posner, B.I. Insulin Signalling: The Inside Story. Can. J. Diabetes 2017, 41, 108–113. [Google Scholar] [CrossRef]
- Goetze, S.; Kim, S.; Xi, X.P.; Graf, K.; Yang, D.C.; Fleck, E.; Meehan, W.P.; Hsueh, W.A.; Law, R.E. Troglitazone inhibits mitogenic signaling by insulin in vascular smooth muscle cells. J. Cardiovasc. Pharmacol. 2000, 35, 749–757. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.B.; Nikoulina, S.E.; Ciaraldi, T.P.; Henry, R.R.; Kahn, B.B. Normal insulin-dependent activation of Akt/protein kinase B, with diminished activation of phosphoinositide 3-kinase, in muscle in type 2 diabetes. J. Clin. Investig. 1999, 104, 733–741. [Google Scholar] [CrossRef]
- Yaribeygi, H.; Farrokhi, F.R.; Butler, A.E.; Sahebkar, A. Insulin resistance: Review of the underlying molecular mechanisms. J. Cell Physiol. 2019, 234, 8152–8161. [Google Scholar] [CrossRef]
- Romao, I.; Roth, J. Genetic and environmental interactions in obesity and type 2 diabetes. J. Am. Diet. Assoc. 2008, 108, S24–S28. [Google Scholar] [CrossRef] [PubMed]
- Goodpaster, B.H.; Kelley, D.E.; Wing, R.R.; Meier, A.; Thaete, F.L. Effects of weight loss on regional fat distribution and insulin sensitivity in obesity. Diabetes 1999, 48, 839–847. [Google Scholar] [CrossRef]
- Zac-Varghese, S.; Tan, T.; Bloom, S.R. Hormonal interactions between gut and brain. Discov. Med. 2010, 10, 543–552. [Google Scholar] [PubMed]
- Dresner, A.; Laurent, D.; Marcucci, M.; Griffin, M.E.; Dufour, S.; Cline, G.W.; Slezak, L.A.; Andersen, D.K.; Hundal, R.S.; Rothman, D.L.; et al. Effects of free fatty acids on glucose transport and IRS-1-associated phosphatidylinositol 3-kinase activity. J. Clin. Investig. 1999, 103, 253–259. [Google Scholar] [CrossRef]
- Ahima, R.S.; Lazar, M.A. Adipokines and the peripheral and neural control of energy balance. Mol. Endocrinol. 2008, 22, 1023–1031. [Google Scholar] [CrossRef] [PubMed]
- Olefsky, J.M.; Glass, C.K. Macrophages, inflammation, and insulin resistance. Annu. Rev. Physiol. 2010, 72, 219–246. [Google Scholar] [CrossRef]
- Esposito, K.; Ciotola, M.; Sasso, F.C.; Cozzolino, D.; Saccomanno, F.; Assaloni, R.; Ceriello, A.; Giugliano, D. Effect of a single high-fat meal on endothelial function in patients with the metabolic syndrome: Role of tumor necrosis factor-alpha. Nutr. Metab. Cardiovasc. Dis. 2007, 17, 274–279. [Google Scholar] [CrossRef]
- Bugianesi, E.; Moscatiello, S.; Ciaravella, M.; Marchesini, G. Insulin resistance in nonalcoholic fatty liver disease. Curr. Pharm. Des. 2010, 16, 1941–1951. [Google Scholar] [CrossRef]
- Jacome-Sosa, M.M.; Parks, E.J. Fatty acid sources and their fluxes as they contribute to plasma triglyceride concentrations and fatty liver in humans. Curr. Opin. Lipidol. 2014, 25, 213–220. [Google Scholar] [CrossRef]
- Musso, G.; Gambino, R.; Cassader, M. Cholesterol metabolism and the pathogenesis of non-alcoholic steatohepatitis. Prog. Lipid Res. 2013, 52, 175–191. [Google Scholar] [CrossRef]
- Lambert, J.E.; Ramos-Roman, M.A.; Browning, J.D.; Parks, E.J. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology 2014, 146, 726–735. [Google Scholar] [CrossRef] [PubMed]
- George, J.; Liddle, C. Nonalcoholic fatty liver disease: Pathogenesis and potential for nuclear receptors as therapeutic targets. Mol. Pharm. 2008, 5, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Schultz, J.R.; Tu, H.; Luk, A.; Repa, J.J.; Medina, J.C.; Li, L.; Schwendner, S.; Wang, S.; Thoolen, M.; Mangelsdorf, D.J.; et al. Role of LXRs in control of lipogenesis. Genes Dev. 2000, 14, 2831–2838. [Google Scholar] [CrossRef] [PubMed]
- Stefan, N.; Kantartzis, K.; Häring, H.U. Causes and metabolic consequences of Fatty liver. Endocr. Rev. 2008, 29, 939–960. [Google Scholar] [CrossRef] [PubMed]
- Postic, C.; Girard, J. Contribution of de novo fatty acid synthesis to hepatic steatosis and insulin resistance: Lessons from genetically engineered mice. J. Clin. Investig. 2008, 118, 829–838. [Google Scholar] [CrossRef]
- Ursell, L.K.; Clemente, J.C.; Rideout, J.R.; Gevers, D.; Caporaso, J.G.; Knight, R. The interpersonal and intrapersonal diversity of human-associated microbiota in key body sites. J. Allergy Clin. Immunol. 2012, 129, 1204–1208. [Google Scholar] [CrossRef]
- Neish, A.S. Microbes in gastrointestinal health and disease. Gastroenterology 2009, 136, 65–80. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, Y.; Zhu, J.; Zhang, M.; Ho, C.T.; Huang, Q.; Cao, J. Metagenomics Analysis of Gut Microbiota in a High Fat Diet-Induced Obesity Mouse Model Fed with (-)-Epigallocatechin 3-O-(3-O-Methyl) Gallate (EGCG3″Me). Mol. Nutr. Food Res. 2018, 62, e1800274. [Google Scholar] [CrossRef]
- Devillard, E.; McIntosh, F.M.; Duncan, S.H.; Wallace, R.J. Metabolism of linoleic acid by human gut bacteria: Different routes for biosynthesis of conjugated linoleic acid. J. Bacteriol. 2007, 189, 2566–2570. [Google Scholar] [CrossRef]
- Li, C.Y.; Dempsey, J.L.; Wang, D.; Lee, S.; Weigel, K.M.; Fei, Q.; Bhatt, D.K.; Prasad, B.; Raftery, D.; Gu, H.; et al. PBDEs Altered Gut Microbiome and Bile Acid Homeostasis in Male C57BL/6 Mice. Drug Metab. Dispos. 2018, 46, 1226–1240. [Google Scholar] [CrossRef]
- Zununi Vahed, S.; Moghaddas Sani, H.; Rahbar Saadat, Y.; Barzegari, A.; Omidi, Y. Type 1 diabetes: Through the lens of human genome and metagenome interplay. Biomed. Pharmacother. 2018, 104, 332–342. [Google Scholar] [CrossRef] [PubMed]
- Baddini Feitoza, A.; Fernandes Pereira, A.; Ferreira da Costa, N.; Gonçalves Ribeiro, B. Conjugated linoleic acid (CLA): Effect modulation of body composition and lipid profile. Nutr. Hosp. 2009, 24, 422–428. [Google Scholar] [PubMed]
- Zhang, C.; Björkman, A.; Cai, K.; Liu, G.; Wang, C.; Li, Y.; Xia, H.; Sun, L.; Kristiansen, K.; Wang, J.; et al. Impact of a 3-Months Vegetarian Diet on the Gut Microbiota and Immune Repertoire. Front. Immunol. 2018, 9, 908. [Google Scholar] [CrossRef] [PubMed]
- Shin, N.R.; Whon, T.W.; Bae, J.W. Proteobacteria: Microbial signature of dysbiosis in gut microbiota. Trends Biotechnol. 2015, 33, 496–503. [Google Scholar] [CrossRef]
- Leung, C.; Rivera, L.; Furness, J.B.; Angus, P.W. The role of the gut microbiota in NAFLD. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 412–425. [Google Scholar] [CrossRef]
- Membrez, M.; Blancher, F.; Jaquet, M.; Bibiloni, R.; Cani, P.D.; Burcelin, R.G.; Corthesy, I.; Macé, K.; Chou, C.J. Gut microbiota modulation with norfloxacin and ampicillin enhances glucose tolerance in mice. FASEB J. 2008, 22, 2416–2426. [Google Scholar] [CrossRef]
