New Insights into the Role of Neuron-Specific Enolase in Neuro-Inflammation, Neurodegeneration, and Neuroprotection


{kind=link}
Abstract
1. Introduction
2. NSE in Neurons and Glia
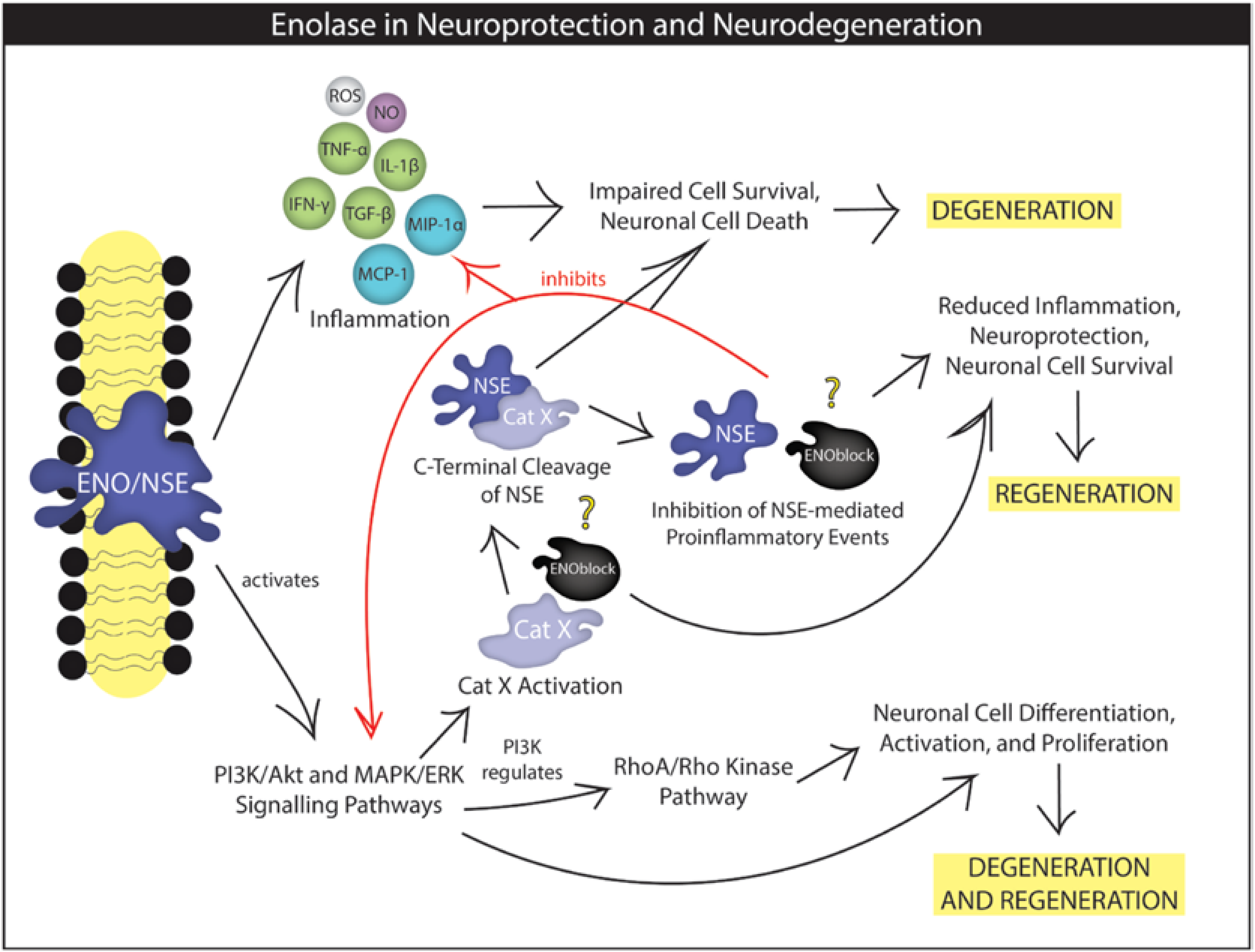
3. NSE as Prognostic Factors in Patients during Neuroinflammation
4. NSE in Neuronal and Glial Cell Activation, Differentiation, and Migration
5. NSE in Neurodegeneration and Neuroprotection
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Shimizu, A.; Suzuki, F.; Kato, K. Characterization of alpha alpha, beta beta, gamma gamma and alpha gamma human enolase isozymes, and preparation of hybrid enolases (alpha gamma, beta gamma and alpha beta) from homodimeric forms. Biochim. Biophys. Acta 1983, 748, 278–284. [Google Scholar] [CrossRef]
- Merkulova, T.; Dehaupas, M.; Nevers, M.C.; Creminon, C.; Alameddine, H.; Keller, A. Differential modulation of alpha, beta and gamma enolase isoforms in regenerating mouse skeletal muscle. Eur. J. Biochem. 2000, 267, 3735–3743. [Google Scholar] [CrossRef] [PubMed]
- Piast, M.; Kustrzeba-Wojcicka, I.; Matusiewicz, M.; Banas, T. Molecular evolution of enolase. Acta Biochim. Pol. 2005, 52, 507–513. [Google Scholar] [PubMed]
- Deloulme, J.C.; Helies, A.; Ledig, M.; Lucas, M.; Sensenbrenner, M. A comparative study of the distribution of alpha- and gamma-enolase subunits in cultured rat neural cells and fibroblasts. Int. J. Dev. Neurosci. 1997, 15, 183–194. [Google Scholar] [CrossRef]
- Hafner, A.; Glavan, G.; Obermajer, N.; Zivin, M.; Schliebs, R.; Kos, J. Neuroprotective role of gamma-enolase in microglia in a mouse model of Alzheimer’s disease is regulated by cathepsin X. Aging Cell 2013, 12, 604–614. [Google Scholar] [CrossRef] [PubMed]
- Ergun, R.; Bostanci, U.; Akdemir, G.; Beskonakli, E.; Kaptanoglu, E.; Gursoy, F.; Taskin, Y. Prognostic value of serum neuron-specific enolase levels after head injury. Neurol. Res. 1998, 20, 418–420. [Google Scholar] [CrossRef] [PubMed]
- Kleine, T.O.; Benes, L.; Zofel, P. Studies of the brain specificity of S100B and neuron-specific enolase (NSE) in blood serum of acute care patients. Brain Res. Bull. 2003, 61, 265–279. [Google Scholar] [CrossRef]
- Streitburger, D.P.; Arelin, K.; Kratzsch, J.; Thiery, J.; Steiner, J.; Villringer, A.; Mueller, K.; Schroeter, M.L. Validating serum S100B and neuron-specific enolase as biomarkers for the human brain—A combined serum, gene expression and MRI study. PLoS ONE 2012, 7, e43284. [Google Scholar] [CrossRef] [PubMed]
- Egea-Guerrero, J.J.; Murillo-Cabezas, F.; Rodriguez-Rodriguez, A.; Gordillo-Escobar, E.; Revuelto-Rey, J.; Munoz-Sanchez, M.A.; Leon-Justel, A.; Vilches-Arenas, A. (An experimental model of mass-type brain damage in the rat: Expression of brain damage based on neurospecific enolase and protein S100B). Med. Intensiv. 2014, 38, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Johnsson, P.; Blomquist, S.; Luhrs, C.; Malmkvist, G.; Alling, C.; Solem, J.O.; Stahl, E. Neuron-specific enolase increases in plasma during and immediately after extracorporeal circulation. Ann. Thorac. Surg. 2000, 69, 750–754. [Google Scholar] [CrossRef]
