Aging and Central Auditory Disinhibition: Is It a Reflection of Homeostatic Downregulation or Metabolic Vulnerability?


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Synaptic Inhibition in the Aging Central Auditory System
2.1. Auditory Brainstem Changes
2.2. Auditory Midbrain Changes
2.3. Auditory Thalamus and Cortex Changes
3. Aging and Mitochondrial Energetics
4. GABAergic Neurons are More Vulnerable to Energy Insults
The Impact of the Vulnerability of GABAergic Cells on Brain Pathology
5. The Dilemma of Inhibitory Downregulation in the Aging Central Auditory System
6. Future Directions
Funding
Acknowledgments
Conflicts of Interest
References
- Lin, F.R.; Thorpe, R.; Gordon-Salant, S.; Ferrucci, L. Hearing loss prevalence and risk factors among older adults in the United States. J. Gerontol. A Biol. Sci. Med. Sci. 2011, 66, 582–590. [Google Scholar] [CrossRef] [PubMed]
- Cruickshanks, K.J.; Zhan, W.; Zhong, W. Epidemiology of Age-Related Hearing Impairment. In The Aging Auditory System; Gordon-Salant, S., Frisina, R.D., Popper, A.N.F., Richard, R., Eds.; Springer: New York, NY, USA, 2010; Volume 34, pp. 259–274. [Google Scholar]
- Ortman, J.M.; Velkoff, V.A.; Hogan, H. An Aging Nation: The Older Population in the United States; US Census Bureau: Washington, DC, USA, 2014; pp. 25–1140.
- Helfer, K.S.; Wilber, L.A. Hearing loss, aging, and speech perception in reverberation and noise. J. Speech Lang. Hear. Res. 1990, 33, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.S.; Jerger, J.F. Some effects of aging on central auditory processing. J. Rehabil. Res. Dev. 2005, 42, 25–44. [Google Scholar] [CrossRef] [PubMed]
- Pichora-Fuller, M.K.; Souza, P.E. Effects of aging on auditory processing of speech. Int. J. Audiol. 2003, 42, 11–16. [Google Scholar] [CrossRef]
- Jayakody, D.M.P.; Friedland, P.L.; Martins, R.N.; Sohrabi, H.R. Impact of Aging on the Auditory System and Related Cognitive Functions: A Narrative Review. Front. Neurosci. 2018, 12, 125. [Google Scholar] [CrossRef] [PubMed]
- Ouda, L.; Profant, O.; Syka, J. Age-related changes in the central auditory system. Cell Tissue Res. 2015, 361, 337–358. [Google Scholar] [CrossRef] [PubMed]
- Frisina, R.D. Age-related hearing loss: Ear and brain mechanisms. Ann. N. Y. Acad. Sci. 2009, 1170, 708–717. [Google Scholar] [CrossRef]
- Caspary, D.M.; Llano, D.A. Aging Processes in the Subcortical Auditory System. In The Oxford Handbook of the Auditory Brainstem; Kandler, K., Ed.; Oxford University Press: New York, NY, USA, 2019. [Google Scholar] [CrossRef]
- Wang, H.; Brozoski, T.J.; Caspary, D.M. Inhibitory neurotransmission in animal models of tinnitus: Maladaptive plasticity. Hear. Res. 2011, 279, 111–117. [Google Scholar] [CrossRef]
- Oberem, J.; Koch, I.; Fels, J. Intentional switching in auditory selective attention: Exploring age-related effects in a spatial setup requiring speech perception. Acta Psychol. 2017, 177, 36–43. [Google Scholar] [CrossRef]
- Chao, L.L.; Knight, R.T. Prefrontal deficits in attention and inhibitory control with aging. Cereb. Cortex 1997, 7, 63–69. [Google Scholar] [CrossRef]
- Turrigiano, G. Homeostatic synaptic plasticity: Local and global mechanisms for stabilizing neuronal function. Cold Spring Harb. Perspect. Biol. 2012, 4, a005736. [Google Scholar] [CrossRef] [PubMed]
- Schaette, R.; Kempter, R. Development of tinnitus-related neuronal hyperactivity through homeostatic plasticity after hearing loss: A computational model. Eur. J. Neurosci. 2006, 23, 3124–3138. [Google Scholar] [CrossRef] [PubMed]
- Schaette, R.; Kempter, R. Development of hyperactivity after hearing loss in a computational model of the dorsal cochlear nucleus depends on neuron response type. Hear. Res. 2008, 240, 57–72. [Google Scholar] [CrossRef] [PubMed]
- Schaette, R.; Turtle, C.; Munro, K.J. Reversible induction of phantom auditory sensations through simulated unilateral hearing loss. PLoS ONE 2012, 7, e35238. [Google Scholar] [CrossRef] [PubMed]
- Caspary, D.M.; Hughes, L.F.; Schatteman, T.A.; Turner, J.G. Age-related changes in the response properties of cartwheel cells in rat dorsal cochlear nucleus. Hear. Res. 2006, 216, 207–215. [Google Scholar] [CrossRef]
- Caspary, D.M.; Schatteman, T.A.; Hughes, L.F. Age-related changes in the inhibitory response properties of dorsal cochlear nucleus output neurons: Role of inhibitory inputs. J. Neurosci. 2005, 25, 10952–10959. [Google Scholar] [CrossRef]
- Milbrandt, J.C.; Caspary, D.M. Age-related reduction of [3H] strychnine binding sites in the cochlear nucleus of the Fischer 344 rat. Neuroscience 1995, 67, 713–719. [Google Scholar] [CrossRef]
- Wang, H.; Turner, J.G.; Ling, L.; Parrish, J.L.; Hughes, L.F.; Caspary, D.M. Age-related changes in glycine receptor subunit composition and binding in dorsal cochlear nucleus. Neuroscience 2009, 160, 227–239. [Google Scholar] [CrossRef]
- Krenning, J.; Hughes, L.F.; Caspary, D.M.; Helfert, R.H. Age-related glycine receptor subunit changes in the cochlear nucleus of Fischer-344 rats. Laryngoscope 1998, 108, 26–31. [Google Scholar] [CrossRef]
