Neurodegeneration and Sensorimotor Deficits in the Mouse Model of Traumatic Brain Injury

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Fluid Percussion Injury
2.3. Western Blotting
2.4. Cell Death Analysis
2.5. Behavioral Testing
2.6. Statistical Analyses
3. Results
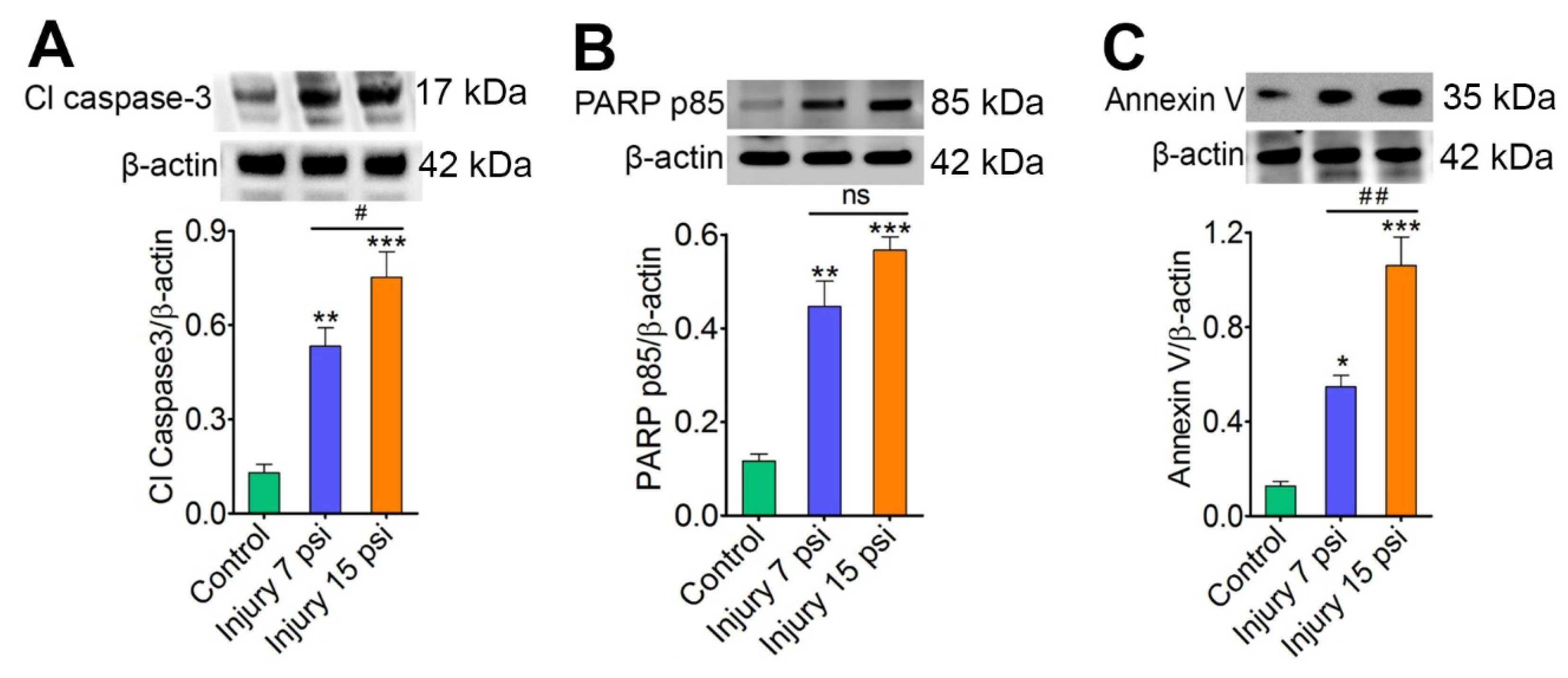
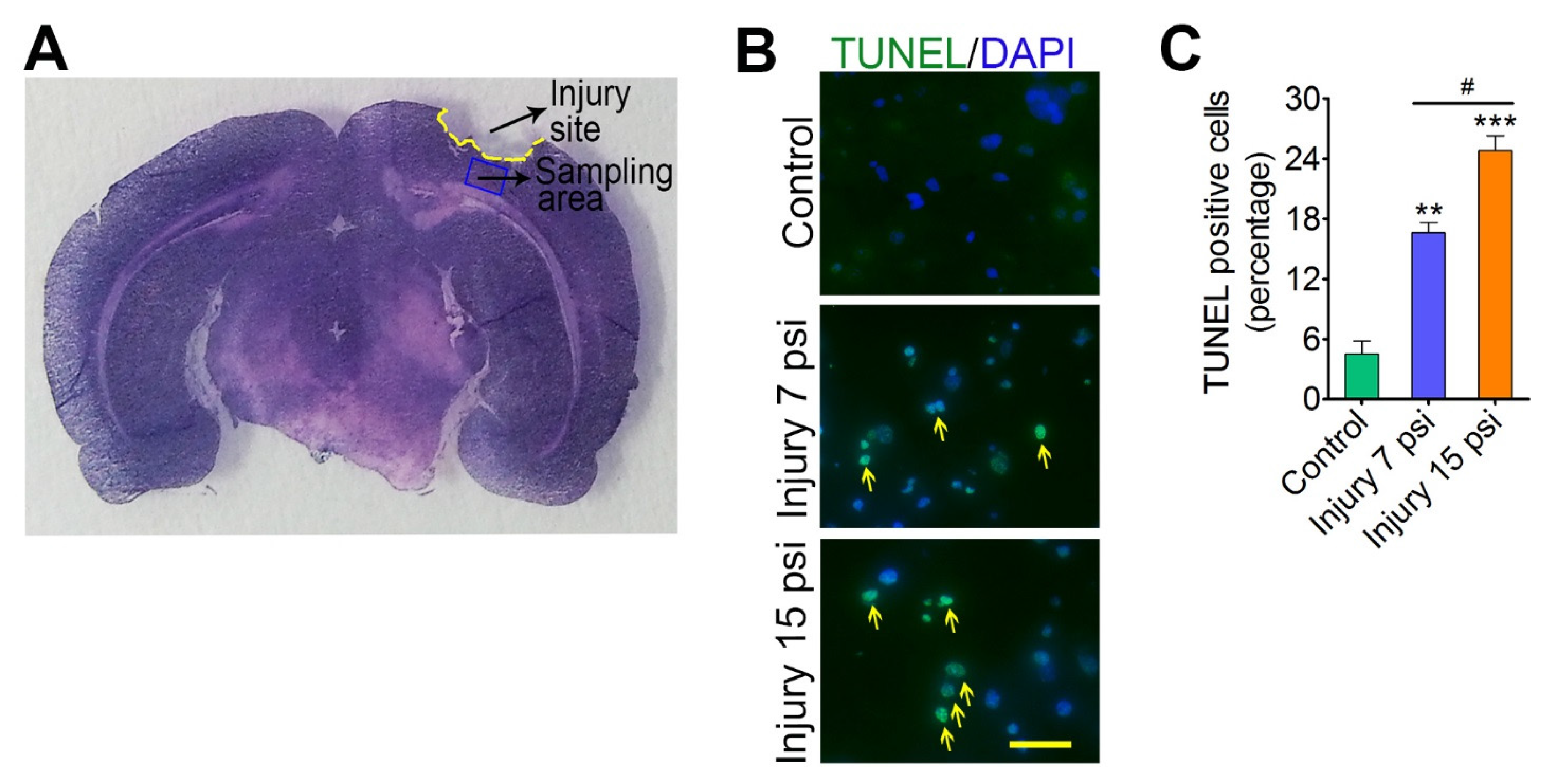
3.1. Mild and Moderate TBI Leads to Progression of Neurodegeneration
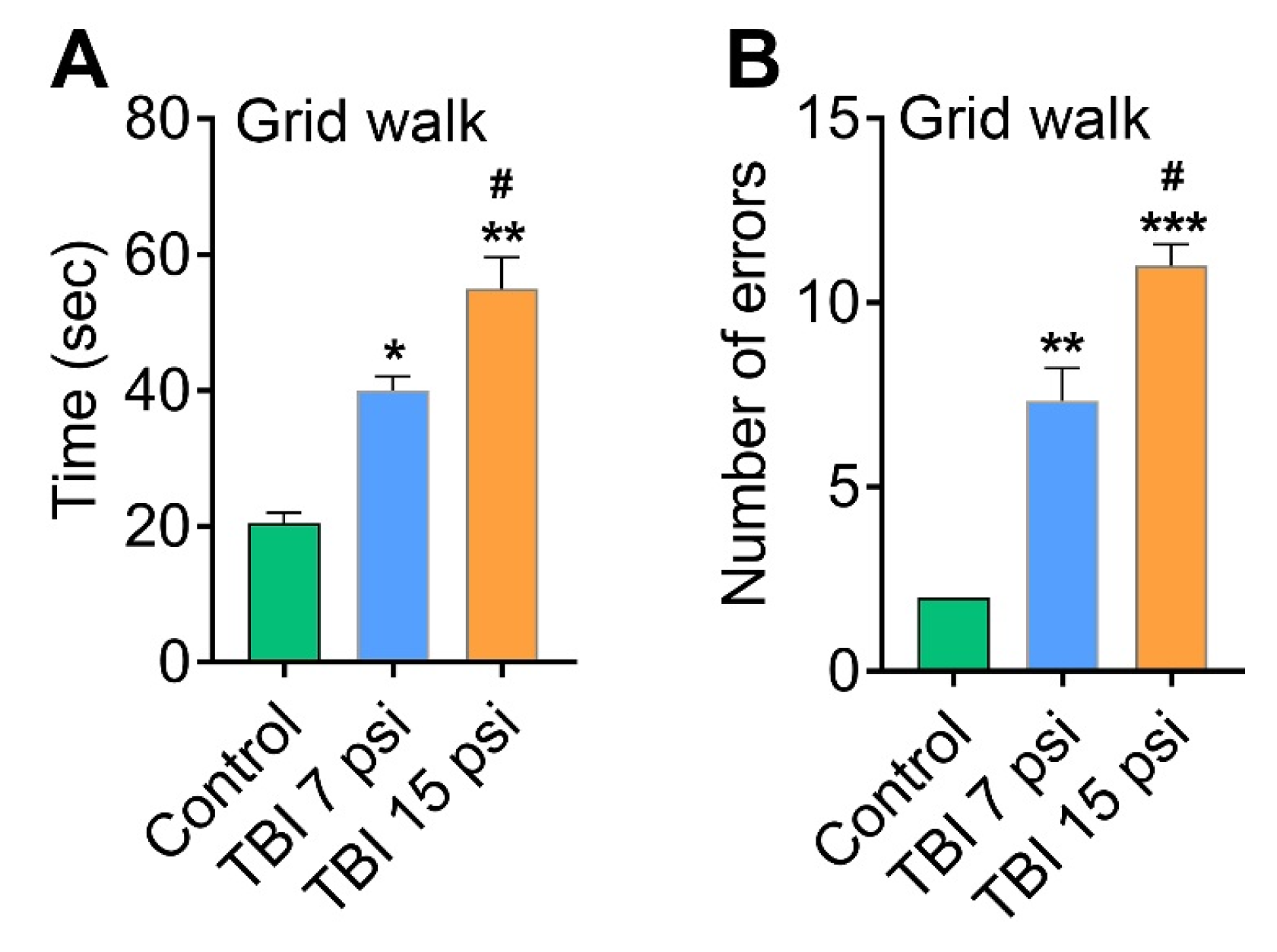
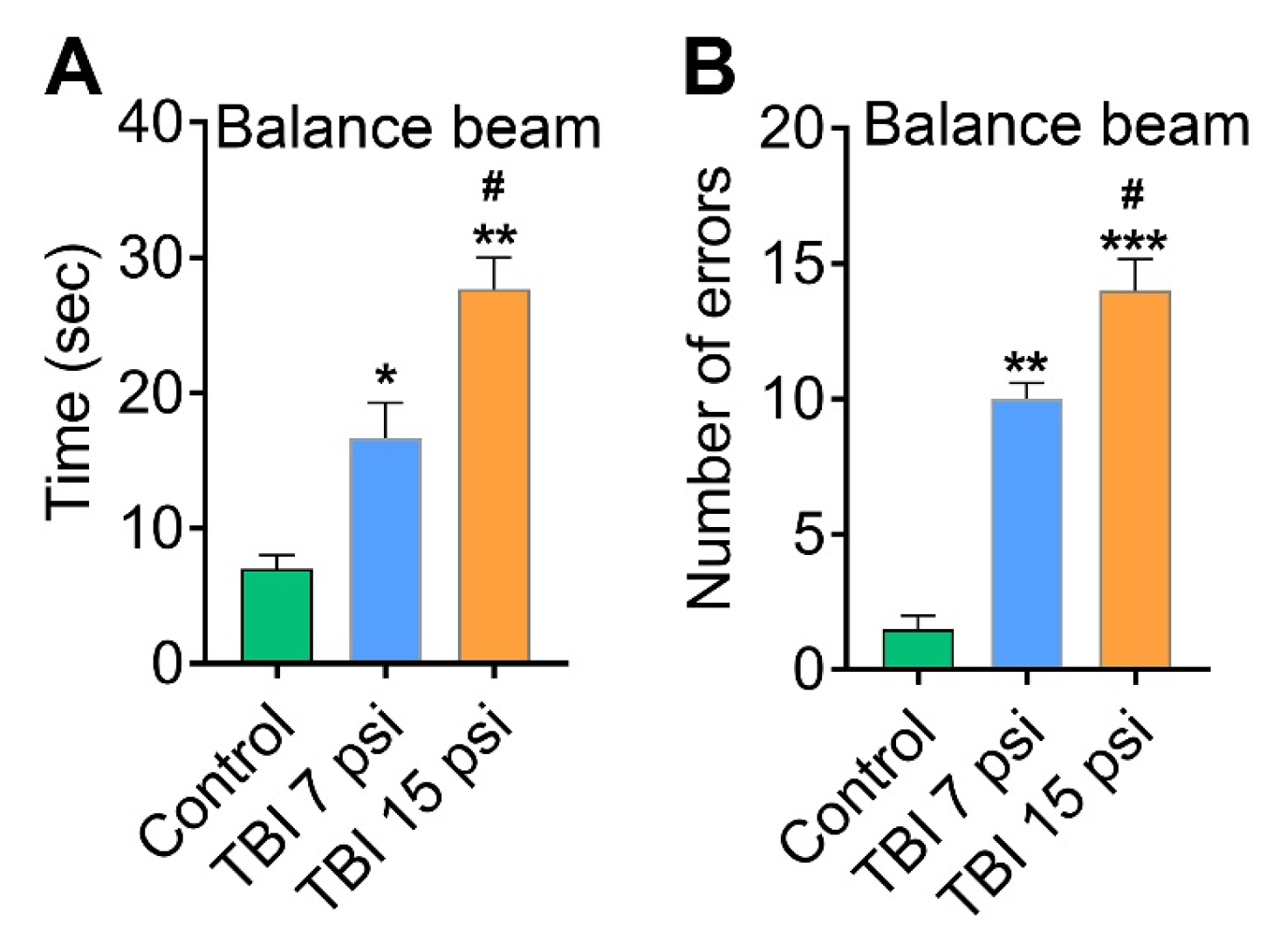
3.2. Mild and Moderate TBI Causes Sensorimotor Deficits Concomitant with Neurodegeneration
4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Chen, Y.H.; Kang, J.H.; Lin, H.C. Patients with traumatic brain injury: Population-based study suggests increased risk of stroke. Stroke J. Cereb. Circ. 2011, 42, 2733–2739. [Google Scholar] [CrossRef] [PubMed]
- Del Zoppo, G.J.; Mabuchi, T. Cerebral microvessel responses to focal ischemia. J. Cereb. Blood Flow Metab. 2003, 23, 879–894. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.A.; Bell, J.M.; Breiding, M.J.; Xu, L. Traumatic Brain Injury-Related Emergency Department Visits, Hospitalizations, and Deaths—United States, 2007 and 2013. MMWR Surveill. Summ. 2017, 66, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Teasdale, G.; Jennett, B. Assessment of coma and impaired consciousness: A practical scale. Lancet 1974, 2, 81–84. [Google Scholar] [CrossRef]
- Goldstein, L.E.; Fisher, A.M.; Tagge, C.A.; Zhang, X.L.; Velisek, L.; Sullivan, J.A.; Upreti, C.; Kracht, J.M.; Ericsson, M.; Wojnarowicz, M.W.; et al. Chronic traumatic encephalopathy in blast-exposed military veterans and a blast neurotrauma mouse model. Sci. Transl. Med. 2012, 4, 134–160. [Google Scholar]