- Sahu, S.; Nag, D.S.; Swain, A.; Samaddar, D.P. Biochemical changes in the injured brain. World J. Biol. Chem. 2017, 8, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, R.; Scarna, H.; Pujol, J.F. Neuron-specific enolase in cerebrospinal fluid: A possible indicator of neuronal damage in kainic acid lesions. Neurosci. Lett. 1984, 45, 147–150. [Google Scholar] [CrossRef]
- Kwon, B.K.; Tetzlaff, W.; Grauer, J.N.; Beiner, J.; Vaccaro, A.R. Pathophysiology and pharmacologic treatment of acute spinal cord injury. Spine J. 2004, 4, 451–464. [Google Scholar] [CrossRef] [PubMed]
- McDonald, J.W.; Sadowsky, C. Spinal-cord injury. Lancet 2002, 359, 417–425. [Google Scholar] [CrossRef]
- Ondruschka, B.; Pohlers, D.; Sommer, G.; Schober, K.; Teupser, D.; Franke, H.; Dressler, J. S100B and NSE as useful postmortem biochemical markers of traumatic brain injury in autopsy cases. J. Neurotrauma 2013, 30, 1862–1871. [Google Scholar] [CrossRef] [PubMed]
- Cao, F.; Yang, X.F.; Liu, W.G.; Hu, W.W.; Li, G.; Zheng, X.J.; Shen, F.; Zhao, X.Q.; Lv, S.T. Elevation of neuron-specific enolase and S-100beta protein level in experimental acute spinal cord injury. J. Clin. Neurosci. 2008, 15, 541–544. [Google Scholar] [CrossRef] [PubMed]
- Sawhney, S.; Hood, K.; Shaw, A.; Braithwaite, A.W.; Stubbs, R.; Hung, N.A.; Royds, J.A.; Slatter, T.L. Alpha-enolase is upregulated on the cell surface and responds to plasminogen activation in mice expressing a 133p53alpha mimic. PLoS ONE 2015, 10, e0116270. [Google Scholar] [CrossRef] [PubMed]
- Hafner, A.; Obermajer, N.; Kos, J. gamma-Enolase C-terminal peptide promotes cell survival and neurite outgrowth by activation of the PI3K/Akt and MAPK/ERK signalling pathways. Biochem. J. 2012, 443, 439–450. [Google Scholar] [CrossRef] [PubMed]
- Bae, S.; Kim, H.; Lee, N.; Won, C.; Kim, H.R.; Hwang, Y.I.; Song, Y.W.; Kang, J.S.; Lee, W.J. Alpha-Enolase expressed on the surfaces of monocytes and macrophages induces robust synovial inflammation in rheumatoid arthritis. J. Immunol. 2012, 189, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Polcyn, R.; Capone, M.; Hossain, A.; Matzelle, D.; Banik, N.L.; Haque, A. Enolase and acute spinal cord injury. J. Clin. Cell. Immunol. 2017, 8, 536. [Google Scholar] [CrossRef] [PubMed]
- Qian, J.; Herrera, J.J.; Narayana, P.A. Neuronal and axonal degeneration in experimental spinal cord injury: In vivo proton magnetic resonance spectroscopy and histology. J. Neurotrauma 2010, 27, 599–610. [Google Scholar] [CrossRef] [PubMed]
- Whalley, K.; O’Neill, P.; Ferretti, P. Changes in response to spinal cord injury with development: Vascularization, hemorrhage and apoptosis. Neuroscience 2006, 137, 821–832. [Google Scholar] [CrossRef] [PubMed]
- Varma, A.K.; Das, A.; Wallace, G.t.; Barry, J.; Vertegel, A.A.; Ray, S.K.; Banik, N.L. Spinal cord injury: A review of current therapy, future treatments, and basic science frontiers. Neurochem. Res. 2013, 38, 895–905. [Google Scholar] [CrossRef] [PubMed]