- Chen, Q.C.; Jen, P.H. Bicuculline application affects discharge patterns, rate-intensity functions, and frequency tuning characteristics of bat auditory cortical neurons. Hear. Res. 2000, 150, 161–174. [Google Scholar] [CrossRef]
- Evans, E.F.; Zhao, W. Varieties of inhibition in the processing and control of processing in the mammalian cochlear nucleus. Prog. Brain Res. 1993, 97, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Pollak, G.D.; Resler, C. GABAergic circuits sharpen tuning curves and modify response properties in the mustache bat inferior colliculus. J. Neurophysiol. 1992, 68, 1760–1774. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Suga, N. Lateral inhibition for center-surround reorganization of the frequency map of bat auditory cortex. J. Neurophysiol. 2004, 92, 3192–3199. [Google Scholar] [CrossRef] [PubMed]
- Suga, N.; Zhang, Y.; Yan, J. Sharpening of frequency tuning by inhibition in the thalamic auditory nucleus of the mustached bat. J. Neurophysiol. 1997, 77, 2098–2114. [Google Scholar] [CrossRef][Green Version]
- Jen, P.H.; Feng, R.B. Bicuculline application affects discharge pattern and pulse-duration tuning characteristics of bat inferior collicular neurons. J. Comp. Physiol. A 1999, 184, 185–194. [Google Scholar] [CrossRef]
- Fuzessery, Z.M.; Hall, J.C. Sound duration selectivity in the pallid bat inferior colliculus. Hear. Res. 1999, 137, 137–154. [Google Scholar] [CrossRef]
- Casseday, J.H.; Ehrlich, D.; Covey, E. Neural tuning for sound duration: Role of inhibitory mechanisms in the inferior colliculus. Science 1994, 264, 847–850. [Google Scholar] [CrossRef]
- Galazyuk, A.V.; Lin, W.; Llano, D.; Feng, A.S. Leading inhibition to neural oscillation is important for time-domain processing in the auditory midbrain. J. Neurophysiol. 2005, 94, 314–326. [Google Scholar] [CrossRef][Green Version]
- Davis, K.A.; Ramachandran, R.; May, B.J. Auditory processing of spectral cues for sound localization in the inferior colliculus. J. Assoc. Res. Otolaryngol. 2003, 4, 148–163. [Google Scholar] [CrossRef]
- Magnusson, A.K.; Park, T.J.; Pecka, M.; Grothe, B.; Koch, U. Retrograde GABA signaling adjusts sound localization by balancing excitation and inhibition in the brainstem. Neuron 2008, 59, 125–137. [Google Scholar] [CrossRef]
- Rabang, C.F.; Parthasarathy, A.; Venkataraman, Y.; Fisher, Z.L.; Gardner, S.M.; Bartlett, E.L. A computational model of inferior colliculus responses to amplitude modulated sounds in young and aged rats. Front. Neural Circuits 2012, 6, 77. [Google Scholar] [CrossRef] [PubMed]
- Caspary, D.M.; Palombi, P.S.; Hughes, L.F. GABAergic inputs shape responses to amplitude modulated stimuli in the inferior colliculus. Hear. Res. 2002, 168, 163–173. [Google Scholar] [CrossRef]
- Zhang, H.; Kelly, J.B. Glutamatergic and GABAergic regulation of neural responses in inferior colliculus to amplitude-modulated sounds. J. Neurophysiol. 2003, 90, 477–490. [Google Scholar] [CrossRef] [PubMed]
- Llano, D.A.; Feng, A.S. Computational models of temporal processing in the auditory thalamus. Biol. Cybern. 2000, 83, 419–433. [Google Scholar] [CrossRef] [PubMed]
- Abel, S.M.; Giguère, C.; Consoli, A.; Papsin, B.C. The effect of aging on horizontal plane sound localization. J. Acoust. Soc. Am. 2000, 108, 743–752. [Google Scholar] [CrossRef]
- Dobreva, M.S.; O’Neill, W.E.; Paige, G.D. Influence of aging on human sound localization. J. Neurophysiol. 2011, 105, 2471–2486. [Google Scholar] [CrossRef]
- König, E. Pitch discrimination and age. Acta Oto-laryngol. 1957, 48, 475–489. [Google Scholar] [CrossRef]
- Clinard, C.G.; Tremblay, K.L.; Krishnan, A.R. Aging alters the perception and physiological representation of frequency: Evidence from human frequency-following response recordings. Hear. Res. 2010, 264, 48–55. [Google Scholar] [CrossRef]
- Elliott, L.L.; Busse, L.A.; Bailet, L.L. Identification and discrimination of consonant-vowel syllables by younger and older adults. Percept. Psychophys. 1985, 37, 307–314. [Google Scholar] [CrossRef]
- Helfer, K.S.; Huntley, R.A. Aging and consonant errors in reverberation and noise. J. Acoust. Soc. Am. 1991, 90, 1786–1796. [Google Scholar] [CrossRef]
- Zheng, Q.Y.; Johnson, K.R.; Erway, L.C. Assessment of hearing in 80 inbred strains of mice by ABR threshold analyses. Hear. Res. 1999, 130, 94–107. [Google Scholar] [CrossRef]
- Di Palma, F.; Holme, R.H.; Bryda, E.C.; Belyantseva, I.A.; Pellegrino, R.; Kachar, B.; Steel, K.P.; Noben-Trauth, K. Mutations in Cdh23, encoding a new type of cadherin, cause stereocilia disorganization in waltzer, the mouse model for Usher syndrome type 1D. Nat. Genet. 2001, 27, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Popelar, J.; Groh, D.; Pelánová, J.; Canlon, B.; Syka, J. Age-related changes in cochlear and brainstem auditory functions in Fischer 344 rats. Neurobiol. Aging 2006, 27, 490–500. [Google Scholar] [CrossRef] [PubMed]