- McInnes, K.; Friesen, C.L.; MacKenzie, D.E.; Westwood, D.A.; Boe, S.G. Mild Traumatic Brain Injury (mTBI) and chronic cognitive impairment: A scoping review. PLoS ONE 2017, 12, e0174847. [Google Scholar] [CrossRef] [PubMed]
- Prins, M.; Greco, T.; Alexander, D.; Giza, C.C. The pathophysiology of traumatic brain injury at a glance. Dis. Model. Mech. 2013, 6, 1307–1315. [Google Scholar] [CrossRef] [PubMed]
- Abdul-Muneer, P.M.; Chandra, N.; Haorah, J. Interactions of oxidative stress and neurovascular inflammation in the pathogenesis of traumatic brain injury. Mol. Neurobiol. 2015, 51, 966–979. [Google Scholar] [CrossRef] [PubMed]
- Abdul-Muneer, P.M.; Conte, A.A.; Haldar, D.; Long, M.; Patel, R.K.; Santhakumar, V.; Overall, C.M.; Pfister, B.J. Traumatic brain injury induced matrix metalloproteinase2 cleaves CXCL12alpha (stromal cell derived factor 1alpha) and causes neurodegeneration. Brain Behav. Immun. 2017, 59, 190–199. [Google Scholar] [CrossRef] [PubMed]
- Abdul-Muneer, P.M.; Long, M.; Conte, A.A.; Santhakumar, V.; Pfister, B.J. High Ca2+ Influx during Traumatic Brain Injury Leads to Caspase-1-Dependent Neuroinflammation and Cell Death. Mol. Neurobiol. 2017, 54, 3964–3975. [Google Scholar] [CrossRef] [PubMed]
- Abdul-Muneer, P.M.; Pfister, B.J.; Haorah, J.; Chandra, N. Role of Matrix Metalloproteinases in the Pathogenesis of Traumatic Brain Injury. Mol. Neurobiol. 2016, 53, 6106–6123. [Google Scholar] [CrossRef] [PubMed]
- Abdul-Muneer, P.M.; Schuetz, H.; Wang, F.; Skotak, M.; Jones, J.; Gorantla, S.; Zimmerman, M.C.; Chandra, N.; Haorah, J. Induction of oxidative and nitrosative damage leads to cerebrovascular inflammation in an animal model of mild traumatic brain injury induced by primary blast. Free Radic. Biol. Med. 2013, 60, 282–291. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.K.; Prasad, N.; Kuwar, R.; Haldar, D.; Abdul-Muneer, P.M. Transforming growth factor-beta 1 signaling regulates neuroinflammation and apoptosis in mild traumatic brain injury. Brain Behav. Immun. 2017, 64, 244–258. [Google Scholar] [CrossRef] [PubMed]