- Haque, A.; Capone, M.; Matzelle, D.; Cox, A.; Banik, N.L. Targeting Enolase in Reducing Secondary Damage in Acute Spinal Cord Injury in Rats. Neurochem. Res. 2017, 42, 2777–2787. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Liang, J.; Deng, X.; Chen, X.; Wu, F.; Zhao, X.; Luo, Y.; Fu, L.; Jiang, Z. Mitogen activated protein kinase signaling pathways participate in the active principle region of Buyang Huanwu decoction-induced differentiation of bone marrow mesenchymal stem cells. Neural Regen. Res. 2012, 7, 1370–1377. [Google Scholar] [PubMed]
- Polcyn, R.; Capone, M.; Hossain, A.; Matzelle, D.; Banik, N.L.; Haque, A. Neuron specific enolase is a potential target for regulating neuronal cell survival and death: Implications in neurodegeneration and regeneration. Neuroimmunol. Neuroinflamm. 2017, 4, 254–257. [Google Scholar] [CrossRef] [PubMed]
- Marangos, P.J.; Schmechel, D.E.; Parma, A.M.; Goodwin, F.K. Developmental profile of neuron-specific (NSE) and non-neuronal (NNE) enolase. Brain Res. 1980, 190, 185–193. [Google Scholar] [CrossRef]
- Kirino, T.; Brightman, M.W.; Oertel, W.H.; Schmechel, D.E.; Marangos, P.J. Neuron-specific enolase as an index of neuronal regeneration and reinnervation. J. Neurosci. 1983, 3, 915–923. [Google Scholar] [PubMed]
- Schmechel, D.E.; Brightman, M.W.; Marangos, P.J. Neurons switch from non-neuronal enolase to neuron-specific enolase during differentiation. Brain Res. 1980, 190, 195–214. [Google Scholar] [CrossRef]
- Pislar, A.; Bozic, B.; Zidar, N.; Kos, J. Inhibition of cathepsin X reduces the strength of microglial-mediated neuroinflammation. Neuropharmacology 2017, 114, 88–100. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Leak, R.K.; Shi, Y.; Suenaga, J.; Gao, Y.; Zheng, P.; Chen, J. Microglial and macrophage polarization-new prospects for brain repair. Nat. Rev. Neurol. 2015, 11, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Deloulme, J.C.; Lucas, M.; Gaber, C.; Bouillon, P.; Keller, A.; Eclancher, F.; Sensenbrenner, M. Expression of the neuron-specific enolase gene by rat oligodendroglial cells during their differentiation. J. Neurochem. 1996, 66, 936–945. [Google Scholar] [CrossRef] [PubMed]
- Vinores, S.A.; Rubinstein, L.J. Simultaneous expression of glial fibrillary acidic (GFA) protein and neuron-specific enolase (NSE) by the same reactive or neoplastic astrocytes. Neuropathol. Appl. Neurobiol. 1985, 11, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Sensenbrenner, M.; Lucas, M.; Deloulme, J.C. Expression of two neuronal markers, growth-associated protein 43 and neuron-specific enolase, in rat glial cells. J. Mol. Med. 1997, 75, 653–663. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, C.; Kawana, E. Immunohistochemical detection of NSE and S-100 protein in the thalamic VB nucleus after ablation of somatosensory cortex in the rat. Okajimas Folia Anat. Jpn. 1991, 67, 417–428. [Google Scholar] [CrossRef] [PubMed]