- Cai, R.; Montgomery, S.C.; Graves, K.A.; Caspary, D.M.; Cox, B.C. The FBN rat model of aging: Investigation of ABR waveforms and ribbon synapse changes. Neurobiol. Aging 2018, 62, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Godfrey, D.A.; Chen, K.; O’Toole, T.R.; Mustapha, A.I.A.A. Amino acid and acetylcholine chemistry in the central auditory system of young, middle-aged and old rats. Hear. Res. 2017, 350, 173–188. [Google Scholar] [CrossRef] [PubMed]
- Whiting, B.; Moiseff, A.; Rubio, M.E. Cochlear nucleus neurons redistribute synaptic AMPA and glycine receptors in response to monaural conductive hearing loss. Neuroscience 2009, 163, 1264–1276. [Google Scholar] [CrossRef]
- Willott, J.F.; Bross, L.S. Morphology of the octopus cell area of the cochlear nucleus in young and aging C57BL/6J and CBA/J mice. J. Comp. neurol. 1990, 300, 61–81. [Google Scholar] [CrossRef]
- Casey, M.A.; Feldman, M.L. Age-related loss of synaptic terminals in the rat medial nucleus of the trapezoid body. Neuroscience 1988, 24, 189–194. [Google Scholar] [CrossRef]
- Casey, M.A.; Feldman, M.L. Aging in the rat medial nucleus of the trapezoid body. II. Electron microscopy. J. Comp. Neurol. 1985, 232, 401–413. [Google Scholar] [CrossRef]
- Casey, M.A.; Feldman, M.L. Aging in the rat medial nucleus of the trapezoid body. I. Light microscopy. Neurobiol. Aging 1982, 3, 187–195. [Google Scholar] [CrossRef]
- Raza, A.; Milbrandt, J.C.; Arneric, S.P.; Caspary, D.M. Age-related changes in brainstem auditory neurotransmitters: Measures of GABA and acetylcholine function. Hear. Res. 1994, 77, 221–230. [Google Scholar] [CrossRef]
- Burianova, J.; Ouda, L.; Profant, O.; Syka, J. Age-related changes in GAD levels in the central auditory system of the rat. Exp. Gerontol. 2009, 44, 161–169. [Google Scholar] [CrossRef]
- Banay-Schwartz, M.; Lajtha, A.; Palkovits, M. Changes with aging in the levels of amino acids in rat CNS structural elements. I. Glutamate and related amino acids. Neurochem. Res. 1989, 14, 555–562. [Google Scholar] [CrossRef] [PubMed]
- Banay-Schwartz, M.; Palkovits, M.; Lajtha, A. Heterogeneous distribution of functionally important amino acids in brain areas of adult and aging humans. Neurochem. Res. 1993, 18, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Milbrandt, J.C.; Albin, R.L.; Caspary, D.M. Age-related decrease in GABAB receptor binding in the Fischer 344 rat inferior colliculus. Neurobiol. Aging 1994, 15, 699–703. [Google Scholar] [CrossRef][Green Version]
- Milbrandt, J.C.; Albin, R.L.; Turgeon, S.M.; Caspary, D.M. GABAA receptor binding in the aging rat inferior colliculus. Neuroscience 1996, 73, 449–458. [Google Scholar] [CrossRef]
- Milbrandt, J.C.; Hunter, C.; Caspary, D.M. Alterations of GABAA receptor subunit mRNA levels in the aging Fischer 344 rat inferior colliculus. J. Comp. Neurol. 1997, 379, 455–465. [Google Scholar] [CrossRef]
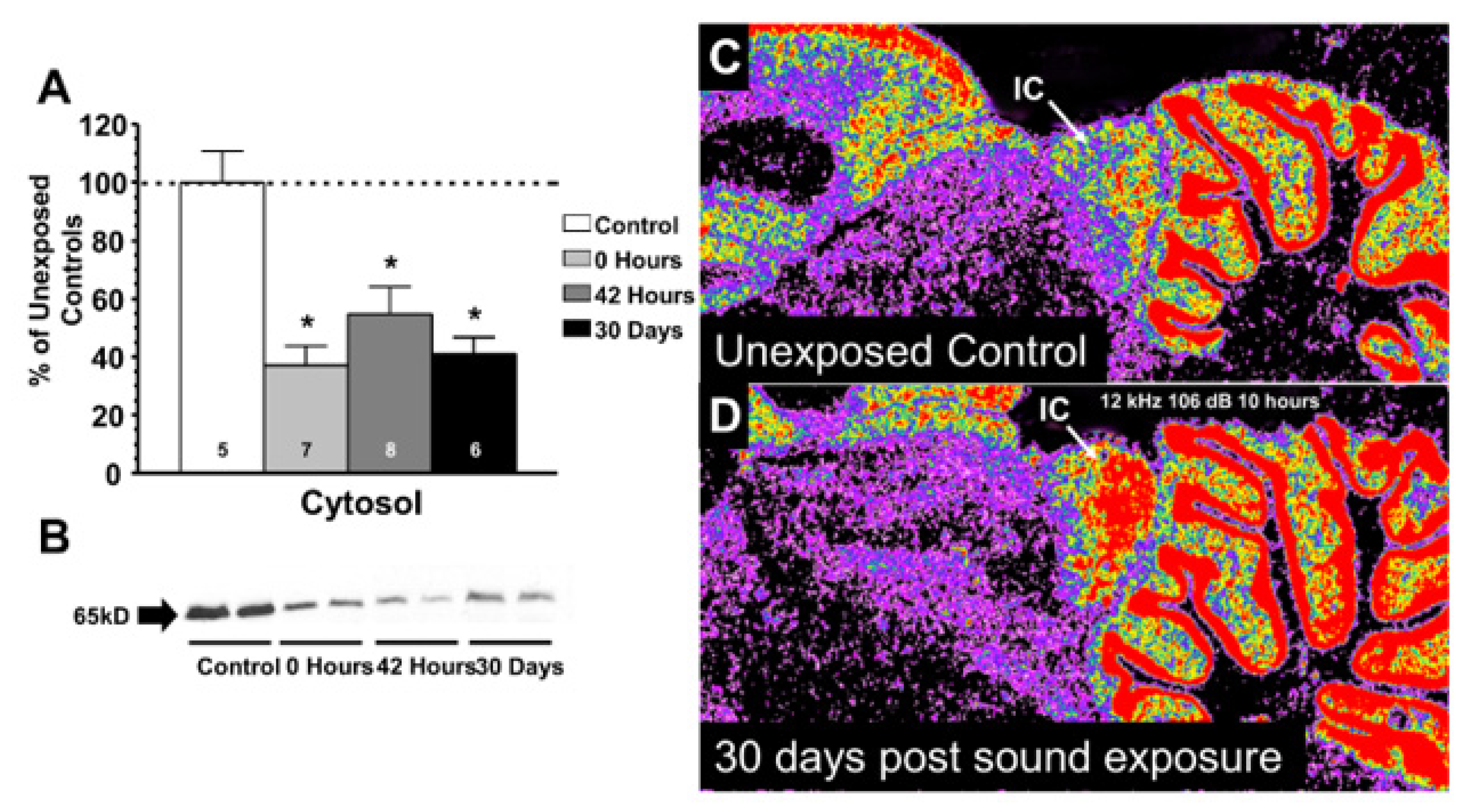
- Milbrandt, J.C.; Holder, T.M.; Wilson, M.C.; Salvi, R.J.; Caspary, D.M. GAD levels and muscimol binding in rat inferior colliculus following acoustic trauma. Hear. Res. 2000, 147, 251–260. [Google Scholar] [CrossRef]
- Pouyatos, B.; Morel, G.; Lambert-Xolin, A.M.; Maguin, K.; Campo, P. Consequences of noise- or styrene-induced cochlear damages on glutamate decarboxylase levels in the rat inferior colliculus. Hear. Res. 2004, 189, 83–91. [Google Scholar] [CrossRef]
- Vale, C.; Sanes, D.H. The effect of bilateral deafness on excitatory and inhibitory synaptic strength in the inferior colliculus. Eur. J. Neurosci. 2002, 16, 2394–2404. [Google Scholar] [CrossRef]
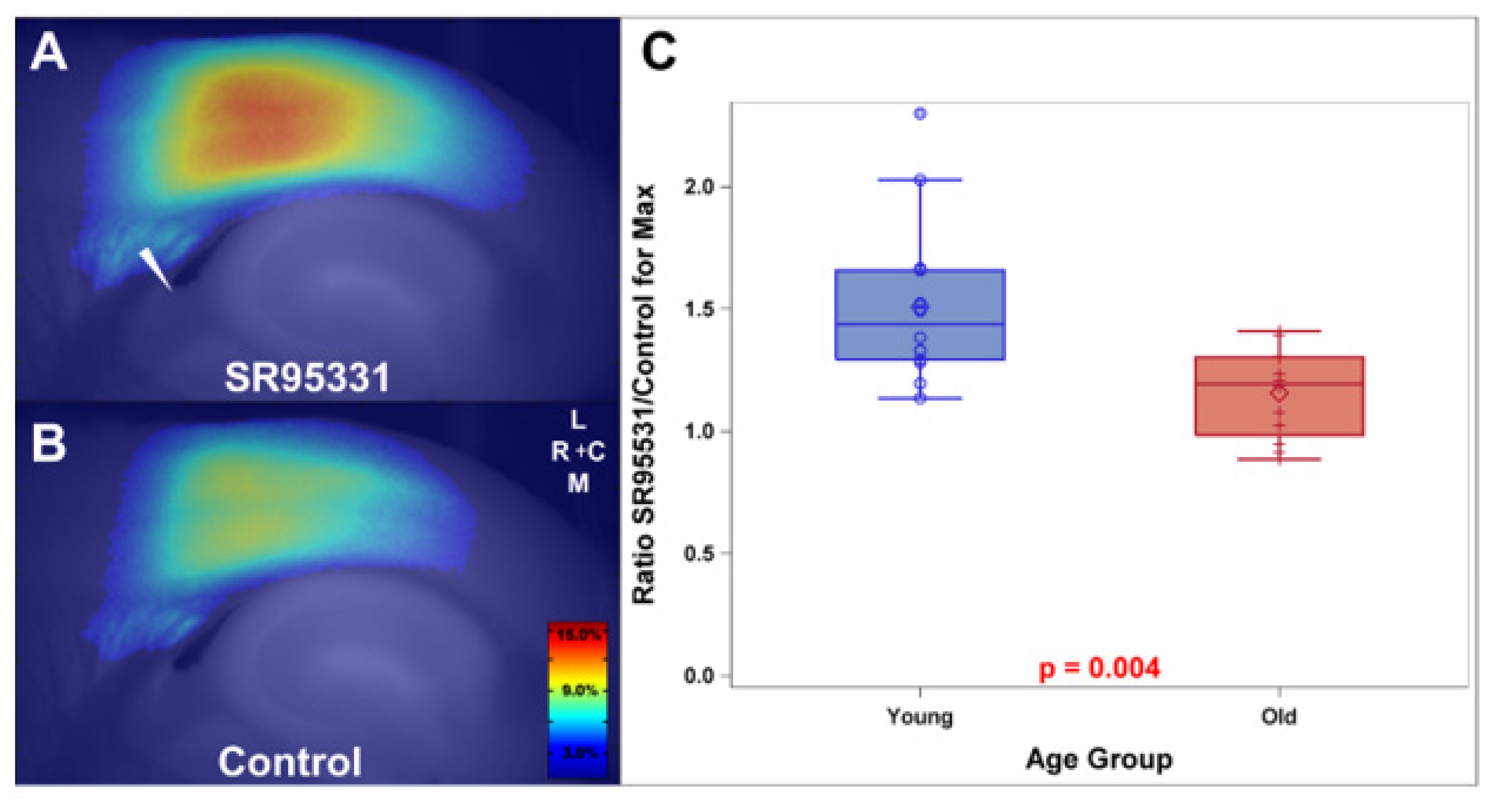
- Richardson, B.D.; Ling, L.L.; Uteshev, V.V.; Caspary, D.M. Reduced GABA(A) receptor-mediated tonic inhibition in aged rat auditory thalamus. J. Neurosci. 2013, 33, 1218–1227. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ouda, L.; Druga, R.; Syka, J. Changes in parvalbumin immunoreactivity with aging in the central auditory system of the rat. Exp. Gerontol. 2008, 43, 782–789. [Google Scholar] [CrossRef] [PubMed]
- Ouellet, L.; de Villers-Sidani, E. Trajectory of the main GABAergic interneuron populations from early development to old age in the rat primary auditory cortex. Front. Neuroanat. 2014, 8, 40. [Google Scholar] [CrossRef] [PubMed]
- Martin del Campo, H.N.; Measor, K.R.; Razak, K.A. Parvalbumin immunoreactivity in the auditory cortex of a mouse model of presbycusis. Hear. Res. 2012, 294, 31–39. [Google Scholar] [CrossRef]
- Brewton, D.H.; Kokash, J.; Jimenez, O.; Pena, E.R.; Razak, K.A. Age-Related Deterioration of Perineuronal Nets in the Primary Auditory Cortex of Mice. Front. Aging Neurosci. 2016, 8, 270. [Google Scholar] [CrossRef]
- Stebbings, K.A.; Choi, H.W.; Ravindra, A.; Caspary, D.M.; Turner, J.G.; Llano, D.A. Ageing-related changes in GABAergic inhibition in mouse auditory cortex, measured using in vitro flavoprotein autofluorescence imaging. J. Physiol. 2016, 594, 207–221. [Google Scholar] [CrossRef]