- Abdul-Muneer, P.M.; Bhowmick, S.; Briski, N. Angiotensin II Causes Neuronal Damage in Stretch-Injured Neurons: Protective Effects of Losartan, an Angiotensin T1 Receptor Blocker. Mol. Neurobiol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Rivera, S.; Monclus, E.A.; Synenki, L.; Zirk, A.; Eisenbart, J.; Feghali-Bostwick, C.; Mutlu, G.M.; Budinger, G.R.; Chandel, N.S. Mitochondrial reactive oxygen species regulate transforming growth factor-β signaling. J. Biol. Chem. 2013, 288, 770–777. [Google Scholar] [CrossRef] [PubMed]
- Krstic, J.; Trivanovic, D.; Mojsilovic, S.; Santibanez, J.F. Transforming Growth Factor-Beta and Oxidative Stress Interplay: Implications in Tumorigenesis and Cancer Progression. Oxid. Med. Cell. Longev. 2015, 2015, 654594. [Google Scholar] [CrossRef] [PubMed]
- Lazebnik, Y.A.; Kaufmann, S.H.; Desnoyers, S.; Poirier, G.G.; Earnshaw, W.C. Cleavage of poly(ADP-ribose) polymerase by a proteinase with properties like ICE. Nature 1994, 371, 346–347. [Google Scholar] [CrossRef] [PubMed]
- Chaitanya, G.V.; Steven, A.J.; Babu, P.P. PARP-1 cleavage fragments: Signatures of cell-death proteases in neurodegeneration. Cell Commun. Signal. 2010, 8, 31. [Google Scholar] [CrossRef] [PubMed]
- Enari, M.; Sakahira, H.; Yokoyama, H.; Okawa, K.; Iwamatsu, A.; Nagata, S. A caspase-activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD. Nature 1998, 391, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Sakahira, H.; Enari, M.; Nagata, S. Cleavage of CAD inhibitor in CAD activation and DNA degradation during apoptosis. Nature 1998, 391, 96–99. [Google Scholar] [CrossRef] [PubMed]