- Steiner, J.; Bielau, H.; Bernstein, H.G.; Bogerts, B.; Wunderlich, M.T. Increased cerebrospinal fluid and serum levels of S100B in first-onset schizophrenia are not related to a degenerative release of glial fibrillar acidic protein, myelin basic protein and neurone-specific enolase from glia or neurones. J. Neurol. Neurosurg. Psychiatry 2006, 77, 1284–1287. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Munch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Cahoy, J.D.; Emery, B.; Kaushal, A.; Foo, L.C.; Zamanian, J.L.; Christopherson, K.S.; Xing, Y.; Lubischer, J.L.; Krieg, P.A.; Krupenko, S.A.; et al. A transcriptome database for astrocytes, neurons, and oligodendrocytes: A new resource for understanding brain development and function. J. Neurosci. 2008, 28, 264–278. [Google Scholar] [CrossRef] [PubMed]
- Srodulski, S.; Sharma, S.; Bachstetter, A.B.; Brelsfoard, J.M.; Pascual, C.; Xie, X.S.; Saatman, K.E.; van Eldik, L.J.; Despa, F. Neuroinflammation and neurologic deficits in diabetes linked to brain accumulation of amylin. Mol. Neurodegener. 2014, 9, 30. [Google Scholar] [CrossRef] [PubMed]
- Kadlubowska, J.; Malaguarnera, L.; Waz, P.; Zorena, K. Neurodegeneration and Neuroinflammation in Diabetic Retinopathy: Potential Approaches to Delay Neuronal Loss. Curr. Neuropharmacol. 2016, 14, 831–839. [Google Scholar] [CrossRef] [PubMed]
- Nayak, A.R.; Badar, S.R.; Lande, N.; Kawle, A.P.; Kabra, D.P.; Chandak, N.H.; Raje, D.V.; Singh, L.R.; Daginawala, H.F.; Kashyap, R.S. Prediction of Outcome in Diabetic Acute Ischemic Stroke Patients: A Hospital-Based Pilot Study Report. Ann. Neurosci. 2016, 23, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Chao, C.; Williams, S.G.; Xu, L.; Chen, J.; Wallner, L.P.; Porter, K.R.; Jacobsen, S.J. Statin therapy is not associated with prostate cancer recurrence among patients who underwent radiation therapy. Cancer Lett. 2013, 335, 214–218. [Google Scholar] [CrossRef] [PubMed]
- Ciancarelli, I.; de Amicis, D.; di Massimo, C.; Sandrini, G.; Pistarini, C.; Carolei, A.; Ciancarelli, M.G.T. Influence of intensive multifunctional neurorehabilitation on neuronal oxidative damage in patients with Huntington’s disease. Funct. Neurol. 2015, 30, 47–52. [Google Scholar] [PubMed]
- Dincel, G.C.; Atmaca, H.T. Role of oxidative stress in the pathophysiology of Toxoplasma gondii infection. Int. J. Immunopathol. Pharmacol. 2016, 29, 226–240. [Google Scholar] [CrossRef] [PubMed]
- Vasiljevic, B.; Maglajlic-Djukic, S.; Gojnic, M.; Stankovic, S. The role of oxidative stress in perinatal hypoxic-ischemic brain injury. Srp. Arh. Celok. Lek. 2012, 140, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Schon, E.A.; Manfredi, G. Neuronal degeneration and mitochondrial dysfunction. J. Clin. Investig. 2003, 111, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Constantinescu, R.; Zetterberg, H.; Holmberg, B.; Rosengren, L. Levels of brain related proteins in cerebrospinal fluid: An aid in the differential diagnosis of parkinsonian disorders. Parkinsonism Relat. Disord. 