- Vrieze, A.; Van Nood, E.; Holleman, F.; Salojärvi, J.; Kootte, R.S.; Bartelsman, J.F.; Dallinga-Thie, G.M.; Ackermans, M.T.; Serlie, M.J.; Oozeer, R.; et al. Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology 2012, 143, 913–916.e7. [Google Scholar] [CrossRef]
- Ley, R.E.; Turnbaugh, P.J.; Klein, S.; Gordon, J.I. Microbial ecology: Human gut microbes associated with obesity. Nature 2006, 444, 1022–1023. [Google Scholar] [CrossRef]
- Clemente, M.G.; Mandato, C.; Poeta, M.; Vajro, P. Pediatric non-alcoholic fatty liver disease: Recent solutions, unresolved issues, and future research directions. World J. Gastroenterol. 2016, 22, 8078–8093. [Google Scholar] [CrossRef] [PubMed]
- Zorn, A.M.; Wells, J.M. Vertebrate endoderm development and organ formation. Annu. Rev. Cell. Dev. Biol. 2009, 25, 221–251. [Google Scholar] [CrossRef] [PubMed]
- Compare, D.; Coccoli, P.; Rocco, A.; Nardone, O.M.; De Maria, S.; Cartenì, M.; Nardone, G. Gut–liver axis: The impact of gut microbiota on Non Alcoholic fatty liver disease. Nutr. Metab. Cardiovasc. Dis. 2012, 22, 471–476. [Google Scholar] [CrossRef] [PubMed]
- Zeisel, S.H.; Wishnok, J.S.; Blusztajn, J.K. Formation of methylamines from ingested choline and lecithin. J. Pharmacol. Exp. Ther. 1983, 225, 320–324. [Google Scholar]
- Spencer, M.D.; Hamp, T.J.; Reid, R.W.; Fischer, L.M.; Zeisel, S.H.; Fodor, A.A. Association between composition of the human gastrointestinal microbiome and development of fatty liver with choline deficiency. Gastroenterology 2011, 140, 976–986. [Google Scholar] [CrossRef] [PubMed]
- Tremaroli, V.; Bäckhed, F. Functional interactions between the gut microbiota and host metabolism. Nature 2012, 489, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Yu, Y.; Cai, J.; Li, H. Emerging Molecular Targets for Treatment of Nonalcoholic Fatty Liver Disease. Trends Endocrinol. Metab. 2019, 30, 903–914. [Google Scholar] [CrossRef] [PubMed]
- Rolo, A.P.; Teodoro, J.S.; Palmeira, C.M. Role of oxidative stress in the pathogenesis of nonalcoholic steatohepatitis. Free Radic. Biol. Med. 2012, 52, 59–69. [Google Scholar] [CrossRef]
- Serviddio, G.; Bellanti, F.; Vendemiale, G. Free radical biology for medicine: Learning from nonalcoholic fatty liver disease. Free Radic. Biol. Med. 2013, 65, 952–968. [Google Scholar] [CrossRef]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef]
- Takaki, A.; Kawai, D.; Yamamoto, K. Multiple hits, including oxidative stress, as pathogenesis and treatment target in non-alcoholic steatohepatitis (NASH). Int. J. Mol. Sci. 2013, 14, 20704–20728. [Google Scholar] [CrossRef]
- Masarone, M.; Rosato, V.; Dallio, M.; Gravina, A.G.; Aglitti, A.; Loguercio, C.; Federico, A.; Persico, M. Role of Oxidative Stress in Pathophysiology of Nonalcoholic Fatty Liver Disease. Oxid. Med. Cell. Longev. 2018, 11, 9547613. [Google Scholar] [CrossRef]
- Forrester, S.J.; Kikuchi, D.S.; Hernandes, M.S.; Xu, Q.; Griendling, K.K. Reactive Oxygen Species in Metabolic and Inflammatory Signaling. Circ. Res. 2018, 122, 877–902. [Google Scholar] [CrossRef]
- Van Der Vliet, A.; Janssen-Heininger, Y.M.; Anathy, V. Oxidative stress in chronic lung disease: From mitochondrial dysfunction to dysregulated redox signaling. Mol. Aspects Med. 2018, 63, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Mansouri, A.; Gattolliat, C.H.; Asselah, T. Mitochondrial Dysfunction and Signaling in Chronic Liver Diseases. Gastroenterology 2018, 155, 629–647. [Google Scholar] [CrossRef]
- Petta, S.; Muratore, C.; Craxì, A. Non-alcoholic fatty liver disease pathogenesis: The present and the future. Dig Liver Dis. 2009, 41, 615–625. [Google Scholar] [CrossRef]
- Begriche, K.; Igoudjil, A.; Pessayre, D.; Fromenty, B. Mitochondrial dysfunction in NASH: Causes, consequences and possible means to prevent it. Mitochondrion 2006, 6, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.S.; Buck, M.; Houglum, K.; Chojkier, M. Activation of hepatic stellate cells by TGF alpha and collagen type I is mediated by oxidative stress through c-myb expression. J. Clin. Investig. 1995, 96, 2461–2468. [Google Scholar] [CrossRef]
- Armutcu, F.; Coskun, O.; Gürel, A.; Kanter, M.; Can, M.; Ucar, F.; Unalacak, M. Thymosin alpha 1 attenuates lipid peroxidation and improves fructose-induced steatohepatitis in rats. Clin. Biochem. 2005, 38, 540–547. [Google Scholar] [CrossRef] [PubMed]
- Uysal, S.; Armutcu, F.; Aydogan, T.; Akin, K.; Ikizek, M.; Yigitoglu, M.R. Some inflammatory cytokine levels, iron metabolism and oxidan stress markers in subjects with nonalcoholic steatohepatitis. Clin. Biochem. 2011, 44, 1375–1379. [Google Scholar] [CrossRef]
- Malaguarnera, M.; Di Rosa, M.; Nicoletti, F.; Malaguarnera, L. Molecular mechanisms involved in NAFLD progression. J. Mol. Med. 2009, 87, 679–695. [Google Scholar] [CrossRef]
- Syn, W.K.; Choi, S.S.; Diehl, A.M. Apoptosis and cytokines in non-alcoholic steatohepatitis. Clin. Liver Dis. 2009, 13, 565–580. [Google Scholar] [CrossRef][Green Version]
- Diehl, A.M. Nonalcoholic steatosis and steatohepatitis IV. Nonalcoholic fatty liver disease abnormalities in macrophage function and cytokines. Am. J. Physiol. Gastrointest Liver Physiol. 2002, 282, G1–G5. [Google Scholar] [CrossRef] [PubMed]
- Feldstein, A.E.; Canbay, A.; Angulo, P.; Taniai, M.; Burgart, L.J.; Lindor, K.D.; Gores, G.J. Hepatocyte apoptosis and fas expression are prominent features of human nonalcoholic steatohepatitis. Gastroenterology 2003, 125, 437–443. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Inflammation and metabolic disorders. Nature 2006, 444, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Wullaert, A.; van Loo, G.; Heyninck, K.; Beyaert, R. Hepatic tumor necrosis factor signaling and nuclear factor-kappaB: Effects on liver homeostasis and beyond. Endocr. Rev. 2007, 28, 365–386. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, P.S.; Cortez-Pinto, H.; Solá, S.; Castro, R.E.; Ramalho, R.M.; Baptista, A.; Moura, M.C.; Camilo, M.E.; Rodrigues, C.M. Hepatocyte apoptosis, expression of death receptors, and activation of NF-kappaB in the liver of nonalcoholic and alcoholic steatohepatitis patients. Am. J. Gastroenterol. 2004, 99, 1708–1717. [Google Scholar] [CrossRef] [PubMed]