- Cardo-Vila, M.; Arap, W.; Pasqualini, R. Alpha v beta 5 integrin-dependent programmed cell death triggered by a peptide mimic of annexin V. Mol. Cell 2003, 11, 1151–1162. [Google Scholar] [CrossRef]
- Kabadi, S.V.; Hilton, G.D.; Stoica, B.A.; Zapple, D.N.; Faden, A.I. Fluid-percussion-induced traumatic brain injury model in rats. Nat. Protoc. 2010, 5, 1552–1563. [Google Scholar] [CrossRef] [PubMed]
- Faul, F.; Erdfelder, E.; Lang, A.G.; Buchner, A. G*Power 3: A flexible statistical power analysis program for the social, behavioral, and biomedical sciences. Behav. Res. Methods 2007, 39, 175–191. [Google Scholar] [CrossRef] [PubMed]
- Abdul-Muneer, P.M.; Alikunju, S.; Szlachetka, A.M.; Murrin, L.C.; Haorah, J. Impairment of brain endothelial glucose transporter by methamphetamine causes blood-brain barrier dysfunction. Mol. Neurodegener. 2011, 6, 23. [Google Scholar] [CrossRef] [PubMed]
- Abdul-Muneer, P.M.; Alikunju, S.; Schuetz, H.; Szlachetka, A.M.; Ma, X.; Haorah, J. Impairment of Thiamine Transport at the GUT-BBB-AXIS Contributes to Wernicke’s Encephalopathy. Mol. Neurobiol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Abdul-Muneer, P.M.; Alikunju, S.; Szlachetka, A.M.; Haorah, J. The mechanisms of cerebral vascular dysfunction and neuroinflammation by MMP-mediated degradation of VEGFR-2 in alcohol ingestion. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1167–1177. [Google Scholar] [CrossRef] [PubMed]
- Shin, Y.S.; Hwang, H.S.; Kang, S.U.; Chang, J.W.; Oh, Y.T.; Kim, C.H. Inhibition of p38 mitogen-activated protein kinase ameliorates radiation-induced ototoxicity in zebrafish and cochlea-derived cell lines. Neurotoxicology 2014, 40, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Onyszchuk, G.; Al-Hafez, B.; He, Y.Y.; Bilgen, M.; Berman, N.E.; Brooks, W.M. A