Maturational Changes in Prefrontal and Amygdala Circuits in Adolescence: Implications for Understanding Fear Inhibition during a Vulnerable Period of Development


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
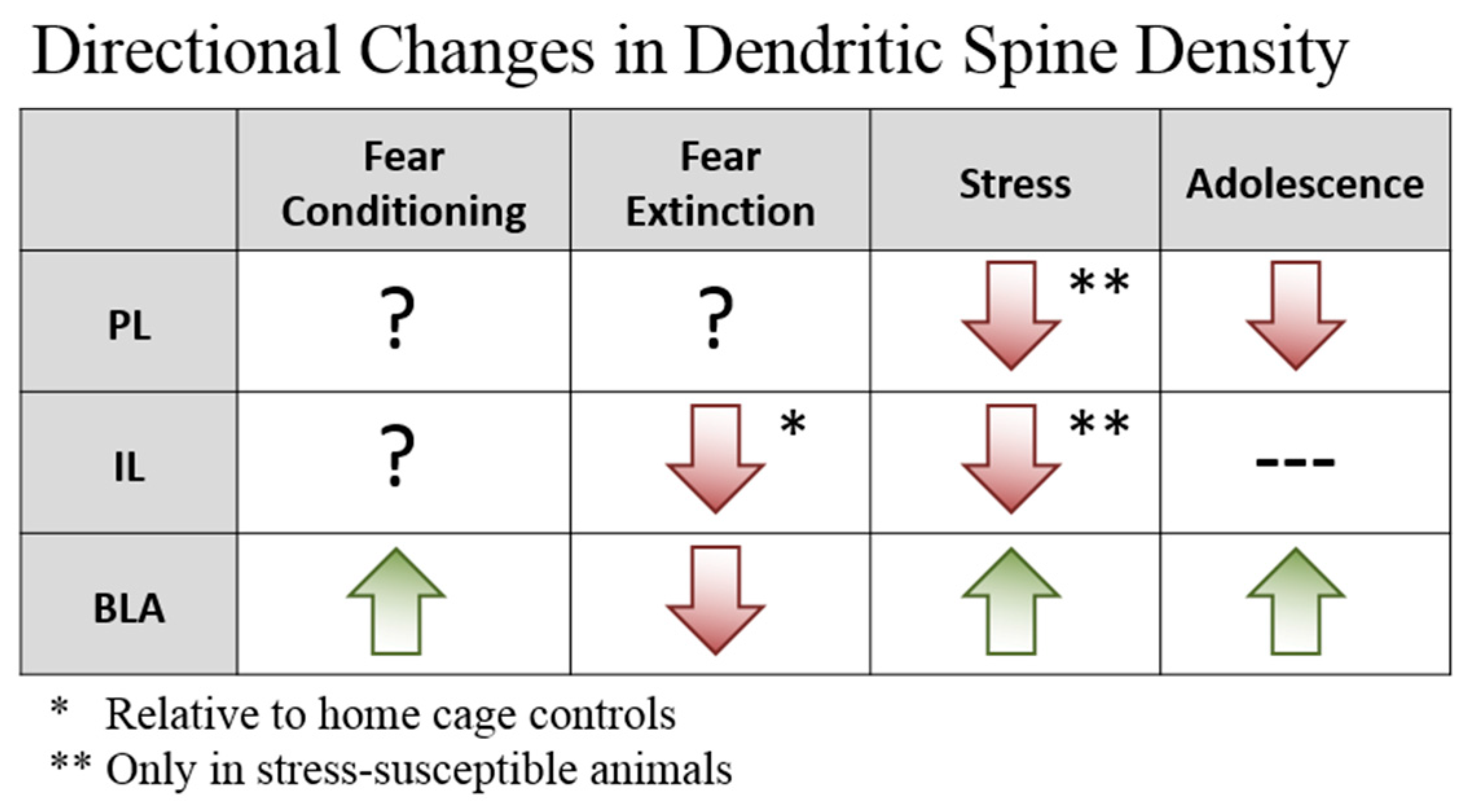
2. Plasticity—Dendritic Spines
2.1. Prefrontal Dendritic Spines
2.2. BLA Dendritic Spines
2.3. Implications for Fear Learning and Inhibition
3. Plasticity—Learning-Dependent Changes in Neural Activity and Excitatory Transmission
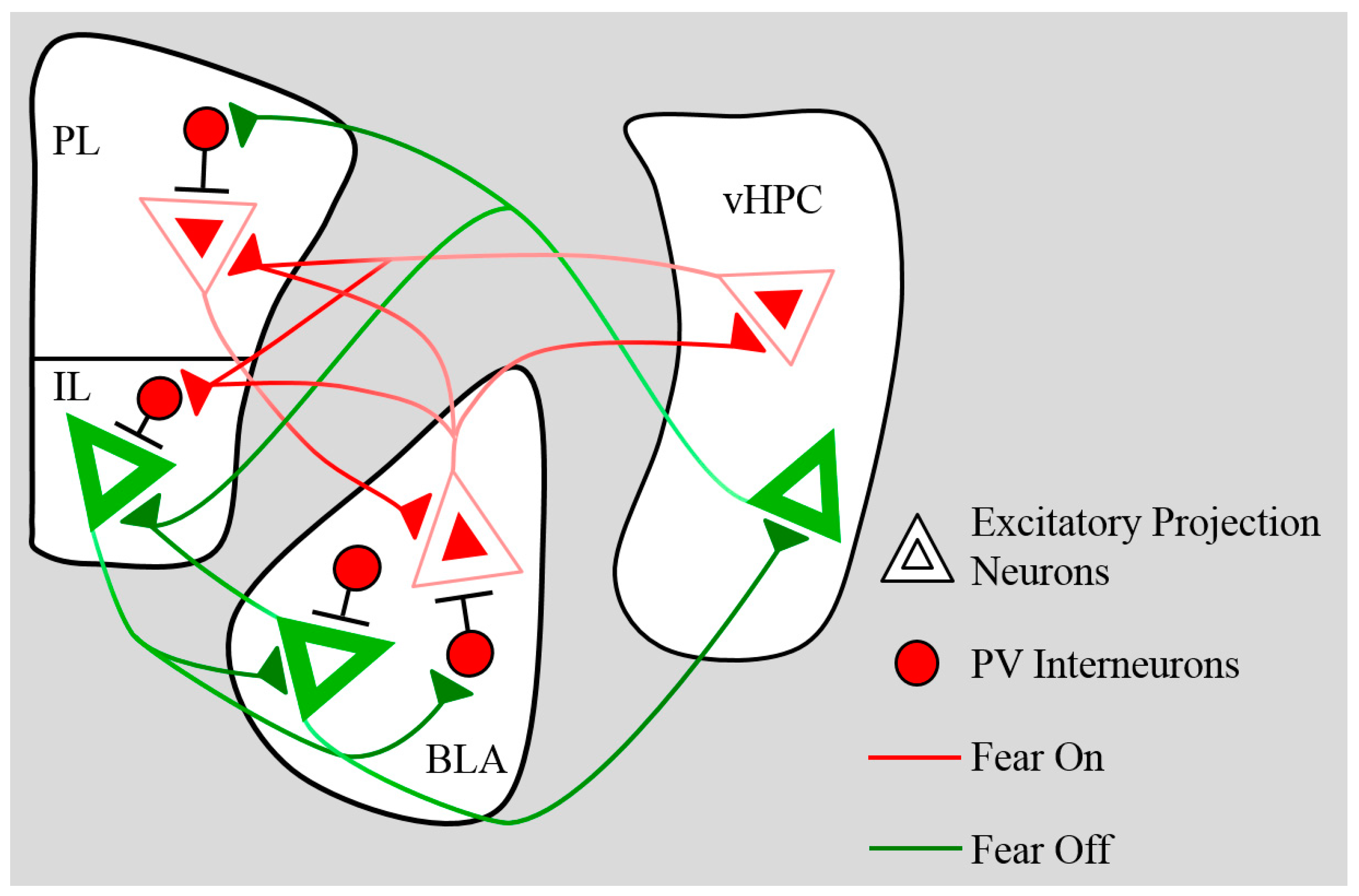
4. Development of Inhibitory Networks
4.1. Prefrontal Inhibition
4.2. BLA Inhibition
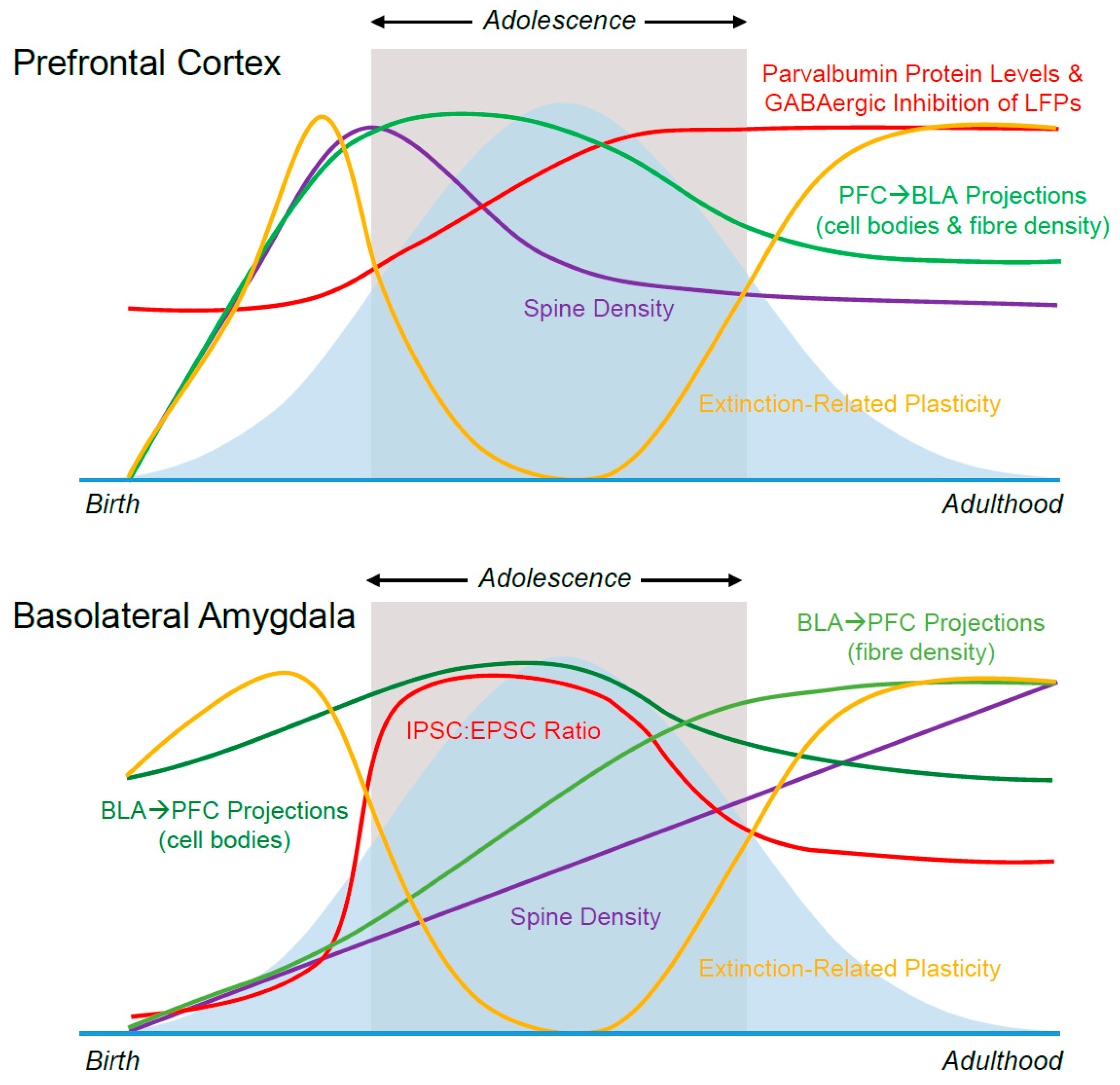
5. Connectivity
5.1. BLA→mPFC
5.1.1. Anatomical
5.1.2. Functional
5.2. mPFC→BLA
5.2.1. Anatomical
5.2.2. Functional
5.3. Ventral Hippocampus→mPFC
6. Disruption by Chronic Stress
7. Conclusions
Funding
Conflicts of Interest
Abbreviations
| BDNF | Brain-Derived Neurotrophic Factor |
| CS | Conditioned stimulus |
| (m)PFC | (medial) Prefrontal Cortex |
| IL | Infralimbic |
| PL | Prelimbic |
| OFC | Orbitofrontal Cortex |
| dlPFC | Dorsolateral Prefrontal Cortex |
| BLA | Basolateral Nucleus of the Amygdala |
| vHPC | Ventral Hippocampus |
| PV | Parvalbumin |
| LTP | Long-Term Potentiation |
| LTD | Long-Term Depression |
| (s)EPSC | (spontaneous) Excitatory Postsynaptic Current |
| (s)IPSC | (spontaneous) Inhibitory Postsynaptic Current |
| AMPA | Alpha-Amino-3-Hydroxy-5-Methyl-4-Isoxazolepropionic Acid |
| NMDA | N-Methyl-D-aspartatic acid |
| DCS | D-Cycloserine |
| GABA | Gamma-Aminobutyric Acid |
| P | Postnatal Day |
| PNN | Perineuronal Nets |
| UCS | Unconditioned stimulus |
Appendix A. Guide to Rodent Development in (Approximate) Postnatal Days
References
- Malter Cohen, M.; Tottenham, N.; Casey, B.J. Translational developmental studies of stress on brain and behavior: Implications for adolescent mental health and illness? Neuroscience 2013, 249, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Tottenham, N.; Galvan, A. Stress and the adolescent brain: Amygdala-prefrontal cortex circuitry and ventral striatum as developmental targets. Neurosci. Biobehav. Rev. 2016, 70, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Casey, B.J.; Duhoux, S.; Malter Cohen, M. Adolescence: What Do Transmission, Transition, and Translation Have to Do with It? Neuron 2010, 67, 749–760. [Google Scholar] [CrossRef]
- Lee, F.S.; Heimer, H.; Giedd, J.N.; Lein, E.S.; Šestan, N.; Weinberger, D.R.; Casey, B.J. Adolescent mental health-Opportunity and obligation. Science 2014, 346, 547–549. [Google Scholar] [CrossRef]
- DiMauro, J.; Domingues, J.; Fernandez, G.; Tolin, D.F. Long-term effectiveness of CBT for anxiety disorders in an adult outpatient clinic sample: A follow-up study. Behav. Res. Ther. 2013, 51, 82–86. [Google Scholar] [CrossRef]
- Ginsburg, G.S.; Becker, E.M.; Keeton, C.P.; Sakolsky, D.; Piacentini, J.; Albano, A.M.; Compton, S.N.; Iyengar, S.; Sullivan, K.; Caporino, N.; et al. Naturalistic follow-up of youths treated for pediatric anxiety disorders. JAMA Psychiatry 2014, 71, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Craske, M.G.; Stein, M.B. Anxiety. Lancet 2016, 388, 3048–3059. [Google Scholar] [CrossRef]
- Casey, B.J.; Glatt, C.E.; Lee, F.S. Treating the developing versus developed brain: Translating preclinical mouse and human studies. Neuron 2015, 86, 1358–1368. [Google Scholar] [CrossRef]
- Hefner, K.; Holmes, A. Ontogeny of fear-, anxiety- and depression-related behavior across adolescence in C57BL/6J mice. Behav. Brain Res. 2007, 176, 210–215. [Google Scholar] [CrossRef]