2009, 15, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Schaf, D.V.; Tort, A.B.; Fricke, D.; Schestatsky, P.; Portela, L.V.; Souza, D.O.; Rieder, C.R. S100B and NSE serum levels in patients with Parkinson’s disease. Parkinsonism Relat. Disord. 2005, 11, 39–43. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, F.M.; Mergl, R.; Stach, B.; Jahn, I.; Gertz, H.J.; Schonknecht, P. Elevated levels of cerebrospinal fluid neuron-specific enolase (NSE) in Alzheimer’s disease. Neurosci. Lett. 2014, 570, 81–85. [Google Scholar] [CrossRef] [PubMed]
- Chaves, M.L.; Camozzato, A.L.; Ferreira, E.D.; Piazenski, I.; Kochhann, R.; Dall’Igna, O.; Mazzini, G.S.; Souza, D.O.; Portela, L.V. Serum levels of S100B and NSE proteins in Alzheimer’s disease patients. J. Neuroinflamm. 2010, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Streitberger, K.J.; Leithner, C.; Wattenberg, M.; Tonner, P.H.; Hasslacher, J.; Joannidis, M.; Pellis, T.; di Luca, E.; Fodisch, M.; Krannich, A.; et al. Neuron-Specific Enolase Predicts Poor Outcome After Cardiac Arrest and Targeted Temperature Management: A Multicenter Study on 1053 Patients. Crit. Care Med. 2017, 45, 1145–1151. [Google Scholar] [CrossRef] [PubMed]
- Wiberg, S.; Hassager, C.; Stammet, P.; Winther-Jensen, M.; Thomsen, J.H.; Erlinge, D.; Wanscher, M.; Nielsen, N.; Pellis, T.; Aneman, A.; et al. Single versus Serial Measurements of Neuron-Specific Enolase and Prediction of Poor Neurological Outcome in Persistently Unconscious Patients after Out-Of-Hospital Cardiac Arrest—A TTM-Trial Substudy. PLoS ONE 2017, 12, e0168894. [Google Scholar] [CrossRef] [PubMed]
- Sogut, O.; Guloglu, C.; Orak, M.; Sayhan, M.B.; Gokdemir, M.T.; Ustundag, M.; Akkus, Z. Trauma scores and neuron-specific enolase, cytokine and C-reactive protein levels as predictors of mortality in patients with blunt head trauma. J. Int. Med. Res. 2010, 38, 1708–1720. [Google Scholar] [CrossRef] [PubMed]
- Dunker, S.; Sadun, A.A.; Sebag, J. Neuron specific enolase in retinal detachment. Curr. Eye Res. 2001, 23, 382–385. [Google Scholar] [CrossRef] [PubMed]
- Quintyn, J.C.; Pereira, F.; Hellot, M.F.; Brasseur, G.; Coquerel, A. Concentration of neuron-specific enolase and S100 protein in the subretinal fluid of rhegmatogenous retinal detachment. Graefes Arch. Clin. Exp. Ophthalmol. 2005, 243, 1167–1174. [Google Scholar] [CrossRef] [PubMed]
- Yee, K.M.; Ross-Cisneros, F.N.; Lee, J.G.; da Rosa, A.B.; Salomao, S.R.; Berezovsky, A.; Belfort, R., Jr.; Chicani, F.; Moraes-Filho, M.; Sebag, J.; et al. Neuron-specific enolase is elevated in asymptomatic carriers of Leber’s hereditary optic neuropathy. Investig. Ophthalmol. Vis. Sci. 2012, 53, 6389–6392. [Google Scholar] [CrossRef] [PubMed]
- Bharosay, A.; Bharosay, V.V.; Varma, M.; Saxena, K.; Sodani, A.; Saxena, R. Correlation of Brain Biomarker Neuron Specific Enolase (NSE) with Degree of Disability and Neurological Worsening in Cerebrovascular Stroke. Indian J. Clin. Biochem. 2012, 27, 186–190. [Google Scholar] [CrossRef] [PubMed]