mouse model of sensorimotor controlled cortical impact: Characterization using longitudinal magnetic resonance imaging, behavioral assessments and histology. J. Neurosci. Methods 2007, 160, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Ohtake, Y.; Park, D.; Abdul-Muneer, P.M.; Li, H.; Xu, B.; Sharma, K.; Smith, G.M.; Selzer, M.E.; Li, S. The effect of systemic PTEN antagonist peptides on axon growth and functional recovery after spinal cord injury. Biomaterials 2014, 35, 4610–4626. [Google Scholar] [CrossRef] [PubMed]
- Fleming, S.M.; Ekhator, O.R.; Ghisays, V. Assessment of sensorimotor function in mouse models of Parkinson’s disease. J. Vis. Exp. 2013. [Google Scholar] [CrossRef] [PubMed]
- Luong, T.N.; Carlisle, H.J.; Southwell, A.; Patterson, P.H. Assessment of motor balance and coordination in mice using the balance beam. J. Vis. Exp. 2011, 10. [Google Scholar] [CrossRef] [PubMed]
- Deacon, R.M. Measuring the strength of mice. J. Vis. Exp. 2013. [Google Scholar] [CrossRef] [PubMed]
- Brooks, S.P.; Dunnett, S.B. Tests to assess motor phenotype in mice: A user’s guide. Nat. Rev. Neurosci. 2009, 10, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Protais, P.; Costentin, J.; Schwartz, J.C. Climbing behavior induced by apomorphine in mice: A simple test for the study of dopamine receptors in striatum. Psychopharmacology 1976, 50, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, W.; Yan, Q.; Zhang, Y.; Fan, Z. Moderate injury in motor-sensory cortex causes behavioral deficits accompanied by electrophysiological changes in mice adulthood. PLoS ONE 2017, 12, e0171976. [Google Scholar] [CrossRef] [PubMed]
- Simon-O’Brien, E.; Gauthier, D.; Riban, V.; Verleye, M. Etifoxine improves sensorimotor deficits and reduces glial activation, neuronal degeneration, and neuroinflammation in a rat model of traumatic brain injury. J. Neuroinflamm. 2016, 13, 203. [Google Scholar] [CrossRef] [PubMed]