- Kamal, B.; Holman, C.; de Villers-Sidani, E. Shaping the aging brain: Role of auditory input patterns in the emergence of auditory cortical impairments. Front. Syst. Neurosci. 2013, 7, 52. [Google Scholar] [CrossRef]
- Kotak, V.C.; Takesian, A.E.; Sanes, D.H. Hearing loss prevents the maturation of GABAergic transmission in the auditory cortex. Cereb. Cortex 2008, 18, 2098–2108. [Google Scholar] [CrossRef]
- Yarian, C.S.; Toroser, D.; Sohal, R.S. Aconitase is the main functional target of aging in the citric acid cycle of kidney mitochondria from mice. Mech. Ageing Dev. 2006, 127, 79–84. [Google Scholar] [CrossRef]
- Lam, P.Y.; Yin, F.; Hamilton, R.T.; Boveris, A.; Cadenas, E. Elevated neuronal nitric oxide synthase expression during ageing and mitochondrial energy production. Free Radic. Res. 2009, 43, 431–439. [Google Scholar] [CrossRef]
- Beckman, K.B.; Ames, B.N. The free radical theory of aging matures. Physiol. Rev. 1998, 78, 547–581. [Google Scholar] [CrossRef] [PubMed]
- Navarro, A.; Boveris, A. The mitochondrial energy transduction system and the aging process. Am. J. Physiol. Cell Physiol. 2007, 292, C670–C686. [Google Scholar] [CrossRef] [PubMed]
- Kwong, L.K.; Sohal, R.S. Age-related changes in activities of mitochondrial electron transport complexes in various tissues of the mouse. Arch. Biochem. Biophys. 2000, 373, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Navarro, A.; Boveris, A. Rat brain and liver mitochondria develop oxidative stress and lose enzymatic activities on aging. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2004, 287, R1244–R1249. [Google Scholar] [CrossRef] [PubMed]
- Navarro, A.; Boveris, A. Brain mitochondrial dysfunction in aging: Conditions that improve survival, neurological performance and mitochondrial function. Front. Biosci. 2007, 12, 1154–1163. [Google Scholar] [CrossRef] [PubMed]
- Rebrin, I.; Kamzalov, S.; Sohal, R.S. Effects of age and caloric restriction on glutathione redox state in mice. Free Radic. Biol. Med. 2003, 35, 626–635. [Google Scholar] [CrossRef]
- Rebrin, I.; Forster, M.J.; Sohal, R.S. Effects of age and caloric intake on glutathione redox state in different brain regions of C57BL/6 and DBA/2 mice. Brain Res. 2007, 1127, 10–18. [Google Scholar] [CrossRef]
- Stebbings, K.A.; Choi, H.W.; Ravindra, A.; Llano, D.A. The impact of aging, hearing loss, and body weight on mouse hippocampal redox state, measured in brain slices using fluorescence imaging. Neurobiol. Aging 2016, 42, 101–109. [Google Scholar] [CrossRef]
- Alle, H.; Roth, A.; Geiger, J.R. Energy-efficient action potentials in hippocampal mossy fibers. Science 2009, 325, 1405–1408. [Google Scholar] [CrossRef]
- Attwell, D.; Laughlin, S.B. An energy budget for signaling in the grey matter of the brain. J. Cereb. Blood Flow Metab. 2001, 21, 1133–1145. [Google Scholar] [CrossRef]
- Sibson, N.R.; Shen, J.; Mason, G.F.; Rothman, D.L.; Behar, K.L.; Shulman, R.G. Functional energy metabolism: In vivo 13C-NMR spectroscopy evidence for coupling of cerebral glucose consumption and glutamatergic neuronal activity. Dev. Neurosci. 1998, 20, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Hyder, F.; Patel, A.B.; Gjedde, A.; Rothman, D.L.; Behar, K.L.; Shulman, R.G. Neuronal-glial glucose oxidation and glutamatergic-GABAergic function. J. Cereb. Blood Flow Metab. 2006, 26, 865–877. [Google Scholar] [CrossRef] [PubMed]
- Cobb, S.R.; Buhl, E.H.; Halasy, K.; Paulsen, O.; Somogyi, P. Synchronization of neuronal activity in hippocampus by individual GABAergic interneurons. Nature 1995, 378, 75–78. [Google Scholar] [CrossRef] [PubMed]
- Livingstone, M.S. Oscillatory firing and interneuronal correlations in squirrel monkey striate cortex. J. Neurophysiol. 1996, 75, 2467–2485. [Google Scholar] [CrossRef] [PubMed]
- Lytton, W.W.; Sejnowski, T.J. Simulations of cortical pyramidal neurons synchronized by inhibitory interneurons. J. Neurophysiol. 1991, 66, 1059–1079. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, R.; Lee, S.; Rudy, B. GABAergic Interneurons in the Neocortex: From Cellular Properties to Circuits. Neuron 2016, 91, 260–292. [Google Scholar] [CrossRef]
- Gonzalez-Burgos, G.; Lewis, D.A. GABA neurons and the mechanisms of network oscillations: Implications for understanding cortical dysfunction in schizophrenia. Schizophr. Bull. 2008, 34, 944–961. [Google Scholar] [CrossRef]
- Kann, O.; Papageorgiou, I.E.; Draguhn, A. Highly energized inhibitory interneurons are a central element for information processing in cortical networks. J. Cereb. Blood Flow Metab. 2014, 34, 1270–1282. [Google Scholar] [CrossRef]
- Sederberg, P.B.; Schulze-Bonhage, A.; Madsen, J.R.; Bromfield, E.B.; McCarthy, D.C.; Brandt, A.; Tully, M.S.; Kahana, M.J. Hippocampal and neocortical gamma oscillations predict memory formation in humans. Cereb. Cortex 2007, 17, 1190–1196. [Google Scholar] [CrossRef]