- Cai, D.; Yuan, M.; Frantz, D.F.; Melendez, P.A.; Hansen, L.; Lee, J.; Shoelson, S.E. Local and systemic insulin resistance resulting from hepatic activation of IKK-beta and NF-kappaB. Nat. Med. 2005, 11, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Tomita, K.; Tamiya, G.; Ando, S.; Ohsumi, K.; Chiyo, T.; Mizutani, A.; Kitamura, N.; Toda, K.; Kaneko, T.; Horie, Y.; et al. Tumour necrosis factor alpha signalling through activation of Kupffer cells plays an essential role in liver fibrosis of non-alcoholic steatohepatitis in mice. Gut 2006, 55, 415–424. [Google Scholar] [CrossRef]
- Fernandez-Real, J.M.; Vayreda, M.; Richart, C.; Gutierrez, C.; Broch, M.; Vendrell, J.; Ricart, W. Circulating interleukin 6 levels, blood pressure, and insulin sensitivity in apparently healthy men and women. J. Clin. Endocrinol. Metab. 2001, 86, 1154–1159. [Google Scholar] [CrossRef]
- Crespo, J.; Cayón, A.; Fernández-Gil, P.; Hernández-Guerra, M.; Mayorga, M.; Domínguez-Díez, A.; Fernández-Escalante, J.C.; Pons-Romero, F. Gene expression of tumor necrosis factor alpha and TNF-receptors, p55 and p75, in nonalcoholic steatohepatitis patients. Hepatology 2001, 34, 1158–1163. [Google Scholar] [CrossRef]
- Romero-Gómez, M.; Zelber-Sagi, S.; Trenell, M. Treatment of NAFLD with diet, physical activity and exercise. J. Hepatol. 2017, 67, 829–846. [Google Scholar] [CrossRef]
- Marchesini, G.; Petta, S.; Dalle Grave, R. Diet, weight loss, and liver health in nonalcoholic fatty liver disease: Pathophysiology, evidence, and practice. Hepatology 2016, 63, 2032–2043. [Google Scholar] [CrossRef]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018, 67, 328–357. [Google Scholar] [CrossRef]
- Vilar-Gomez, E.; Martinez-Perez, Y.; Calzadilla-Bertot, L.; Torres-Gonzalez, A.; Gra-Oramas, B.; Gonzalez-Fabian, L.; Friedman, S.L.; Diago, M.; Romero-Gomez, M. Weight Loss Through Lifestyle Modification Significantly Reduces Features of Nonalcoholic Steatohepatitis. Gastroenterology 2015, 149, 367–378.e5. [Google Scholar] [CrossRef]
- Ahn, J.; Jun, D.W.; Lee, H.Y.; Moon, J.H. Critical appraisal for low-carbohydrate diet in nonalcoholic fatty liver disease: Review and meta-analyses. Clin. Nutr. 2019, 38, 2023–2030. [Google Scholar] [CrossRef]
- Di Francia, R.; Rinaldi, L.; Cillo, M.; Varriale, E.; Facchini, G.; D’Aniello, C.; Marotta, G.; Berretta, M. Antioxidant diet and genotyping as tools for the prevention of liver disease. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 5155–5163. [Google Scholar] [PubMed]
- Anania, C.; Perla, F.M.; Olivero, F.; Pacifico, L.; Chiesa, C. Mediterranean diet and nonalcoholic fatty liver disease. World J. Gastroenterol. 2018, 24, 2083–2094. [Google Scholar] [CrossRef] [PubMed]
- Plaz Torres, M.C.; Aghemo, A.; Lleo, A.; Bodini, G.; Furnari, M.; Marabotto, E.; Miele, L.; Giannini, E.G. Mediterranean Diet and NAFLD: What We Know and Questions That Still Need to Be Answered. Nutrients 2019, 11, 2971. [Google Scholar] [CrossRef] [PubMed]
- Abenavoli, L.; Boccuto, L.; Federico, A.; Dallio, M.; Loguercio, C.; Di Renzo, L.; De Lorenzo, A. Diet and Non-Alcoholic Fatty Liver Disease: The Mediterranean Way. Int. J. Environ. Res. Public Health 2019, 16, 3011. [Google Scholar] [CrossRef] [PubMed]
- Moosavian, S.P.; Arab, A.; Paknahad, Z. The effect of a Mediterranean diet on metabolic parameters in patients with non-alcoholic fatty liver disease: A systematic review of randomized controlled trials. Clin. Nutr. ESPEN 2020, 35, 40–46. [Google Scholar] [CrossRef]
- Berná, G.; Romero-Gomez, M. The role of nutrition in non-alcoholic fatty liver disease: Pathophysiology and management. Liver Int. 2020, 40 (Suppl. 1), 102–108. [Google Scholar] [CrossRef]
- Di Francia, R.; Rinaldi, L.; Troisi, A.; Di Benedetto, F.; Berretta, M. Effect of anti-oxidant agents in patients with hepatocellular diseases. Eur. Rev. Med. Pharmacol. Sci. 2015, 19, 3993–3995. [Google Scholar] [PubMed]
- Tosti, V.; Bertozzi, B.; Fontana, L. Health Benefits of the Mediterranean Diet: Metabolic and Molecular Mechanisms. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2018, 73, 318–326. [Google Scholar] [CrossRef] [PubMed]
- Gelli, C.; Tarocchi, M.; Abenavoli, L.; Di Renzo, L.; Galli, A.; De Lorenzo, A. Effect of a counseling-supported treatment with the Mediterranean diet and physical activity on the severity of the non-alcoholic fatty liver disease. World J. Gastroenterol. 2017, 23, 3150–3162. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, K.M.B.; Ronca, D.B.; Michels, N.; Huybrechts, I.; Cuenca-Garcia, M.; Marcos, A.; Molnár, D.; Dallongeville, J.; Manios, Y.; Schaan, B.D.; et al. Does the Mediterranean Diet Protect against Stress-Induced Inflammatory Activation in European Adolescents? The HELENA Study. Nutrients 2018, 10, 1770. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Lista, J.; Perez-Martinez, P.; Garcia-Rios, A.; Díaz, J.F.A.; Perez-Caballero, A.I.; Delgado-Lista, J.; Fuentes, F.; Quintana-Navarro, G.; Lopez-Segura, F.; Ortiz-Morales, A.M.; et al. CORonary Diet Intervention with Olive oil and cardiovascular PREVention study (the CORDIOPREV study): Rationale, methods, and baseline characteristics: A clinical trial comparing the efficacy of a Mediterranean diet rich in olive oil versus a low-fat diet on cardiovascular disease in coronary patients. Am. Heart J. 2016, 177, 42–50. [Google Scholar] [CrossRef]
- Misciagna, G.; Del Pilar Díaz, M.; Caramia, D.V.; Bonfiglio, C.; Franco, I.; Noviello, M.R.; Chiloiro, M.; Abbrescia, D.I.; Mirizzi, A.; Tanzi, M.; et al. Effect of a Low Glycemic Index Mediterranean Diet on Non-Alcoholic Fatty Liver Disease. A Randomized Controlled Clinici Trial. J. Nutr. Health Aging 2017, 21, 404–412. [Google Scholar] [CrossRef] [PubMed]
- Baratta, F.; Pastori, D.; Polimeni, L.; Bucci, T.; Ceci, F.; Calabrese, C.; Ernesti, I.; Pannitteri, G.; Violi, F.; Angelico, F.; et al. Adherence to Mediterranean Diet and Non-Alcoholic Fatty Liver Disease: Effect on Insulin Resistance. Am. J. Gastroenterol. 2017, 112, 1832–1839. [Google Scholar] [CrossRef] [PubMed]
- Estruch, R.; Ros, E.; Salas-Salvadó, J.; Covas, M.-I.; Corella, D.; Arós, F.; Gómez-Gracia, E.; Ruiz-Gutiérrez, V.; Fiol, M.; Lapetra, J.; et al. Primary Prevention of Cardiovascular Disease with a Mediterranean Diet Supplemented with Extra-Virgin Olive Oil or Nuts. N. Engl. J. Med. 2018, 378, e34. [Google Scholar] [CrossRef] [PubMed]
- Kesse-Guyot, E.; Ahluwalia, N.; Lassale, C.; Hercberg, S.; Fezeu, L.; Lairon, D. Adherence to Mediterranean diet reduces the risk of metabolic syndrome: A 6-year prospective study. Nutr. Metab. Cardiovasc. Dis. 2013, 23, 677–683. [Google Scholar] [CrossRef] [PubMed]