- Bao, F.; Shultz, S.R.; Hepburn, J.D.; Omana, V.; Weaver, L.C.; Cain, D.P.; Brown, A. A CD11d monoclonal antibody treatment reduces tissue injury and improves neurological outcome after fluid percussion brain injury in rats. J. Neurotrauma 2012, 29, 2375–2392. [Google Scholar] [CrossRef] [PubMed]
- Shultz, S.R.; Tan, X.L.; Wright, D.K.; Liu, S.J.; Semple, B.D.; Johnston, L.; Jones, N.C.; Cook, A.D.; Hamilton, J.A.; O’Brien, T.J. Granulocyte-macrophage colony-stimulating factor is neuroprotective in experimental traumatic brain injury. J. Neurotrauma 2014, 31, 976–983. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, V.P.; Wright, D.K.; Wong, K.; O’Brien, T.J.; Rajan, R.; Shultz, S.R. Experimental Traumatic Brain Injury Results in Long-Term Recovery of Functional Responsiveness in Sensory Cortex but Persisting Structural Changes and Sensorimotor, Cognitive, and Emotional Deficits. J. Neurotrauma 2015, 32, 1333–1346. [Google Scholar] [CrossRef] [PubMed]
- Wright, D.K.; Trezise, J.; Kamnaksh, A.; Bekdash, R.; Johnston, L.A.; Ordidge, R.; Semple, B.D.; Gardner, A.J.; Stanwell, P.; O’Brien, T.J.; et al. Behavioral, blood, and magnetic resonance imaging biomarkers of experimental mild traumatic brain injury. Sci. Rep. 2016, 6, 28713. [Google Scholar] [CrossRef] [PubMed]
- Wright, D.K.; Liu, S.; van der Poel, C.; McDonald, S.J.; Brady, R.D.; Taylor, L.; Yang, L.; Gardner, A.J.; Ordidge, R.; O’Brien, T.J.; et al. Traumatic Brain Injury Results in Cellular, Structural and Functional Changes Resembling Motor Neuron Disease. Cereb. Cortex 2017, 27, 4503–4515. [Google Scholar] [CrossRef] [PubMed]