- Chrobak, J.J.; Buzsáki, G. Gamma oscillations in the entorhinal cortex of the freely behaving rat. J. Neurosci. 1998, 18, 388–398. [Google Scholar] [CrossRef]
- Bragin, A.; Jandó, G.; Nádasdy, Z.; Hetke, J.; Wise, K.; Buzsáki, G. Gamma (40-100 Hz) oscillation in the hippocampus of the behaving rat. J. Neurosci. 1995, 15, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Kann, O.; Huchzermeyer, C.; Kovács, R.; Wirtz, S.; Schuelke, M. Gamma oscillations in the hippocampus require high complex I gene expression and strong functional performance of mitochondria. Brain 2011, 134, 345–358. [Google Scholar] [CrossRef] [PubMed]
- Nishida, M.; Juhász, C.; Sood, S.; Chugani, H.T.; Asano, E. Cortical glucose metabolism positively correlates with gamma-oscillations in nonlesional focal epilepsy. Neuroimage 2008, 42, 1275–1284. [Google Scholar] [CrossRef] [PubMed]
- Lord, L.D.; Expert, P.; Huckins, J.F.; Turkheimer, F.E. Cerebral energy metabolism and the brain’s functional network architecture: An integrative review. J. Cereb. Blood Flow Metab. 2013, 33, 1347–1354. [Google Scholar] [CrossRef]
- Jonas, P.; Bischofberger, J.; Fricker, D.; Miles, R. Interneuron Diversity series: Fast in, fast out--temporal and spatial signal processing in hippocampal interneurons. Trends Neurosci. 2004, 27, 30–40. [Google Scholar] [CrossRef]
- Hu, H.; Jonas, P. A supercritical density of Na(+) channels ensures fast signaling in GABAergic interneuron axons. Nat. Neurosci. 2014, 17, 686–693. [Google Scholar] [CrossRef]
- Kraushaar, U.; Jonas, P. Efficacy and stability of quantal GABA release at a hippocampal interneuron-principal neuron synapse. J. Neurosci. 2000, 20, 5594–5607. [Google Scholar] [CrossRef]
- Gulyás, A.I.; Buzsáki, G.; Freund, T.F.; Hirase, H. Populations of hippocampal inhibitory neurons express different levels of cytochrome c. Eur. J. Neurosci. 2006, 23, 2581–2594. [Google Scholar] [CrossRef]
- Cserép, C.; Pósfai, B.; Schwarcz, A.D.; Dénes, Á. Mitochondrial Ultrastructure Is Coupled to Synaptic Performance at Axonal Release Sites. eNeuro 2018, 5. [Google Scholar] [CrossRef]
- Li, Y.; Park, J.S.; Deng, J.H.; Bai, Y. Cytochrome c oxidase subunit IV is essential for assembly and respiratory function of the enzyme complex. J. Bioenerg. Biomembr. 2006, 38, 283–291. [Google Scholar] [CrossRef]
- Srinivasan, S.; Avadhani, N.G. Cytochrome c oxidase dysfunction in oxidative stress. Free Radic. Biol. Med. 2012, 53, 1252–1263. [Google Scholar] [CrossRef] [PubMed]
- Kolyva, C.; Ghosh, A.; Tachtsidis, I.; Highton, D.; Cooper, C.E.; Smith, M.; Elwell, C.E. Cytochrome c oxidase response to changes in cerebral oxygen delivery in the adult brain shows higher brain-specificity than haemoglobin. Neuroimage 2014, 85 Pt 1, 234–244. [Google Scholar] [CrossRef]
- Holper, L.; Mann, J.J. Test-retest reliability of brain mitochondrial cytochrome-c-oxidase assessed by functional near-infrared spectroscopy. J. Biomed. Opt. 2018, 23, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.L.; Economides, J.R.; Horton, J.C. Co-localization of glutamic acid decarboxylase and vesicular GABA transporter in cytochrome oxidase patches of macaque striate cortex. Vis. Neurosci. 2015, 32, E026. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.G.; Hevner, R.F.; Wong-Riley, M.T. Double labeling of cytochrome oxidase and gamma-aminobutyric acid in central nervous system neurons of adult cats. J. Neurosci. Methods 1989, 30, 189–195. [Google Scholar] [CrossRef]
- Lesicko, A.M.; Hristova, T.S.; Maigler, K.C.; Llano, D.A. Connectional Modularity of Top-Down and Bottom-Up Multimodal Inputs to the Lateral Cortex of the Mouse Inferior Colliculus. J. Neurosci. 2016, 36, 11037–11050. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, R.F.; Finch, D.M.; Babb, T.L.; Engel, J. Increased glucose metabolism during long-duration recurrent inhibition of hippocampal pyramidal cells. J. Neurosci. 1984, 4, 251–264. [Google Scholar] [CrossRef] [PubMed]
- McCasland, J.S.; Hibbard, L.S. GABAergic neurons in barrel cortex show strong, whisker-dependent metabolic activation during normal behavior. J. Neurosci. 1997, 17, 5509–5527. [Google Scholar] [CrossRef]
- Wang, J.H. Short-term cerebral ischemia causes the dysfunction of interneurons and more excitation of pyramidal neurons in rats. Brain Res. Bull. 2003, 60, 53–58. [Google Scholar] [CrossRef]
- Huchzermeyer, C.; Albus, K.; Gabriel, H.J.; Otáhal, J.; Taubenberger, N.; Heinemann, U.; Kovács, R.; Kann, O. Gamma oscillations and spontaneous network activity in the hippocampus are highly sensitive to decreases in pO2 and concomitant changes in mitochondrial redox state. J. Neurosci. 2008, 28, 1153–1162. [Google Scholar] [CrossRef]
- Barth, A.M.; Mody, I. Changes in hippocampal neuronal activity during and after unilateral selective hippocampal ischemia in vivo. J. Neurosci. 2011, 31, 851–860. [Google Scholar] [CrossRef] [PubMed]