- Pattwell, S.S.; Duhoux, S.; Hartley, C.A.; Johnson, D.C.; Jing, D.; Elliott, M.D.; Ruberry, E.J.; Powers, A.; Mehta, N.; Yang, R.R.; et al. Altered fear learning across development in both mouse and human. Proc. Natl. Acad. Sci. USA 2012, 109, 16318–16323. [Google Scholar] [CrossRef]
- McCallum, J.; Kim, J.H.; Richardson, R. Impaired extinction retention in adolescent rats: Effects of D-cycloserine. Neuropsychopharmacology 2010, 35, 2134–2142. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Li, S.; Richardson, R. Immunohistochemical analyses of long-term extinction of conditioned fear in adolescent rats. Cereb. Cortex 2011, 21, 530–538. [Google Scholar] [CrossRef] [PubMed]
- Baker, K.D.; Richardson, R. Forming competing fear learning and extinction memories in adolescence makes fear difficult to inhibit. Learn. Mem. 2015, 22, 537–543. [Google Scholar] [CrossRef]
- Sevenster, D.; Visser, R.M.; D’Hooge, R. A translational perspective on neural circuits of fear extinction: Current promises and challenges. Neurobiol. Learn. Mem. 2018, 155, 113–126. [Google Scholar] [CrossRef] [PubMed]
- Orsini, C.A.; Maren, S. Neural and cellular mechanisms of fear and extinction memory formation. Neurosci. Biobehav. Rev. 2012, 36, 1773–1802. [Google Scholar] [CrossRef] [PubMed]
- Maren, S. Out with the old and in with the new: Synaptic mechanisms of extinction in the amygdala. Brain Res. 2015, 1621, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Marek, R.; Sun, Y.; Sah, P. Neural circuits for a top-down control of fear and extinction. Psychopharmacology 2018. [Google Scholar] [CrossRef] [PubMed]
- Milad, M.R.; Quirk, G.J. Fear extinction as a model for translational neuroscience: Ten years of progress. Annu. Rev. Psychol. 2012, 63, 129–151. [Google Scholar] [CrossRef]
- Arruda-Carvalho, M.; Clem, R.L. Prefrontal-amygdala fear networks come into focus. Front. Syst. Neurosci. 2015, 9, 145. [Google Scholar] [CrossRef]
- Krabbe, S.; Grundemann, J.; Luthi, A. Amygdala Inhibitory Circuits Regulate Associative Fear Conditioning. Biol. Psychiatry 2017. [Google Scholar] [CrossRef] [PubMed]
- Caballero, A.; Granberg, R.; Tseng, K.Y. Mechanisms contributing to prefrontal cortex maturation during adolescence. Neurosci. Biobehav. Rev. 2016, 70, 4–12. [Google Scholar] [CrossRef]
- Jalbrzikowski, M.; Larsen, B.; Hallquist, M.N.; Foran, W.; Calabro, F.; Luna, B. Development of White Matter Microstructure and Intrinsic Functional Connectivity Between the Amygdala and Ventromedial Prefrontal Cortex: Associations With Anxiety and Depression. Biol. Psychiatry 2017, 82, 511–521. [Google Scholar] [CrossRef] [PubMed]
- Koss, W.A.; Belden, C.E.; Hristov, A.D.; Juraska, J.M. Dendritic remodeling in the adolescent medial prefrontal cortex and the basolateral amygdala of male and female rats. Synapse 2014, 68, 61–72. [Google Scholar] [CrossRef]
- Oberman, L.; Pascual-Leone, A. Changes in plasticity across the lifespan: Cause of disease and target for intervention. Prog. Brain Res. 2013, 207, 91–120. [Google Scholar] [CrossRef]
- Gogtay, N.; Giedd, J.N.; Lusk, L.; Hayashi, K.M.; Greenstein, D.; Vaituzis, A.C.; Nugent, T.F.; Herman, D.H.; Clasen, L.S.; Toga, A.W.; et al. Dynamic mapping of human cortical development during childhood through early adulthood. Proc. Natl. Acad. Sci. USA 2004, 101, 8174–8179. [Google Scholar] [CrossRef]
- Giedd, J.N.; Raznahan, A.; Alexander-Bloch, A.; Schmitt, E.; Gogtay, N.; Rapoport, J.L. Child Psychiatry Branch of the National Institute of Mental Health Longitudinal Structural Magnetic Resonance Imaging Study of Human Brain Development. Neuropsychopharmacology 2015, 40, 43–49. [Google Scholar] [CrossRef]