- Pandey, A.; Shrivastava, A.K.; Saxena, K. Neuron specific enolase and c-reactive protein levels in stroke and its subtypes: Correlation with degree of disability. Neurochem. Res. 2014, 39, 1426–1432. [Google Scholar] [CrossRef] [PubMed]
- Goksuluk, H.; Gulec, S.; Ozcan, O.U.; Gerede, M.; Vurgun, V.K.; Ozyuncu, N.; Erol, C. Usefulness of Neuron-Specific Enolase to Detect Silent Neuronal Ischemia After Percutaneous Coronary Intervention. Am. J. Cardiol. 2016, 117, 1917–1920. [Google Scholar] [CrossRef] [PubMed]
- Rech, T.H.; Vieira, S.R.; Nagel, F.; Brauner, J.S.; Scalco, R. Serum neuron-specific enolase as early predictor of outcome after in-hospital cardiac arrest: A cohort study. Crit. Care 2006, 10, R133. [Google Scholar] [CrossRef] [PubMed]
- Rech, T.H.; Vieira, S.R.; Brauner, J.S. Serum neuron-specific enolase as a prognostic marker after a cardiac arrest. Rev. Bras. Ter. Intensiv. 2006, 18, 396–401. [Google Scholar]
- Yan, T.; Skaftnesmo, K.O.; Leiss, L.; Sleire, L.; Wang, J.; Li, X.; Enger, P.O. Neuronal markers are expressed in human gliomas and NSE knockdown sensitizes glioblastoma cells to radiotherapy and temozolomide. BMC Cancer 2011, 11, 524. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.D.; Zhen, Y.Y.; Lin, J.W.; Lin, T.K.; Huang, C.W.; Liou, C.W.; Chan, S.H.; Chuang, Y.C. Dynamin-Related Protein 1 Promotes Mitochondrial Fission and Contributes to The Hippocampal Neuronal Cell Death Following Experimental Status Epilepticus. CNS Neurosci. Ther. 2016, 22, 988–999. [Google Scholar] [CrossRef] [PubMed]
- Anderson, B.J.; Reilly, J.P.; Shashaty, M.G.S.; Palakshappa, J.A.; Wysoczanski, A.; Dunn, T.G.; Kazi, A.; Tommasini, A.; Mikkelsen, M.E.; Schweickert, W.D.; et al. Admission plasma levels of the neuronal injury marker neuron-specific enolase are associated with mortality and delirium in sepsis. J. Crit. Care 2016, 36, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Koch, M.; Mostert, J.; Heersema, D.; Teelken, A.; de Keyser, J. Plasma S100beta and NSE levels and progression in multiple sclerosis. J. Neurol. Sci. 2007, 252, 154–158. [Google Scholar] [CrossRef] [PubMed]
- Hajdukova, L.; Sobek, O.; Prchalova, D.; Bilkova, Z.; Koudelkova, M.; Lukaskova, J.; Matuchova, I. Biomarkers of Brain Damage: S100B and NSE Concentrations in Cerebrospinal Fluid—A Normative Study. Biomed. Res. Int. 2015, 2015, 379071. [Google Scholar] [CrossRef] [PubMed]
- Ilzecki, M.; Ilzecka, J.; Przywara, S.; Terlecki, P.; Grabarska, A.; Stepulak, A.; Zubilewicz, T. Serum Neuron-Specific Enolase as a Marker of Brain Ischemia-Reperfusion Injury in Patients Undergoing Carotid Endarterectomy. Acta Clin. Croat. 2016, 55, 579–584. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Rodriguez, A.; Egea-Guerrero, J.J.; Gordillo-Escobar, E.; Enamorado-Enamorado, J.; Hernandez-Garcia, C.; de Azua-Lopez, Z.R.; Vilches-Arenas, A.; Guerrero, J.M.; Murillo-Cabezas, F. S100B and Neuron-Specific Enolase as mortality predictors in patients with severe traumatic brain injury. Neurol. Res. 2016, 38, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Isgro, M.A.; Bottoni, P.; Scatena, R. Neuron-Specific Enolase as a Biomarker: Biochemical and Clinical Aspects. Adv. Exp. Med. Biol. 2015, 867, 125–143. [Google Scholar] [PubMed]