- Whittaker, R.G.; Turnbull, D.M.; Whittington, M.A.; Cunningham, M.O. Impaired mitochondrial function abolishes gamma oscillations in the hippocampus through an effect on fast-spiking interneurons. Brain 2011, 134, e180. [Google Scholar] [CrossRef] [PubMed]
- Calabresi, P.; Centonze, D.; Bernardi, G. Cellular factors controlling neuronal vulnerability in the brain: A lesson from the striatum. Neurology 2000, 55, 1249–1255. [Google Scholar] [CrossRef] [PubMed]
- Pisani, A.; Bonsi, P.; Centonze, D.; Giacomini, P.; Calabresi, P. Involvement of intracellular calcium stores during oxygen/glucose deprivation in striatal large aspiny interneurons. J. Cereb. Blood Flow Metab. 2000, 20, 839–846. [Google Scholar] [CrossRef]
- Lewerenz, J.; Maher, P. Chronic Glutamate Toxicity in Neurodegenerative Diseases-What is the Evidence? Front. Neurosci. 2015, 9, 469. [Google Scholar] [CrossRef] [PubMed]
- Beal, M.F. Aging, energy, and oxidative stress in neurodegenerative diseases. Ann. Neurol. 1995, 38, 357–366. [Google Scholar] [CrossRef]
- Camandola, S.; Mattson, M.P. Brain metabolism in health, aging, and neurodegeneration. EMBO J. 2017, 36, 1474–1492. [Google Scholar] [CrossRef]
- Mark, L.P.; Prost, R.W.; Ulmer, J.L.; Smith, M.M.; Daniels, D.L.; Strottmann, J.M.; Brown, W.D.; Hacein-Bey, L. Pictorial review of glutamate excitotoxicity: Fundamental concepts for neuroimaging. AJNR Am. J. Neuroradiol. 2001, 22, 1813–1824. [Google Scholar]
- Steullet, P.; Cabungcal, J.H.; Kulak, A.; Kraftsik, R.; Chen, Y.; Dalton, T.P.; Cuenod, M.; Do, K.Q. Redox dysregulation affects the ventral but not dorsal hippocampus: Impairment of parvalbumin neurons, gamma oscillations, and related behaviors. J. Neurosci. 2010, 30, 2547–2558. [Google Scholar] [CrossRef]
- Cowell, R.M.; Blake, K.R.; Russell, J.W. Localization of the transcriptional coactivator PGC-1alpha to GABAergic neurons during maturation of the rat brain. J. Comp. Neurol. 2007, 502, 1–18. [Google Scholar] [CrossRef]
- Iacovelli, J.; Rowe, G.C.; Khadka, A.; Diaz-Aguilar, D.; Spencer, C.; Arany, Z.; Saint-Geniez, M. PGC-1α Induces Human RPE Oxidative Metabolism and Antioxidant Capacity. Investig. Ophthalmol. Vis. Sci. 2016, 57, 1038–1051. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Handschin, C.; Spiegelman, B.M. Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab. 2005, 1, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Austin, S.; St-Pierre, J. PGC1α and mitochondrial metabolism--emerging concepts and relevance in ageing and neurodegenerative disorders. J. Cell Sci. 2012, 125, 4963–4971. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Rompala, G.R.; Zhang, S.; Cowell, R.M.; Nakazawa, K. Social isolation exacerbates schizophrenia-like phenotypes via oxidative stress in cortical interneurons. Biol. Psychiatry 2013, 73, 1024–1034. [Google Scholar] [CrossRef]
- Cabungcal, J.H.; Steullet, P.; Kraftsik, R.; Cuenod, M.; Do, K.Q. Early-life insults impair parvalbumin interneurons via oxidative stress: Reversal by N-acetylcysteine. Biol. Psychiatry 2013, 73, 574–582. [Google Scholar] [CrossRef]
- Shetty, A.K.; Hattiangady, B.; Rao, M.S. Vulnerability of hippocampal GABA-ergic interneurons to kainate-induced excitotoxic injury during old age. J. Cell Mol. Med. 2009, 13, 2408–2423. [Google Scholar] [CrossRef]
- Cabungcal, J.H.; Nicolas, D.; Kraftsik, R.; Cuénod, M.; Do, K.Q.; Hornung, J.P. Glutathione deficit during development induces anomalies in the rat anterior cingulate GABAergic neurons: Relevance to schizophrenia. Neurobiol. Dis. 2006, 22, 624–637. [Google Scholar] [CrossRef]
- Dong, X.X.; Wang, Y.; Qin, Z.H. Molecular mechanisms of excitotoxicity and their relevance to pathogenesis of neurodegenerative diseases. Acta Pharmacol. Sin. 2009, 30, 379–387. [Google Scholar] [CrossRef]
- Hassel, B.; Sonnewald, U. Selective inhibition of the tricarboxylic acid cycle of GABAergic neurons with 3-nitropropionic acid in vivo. J. Neurochem. 1995, 65, 1184–1191. [Google Scholar] [CrossRef]
- Steullet, P.; Cabungcal, J.H.; Coyle, J.; Didriksen, M.; Gill, K.; Grace, A.A.; Hensch, T.K.; LaMantia, A.S.; Lindemann, L.; Maynard, T.M.; et al. Oxidative stress-driven parvalbumin interneuron impairment as a common mechanism in models of schizophrenia. Mol. Psychiatry 2017, 22, 936–943. [Google Scholar] [CrossRef]
- Torrey, E.F.; Barci, B.M.; Webster, M.J.; Bartko, J.J.; Meador-Woodruff, J.H.; Knable, M.B. Neurochemical markers for schizophrenia, bipolar disorder, and major depression in postmortem brains. Biol. Psychiatry 2005, 57, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Akbarian, S.; Kim, J.J.; Potkin, S.G.; Hagman, J.O.; Tafazzoli, A.; Bunney, W.E.; Jones, E.G. Gene expression for glutamic acid decarboxylase is reduced without loss of neurons in prefrontal cortex of schizophrenics. Arch. Gen. Psychiatry 1995, 52, 258–266. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.Y.; Lohmann, K.M.; Yang, C.K.; Zimmerman, E.I.; Pantazopoulos, H.; Herring, N.; Berretta, S.; Heckers, S.; Konradi, C. Bipolar disorder type 1 and schizophrenia are accompanied by decreased density of parvalbumin-and somatostatin-positive interneurons in the parahippocampal region. Acta neuropathol. 2011, 122, 615. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.-C.; Sibille, E. Reduced brain somatostatin in mood disorders: A common pathophysiological substrate and drug target? Front. Pharmacol. 2013, 4, 110. [Google Scholar] [CrossRef] [PubMed]