- Alvarez, V.A.; Sabatini, B.L. Anatomical and physiological plasticity of dendritic spines. Annu. Rev. Neurosci. 2007, 30, 79–97. [Google Scholar] [CrossRef] [PubMed]
- Segal, M. Dendritic spines: Morphological building blocks of memory. Neurobiol. Learn. Mem. 2017, 138, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Moyer, C.E.; Zuo, Y. Cortical dendritic spine development and plasticity: Insights from in vivo imaging. Curr. Opin. Neurobiol. 2018, 53, 76–82. [Google Scholar] [CrossRef]
- Spear, L.P. The adolescent brain and age-related behavioral manifestations. Neurosci. Biobehav. Rev. 2000, 24, 417–463. [Google Scholar] [CrossRef]
- Huttenlocher, P.R. Synaptic density in human frontal cortex — Developmental changes and effects of aging. Brain Res. 1979, 163, 195–205. [Google Scholar] [CrossRef]
- Petanjek, Z.; Judaš, M.; Šimić, G.; Rašin, M.R.; Uylings, H.B.M.; Rakic, P.; Kostović, I. Extraordinary neoteny of synaptic spines in the human prefrontal cortex. Proc. Natl. Acad. Sci. USA 2011, 108, 13281–13286. [Google Scholar] [CrossRef]
- Bourgeois, J.-P.; Goldman-Rakic, P.S.; Rakic, P. Synaptogenesis in the Prefrontal Cortex of Rhesus Monkeys. Cereb. Cortex 1994, 4, 78–96. [Google Scholar] [CrossRef]
- Rakic, P.; Bourgeois, J.-P.; Goldman-Rakic, P.S. Synaptic development of the cerebral cortex: Implications for learning, memory, and mental illness. In Progress in Brain Research; Van Pelt, J., Corner, M.A., Uylings, H.B.M., Lopes Da Silva, F.H., Eds.; Elsevier: Amsterdam, the Netherlands, 1994; Volume 102, pp. 227–243. [Google Scholar]
- Anderson, S.A.; Classey, J.D.; Condé, F.; Lund, J.S.; Lewis, D.A. Synchronous development of pyramidal neuron dendritic spines and parvalbumin-immunoreactive chandelier neuron axon terminals in layer III of monkey prefrontal cortex. Neuroscience 1995, 67, 7–22. [Google Scholar] [CrossRef]
- Pattwell, S.S.; Liston, C.; Jing, D.; Ninan, I.; Yang, R.R.; Witztum, J.; Murdock, M.H.; Dincheva, I.; Bath, K.G.; Casey, B.J.; et al. Dynamic changes in neural circuitry during adolescence are associated with persistent attenuation of fear memories. Nat. Commun. 2016, 7, 11475. [Google Scholar] [CrossRef]
- Gourley, S.L.; Olevska, A.; Warren, M.S.; Taylor, J.R.; Koleske, A.J. Arg Kinase Regulates Prefrontal Dendritic Spine Refinement and Cocaine-Induced Plasticity. J. Neurosci. 2012, 32, 2314–2323. [Google Scholar] [CrossRef] [PubMed]
- DePoy, L.M.; Noble, B.; Allen, A.G.; Gourley, S.L. Developmentally divergent effects of Rho-kinase inhibition on cocaine- and BDNF-induced behavioral plasticity. Behav. Brain Res. 2013, 243, 171–175. [Google Scholar] [CrossRef]
- Kraszpulski, M.; Dickerson, P.A.; Salm, A.K. Prenatal stress affects the developmental trajectory of the rat amygdala. Stress 2006, 9, 85–95. [Google Scholar] [CrossRef]
- Rubinow, M.J.; Juraska, J.M. Neuron and glia numbers in the basolateral nucleus of the amygdala from preweaning through old age in male and female rats: A stereological study. J. Comp. Neurol. 2009, 512, 717–725. [Google Scholar] [CrossRef]
- Giedd, J.N.; Vaituzis, A.C.; Hamburger, S.D.; Lange, N.; Rajapakse, J.C.; Kaysen, D.; Vauss, Y.C.; Rapoport, J.L. Quantitative MRI of the temporal lobe, amygdala, and hippocampus in normal human development: Ages 4–18 years. J. Comp. Neurol. 1996, 366, 223–230. [Google Scholar] [CrossRef]
- Hu, S.; Pruessner, J.C.; Coupé, P.; Collins, D.L. Volumetric analysis of medial temporal lobe structures in brain development from childhood to adolescence. NeuroImage 2013, 74, 276–287. [Google Scholar] [CrossRef]