- Ramont, L.; Thoannes, H.; Volondat, A.; Chastang, F.; Millet, M.C.; Maquart, F.X. Effects of hemolysis and storage condition on neuron-specific enolase (NSE) in cerebrospinal fluid and serum: Implications in clinical practice. Clin. Chem. Lab. Med. 2005, 43, 1215–1217. [Google Scholar] [CrossRef] [PubMed]
- Zabel, M.K.; Kirsch, W.M. From development to dysfunction: Microglia and the complement cascade in CNS homeostasis. Ageing Res. Rev. 2013, 12, 749–756. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Chen, C.; Hao, J.; Zhang, J.; Zhang, F. Effect of CLIP3 Upregulation on Astrocyte Proliferation and Subsequent Glial Scar Formation in the Rat Spinal Cord via STAT3 Pathway After Injury. J. Mol. Neurosci. 2018, 64, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Perea, G.; Sur, M.; Araque, A. Neuron-glia networks: Integral gear of brain function. Front. Cell. Neurosci. 2014, 8, 378. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, M.; Haque, A.; Banik, N.L.; Nagarkatti, P.; Nagarkatti, M.; Ray, S.K. Estrogen receptor agonists for attenuation of neuroinflammation and neurodegeneration. Brain Res. Bull. 2014, 109, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Samantaray, S.; Smith, J.A.; Das, A.; Matzelle, D.D.; Varma, A.K.; Ray, S.K.; Banik, N.L. Low dose estrogen prevents neuronal degeneration and microglial reactivity in an acute model of spinal cord injury: Effect of dosing, route of administration, and therapy delay. Neurochem. Res. 2011, 36, 1809–1816. [Google Scholar] [CrossRef] [PubMed]
- Samantaray, S.; Knaryan, V.H.; Shields, D.C.; Cox, A.A.; Haque, A.; Banik, N.L. Inhibition of Calpain Activation Protects MPTP-Induced Nigral and Spinal Cord Neurodegeneration, Reduces Inflammation, and Improves Gait Dynamics in Mice. Mol. Neurobiol. 2015, 52, 1054–1066. [Google Scholar] [CrossRef] [PubMed]
- Gold, M.; el Khoury, J. beta-amyloid, microglia, and the inflammasome in Alzheimer’s disease. Semin. Immunopathol. 2015, 37, 607–611. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Hussain, M.D.; Yan, L.J. Microglia, neuroinflammation, and beta-amyloid protein in Alzheimer’s disease. Int. J. Neurosci. 2014, 124, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Piovesana, R.; Faroni, A.; Magnaghi, V.; Reid, AJ.; Tata, AM. Supplement: GLIA Bilbao 2017: Abstracts Oral Presentations, Posters, Indexes. Glia 2017, 65, E533. [Google Scholar]
- Polazzi, E.; Altamira, L.E.; Eleuteri, S.; Barbaro, R.; Casadio, C.; Contestabile, A.; Monti, B. Neuroprotection of microglial conditioned medium on 6-hydroxydopamine-induced neuronal death: Role of transforming growth factor beta-2. J. Neurochem. 2009, 110, 545–556. [Google Scholar] [CrossRef] [PubMed]