- Fatemi, S.H.; Halt, A.R.; Stary, J.M.; Kanodia, R.; Schulz, S.C.; Realmuto, G.R. Glutamic acid decarboxylase 65 and 67 kDa proteins are reduced in autistic parietal and cerebellar cortices. Biol. Psychiatry 2002, 52, 805–810. [Google Scholar] [CrossRef]
- Buckmaster, P.S.; Jongen-Rêlo, A.L. Highly specific neuron loss preserves lateral inhibitory circuits in the dentate gyrus of kainate-induced epileptic rats. J. Neurosci. 1999, 19, 9519–9529. [Google Scholar] [CrossRef]
- Dinocourt, C.; Petanjek, Z.; Freund, T.F.; Ben-Ari, Y.; Esclapez, M. Loss of interneurons innervating pyramidal cell dendrites and axon initial segments in the CA1 region of the hippocampus following pilocarpine-induced seizures. J. Comp. Neurol. 2003, 459, 407–425. [Google Scholar] [CrossRef]
- Palop, J.J.; Chin, J.; Roberson, E.D.; Wang, J.; Thwin, M.T.; Bien-Ly, N.; Yoo, J.; Ho, K.O.; Yu, G.Q.; Kreitzer, A.; et al. Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer’s disease. Neuron 2007, 55, 697–711. [Google Scholar] [CrossRef]
- Verret, L.; Mann, E.O.; Hang, G.B.; Barth, A.M.; Cobos, I.; Ho, K.; Devidze, N.; Masliah, E.; Kreitzer, A.C.; Mody, I.; et al. Inhibitory interneuron deficit links altered network activity and cognitive dysfunction in Alzheimer model. Cell 2012, 149, 708–721. [Google Scholar] [CrossRef]
- Cardin, J.A.; Carlén, M.; Meletis, K.; Knoblich, U.; Zhang, F.; Deisseroth, K.; Tsai, L.H.; Moore, C.I. Driving fast-spiking cells induces gamma rhythm and controls sensory responses. Nature 2009, 459, 663–667. [Google Scholar] [CrossRef]
- Sohal, V.S.; Zhang, F.; Yizhar, O.; Deisseroth, K. Parvalbumin neurons and gamma rhythms enhance cortical circuit performance. Nature 2009, 459, 698–702. [Google Scholar] [CrossRef] [PubMed]
- Dutton, S.B.; Makinson, C.D.; Papale, L.A.; Shankar, A.; Balakrishnan, B.; Nakazawa, K.; Escayg, A. Preferential inactivation of Scn1a in parvalbumin interneurons increases seizure susceptibility. Neurobiol. Dis. 2013, 49, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Annunziato, L.; Pannaccione, A.; Cataldi, M.; Secondo, A.; Castaldo, P.; Di Renzo, G.; Taglialatela, M. Modulation of ion channels by reactive oxygen and nitrogen species: A pathophysiological role in brain aging? Neurobiol. Aging 2002, 23, 819–834. [Google Scholar] [CrossRef]
- Rubio, S.E.; Vega-Flores, G.; Martínez, A.; Bosch, C.; Pérez-Mediavilla, A.; del Río, J.; Gruart, A.; Delgado-García, J.M.; Soriano, E.; Pascual, M. Accelerated aging of the GABAergic septohippocampal pathway and decreased hippocampal rhythms in a mouse model of Alzheimer’s disease. FASEB J. 2012, 26, 4458–4467. [Google Scholar] [CrossRef] [PubMed]
- Soler, H.; Dorca-Arévalo, J.; González, M.; Rubio, S.E.; Ávila, J.; Soriano, E.; Pascual, M. The GABAergic septohippocampal connection is impaired in a mouse model of tauopathy. Neurobiol. Aging 2017, 49, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Błaszczyk, J.W. Parkinson’s Disease and Neurodegeneration: GABA-Collapse Hypothesis. Front. Neurosci. 2016, 10, 269. [Google Scholar] [CrossRef] [PubMed]
- Moon, H.E.; Paek, S.H. Mitochondrial Dysfunction in Parkinson’s Disease. Exp. Neurobiol. 2015, 24, 103–116. [Google Scholar] [CrossRef]
- Feng, Z.J.; Zhang, X.; Chergui, K. Allosteric modulation of NMDA receptors alters neurotransmission in the striatum of a mouse model of Parkinson’s disease. Exp. Neurol. 2014, 255, 154–160. [Google Scholar] [CrossRef]
- De Jong, P.J.; Lakke, J.P.; Teelken, A.W. CSF GABA levels in Parkinson’s disease. Adv. Neurol. 1984, 40, 427–430. [Google Scholar]
- Firbank, M.J.; Parikh, J.; Murphy, N.; Killen, A.; Allan, C.L.; Collerton, D.; Blamire, A.M.; Taylor, J.P. Reduced occipital GABA in Parkinson disease with visual hallucinations. Neurology 2018, 91, e675–e685. [Google Scholar] [CrossRef] [PubMed]
- Mei, Y.; Gawai, K.R.; Nie, Z.; Ramkumar, V.; Helfert, R.H. Age-related reductions in the activities of antioxidant enzymes in the rat inferior colliculus. Hear. Res. 1999, 135, 169–180. [Google Scholar] [CrossRef]
- Zeng, L.; Yang, Y.; Hu, Y.; Sun, Y.; Du, Z.; Xie, Z.; Zhou, T.; Kong, W. Age-related decrease in the mitochondrial sirtuin deacetylase Sirt3 expression associated with ROS accumulation in the auditory cortex of the mimetic aging rat model. PLoS ONE 2014, 9, e88019. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.D.; He, L.; Tu, C.; Guo, X.A.; Yu, S.; Liu, K.; Gong, S. Mitochondrial DNA 3,860-bp Deletion Increases with Aging in the Auditory Nervous System of C57BL/6J Mice. ORL J. Otorhinolaryngol. Relat. Spec. 2019, 81, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Sanes, D.H.; Rubel, E.W. The ontogeny of inhibition and excitation in the gerbil lateral superior olive. J. Neurosci. 1988, 8, 682–700. [Google Scholar] [CrossRef]