- McEvoy, P.M.; Grove, R.; Slade, T. Epidemiology of anxiety disorders in the Australian general population: Findings of the 2007 Australian National Survey of Mental Health and Wellbeing. Aus. NZJ Psychiatry 2011, 45, 957–967. [Google Scholar] [CrossRef] [PubMed]
- McLean, C.P.; Asnaani, A.; Litz, B.T.; Hofmann, S.G. Gender differences in anxiety disorders: Prevalence, course of illness, comorbidity and burden of illness. J. Psychiatr Res. 2011, 45, 1027–1035. [Google Scholar] [CrossRef] [PubMed]
- Li, S.H.; Graham, B.M. Why are women so vulnerable to anxiety, trauma-related and stress-related disorders? The potential role of sex hormones. Lancet Psychiatry 2017, 4, 73–82. [Google Scholar] [CrossRef]
- Kessler, R.C.; Avenevoli, S.; Costello, E.J.; Georgiades, K.; Green, J.G.; Gruber, M.J.; He, J.-P.; Koretz, D.; McLaughlin, K.A.; Petukhova, M.; et al. Prevalence, persistence, and sociodemographic correlates of DSM-IV disorders in the National Comorbidity Survey Replication Adolescent Supplement. Arch. Gen. Psychiatry 2012, 69, 372–380. [Google Scholar] [CrossRef] [PubMed]
- Gruene, T.M.; Roberts, E.; Thomas, V.; Ronzio, A.; Shansky, R.M. Sex-specific neuroanatomical correlates of fear expression in prefrontal-amygdala circuits. Biol. Psychiatry 2015, 78, 186–193. [Google Scholar] [CrossRef]
- Herry, C.; Ciocchi, S.; Senn, V.; Demmou, L.; Muller, C.; Luthi, A. Switching on and off fear by distinct neuronal circuits. Nature 2008, 454, 600–606. [Google Scholar] [CrossRef]
- Kim, J.; Pignatelli, M.; Xu, S.; Itohara, S.; Tonegawa, S. Antagonistic negative and positive neurons of the basolateral amygdala. Nat. Neurosci. 2016, 19, 1636–1646. [Google Scholar] [CrossRef]
- Lai, C.S.; Franke, T.F.; Gan, W.B. Opposite effects of fear conditioning and extinction on dendritic spine remodelling. Nature 2012, 483, 87–91. [Google Scholar] [CrossRef]
- Heinrichs, S.C.; Leite-Morris, K.A.; Guy, M.D.; Goldberg, L.R.; Young, A.J.; Kaplan, G.B. Dendritic structural plasticity in the basolateral amygdala after fear conditioning and its extinction in mice. Behav. Brain Res. 2013, 248, 80–84. [Google Scholar] [CrossRef]
- Maroun, M.; Ioannides, P.J.; Bergman, K.L.; Kavushansky, A.; Holmes, A.; Wellman, C.L. Fear extinction deficits following acute stress associate with increased spine density and dendritic retraction in basolateral amygdala neurons. Eur. J. Neurosci. 2013, 38, 2611–2620. [Google Scholar] [CrossRef]
- Vyas, A.; Jadhav, S.; Chattarji, S. Prolonged behavioral stress enhances synaptic connectivity in the basolateral amygdala. Neuroscience 2006, 143, 387–393. [Google Scholar] [CrossRef]
- Moench, K.M.; Maroun, M.; Kavushansky, A.; Wellman, C. Alterations in neuronal morphology in infralimbic cortex predict resistance to fear extinction following acute stress. Neurobiol. Stress 2016, 3, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Qu, Y.; Yang, C.; Ren, Q.; Ma, M.; Dong, C.; Hashimoto, K. Regional differences in dendritic spine density confer resilience to chronic social defeat stress. Acta Neuropsychiatr. 2018, 30, 117–122. [Google Scholar] [CrossRef]
- Kim, J.J.; Jung, M.W. Neural circuits and mechanisms involved in Pavlovian fear conditioning: A critical review. Neurosci. Biobehav. Rev. 2006, 30, 188–202. [Google Scholar] [CrossRef] [PubMed]
- Yizhar, O.; Klavir, O. Reciprocal amygdala-prefrontal interactions in learning. Curr. Opin. Neurobiol. 2018, 52, 149–155. [Google Scholar] [CrossRef]
- Pattwell, S.S.; Bath, K.G.; Casey, B.J.; Ninan, I.; Lee, F.S. Selective early-acquired fear memories undergo temporary suppression during adolescence. Proc. Natl. Acad. Sci. USA 2011, 108, 1182–1187. [Google Scholar] [CrossRef] [PubMed]