- Posner, M.I.; Walker, J.A.; Friedrich, F.J.; Rafal, R.D. Effects of parietal injury on covert orienting of attention. J. Neurosci. 1984, 4, 1863–1874. [Google Scholar] [PubMed]
- Han, K.; Chapman, S.B.; Krawczyk, D.C. Disrupted Intrinsic Connectivity among Default, Dorsal Attention, and Frontoparietal Control Networks in Individuals with Chronic Traumatic Brain Injury. J. Int. Neuropsychol. Soc. 2016, 22, 263–279. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Y.; Bakhadirov, K.; Abdi, H.; Devous, M.D., Sr.; de la Plata, C.D.M.; Moore, C.; Madden, C.J.; Diaz-Arrastia, R. Longitudinal changes of structural connectivity in traumatic axonal injury. Neurology 2011, 77, 818–826. [Google Scholar] [CrossRef] [PubMed]
- Farbota, K.D.; Sodhi, A.; Bendlin, B.B.; McLaren, D.G.; Xu, G.; Rowley, H.A.; Johnson, S.C. Longitudinal volumetric changes following traumatic brain injury: A tensor-based morphometry study. J. Int. Neuropsychol. Soc. 2012, 18, 1006–1018. [Google Scholar] [CrossRef] [PubMed]
- Myburgh, J.A.; Cooper, D.J.; Finfer, S.R.; Venkatesh, B.; Jones, D.; Higgins, A.; Bishop, N.; Higlett, T. Australasian Traumatic Brain Injury Study Investigators for the Australian; New Zealand Intensive Care Society Clinical Trials Group. Epidemiology and 12-month outcomes from traumatic brain injury in australia and new zealand. J. Trauma 2008, 64, 854–862. [Google Scholar] [CrossRef] [PubMed]
- Masson, F.; Thicoipe, M.; Aye, P.; Mokni, T.; Senjean, P.; Schmitt, V.; Dessalles, P.H.; Cazaugade, M.; Labadens, P. Aquitaine Group for Severe Brain Injuries Study. Epidemiology of severe brain injuries: A prospective population-based study. J. Trauma 2001, 51, 481–489. [Google Scholar] [PubMed]
- Thompson, H.J.; Lifshitz, J.; Marklund, N.; Grady, M.S.; Graham, D.I.; Hovda, D.A.; McIntosh, T.K. Lateral fluid percussion brain injury: A 15-year review and evaluation. J. Neurotrauma 2005, 22, 42–75. [Google Scholar] [CrossRef] [PubMed]