- Van Ham, T.J.; Brady, C.A.; Kalicharan, R.D.; Oosterhof, N.; Kuipers, J.; Veenstra-Algra, A.; Sjollema, K.A.; Peterson, R.T.; Kampinga, H.H.; Giepmans, B.N. Intravital correlated microscopy reveals differential macrophage and microglial dynamics during resolution of neuroinflammation. Dis. Model Mech. 2014, 7, 857–869. [Google Scholar] [CrossRef] [PubMed]
- Hornik, T.C.; Vilalta, A.; Brown, G.C. Activated microglia cause reversible apoptosis of pheochromocytoma cells, inducing their cell death by phagocytosis. J. Cell Sci. 2016, 129, 65–79. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, A.M.; Delpino, M.V.; Miraglia, M.C.; Franco, M.M.C.; Barrionuevo, P.; Dennis, V.A.; Oliveira, S.C.; Giambartolomei, G.H. Brucella abortus-activated microglia induce neuronal death through primary phagocytosis. Glia 2017, 65, 1137–1151. [Google Scholar] [CrossRef] [PubMed]
- Hamadi, N.; Sheikh, A.; Madjid, N.; Lubbad, L.; Amir, N.; Shehab, S.A.; Khelifi-Touhami, F.; Adem, A. Increased pro-inflammatory cytokines, glial activation and oxidative stress in the hippocampus after short-term bilateral adrenalectomy. BMC Neurosci. 2016, 17, 61. [Google Scholar] [CrossRef] [PubMed]
- Obermajer, N.; Doljak, B.; Jamnik, P.; Fonovic, U.P.; Kos, J. Cathepsin X cleaves the C-terminal dipeptide of alpha- and gamma-enolase and impairs survival and neuritogenesis of neuronal cells. Int. J. Biochem. Cell Biol. 2009, 41, 1685–1696. [Google Scholar] [CrossRef] [PubMed]
- Burke, R.E.; O’Malley, K. Axon degeneration in Parkinson’s disease. Exp. Neurol. 2013, 246, 72–83. [Google Scholar] [CrossRef] [PubMed]
- Coleman, M. Axon degeneration mechanisms: Commonality amid diversity. Nat. Rev. Neurosci. 2005, 6, 889–898. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.T.; Medress, Z.A.; Barres, B.A. Axon degeneration: Molecular mechanisms of a self-destruction pathway. J. Cell Biol. 2012, 196, 7–18. [Google Scholar] [CrossRef] [PubMed]
- De Vos, K.J.; Grierson, A.J.; Ackerley, S.; Miller, C.C. Role of axonal transport in neurodegenerative diseases. Annu. Rev. Neurosci. 2008, 31, 151–173. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haque, A.; Polcyn, R.; Matzelle, D.; Banik, N.L. New Insights into the Role of Neuron-Specific Enolase in Neuro-Inflammation, Neurodegeneration, and Neuroprotection. Brain Sci. 2018, 8, 33. https://doi.org/10.3390/brainsci8020033
Haque A, Polcyn R, Matzelle D, Banik NL. New Insights into the Role of Neuron-Specific Enolase in Neuro-Inflammation, Neurodegeneration, and Neuroprotection. Brain Sciences. 2018; 8(2):33. https://doi.org/10.3390/brainsci8020033
Chicago/Turabian StyleHaque, Azizul, Rachel Polcyn, Denise Matzelle, and Naren L. Banik. 2018. "New Insights into the Role of Neuron-Specific Enolase in Neuro-Inflammation, Neurodegeneration, and Neuroprotection" Brain Sciences 8, no. 2: 33. https://doi.org/10.3390/brainsci8020033
APA StyleHaque, A., Polcyn, R., Matzelle, D., & Banik, N. L. (2018). New Insights into the Role of Neuron-Specific Enolase in Neuro-Inflammation, Neurodegeneration, and Neuroprotection. Brain Sciences, 8(2), 33. https://doi.org/10.3390/brainsci8020033