- Baker, K.D.; Richardson, R. Pharmacological evidence that a failure to recruit NMDA receptors contributes to impaired fear extinction retention in adolescent rats. Neurobiol. Learn. Mem. 2017, 143, 18–26. [Google Scholar] [CrossRef]
- Inoue, S.; Kamiyama, H.; Matsumoto, M.; Yanagawa, Y.; Hiraide, S.; Saito, Y.; Shimamura, K.; Togashi, H. Synaptic Modulation via Basolateral Amygdala on the Rat Hippocampus–Medial Prefrontal Cortex Pathway in Fear Extinction. J. Pharmacol. Sci. 2013, 123, 267–278. [Google Scholar] [CrossRef]
- Lucas, E.K.; Clem, R.L. GABAergic interneurons: The orchestra or the conductor in fear learning and memory? Brain Res. Bull. 2017. [Google Scholar] [CrossRef] [PubMed]
- Courtin, J.; Chaudun, F.; Rozeske, R.R.; Karalis, N.; Gonzalez-Campo, C.; Wurtz, H.; Abdi, A.; Baufreton, J.; Bienvenu, T.C.; Herry, C. Prefrontal parvalbumin interneurons shape neuronal activity to drive fear expression. Nature 2014, 505, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Davis, P.; Zaki, Y.; Maguire, J.; Reijmers, L.G. Cellular and oscillatory substrates of fear extinction learning. Nat. Neurosci. 2017, 20, 1624–1633. [Google Scholar] [CrossRef] [PubMed]
- Caballero, A.; Flores-Barrera, E.; Cass, D.K.; Tseng, K.Y. Differential regulation of parvalbumin and calretinin interneurons in the prefrontal cortex during adolescence. Brain Struct. Funct. 2014, 219, 395–406. [Google Scholar] [CrossRef]
- Caballero, A.; Tseng, K.Y. GABAergic Function as a Limiting Factor for Prefrontal Maturation during Adolescence. Trends Neurosci. 2016, 39, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Baker, K.D.; Gray, A.R.; Richardson, R. The Development of Perineuronal Nets Around Parvalbumin GABAergic Neurons in the Medial Prefrontal Cortex and Basolateral Amygdala of Rats. Behav. Neurosci. 2017, 131, 289–303. [Google Scholar] [CrossRef]
- Brenhouse, H.C.; Andersen, S.L. Nonsteroidal anti-inflammatory treatment prevents delayed effects of early life stress in rats. Biol. Psychiatry 2011, 70, 434–440. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, M.G.; Bhattacharyya, S.; Benes, F.M. Increasing Interaction of Amygdalar Afferents with GABAergic Interneurons between Birth and Adulthood. Cereb. Cortex 2008, 18, 1529–1535. [Google Scholar] [CrossRef] [PubMed]
- Chung, D.W.; Wills, Z.P.; Fish, K.N.; Lewis, D.A. Developmental pruning of excitatory synaptic inputs to parvalbumin interneurons in monkey prefrontal cortex. Proc. Natl. Acad. Sci. USA 2017, 114, E629–E637. [Google Scholar] [CrossRef]
- Thomases, D.R.; Cass, D.K.; Tseng, K.Y. Periadolescent exposure to the NMDA receptor antagonist MK-801 impairs the functional maturation of local GABAergic circuits in the adult prefrontal cortex. J. Neurosci. 2013, 33, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Jia, M.; Travaglia, A.; Pollonini, G.; Fedele, G.; Alberini, C.M. Developmental changes in plasticity, synaptic, glia, and connectivity protein levels in rat medial prefrontal cortex. Learn. Mem. 2018, 25, 533–543. [Google Scholar] [CrossRef]
- Jeevakumar, V.; Kroener, S. Ketamine Administration During the Second Postnatal Week Alters Synaptic Properties of Fast-Spiking Interneurons in the Medial Prefrontal Cortex of Adult Mice. Cereb. Cortex 2016, 26, 1117–1129. [Google Scholar] [CrossRef] [PubMed]
- Murueta-Goyena, A.; Ortuzar, N.; Gargiulo, P.A.; Lafuente, J.V.; Bengoetxea, H. Short-Term Exposure to Enriched Environment in Adult Rats Restores MK-801-Induced Cognitive Deficits and GABAergic Interneuron Immunoreactivity Loss. Mol. Neurobiol. 2018, 55, 26–41. [Google Scholar] [CrossRef] [PubMed]