- Yamaki, T.; Murakami, N.; Iwamoto, Y.; Sakakibara, T.; Kobori, N.; Ueda, S.; Kikuchi, T.; Uwahodo, Y. Evaluation of learning and memory dysfunction and histological findings in rats with chronic stage contusion and diffuse axonal injury. Brain Res. 1997, 752, 151–160. [Google Scholar] [CrossRef]
- Levasseur, J.E.; Patterson, J.L., Jr.; Ghatak, N.R.; Kontos, H.A.; Choi, S.C. Combined effect of respirator-induced ventilation and superoxide dismutase in experimental brain injury. J. Neurosurg. 1989, 71, 573–577. [Google Scholar] [CrossRef] [PubMed]
- Dixon, C.E.; Lyeth, B.G.; Povlishock, J.T.; Findling, R.L.; Hamm, R.J.; Marmarou, A.; Young, H.F.; Hayes, R.L. A fluid percussion model of experimental brain injury in the rat. J. Neurosurg. 1987, 67, 110–119. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhowmick, S.; D‘Mello, V.; Ponery, N.; Muneer, P.M.A. Neurodegeneration and Sensorimotor Deficits in the Mouse Model of Traumatic Brain Injury. Brain Sci. 2018, 8, 11. https://doi.org/10.3390/brainsci8010011
Bhowmick S, D‘Mello V, Ponery N, Muneer PMA. Neurodegeneration and Sensorimotor Deficits in the Mouse Model of Traumatic Brain Injury. Brain Sciences. 2018; 8(1):11. https://doi.org/10.3390/brainsci8010011
Chicago/Turabian StyleBhowmick, Saurav, Veera D‘Mello, Nizmi Ponery, and P. M. Abdul Muneer. 2018. "Neurodegeneration and Sensorimotor Deficits in the Mouse Model of Traumatic Brain Injury" Brain Sciences 8, no. 1: 11. https://doi.org/10.3390/brainsci8010011
APA StyleBhowmick, S., D‘Mello, V., Ponery, N., & Muneer, P. M. A. (2018). Neurodegeneration and Sensorimotor Deficits in the Mouse Model of Traumatic Brain Injury. Brain Sciences, 8(1), 11. https://doi.org/10.3390/brainsci8010011