- Uusitupa, M. Lifestyle changes and cardiovascular risk reduction in diabetes. Lancet Diabetes Endocrinol. 2016, 4, 877–878. [Google Scholar] [CrossRef]
- Zhang, H.J.; He, J.; Pan, L.L.; Ma, Z.M.; Han, C.K.; Chen, C.S.; Chen, Z.; Han, H.W.; Chen, S.; Sun, Q.; et al. Effects of Moderate and Vigorous Exercise on Nonalcoholic Fatty Liver Disease: A Randomized Clinical Trial. JAMA Intern. Med. 2016, 176, 1074–1082. [Google Scholar] [CrossRef] [PubMed]
- Ryu, S.; Chang, Y.; Jung, H.S.; Yun, K.E.; Kwon, M.J.; Choi, Y.; Kim, C.W.; Cho, J.; Suh, B.S.; Cho, Y.K.; et al. Relationship of sitting time and physical activity with non-alcoholic fatty liver disease. J. Hepatol. 2015, 63, 1229–1237. [Google Scholar] [CrossRef]
- Hashida, R.; Kawaguchi, T.; Bekki, M.; Omoto, M.; Matsuse, H.; Nago, T.; Takano, Y.; Ueno, T.; Koga, H.; George, J.; et al. Aerobic vs. resistance exercise in non-alcoholic fatty liver disease: A systematic review. J. Hepatol. 2017, 66, 142–152. [Google Scholar] [CrossRef]
- Shojaee-Moradie, F.; Cuthbertson, D.J.; Barrett, M.; Jackson, N.C.; Herring, R.; Thomas, E.L.; Bell, J.; Kemp, G.J.; Wright, J.; Umpleby, A.M. Exercise Training Reduces Liver Fat and Increases Rates of VLDL Clearance But Not VLDL Production in NAFLD. J. Clin. Endocrinol. Metab. 2016, 101, 4219–4228. [Google Scholar] [CrossRef] [PubMed]
- Keating, S.E.; Hackett, D.A.; Parker, H.M.; O’Connor, H.T.; Gerofi, J.A.; Sainsbury, A.; Baker, M.K.; Chuter, V.H.; Caterson, I.D.; George, J.; et al. Effect of aerobic exercise training dose on liver fat and visceral adiposity. J. Hepatol. 2015, 63, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Bullón-Vela, V.; Abete, I.; Tur, J.A.; Pintó, X.; Corbella, E.; Martínez-González, M.A.; Toledo, E.; Corella, D.; Macías, M.; Tinahones, F.; et al. Influence of lifestyle factors and staple foods from the Mediterranean diet on non-alcoholic fatty liver disease among older individuals with metabolic syndrome features. Nutrition 2020, 71, 110620. [Google Scholar] [CrossRef] [PubMed]
- Sookoian, S.; Pirola, C.J. Meta-analysis of the influence of I148M variant of patatin-like phospholipase domain containing 3 gene (PNPLA3) on the susceptibility and histological severity of nonalcoholic fatty liver disease. Hepatology 2011, 53, 1883–1894. [Google Scholar] [CrossRef]
- Hyysalo, J.; Stojkovic, I.; Kotronen, A.; Hakkarainen, A.; Sevastianova, K.; Makkonen, J.; Lundbom, N.; Rissanen, A.; Krauss, R.M.; Melander, O.; et al. Genetic variation in PNPLA3 but not APOC3 influences liver fat in non-alcoholic fatty liver disease. J. Gastroenterol. Hepatol. 2012, 27, 951–956. [Google Scholar] [CrossRef]
- He, S.; McPhaul, C.; Li, J.Z.; Garuti, R.; Kinch, L.; Grishin, N.V.; Cohen, J.C.; Hobbs, H.H. A sequence variation (I148M) in PNPLA3 associated with nonalcoholic fatty liver disease disrupts triglyceride hydrolysis. J. Biol. Chem. 2010, 285, 6706–6715. [Google Scholar] [CrossRef]
- BasuRay, S.; Wang, Y.; Smagris, E.; Cohen, J.C.; Hobbs, H.H. Accumulation of PNPLA3 on lipid droplets is the basis of associated hepatic steatosis. Proc. Natl. Acad. Sci. USA 2019, 116, 9521–9526. [Google Scholar] [CrossRef]
- Pirazzi, C.; Adiels, M.; Burza, M.A.; Mancina, R.M.; Levin, M.; Ståhlman, M.; Taskinen, M.R.; Orho-Melander, M.; Perman, J.; Pujia, A.; et al. Patatin-like phospholipase domain-containing 3 (PNPLA3) I148M (rs738409) affects hepatic VLDL secretion in humans and in vitro. J. Hepatol. 2012, 57, 1276–1282. [Google Scholar] [CrossRef] [PubMed]
- Pingitore, P.; Dongiovanni, P.; Motta, B.M.; Meroni, M.; Lepore, S.M.; Mancina, R.M.; Pelusi, S.; Russo, C.; Caddeo, A.; Rossi, G.; et al. PNPLA3 overexpression results in reduction of proteins predisposing to fibrosis. Hum. Mol. Genet. 2016, 25, 5212–5222. [Google Scholar] [CrossRef]
- Mondul, A.M.; Mancina, R.M.; Merlo, A.; Dongiovanni, P.; Rametta, R.; Montalcini, T.; Valenti, L.; Albanes, D.; Romeo, S. PNPLA3 I148M Variant Influences Circulating Retinol in Adults with Nonalcoholic Fatty Liver Disease or Obesity. J. Nutr. 2015, 145, 1687–1691. [Google Scholar] [CrossRef] [PubMed]
- BasuRay, S.; Smagris, E.; Cohen, J.C.; Hobbs, H.H. The PNPLA3 variant associated with fatty liver disease (I148M) accumulates on lipid droplets by evading ubiquitylation. Hepatology 2017, 66, 1111–1124. [Google Scholar] [CrossRef] [PubMed]
- Donati, B.; Motta, B.M.; Pingitore, P.; Meroni, M.; Pietrelli, A.; Alisi, A.; Petta, S.; Xing, C.; Dongiovanni, P.; del Menico, B.; et al. The rs2294918 E434K variant modulates patatin-like phospholipase domain-containing 3 expression and liver damage. Hepatology 2016, 63, 787–798. [Google Scholar] [CrossRef]
- Dongiovanni, P.; Donati, B.; Fares, R.; Lombardi, R.; Mancina, R.M.; Romeo, S.; Valenti, L. PNPLA3 I148M polymorphism and progressive liver disease. World J. Gastroenterol. 2013, 19, 6969–6978. [Google Scholar] [CrossRef]
- Valenti, L.; Motta, B.M.; Soardo, G.; Iavarone, M.; Donati, B.; SanGiovanni, A.; Carnelutti, A.; Dongiovanni, P.; Rametta, R.; Bertelli, C.; et al. PNPLA3 I148M polymorphism, clinical presentation, and survival in patients with hepatocellular carcinoma. PLoS ONE 2013, 8, e75982. [Google Scholar] [CrossRef]
- Pirazzi, C.; Valenti, L.; Motta, B.M.; Pingitore, P.; Hedfalk, K.; Mancina, R.M.; Burza, M.A.; Indiveri, C.; Ferro, Y.; Montalcini, T.; et al. PNPLA3 has retinyl-palmitate lipase activity in human hepatic stellate cells. Hum. Mol. Genet. 2014, 23, 4077–4085. [Google Scholar] [CrossRef]
- Carim-Todd, L.; Escarceller, M.; Estivill, X.; Sumoy, L. Cloning of the novel gene TM6SF1 reveals conservation of clusters of paralogous genes between human chromosomes 15q24→q26 and 19p13.3→p12. Cytogenet. Cell Genet. 2000, 90, 255–260. [Google Scholar] [CrossRef]
- Mahdessian, H.; Taxiarchis, A.; Popov, S.; Silveira, A.; Franco-Cereceda, A.; Hamsten, A.; Eriksson, P.; Hooft, F.V. TM6SF2 is a regulator of liver fat metabolism influencing triglyceride secretion and hepatic lipid droplet content. Proc. Natl. Acad. Sci. USA 2014, 111, 8913–8918. [Google Scholar] [CrossRef]
- Kozlitina, J.; Smagris, E.; Stender, S.; Nordestgaard, B.G.; Zhou, H.H.; Tybjærg-Hansen, A.; Vogt, T.F.; Hobbs, H.H.; Cohen, J.C. Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2014, 46, 352–356. [Google Scholar] [CrossRef]