- Bissonette, G.B.; Bae, M.H.; Suresh, T.; Jaffe, D.E.; Powell, E.M. Prefrontal cognitive deficits in mice with altered cerebral cortical GABAergic interneurons. Behav. Brain Res. 2014, 259, 143–151. [Google Scholar] [CrossRef]
- Berdel, B.; Moryś, J. Expression of calbindin-D28k and parvalbumin during development of rat’s basolateral amygdaloid complex. Int. J. Dev. Neurosci. 2000, 18, 501–513. [Google Scholar] [CrossRef]
- Ehrlich, D.E.; Ryan, S.J.; Hazra, R.; Guo, J.D.; Rainnie, D.G. Postnatal maturation of GABAergic transmission in the rat basolateral amygdala. J. Neurophysiol. 2013, 110, 926–941. [Google Scholar] [CrossRef]
- Arruda-Carvalho, M.; Wu, W.C.; Cummings, K.A.; Clem, R.L. Optogenetic Examination of Prefrontal-Amygdala Synaptic Development. J. Neurosci. 2017, 37, 2976–2985. [Google Scholar] [CrossRef] [PubMed]
- Bosch, D.; Ehrlich, I. Postnatal maturation of GABAergic modulation of sensory inputs onto lateral amygdala principal neurons. J. Physiol. 2015, 593, 4387–4409. [Google Scholar] [CrossRef]
- Cunningham, M.G.; Bhattacharyya, S.; Benes, F.M. Amygdalo-cortical sprouting continues into early adulthood: Implications for the development of normal and abnormal function during adolescence. J. Comp. Neurol. 2002, 453, 116–130. [Google Scholar] [CrossRef]
- Caballero, A.; Thomases, D.R.; Flores-Barrera, E.; Cass, D.K.; Tseng, K.Y. Emergence of GABAergic-dependent regulation of input-specific plasticity in the adult rat prefrontal cortex during adolescence. Psychopharmacology 2014, 231, 1789–1796. [Google Scholar] [CrossRef]
- Ishikawa, A.; Nakamura, S. Convergence and Interaction of Hippocampal and Amygdalar Projections within the Prefrontal Cortex in the Rat. J. Neurosci. 2003, 23, 9987–9995. [Google Scholar] [CrossRef] [PubMed]
- Little, J.P.; Carter, A.G. Synaptic mechanisms underlying strong reciprocal connectivity between the medial prefrontal cortex and basolateral amygdala. J. Neurosci. 2013, 33, 15333–15342. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, A.N.; Yousuf, H.; Dalton, S.; Sheets, P.L. Highly differentiated cellular and circuit properties of infralimbic pyramidal neurons projecting to the periaqueductal gray and amygdala. Front. Cell Neurosci. 2015, 9. [Google Scholar] [CrossRef] [PubMed]
- Bouwmeester, H.; Smits, K.; Van Ree, J.M. Neonatal development of projections to the basolateral amygdala from prefrontal and thalamic structures in rat. J. Comp. Neurol. 2002, 450, 241–255. [Google Scholar] [CrossRef] [PubMed]
- Gabbott, P.L.; Warner, T.A.; Jays, P.R.; Salway, P.; Busby, S.J. Prefrontal cortex in the rat: Projections to subcortical autonomic, motor, and limbic centers. J. Comp. Neurol. 2005, 492, 145–177. [Google Scholar] [CrossRef] [PubMed]
- Hirai, Y.; Morishima, M.; Karube, F.; Kawaguchi, Y. Specialized Cortical Subnetworks Differentially Connect Frontal Cortex to Parahippocampal Areas. J. Neurosci. 2012, 32, 1898–1913. [Google Scholar] [CrossRef] [PubMed]
- Marek, R.; Jin, J.; Goode, T.D.; Giustino, T.F.; Wang, Q.; Acca, G.M.; Holehonnur, R.; Ploski, J.E.; Fitzgerald, P.J.; Lynagh, T.; et al. Hippocampus-driven feed-forward inhibition of the prefrontal cortex mediates relapse of extinguished fear. Nat. Neurosci. 2018, 21, 384–392. [Google Scholar] [CrossRef] [PubMed]
- Cressman, V.L.; Balaban, J.; Steinfeld, S.; Shemyakin, A.; Graham, P.; Parisot, N.; Moore, H. Prefrontal cortical inputs to the basal amygdala undergo pruning during late adolescence in the rat. J. Comp. Neurol. 2010, 518, 2693–2709. [Google Scholar] [CrossRef] [PubMed]