- Wu, S.H.; Kelly, J.B. Response of neurons in the lateral superior olive and medial nucleus of the trapezoid body to repetitive stimulation: Intracellular and extracellular recordings from mouse brain slice. Hear. Res. 1993, 68, 189–201. [Google Scholar] [CrossRef]
- Brosel, S.; Grothe, B.; Kunz, L. An auditory brainstem nucleus as a model system for neuronal metabolic demands. Eur. J. Neurosci. 2018, 47, 222–235. [Google Scholar] [CrossRef] [PubMed]
- Caspary, D.M.; Llano, D.A. Auditory thalamic circuits and GABA. Hear. Res. 2017, 349, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Llano, D.A.; Turner, J.; Caspary, D.M. Diminished cortical inhibition in an aging mouse model of chronic tinnitus. J. Neurosci. 2012, 32, 16141–16148. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hoffman, H.; Reed, G. Epidemiology of tinnitus. In Tinnitus: Theory and Management; Snow, J., Ed.; BC Decker Inc.: New York, NY, USA, 2014; pp. 16–41. [Google Scholar]
- Guest, H.; Munro, K.J.; Prendergast, G.; Howe, S.; Plack, C.J. Tinnitus with a normal audiogram: Relation to noise exposure but no evidence for cochlear synaptopathy. Hear. Res. 2017, 344, 265–274. [Google Scholar] [CrossRef]
- Sanchez, T.G.; Medeiros, I.R.; Levy, C.P.; Ramalho, J.a.R.; Bento, R.F. Tinnitus in normally hearing patients: Clinical aspects and repercussions. Braz. J. Otorhinolaryngol. 2005, 71, 427–431. [Google Scholar] [CrossRef]
- Barnea, G.; Attias, J.; Gold, S.; Shahar, A. Tinnitus with normal hearing sensitivity: Extended high-frequency audiometry and auditory-nerve brain-stem-evoked responses. Audiology 1990, 29, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Jiang, Q.; Wu, X.; Xie, S.; Feng, Y.; Sun, H. The Influence of Metabolic Syndrome on the Prognosis of Idiopathic Sudden Sensorineural Hearing Loss. Otol. Neurotol. 2019, 40, 994–997. [Google Scholar] [CrossRef] [PubMed]
- Gosselin, E.J.; Yanick, P. Audiologic and metabolic findings in 90 patients with fluctuant hearing loss. J. Am. Audiol. Soc. 1976, 2, 15–18. [Google Scholar] [PubMed]
- Kim, H.J.; Lee, H.J.; An, S.Y.; Sim, S.; Park, B.; Kim, S.W.; Lee, J.S.; Hong, S.K.; Choi, H.G. Analysis of the prevalence and associated risk factors of tinnitus in adults. PLoS ONE 2015, 10, e0127578. [Google Scholar] [CrossRef]
- Wang, J.P.; Fan, R.H.; Wang, Y.; Mei, Y. Treatment of hyperlipoidemia by xiaozhi capsule: A clinical efficacy research]. Chin. J. Integr. Tradit. West. Med. 2013, 33, 736–740. [Google Scholar]
- Canis, M.; Olzowy, B.; Welz, C.; Suckfüll, M.; Stelter, K. Simvastatin and Ginkgo biloba in the treatment of subacute tinnitus: A retrospective study of 94 patients. Am. J. Otolaryngol. 2011, 32, 19–23. [Google Scholar] [CrossRef]
- Larsson, M.; Lietzau, G.; Nathanson, D.; Östenson, C.G.; Mallard, C.; Johansson, M.E.; Nyström, T.; Patrone, C.; Darsalia, V. Diabetes negatively affects cortical and striatal GABAergic neurons: An effect that is partially counteracted by exendin-4. Biosci. Rep. 2016, 36. [Google Scholar] [CrossRef]
- Castillo-Gómez, E.; Coviello, S.; Perez-Rando, M.; Curto, Y.; Carceller, H.; Salvador, A.; Nacher, J. Streptozotocin diabetic mice display depressive-like behavior and alterations in the structure, neurotransmission and plasticity of medial prefrontal cortex interneurons. Brain Res. Bull. 2015, 116, 45–56. [Google Scholar] [CrossRef]
- Vasilyeva, O.N.; Frisina, S.T.; Zhu, X.; Walton, J.P.; Frisina, R.D. Interactions of hearing loss and diabetes mellitus in the middle age CBA/CaJ mouse model of presbycusis. Hear. Res. 2009, 249, 44–53. [Google Scholar] [CrossRef]
- Inan, M.; Zhao, M.; Manuszak, M.; Karakaya, C.; Rajadhyaksha, A.M.; Pickel, V.M.; Schwartz, T.H.; Goldstein, P.A.; Manfredi, G. Energy deficit in parvalbumin neurons leads to circuit dysfunction, impaired sensory gating and social disability. Neurobiol. Dis. 2016, 93, 35–46. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ibrahim, B.A.; Llano, D.A. Aging and Central Auditory Disinhibition: Is It a Reflection of Homeostatic Downregulation or Metabolic Vulnerability? Brain Sci. 2019, 9, 351. https://doi.org/10.3390/brainsci9120351
Ibrahim BA, Llano DA. Aging and Central Auditory Disinhibition: Is It a Reflection of Homeostatic Downregulation or Metabolic Vulnerability? Brain Sciences. 2019; 9(12):351. https://doi.org/10.3390/brainsci9120351
Chicago/Turabian StyleIbrahim, Baher A., and Daniel A. Llano. 2019. "Aging and Central Auditory Disinhibition: Is It a Reflection of Homeostatic Downregulation or Metabolic Vulnerability?" Brain Sciences 9, no. 12: 351. https://doi.org/10.3390/brainsci9120351
APA StyleIbrahim, B. A., & Llano, D. A. (2019). Aging and Central Auditory Disinhibition: Is It a Reflection of Homeostatic Downregulation or Metabolic Vulnerability? Brain Sciences, 9(12), 351. https://doi.org/10.3390/brainsci9120351