- Fan, Y.; Lu, H.; Guo, Y.; Zhu, T.; Garcia-Barrio, M.T.; Jiang, Z.; Willer, C.J.; Zhang, J.; Chen, Y.E. Hepatic Transmembrane 6 Superfamily Member 2 Regulates Cholesterol Metabolism in Mice. Gastroenterology 2016, 150, 1208–1218. [Google Scholar] [CrossRef]
- Smagris, E.; Gilyard, S.; BasuRay, S.; Cohen, J.C.; Hobbs, H.H. Inactivation of Tm6sf2, a Gene Defective in Fatty Liver Disease, Impairs Lipidation but Not Secretion of Very Low Density Lipoproteins. J. Biol. Chem. 2016, 291, 10659–10676. [Google Scholar] [CrossRef] [PubMed]
- Hirata, Y.; Yamamori, N.; Kono, N.; Lee, H.C.; Inoue, T.; Arai, H. Identification of small subunit of serine palmitoyltransferase a as a lysophosphatidylinositol acyltransferase 1-interacting protein. Genes Cells 2013, 18, 397–409. [Google Scholar] [CrossRef] [PubMed]
- Zarini, S.; Hankin, J.A.; Murphy, R.C.; Gijón, M.A. Lysophospholipid acyltransferases and eicosanoid biosynthesis in zebrafish myeloid cells. Prostaglandins Other Lipid Mediat. 2014, 113–115, 52–61. [Google Scholar] [CrossRef][Green Version]
- Tanaka, Y.; Shimanaka, Y.; Caddeo, A.; Kubo, T.; Mao, Y.; Kubota, T.; Kubota, N.; Yamauchi, T.; Mancina, R.M.; Baselli, G.; et al. LPIAT1/MBOAT7 depletion increases triglyceride synthesis fueled by high phosphatidylinositol turnover. Gut 2021, 70, 180–193. [Google Scholar] [CrossRef] [PubMed]
- Helsley, R.N.; Varadharajan, V.; Brown, A.L.; Gromovsky, A.D.; Schugar, R.C.; Ramachandiran, I.; Fung, K.; Kabbany, M.N.; Banerjee, R.; Neumann, C.K.; et al. Obesity-linked suppression of membrane-bound O-acyltransferase 7 (MBOAT7) drives non-alcoholic fatty liver disease. Elife 2019, 8, e49882. [Google Scholar] [CrossRef]
- Wu, Q.; Ortegon, A.M.; Tsang, B.; Doege, H.; Feingold, K.R.; Stahl, A. FATP1 is an insulin-sensitive fatty acid transporter involved in diet-induced obesity. Mol. Cell Biol. 2006, 26, 3455–3467. [Google Scholar] [CrossRef] [PubMed]
- Beer, N.L.; Tribble, N.D.; McCulloch, L.J.; Roos, C.; Johnson, P.R.; Orho-Melander, M.; Gloyn, A.L. The P446L variant in GCKR associated with fasting plasma glucose and triglyceride levels exerts its effect through increased glucokinase activity in liver. Hum. Mol. Genet. 2009, 18, 4081–4088. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.J.; Zhang, H.W.; Zhou, J.Y.; Liu, Y.; Yang, Y.; Chen, X.L.; Zhu, C.H.; Zheng, R.D.; Ling, W.H.; Zhu, H.L. Betaine attenuates hepatic steatosis by reducing methylation of the MTTP promoter and elevating genomic methylation in mice fed a high-fat diet. J. Nutr. Biochem. 2014, 25, 329–336. [Google Scholar] [CrossRef]
- Pooya, S.; Blaise, S.; Moreno Garcia, M.; Giudicelli, J.; Alberto, J.M.; Guéant-Rodriguez, R.M.; Jeannesson, E.; Gueguen, N.; Bressenot, A.; Nicolas, B.; et al. Methyl donor deficiency impairs fatty acid oxidation through PGC-1α hypomethylation and decreased ER-α, ERR-α, and HNF-4α in the rat liver. J. Hepatol. 2012, 57, 344–351. [Google Scholar] [CrossRef]
- Zeybel, M.; Hardy, T.; Robinson, S.M.; Fox, C.; Anstee, Q.M.; Ness, T.; Masson, S.; Mathers, J.C.; French, J.; White, S.; et al. Differential DNA methylation of genes involved in fibrosis progression in non-alcoholic fatty liver disease and alcoholic liver disease. Clin. Epigenetics 2015, 7, 25. [Google Scholar] [CrossRef]
- Cordero, P.; Milagro, F.I.; Campion, J.; Martinez, J.A. Maternal methyl donors supplementation during lactation prevents the hyperhomocysteinemia induced by a high-fat-sucrose intake by dams. Int. J. Mol. Sci. 2013, 14, 24422–24437. [Google Scholar] [CrossRef] [PubMed]
- Whayne, T.F. Epigenetics in the development, modification, and prevention of cardiovascular disease. Mol. Biol. Rep. 2015, 42, 765–776. [Google Scholar] [CrossRef] [PubMed]
- Marfella, R.; Ferraraccio, F.; Rizzo, M.R.; Portoghese, M.; Barbieri, M.; Basilio, C.; Nersita, R.; Siniscalchi, L.I.; Sasso, F.C.; Ambrosino, I.; et al. Innate immune activity in plaque of patients with untreated and L-thyroxine-treated subclinical hypothyroidism. J. Clin. Endocrinol. Metab. 2011, 96, 1015–1020. [Google Scholar] [CrossRef]
- Chang, H.C.; Guarente, L. SIRT1 and other sirtuins in metabolism. Trends Endocrinol. Metab. 2014, 25, 138–145. [Google Scholar] [CrossRef]
- Moschen, A.R.; Wieser, V.; Gerner, R.R.; Bichler, A.; Enrich, B.; Moser, P.; Ebenbichler, C.F.; Kaser, S.; Tilg, H. Adipose tissue and liver expression of SIRT1, 3, and 6 increase after extensive weight loss in morbid obesity. J. Hepatol. 2013, 59, 1315–1322. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Friso, S.; Choi, S.W. Epigenetic mechanisms underlying the link between non-alcoholic fatty liver diseases and nutrition. Nutrients 2014, 6, 3303–3325. [Google Scholar] [CrossRef]
- Tan, J.; McKenzie, C.; Potamitis, M.; Thorburn, A.N.; Mackay, C.R.; Macia, L. The role of short-chain fatty acids in health and disease. Adv. Immunol. 2014, 121, 91–119. [Google Scholar] [CrossRef]
- Cheung, O.; Puri, P.; Eicken, C.; Contos, M.J.; Mirshahi, F.; Maher, J.W.; Kellum, J.M.; Min, H.; Luketic, V.A.; Sanyal, A.J. Nonalcoholic steatohepatitis is associated with altered hepatic MicroRNA expression. Hepatology 2008, 48, 1810–1820. [Google Scholar] [CrossRef]
- Takaki, Y.; Saito, Y.; Takasugi, A.; Toshimitsu, K.; Yamada, S.; Muramatsu, T.; Kimura, M.; Sugiyama, K.; Suzuki, H.; Arai, E.; et al. Silencing of microRNA-122 is an early event during hepatocarcinogenesis from non-alcoholic steatohepatitis. Cancer Sci. 2014, 105, 1254–1260. [Google Scholar] [CrossRef] [PubMed]
- Long, J.K.; Dai, W.; Zheng, Y.W.; Zhao, S.P. miR-122 promotes hepatic lipogenesis via inhibiting the LKB1/AMPK pathway by targeting Sirt1 in non-alcoholic fatty liver disease. Mol. Med. 2019, 25, 26. [Google Scholar] [CrossRef] [PubMed]
- Brea, A.; Puzo, J. Non-alcoholic fatty liver disease and cardiovascular risk. Int. J. Cardiol. 2013, 167, 1109–1117. [Google Scholar] [CrossRef] [PubMed]
- Santoliquido, A.; Di Campli, C.; Miele, L.; Gabrieli, M.L.; Forgione, A.; Zocco, M.A.; Lupascu, A.; Di Giorgio, A.; Flore, R.; Pola, P.; et al. Hepatic steatosis and vascular disease. Eur. Rev. Med. Pharmacol. Sci. 2005, 9, 269–271. [Google Scholar] [PubMed]
- Calabrò, P.; Moscarella, E.; Gragnano, F.; Cesaro, A.; Pafundi, P.C.; Patti, G.; Cavallari, I.; Antonucci, E.; Cirillo, P.; Pignatelli, P.; et al. Effect of Body Mass Index on Ischemic and Bleeding Events in Patients Presenting With Acute Coronary Syndromes (from the START-ANTIPLATELET Registry). Am. J. Cardiol. 2019, 124, 1662–1668. [Google Scholar] [CrossRef]