- Selleck, R.A.; Zhang, W.; Samberg, H.D.; Padival, M.; Rosenkranz, J.A. Limited prefrontal cortical regulation over the basolateral amygdala in adolescent rats. Sci. Rep. 2018, 8, 17171. [Google Scholar] [CrossRef] [PubMed]
- Gee, D.G.; Humphreys, K.L.; Flannery, J.; Goff, B.; Telzer, E.H.; Shapiro, M.; Hare, T.A.; Bookheimer, S.Y.; Tottenham, N. A Developmental Shift from Positive to Negative Connectivity in Human Amygdala–Prefrontal Circuitry. J. Neurosci. 2013, 33, 4584–4593. [Google Scholar] [CrossRef]
- Thomases, D.R.; Cass, D.K.; Meyer, J.D.; Caballero, A.; Tseng, K.Y. Early adolescent MK-801 exposure impairs the maturation of ventral hippocampal control of basolateral amygdala drive in the adult prefrontal cortex. J. Neurosci. 2014, 34, 9059–9066. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Carter, A.G. Ventral Hippocampal Inputs Preferentially Drive Corticocortical Neurons in the Infralimbic Prefrontal Cortex. J. Neurosci. 2018, 38, 7351–7363. [Google Scholar] [CrossRef] [PubMed]
- Rosas-Vidal, L.E.; Do-Monte, F.H.; Sotres-Bayon, F.; Quirk, G.J. Hippocampal-prefrontal BDNF and memory for fear extinction. Neuropsychopharmacology 2014, 39, 2161–2169. [Google Scholar] [CrossRef] [PubMed]
- Rosas-Vidal, L.E.; Lozada-Miranda, V.; Cantres-Rosario, Y.; Vega-Medina, A.; Melendez, L.; Quirk, G.J. Alteration of BDNF in the medial prefrontal cortex and the ventral hippocampus impairs extinction of avoidance. Neuropsychopharmacology 2018, 43, 2636–2644. [Google Scholar] [CrossRef] [PubMed]
- Sierra-Mercado, D.; Padilla-Coreano, N.; Quirk, G.J. Dissociable roles of prelimbic and infralimbic cortices, ventral hippocampus, and basolateral amygdala in the expression and extinction of conditioned fear. Neuropsychopharmacology 2011, 36, 529–538. [Google Scholar] [CrossRef]
- Giza, J.I.; Kim, J.; Meyer, H.C.; Anastasia, A.; Dincheva, I.; Zheng, C.I.; Lopez, K.; Bains, H.; Yang, J.; Bracken, C.; et al. The BDNF Val66Met Prodomain Disassembles Dendritic Spines Altering Fear Extinction Circuitry and Behavior. Neuron 2018, 99, 163–178. [Google Scholar] [CrossRef] [PubMed]
- Keenan, K.; Hipwell, A.E.; Class, Q.A.; Mbayiwa, K. Extending the developmental origins of disease model: Impact of preconception stress exposure on offspring neurodevelopment. Dev. Psychol. Biol. 2018, 60, 753–764. [Google Scholar] [CrossRef]
- van den Bergh, B.R.H.; Dahnke, R.; Mennes, M. Prenatal stress and the developing brain: Risks for neurodevelopmental disorders. Dev. Psychopathol. 2018, 30, 743–762. [Google Scholar] [CrossRef] [PubMed]
- Cowan, C.S.; Callaghan, B.L.; Kan, J.M.; Richardson, R. The lasting impact of early-life adversity on individuals and their descendants: Potential mechanisms and hope for intervention. Genes Brain Behav. 2016, 15, 155–168. [Google Scholar] [CrossRef] [PubMed]
- McCormick, C.M.; Mongillo, D.L.; Simone, J.J. Age and adolescent social stress effects on fear extinction in female rats. Stress 2013, 16, 678–688. [Google Scholar] [CrossRef] [PubMed]
- Negrón-Oyarzo, I.; Pérez, M.Á.; Terreros, G.; Muñoz, P.; Dagnino-Subiabre, A. Effects of chronic stress in adolescence on learned fear, anxiety, and synaptic transmission in the rat prelimbic cortex. Behav. Brain Res. 2014, 259, 342–353. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Rosenkranz, J.A. Repeated restraint stress enhances cue-elicited conditioned freezing and impairs acquisition of extinction in an age-dependent manner. Behav. Brain Res. 2013, 248, 12–24. [Google Scholar] [CrossRef]
- Stylianakis, A.A.; Richardson, R.; Baker, K.D. Timing is everything: Developmental differences in the effect of a chronic stressor on extinction retention. Behav. Neurosci. under review.