- Wang, Z.; Xia, B.; Ma, C.; Hu, Z.; Chen, X.; Cao, P. Prevalence and risk factors of fatty liver disease in the Shuiguohu district of Wuhan city, central China. Postgrad. Med. J. 2007, 83, 192–195. [Google Scholar] [CrossRef][Green Version]
- Anderson, T.J. Nitric oxide, atherosclerosis and the clinical relevance of endothelial dysfunction. Heart Fail. Rev. 2003, 8, 71–86. [Google Scholar] [CrossRef]
- Sima, A.V.; Stancu, C.S.; Simionescu, M. Vascular endothelium in atherosclerosis. Cell Tissue Res. 2009, 335, 191–203. [Google Scholar] [CrossRef] [PubMed]
- Lüscher, T.F.; Vanhoutte, P.M. The Endothelium: Modulator of Cardiovascular Function: Modulator of Cardiovascular Function, 1st ed.; CRC Press: Boca Raton, FL, USA, 1990. [Google Scholar] [CrossRef]
- Fracanzani, A.L.; Burdick, L.; Raselli, S.; Pedotti, P.; Grigore, L.; Santorelli, G.; Valenti, L.; Maraschi, A.; Catapano, A.; Fargion, S. Carotid artery intima-media thickness in nonalcoholic fatty liver disease. Am. J. Med. 2008, 121, 72–78. [Google Scholar] [CrossRef]
- Ozturk, K.; Uygun, A.; Guler, A.K.; Demirci, H.; Ozdemir, C.; Cakir, M.; Sakin, Y.S.; Turker, T.; Sari, S.; Demirbas, S.; et al. Nonalcoholic fatty liver disease is an independent risk factor for atherosclerosis in young adult men. Atherosclerosis. 2015, 240, 380–386. [Google Scholar] [CrossRef]
- Long, M.T.; Wang, N.; Larson, M.G.; Mitchell, G.F.; Palmisano, J.; Vasan, R.S.; Hoffmann, U.; Speliotes, E.K.; Vita, J.A.; Benjamin, E.J.; et al. Nonalcoholic fatty liver disease and vascular function: Cross-sectional analysis in the Framingham heart study. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1284–1291. [Google Scholar] [CrossRef] [PubMed]
- Hamburg, N.M.; Keyes, M.J.; Larson, M.G.; Vasan, R.S.; Schnabel, R.; Pryde, M.M.; Mitchell, G.F.; Sheffy, J.; Vita, J.A.; Benjamin, E.J. Cross-sectional relations of digital vascular function to cardiovascular risk factors in the Framingham Heart Study. Circulation 2008, 117, 2467–2474. [Google Scholar] [CrossRef] [PubMed]
- Neri, S.; Bruno, C.M.; Leotta, C.; D’Amico, R.A.; Pennisi, G.; Ierna, D. Early endothelial alterations in non-insulin-dependent diabetes mellitus. Int. J. Clin. Lab. Res. 1998, 28, 100–103. [Google Scholar] [CrossRef] [PubMed]
- Pasarín, M.; La Mura, V.; Gracia-Sancho, J.; García-Calderó, H.; Rodríguez-Vilarrupla, A.; García-Pagán, J.C.; Bosch, J.; Abraldes, J.G. Sinusoidal endothelial dysfunction precedes inflammation and fibrosis in a model of NAFLD. PLoS ONE 2012, 7, e32785. [Google Scholar] [CrossRef] [PubMed]
- Wanless, I.R.; Shiota, K. The pathogenesis of nonalcoholic steatohepatitis and other fatty liver diseases: A four-step model including the role of lipid release and hepatic venular obstruction in the progression to cirrhosis. Semin. Liver Dis. 2004, 24, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Lassalle, P.; Molet, S.; Janin, A.; Van Der Heyden, J.; Tavernier, J.; Fiers, W.; Devos, R.; Tonnel, A.-B. ESM-1 is a novel human endothelial cell-specific molecule expressed in lung and regulated by cytokines. J. Biol. Chem. 1996, 271, 20458–20464. [Google Scholar] [CrossRef] [PubMed]
- Cross, S.; Buhimschi, I.; Duzyj, C.; Shook, L.; McCarthy, M.; Hardy, J.; Zhao, G.; Buhimschi, C. 654: Endocan (ESM-1): A novel soluble endothelial cell injury marker in preeclampsia (PE) and intrauterine growth restriction (IUGR). Am. J. Obstet. Gynecol. 2013, 208, S276. [Google Scholar] [CrossRef]
- Scherpereel, A.; Depontieu, F.; Grigoriu, B.; Cavestri, B.; Tsicopoulos, A.; Gentina, T.; Jourdain, M.; Pugin, J.; Tonnel, A.B.; Lassalle, P. Endocan, a new endothelial marker in human sepsis. Crit. Care Med. 2006, 34, 532–537. [Google Scholar] [CrossRef] [PubMed]
- Biscetti, F.; Straface, G.; De Cristofaro, R.; Lancellotti, S.; Rizzo, P.; Arena, V.; Stigliano, E.; Pecorini, G.; Egashira, K.; De Angelis, G.; et al. High-mobility group box-1 protein promotes angiogenesis after peripheral ischemia in diabetic mice through a VEGF-dependent mechanism. Diabetes 2010, 59, 1496–1505. [Google Scholar] [CrossRef]
- Kumar, R.; Priyadarshi, R.N.; Anand, U. Non-alcoholic Fatty Liver Disease: Growing Burden, Adverse Outcomes and Associations. J. Clin. Transl. Hepatol. 2020, 8, 76–86. [Google Scholar] [CrossRef]
- Rinaldi, L.; Nascimbeni, F.; Giordano, M.; Masetti, C.; Guerrera, B.; Amelia, A.; Fascione, M.C.; Ballestri, S.; Romagnoli, D.; Zampino, R.; et al. Clinical features and natural history of cryptogenic cirrhosis compared to hepatitis C virus-related cirrhosis. World J. Gastroenterol. 2017, 23, 1458–1468. [Google Scholar] [CrossRef]
- Cotter, T.G.; Rinella, M. Nonalcoholic Fatty Liver Disease 2020: The State of the Disease. Gastroenterology 2020, 158, 1851–1864. [Google Scholar] [CrossRef]
- Coppola, N.; De Pascalis, S.; Messina, V.; Di Caprio, G.; Martini, S.; De Stefano, G.; Starace, M.; Stornaiuolo, G.; Stanzione, M.; Ascione, T.; et al. ITPase activity modulates the severity of anaemia in HCV-related cirrhosis treated with ribavirin-containing interferon-free regimens. Antivir. Ther. 2017, 22, 551–558. [Google Scholar] [CrossRef] [PubMed]
- Marrone, A.; Capoluongo, N.; D’Amore, C.; Pisaturo, M.; Esposito, M.; Guastafierro, S.; Siniscalchi, I.; Macera, M.; Boemio, A.; Onorato, L.; et al. Eighteen-month lamivudine prophylaxis on preventing occult hepatitis B virus infection reactivation in patients with haematological malignancies receiving immunosuppression therapy. J. Viral. Hepat. 2018, 25, 198–204. [Google Scholar] [CrossRef] [PubMed]
- Adinolfi, L.E.; Rinaldi, L.; Marrone, A.; Giordano, M. The effect of sustained virological response by direct-acting antivirals on insulin resistance and diabetes mellitus in patients with chronic hepatitis C. Expert Rev. Anti Infect. Ther. 2018, 16, 595–597. [Google Scholar] [CrossRef] [PubMed]
- Durante-Mangoni, E.; Parrella, A.; Vitrone, M.; Rago, A.; Pafundi, P.C.; Nigro, G.; Utili, R.; Russo, V. Electrophysiological Adverse Effects of Direct Acting Antivirals in Patients With Chronic Hepatitis C. J. Clin. Pharmacol. 2017, 57, 924–930. [Google Scholar] [CrossRef] [PubMed]
- Loomba, R.; Lim, J.K.; Patton, H.; El-Serag, H.B. AGA Clinical Practice Update on Screening and Surveillance for Hepatocellular Carcinoma in Patients With Nonalcoholic Fatty Liver Disease: Expert Review. Gastroenterology 2020, 158, 1822–1830. [Google Scholar] [CrossRef] [PubMed]
- Lenti, M.V.; Pasina, L.; Cococcia, S.; Cortesi, L.; Miceli, E.; Dominioni, C.C.; Pisati, M.; Mengoli, C.; Perticone, F.; Nobili, A.; et al. Mortality rate and risk factors for gastrointestinal bleeding in elderly patients. Eur. J. Intern. Med. 2019, 61, 54–61. [Google Scholar] [CrossRef]
- Zampino, R.; Lebano, R.; Coppola, N.; Macera, M.; Grandone, A.; Rinaldi, L.; De Sio, I.; Tufano, A.; Stornaiuolo, G.; Adinolfi, L.E.; et al. The use of nonselective beta blockers is a risk factor for portal vein thrombosis in cirrhotic patients. Saudi J. Gastroenterol. 2018, 24, 25–29. [Google Scholar] [CrossRef]
- Majumdar, A.; Tsochatzis, E.A. Changing trends of liver transplantation and mortality from non-alcoholic fatty liver disease. Metabolism 2020, 111, 154291. [Google Scholar] [CrossRef]
- Valente, G.; Rinaldi, L.; Sgambato, M.; Piai, G. Conversion from twice-daily to once-daily tacrolimus in stable liver transplant patients: Effectiveness in a real-world setting. Transp. Proc. 2013, 45, 1273–1275. [Google Scholar] [CrossRef] [PubMed]
- Loomba, R.; Wong, R.; Fraysse, J.; Shreay, S.; Li, S.; Harrison, S.; Gordon, S.C. Nonalcoholic fatty liver disease progression rates to cirrhosis and progression of cirrhosis to decompensation and mortality: A real world analysis of Medicare data. Aliment. Pharmacol. Ther. 2020, 51, 1149–1159. [Google Scholar] [CrossRef] [PubMed]
- Rinaldi, L.; Valente, G.; Piai, G. Serial Liver Stiffness Measurements and Monitoring of Liver-Transplanted Patients in a Real-Life Clinical Practice. Hepat. Mon. 2016, 16, e41162. [Google Scholar] [CrossRef]
- Augustin, S.; Ahmed, A.; Alkhouri, N.; Canbay, A.; Corless, L.; Nakajima, A.; Okanoue, T.; Petta, S.; Ratziu, V.; Tsochatzis, E.A.; et al. Identification of Patients with Advanced Fibrosis Due to Nonalcoholic Fatty Liver Disease: Considerations for Best Practice. J. Gastrointest. Liver Dis. 2020, 29, 235–245. [Google Scholar] [CrossRef]
- Sardu, C.; Paolisso, P.; Sacra, C.; Mauro, C.; Minicucci, F.; Portoghese, M.; Rizzo, M.R.; Barbieri, M.; Sasso, F.C.; D’Onofrio, N.; et al. Effects of Metformin Therapy on Coronary Endothelial Dysfunction in Patients with Prediabetes with Stable Angina and Nonobstructive Coronary Artery Stenosis: The CODYCE Multicenter Prospective Study. Diabetes Care 2019, 42, 1946–1955. [Google Scholar] [CrossRef]
- Torella, D.; Ellison, G.M.; Torella, M.; Vicinanza, C.; Aquila, I.; Iaconetti, C.; Scalise, M.; Marino, F.; Henning, B.J.; Lewis, F.C.; et al. Carbonic anhydrase activation is associated with worsened pathological remodeling in human ischemic diabetic cardiomyopathy. J. Am. Heart Assoc. 2014, 3, e000434. [Google Scholar] [CrossRef]
- Della Corte, C.M.; Ciaramella, V.; Di Mauro, C.; Castellone, M.D.; Papaccio, F.; Fasano, M.; Sasso, F.C.; Martinelli, E.; Troiani, T.; De Vita, F.; et al. Metformin increases antitumor activity of MEK inhibitors through GLI1 downregulation in LKB1 positive human NSCLC cancer cells. Oncotarget 2015, 7, 4265–4278. [Google Scholar] [CrossRef] [PubMed]
- Sasso, F.C.; Salvatore, T.; Tranchino, G.; Cozzolino, D.; Caruso, A.A.; Persico, M.; Gentile, S.; Torella, D.; Torella, R. Cochlear dysfunction in type 2 diabetes: A complication independent of neuropathy and acute hyperglycemia. Metabolism 1999, 48, 1346–1350. [Google Scholar] [CrossRef]
- Mangge, H.; Almer, G. Immune-Mediated Inflammation in Vulnerable Atherosclerotic Plaques. Molecules 2019, 24, 3072. [Google Scholar] [CrossRef]
- Marfella, R.; Sasso, F.C.; Siniscalchi, M.; Paolisso, P.; Rizzo, M.R.; Ferraro, F.; Stabile, E.; Sorropago, G.; Calabrò, P.; Carbonara, O.; et al. Peri-procedural tight glycemic control during early percutaneous coronary intervention is associated with a lower rate of in-stent restenosis in patients with acute ST-elevation myocardial infarction. J. Clin. Endocrinol. Metab. 2012, 97, 2862–2871. [Google Scholar] [CrossRef]
- Marfella, R.; D’ Amico, M.; Di Filippo, C.; Siniscalchi, M.; Sasso, F.C.; Ferraraccio, F.; Rossi, F.; Paolisso, G. The possible role of the ubiquitin proteasome system in the development of atherosclerosis in diabetes. Cardiovasc. Diabetol. 2007, 6, 35. [Google Scholar] [CrossRef]
- Cozzolino, D.; Sessa, G.; Salvatore, T.; Sasso, F.C.; Giugliano, D.; Lefebvre, P.J.; Torella, R. The involvement of the opioid system in human obesity: A study in normal weight relatives of obese people. J. Clin. Endocrinol. Metab. 1996, 81, 713–718. [Google Scholar] [CrossRef][Green Version]
- Hsu, P.F.; Wang, Y.W.; Lin, C.C.; Wang, Y.J.; Ding, Y.Z.; Liou, T.L.; Huang, S.S.; Lu, T.M.; Chan, W.L.; Lin, S.J.; et al. The association of the steatosis severity in fatty liver disease with coronary plaque pattern in general population. Liver Int. 2021, 41, 81–90. [Google Scholar] [CrossRef]
- Lee, H.; Lee, Y.H.; Kim, S.U.; Chang Kim, H. Metabolic Dysfunction-Associated Fatty Liver Disease and Incident Cardiovascular Disease Risk: A Nationwide Cohort Study. Clin. Gastroenterol. Hepatol. 2020, 22. [Google Scholar] [CrossRef]
- Ismaiel, A.; Popa, S.L.; Dumitrascu, D.L. Acute Coronary Syndromes and Nonalcoholic Fatty Liver Disease: “Un Affaire de Coeur”. Can. J. Gastroenterol. Hepatol. 2020, 2020, 8825615. [Google Scholar] [CrossRef] [PubMed]
- Targher, G.; Byrne, C.D.; Lonardo, A.; Zoppini, G.; Barbui, C. Non-alcoholic fatty liver disease and risk of incident cardiovascular disease: A meta-analysis. J. Hepatol. 2016, 65, 589–600. [Google Scholar] [CrossRef] [PubMed]
- Motamed, N.; Rabiee, B.; Poustchi, H.; Dehestani, B.; Hemasi, G.R.; Khonsari, M.R.; Maadi, M.; Saeedian, F.S.; Zamani, F. Non-alcoholic fatty liver disease (NAFLD) and 10-year risk of cardiovascular diseases. Clin. Res. Hepatol. Gastroenterol. 2017, 41, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Ekstedt, M.; Hagström, H.; Nasr, P.; Fredrikson, M.; Stål, P.; Kechagias, S.; Hultcrantz, R. Fibrosis stage is the strongest predictor for disease-specific mortality in NAFLD after up to 33 years of follow-up. Hepatology 2015, 61, 1547–1554. [Google Scholar] [CrossRef]
- Valbusa, F.; Agnoletti, D.; Scala, L.; Grillo, C.; Arduini, P.; Bonapace, S.; Calabria, S.; Scaturro, G.; Mantovani, A.; Zoppini, G.; et al. Non-alcoholic fatty liver disease and increased risk of all-cause mortality in elderly patients admitted for acute heart failure. Int. J. Cardiol. 2018, 265, 162–168. [Google Scholar] [CrossRef]