- Den, M.L.; Altmann, S.R.; Richardson, R. A comparison of the short- and long-term effects of cortico- sterone exposure on extinction in adolescence versus adulthood. Behav. Neurosci. 2014, 128, 722–735. [Google Scholar] [CrossRef]
- Yuen, E.Y.; Wei, J.; Liu, W.; Zhong, P.; Li, X.; Yan, Z. Repeated stress causes cognitive impairment by suppressing glutamate receptor expression and function in prefrontal cortex. Neuron 2012, 73, 962–977. [Google Scholar] [CrossRef] [PubMed]
- Gourley, S.L.; Swanson, A.M.; Koleske, A.J. Corticosteroid-induced neural remodeling predicts behavioral vulnerability and resilience. J. Neurosci. 2013, 33, 3107–3112. [Google Scholar] [CrossRef] [PubMed]
- Pinzon-Parra, C.; Vidal-Jimenez, B.; Camacho-Abrego, I.; Flores-Gomez, A.A.; Rodriguez-Moreno, A.; Flores, G. Juvenile stress causes reduced locomotor behavior and dendritic spine density in the prefrontal cortex and basolateral amygdala in Sprague-Dawley rats. Synapse 2019, 73, e22066. [Google Scholar] [CrossRef] [PubMed]
- Vyas, A.; Mitra, R.; Shankaranarayana Rao, B.S.; Chattarji, S. Chronic Stress Induces Contrasting Patterns of Dendritic Remodeling in Hippocampal and Amygdaloid Neurons. J. Neurosci. 2002, 22, 6810–6818. [Google Scholar] [CrossRef]
- Page, C.E.; Coutellier, L. Adolescent Stress Disrupts the Maturation of Anxiety-related Behaviors and Alters the Developmental Trajectory of the Prefrontal Cortex in a Sex- and Age-specific Manner. Neuroscience 2018, 390, 265–277. [Google Scholar] [CrossRef]
- De Araujo Costa Folha, O.A.; Bahia, C.P.; de Aguiar, G.P.S.; Herculano, A.M.; Coelho, N.L.G.; de Sousa, M.B.C.; Shiramizu, V.K.M.; de Menezes Galvao, A.C.; de Carvalho, W.A.; Pereira, A. Effect of chronic stress during adolescence in prefrontal cortex structure and function. Behav. Brain Res. 2017, 326, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.H.; Yan, W.; Han, Y.; Chen, C.; Meng, S.Q.; Sun, C.Y.; Xu, L.Z.; Xue, Y.X.; Gao, X.J.; Chen, N.; et al. Predictable Chronic Mild Stress during Adolescence Promotes Fear Memory Extinction in Adulthood. Sci. Rep. 2017, 7, 7857. [Google Scholar] [CrossRef]
- Cowan, C.S.M.; Richardson, R. A Brief Guide to Studying Fear in Developing Rodents: Important Considerations and Common Pitfalls. Curr. Protoc. Neurosci. 2018, 83, e44. [Google Scholar] [CrossRef] [PubMed]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zimmermann, K.S.; Richardson, R.; Baker, K.D. Maturational Changes in Prefrontal and Amygdala Circuits in Adolescence: Implications for Understanding Fear Inhibition during a Vulnerable Period of Development. Brain Sci. 2019, 9, 65. https://doi.org/10.3390/brainsci9030065
Zimmermann KS, Richardson R, Baker KD. Maturational Changes in Prefrontal and Amygdala Circuits in Adolescence: Implications for Understanding Fear Inhibition during a Vulnerable Period of Development. Brain Sciences. 2019; 9(3):65. https://doi.org/10.3390/brainsci9030065
Chicago/Turabian StyleZimmermann, Kelsey S., Rick Richardson, and Kathryn D. Baker. 2019. "Maturational Changes in Prefrontal and Amygdala Circuits in Adolescence: Implications for Understanding Fear Inhibition during a Vulnerable Period of Development" Brain Sciences 9, no. 3: 65. https://doi.org/10.3390/brainsci9030065
APA StyleZimmermann, K. S., Richardson, R., & Baker, K. D. (2019). Maturational Changes in Prefrontal and Amygdala Circuits in Adolescence: Implications for Understanding Fear Inhibition during a Vulnerable Period of Development. Brain Sciences, 9(3), 65. https://doi.org/10.3390/brainsci9030065