- Lombardi, R.; Airaghi, L.; Targher, G.; Serviddio, G.; Maffi, G.; Mantovani, A.; Maffeis, C.; Colecchia, A.; Villani, R.; Rinaldi, L.; et al. NAFLD fibrosis score (NFS) can be used in outpatient services to identify chronic vascular complications besides advanced liver fibrosis in type 2 diabetes. J. Diabetes Complicat. 2020, 34, 107684. [Google Scholar] [CrossRef]
- Lombardi, R.; Airaghi, L.; Targher, G.; Serviddio, G.; Maffi, G.; Mantovani, A.; Maffeis, C.; Colecchia, A.; Villani, R.; Rinaldi, L.; et al. Liver fibrosis by FibroScan® independently of established cardiovascular risk parameters associates with macrovascular and microvascular complications in patients with type 2 diabetes. Liver Int. 2020, 40, 347–354. [Google Scholar] [CrossRef]
- Bae, Y.S.; Ko, Y.S.; Yun, J.M.; Eo, A.Y.; Kim, H. Association and Prediction of Subclinical Atherosclerosis by Nonalcoholic Fatty Liver Disease in Asymptomatic Patients. Can. J. Gastroenterol. Hepatol. 2020, 2020, 8820445. [Google Scholar] [CrossRef]
- Oni, E.; Budoff, M.J.; Zeb, I.; Li, D.; Veledar, E.; Polak, J.F.; Blankstein, R.; Wong, N.D.; Blaha, M.J.; Agatston, A.; et al. Nonalcoholic Fatty Liver Disease Is Associated With Arterial Distensibility and Carotid Intima-Media Thickness: (from the Multi-Ethnic Study of Atherosclerosis). Am. J. Cardiol. 2019, 124, 534–538. [Google Scholar] [CrossRef]
- Xin, Z.; Zhu, Y.; Wang, S.; Liu, S.; Xu, M.; Wang, T.; Lu, J.; Chen, Y.; Zhao, Z.; Wang, W.; et al. Associations of subclinical atherosclerosis with nonalcoholic fatty liver disease and fibrosis assessed by non-invasive score. Liver Int. 2020, 40, 806–814. [Google Scholar] [CrossRef]
- Alexander, M.; Loomis, A.K.; van der Lei, J.; Duarte-Salles, T.; Prieto-Alhambra, D.; Ansell, D.; Pasqua, A.; Lapi, F.; Rijnbeek, P.; Mosseveld, M.; et al. Non-alcoholic fatty liver disease and risk of incident acute myocardial infarction and stroke: Findings from matched cohort study of 18 million European adults. BMJ 2019, 367, l5367. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.J.; Liu, W.J. Association between non-alcoholic fatty liver and acute cerebral infarction: A protocol of systematic review and meta-analysis. Medicine 2020, 99, e20351. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Dai, L.; Zhang, Y.; Wang, A.; Li, H.; Wang, Y.; Meng, X.; Wu, S.; Wang, Y. Severity of Nonalcoholic Fatty Liver Disease and Risk of Future Ischemic Stroke Events. Stroke 2021, 52, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Webster, A.C.; Nagler, E.V.; Morton, R.L.; Masson, P. Chronic Kidney Disease. Lancet 2017, 389, 1238–1252. [Google Scholar] [CrossRef]
- Giordano, M.; Ciarambino, T.; Castellino, P.; Malatino, L.; Cataliotti, A.; Rinaldi, L.; Paolisso, G.; Adinolfi, L.E. Seasonal variations of hyponatremia in the emergency department: Age-related changes. Am. J. Emerg. Med. 2017, 35, 749–752. [Google Scholar] [CrossRef] [PubMed]
- Esposito, G.; Testa, F.; Zacchia, M.; Crispo, A.A.; Di Iorio, V.; Capolongo, G.; Rinaldi, L.; D’Antonio, M.; Fioretti, T.; Iadicicco, P.; et al. Genetic characterization of Italian patients with Bardet-Biedl syndrome and correlation to ocular, renal and audio-vestibular phenotype: Identification of eleven novel pathogenic sequence variants. BMC Med. Genet. 2017, 18, 10. [Google Scholar] [CrossRef]
- Mantovani, A.; Zusi, C.; Dalbeni, A.; Grani, G.; Buzzetti, E. Risk of Kidney Dysfunction IN Nafld. Curr. Pharm. Des. 2020, 26, 1045–1061. [Google Scholar] [CrossRef] [PubMed]
- Minutolo, R.; Gabbai, F.B.; Provenzano, M.; Chiodini, P.; Borrelli, S.; Garofalo, C.; Sasso, F.C.; Santoro, D.; Bellizzi, V.; Conte, G.; et al. Cardiorenal prognosis by residual proteinuria level in diabetic chronic kidney disease: Pooled analysis of four cohort studies. Nephrol. Dial. Transplant. 2018, 33, 1942–1949. [Google Scholar] [CrossRef] [PubMed]
- Minutolo, R.; Sasso, F.C.; Chiodini, P.; Cianciaruso, B.; Carbonara, O.; Zamboli, P.; Tirino, G.; Pota, A.; Torella, R.; Conte, G.; et al. Management of cardiovascular risk factors in advanced type 2 diabetic nephropathy: A comparative analysis in nephrology, diabetology and primary care settings. J. Hypertens. 2006, 24, 1655–1661. [Google Scholar] [CrossRef]
- Sasso, F.C.; Pafundi, P.C.; Gelso, A.; Bono, V.; Costagliola, C.; Marfella, R.; Sardu, C.; Rinaldi, L.; Galiero, R.; Acierno, C.; et al. Relationship between albuminuric CKD and diabetic retinopathy in a real-world setting of type 2 diabetes: Findings from No blind study. Nutr. Metab. Cardiovasc. Dis. 2019, 29, 923–930. [Google Scholar] [CrossRef]
- Kaps, L.; Labenz, C.; Galle, P.R.; Weinmann-Menke, J.; Kostev, K.; Schattenberg, J.M. Non-alcoholic fatty liver disease increases the risk of incident chronic kidney disease. United Eur. Gastroenterol. J. 2020, 8, 942–948. [Google Scholar] [CrossRef]
- Sun, D.-Q.; Jin, Y.; Wang, T.-Y.; Zheng, K.I.; Rios, R.S.; Zhang, H.-Y.; Targher, G.; Byrne, C.D.; Yuan, W.-J.; Zheng, M.-H. MAFLD and risk of CKD. Metabolism 2021, 115, 154433. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Petracca, G.; Beatrice, G.; Csermely, A.; Lonardo, A.; Schattenberg, J.M.; Tilg, H.; Byrne, C.D.; Targher, G. Non-alcoholic fatty liver disease and risk of incident chronic kidney disease: An updated meta-analysis. Gut 2020. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rinaldi, L.; Pafundi, P.C.; Galiero, R.; Caturano, A.; Morone, M.V.; Silvestri, C.; Giordano, M.; Salvatore, T.; Sasso, F.C. Mechanisms of Non-Alcoholic Fatty Liver Disease in the Metabolic Syndrome. A Narrative Review. Antioxidants 2021, 10, 270. https://doi.org/10.3390/antiox10020270
Rinaldi L, Pafundi PC, Galiero R, Caturano A, Morone MV, Silvestri C, Giordano M, Salvatore T, Sasso FC. Mechanisms of Non-Alcoholic Fatty Liver Disease in the Metabolic Syndrome. A Narrative Review. Antioxidants. 2021; 10(2):270. https://doi.org/10.3390/antiox10020270
Chicago/Turabian StyleRinaldi, Luca, Pia Clara Pafundi, Raffaele Galiero, Alfredo Caturano, Maria Vittoria Morone, Chiara Silvestri, Mauro Giordano, Teresa Salvatore, and Ferdinando Carlo Sasso. 2021. "Mechanisms of Non-Alcoholic Fatty Liver Disease in the Metabolic Syndrome. A Narrative Review" Antioxidants 10, no. 2: 270. https://doi.org/10.3390/antiox10020270
APA StyleRinaldi, L., Pafundi, P. C., Galiero, R., Caturano, A., Morone, M. V., Silvestri, C., Giordano, M., Salvatore, T., & Sasso, F. C. (2021). Mechanisms of Non-Alcoholic Fatty Liver Disease in the Metabolic Syndrome. A Narrative Review. Antioxidants, 10(2), 270. https://doi.org/10.3390/antiox10020270